

Abstract
The transport of charge through the DNA base pair stack offers a route to carry out redox chemistry at a distance. Here we describe characteristics of this chemistry that have been elucidated and how this chemistry may be utilized within the cell. The shallow distance dependence associated with these redox reactions permits DNA-mediated signaling over long molecular distances in the genome and facilitates the activation of redox-sensitive transcription factors globally in response to oxidative stress. The long-range funneling of oxidative damage to sites of low oxidation potential in the genome also may provide a means of protection within the cell. Furthermore, the sensitivity of DNA charge transport to perturbations in base pair stacking, as may arise with base lesions and mismatches, may be used as a route to scan the genome for damage as a first step in DNA repair. Thus, the ability of double helical DNA, in mediating redox chemistry at a distance, provides a natural mechanism for redox sensing and signaling in the genome.
I. Introduction
Over the past several years, our laboratory has focused on chemical studies of DNA. In particular, we have examined how DNA facilitates electron transfer reactions between donors and acceptors that are bound to the double helix.1-4 We have found that well-stacked DNA does indeed mediate redox reactions between electron donors and acceptors well-separated from one another on the DNA duplex and that DNA-bound oxidants can even promote oxidative damage to DNA at sites far from the binding site of the oxidant. Interestingly, these reactions can occur only when the DNA base pairs are well stacked in the duplex. Thus, DNA can facilitate redox reactions at a distance. The DNA duplex, perhaps uniquely, can serve to mediate long-range signaling.5
These studies prompted us to ask whether such chemistry might occur within the cell. Organisms, from bacteria to humans, face a variety of stresses to which they must respond in order to survive. Reactive oxygen species (ROS) represent the chemical threat that constitutes oxidative stress, and various redox-active proteins and small molecules must be activated by the cell to neutralize this threat (Figure 1).
Figure 1.

Illustrative schematic of the generation and detoxification of hydrogen peroxide, an archetypical ROS, and some of its roles in mammalian redox signaling. In this diagram, we see production of hydrogen peroxide in the mitochondria (bottom left) accompanied by its efficient detoxification in both the matrix and the cytosol (center left) and the consequences of excess H2O2 reacting with ferrous iron (bottom left, right). H2O2 is a possible intermediate in the receptor-mediated pathways that rely on NOX activation. (top) Inside the nucleus, many transcription factors require further reduction, such as by APE-1 (right). The blue dashed lines represent the equilibration between the bulk concentration of H2O2 in the cell, and the local concentrations generated as part of signaling or pathological processes; that these processes are functionally distinct illustrates how difficult it is to deconvolute redox sensing and signaling pathways in mammals versus in yeast or prokaryotes. Poorly understood processes are represented as black dashes; in particular, the intermediate species in receptor-initiated ROS signaling are not well-characterized. Abbreviations: ecSOD, extracellular superoxide dismutase; NADPH, nicotinamide adenine dinucleotide phosphate; MAP3K, mitogen-activated protein kinase kinase kinase; MAP2K, mitogen-activated protein kinase kinase; NOX, NADPH oxidase; PTP, protein tyrosine phosphatase; Trx, thioredoxin; ASK-1, apoptosis signaling kinase; Tpx, thioredoxin peroxidase; Cat, catalase; JNK, c-jun N-terminal kinase; Gpx, glutathione peroxidase; Grd, glutathione reductase; MAO, monoamine oxidase; MnSOD, manganese superoxide dismutase; Acn, aconitase; APE-1, apurinic/apyrimidinic endonuclease. These redox pathways and their inter-relationships are discussed in detail in references 8-10, 148, 220, and 221.
But how is oxidative stress signaled within the genome to activate a response? Moreover, how is the genome protected from ROS? In E. coli, glutathione, the redox buffer of the cell, and thioredoxin-related proteins are regulated by ROS-activated transcription factors. OxyR, for example, is transcriptionally activated by H2O2 via oxidation of two cysteine residues to a disulfide.6-8 SoxR is converted to the transcriptionally active, oxidized form by superoxide-generating sensitizers, promoting soxS transcription. SoxS, in turn, promotes superoxide dismutases, cluster repair proteins, and drug efflux enzymes, among others.9 But must ROS be targeted to specific sites within the genome to activate these transcription factors? Are the proteins only activated when the concentrations of ROS are sufficiently high that the proteins and ROS collide? And what other damage to the cell must result under those conditions?
Mammalian redox sensing is still more complicated, with no canonical redox sensor(s). Instead, various signal cascades are activated, turning on DNA repair or inducing apoptosis, and modulating the cellular response depending upon the source and persistence of the cellular stress. Furthermore, in mammalian cells, there is an inherent contradiction between redox sensing and redox signaling. If ROS are used to mediate signaling cascades, as is suspected for potentially dozens of receptor-mediated pathways, this communication cannot be propagated in a specific manner by modulating the global redox state of the cell.8 Chemical specificity in signal transduction, relying on spikes in local concentration of ROS, such as H2O2, allows mutually exclusive pathways to employ redox signaling. In contrast, redox sensing pathways, sensitive to damage to DNA and to the global disulfide/thiol ratio, are activated only when the buffering capacity of catalases, dismutases, glutathione, thioredoxin, and glutaredoxins is overwhelmed.8,10 Thus, any medium that could allow chemical control over the translocation and distribution of oxidative radicals and damage increases the potential sophistication of the cellular machinery.
We have asked whether DNA-mediated charge transport might play some role in the response of the cell to oxidative stress. DNA-mediated charge transport chemistry surely could be utilized to protect the genome from oxidative damage, funneling oxidizing equivalents to regions of the genome that can accommodate higher mutation rates. Indeed, because DNA charge transport chemistry can occur over long molecular distances, might DNA charge transport chemistry be used also as a means to facilitate long-range signaling across the genome to activate the cellular response?
Here we describe recent studies to examine these questions. We intend this report not as an exhaustive review of the field but instead as a means to survey some key characteristics of DNA charge transport chemistry that have been elucidated and offer our perspective on how this unique chemistry might be harnessed advantageously within the cell. We hope to apply a chemical perspective to examine an important biological problem and explore how the cell finds a chemical solution.
II. Redox state of DNA
The critical property of DNA with regard to redox sensing is its ability to mediate charge transport over exceptionally long distances.11-13 In our laboratory, we have observed DNA-mediated charge transport (CT) over as far as 100 base pairs, corresponding to 34 nm;4 other laboratories have observed charge transport over comparable distances,14-16 particularly when oxidation of the DNA is irreversible. Charge migration through DNA occurs through the π-stack of base pairs. The planar, aromatic, hydrophobic, heterocyclic base pairs are stacked upon each other like a pile of coins, protected from solvent by the sugar-phosphate backbone.17 It has been well-established that efficient charge transfer into and out of the π -stack requires direct electronic coupling to the bases, either through stacking interactions18-20 or covalent, electronically conjugated linkage.21-23
We and others have seen long-range charge transport between donors and acceptors bound to DNA, electrochemical oxidation and reduction of DNA-bound redox probes on DNA-modified electrodes, and the generation of oxidative damage to DNA from a distance.3 Figure 2 illustrates some of the classes of DNA assemblies through which long-range charge transport has been documented. The first evidence for long-range DNA-mediated CT was the efficient fluorescence quenching of tethered, intercalated [Ru(phen′)2(dppz)]*/2+ (E ∼ -0.8 V vs NHE) by tethered, intercalated [Rh(phi)2(phen′)]3+ (E ∼ 0 V vs NHE) at long range.24,25 Here, the photoexcited Ru is not a strong enough reductant to add an electron directly to DNA, but reduces the Rh complex on the subnanosecond timescale. Inspired by groups that had found efficient oxidation of guanine by high energy photooxidants,26-28 we demonstrated that a tethered, intercalated, photoexcited [Rh(phi)2(bpy′)]*/3+ (E ∼ 2.0 V vs NHE) could indeed oxidatively damage guanine far from the metal binding site.11,29 It was clear that oxidative radicals could migrate over long distances in DNA, allowing “chemistry at a distance”.
Figure 2.
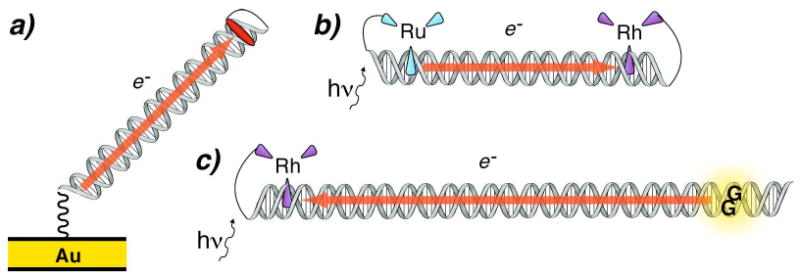
Three types of assemblies employed for studying DNA-mediated CT. An electrode is used to inject an electron and reduce a redox probe attached to the DNA (left). DNA-mediated CT is observed as the quenching mechanism of photoexcited [Ru(phen′)2(dppz)]*/2+ by [Rh(phi)2(phen′)]3+ (top right). Both of these reactions occur at potentials insufficient to reduce or oxidize the DNA. A high energy photoexcited [Rh(phi)2(phen′)]*/3+ is employed that is competent to oxidize all the DNA bases (bottom right). Permanent chemical decomposition products are observed at 5′-GG-3′ steps. Hence, DNA can participate as both a mediator and as a reactant in CT.
When the electronic coupling between a charge donor and acceptor is weak, charge transfer proceeds through superexchange, mediated by the orbitals of the bridge.30 The electronic coupling between the donor and acceptor, and consequently the rate, decays exponentially with increasing bridge length. However, the oxidation yield of guanine by Rh complexes,11 photolytically generated sugar radical,31 and anthraquinone16 was found to be only weakly dependent on bridge length for longer bridges. In some cases, such as for oxidation of deazaguanine by photoexcited ethidium,32,33 this distance dependence represents the distribution of CT-active structures, rather than the inherent decay of the electronic coupling. The rate of guanine oxidation by photoexcited stilbene in bridged DNA hairpins has a far steeper distance dependence, which depends on the driving force in a manner consistent with theory for non-adiabatic electron transfer.34-37
To explain DNA-mediated CT over longer distances, a mechanism was postulated wherein injected charge migrates through DNA by hopping from site to site when it is injected at near or higher energy than the bases.15,31,38-47 The rate of hopping, which is also an important CT mechanism in proteins,48-50 has a geometric dependence with distance, and can proceed over far longer distances than superexchange.51 In the case of stilbene-capped hairpins, the transition between superexchange and hopping has been directly observed, as the rate of hole arrival at the acceptor is longer than the rate of injection for donor-acceptor separation above two A-T base pairs.46 The nature of the photooxidant can have a profound effect on hopping even after injection, due to coulomb attraction between the donor and the hole.52,53 Presumably, positively charged metallointercalators have an opposite effect, promoting migration of an injected hole away from the donor.3
A challenge to the hopping mechanism is the rugged energetic environment of DNA.3 It has been postulated that variations in the energies of neighboring sites are overcome by thermal activation when direct superexchange is inadequate.54 The nature of the hopping intermediates appear to be mixtures of localized and delocalized states,16,55,56 with polaron formation providing a complementary mechanism of overcoming the energetic barrier to hopping.16,57,58 Calculations predict facile polaron delocalization along 4 to 5 adenines59; there is experimental evidence consistent with this delocalization length as well.60,61 Conformational dynamics play a critical role as well, with base motions being required to access CT-active conformations.52,56,60,62-64
We next considered whether large driving forces were necessary for CT to proceed through DNA over long distances. By assembling morphologically well-characterized DNA films on gold and graphite electrodes, we and others have been able to study DNA-mediated CT far below the potentials of the isolated nucleosides.3,4,65,66 Remarkably, DNA can mediate CT over long distances at potentials that should be insufficient for occupation of the bridge. Although the rate has not been characterized, it has been shown that CT through the short alkanethiol linker is rate-limiting.4,23,67
Oxidative damage tends to localize at guanine repeat sites. Although there is substantial spread in the measured and calculated redox potentials of the DNA bases,68-70 all studies support the fact that guanosine is the most readily oxidized nucleoside, followed by adenosine, with cytidine and thymidine having the most negative potential. Furthermore, the oxidation potential of guanosine is more negative in the environment of DNA, due both to base pairing and to stacking interactions, and is also modulated by the conformational sampling of the DNA environment.71 Specifically, 5′ purines decrease the potential of guanosine via interactions with the stacking N7 nitrogen, with the lowest potential site being the 5′ guanine of 5′-GGG-3′.72,73 Hence electron holes equilibrate onto the guanine doublets and triplets on a timescale that is faster than the trapping of irreversible guanine damage products, leading to oxidative damage at the 5′-guanines.
Better understood than the mechanistic aspects of DNA CT are the characteristics of the DNA bridge that affect it. DNA-mediated CT is exquisitely sensitive to the integrity of the π-stack. Damage or binding events that perturb the dynamic stacking will attenuate CT (Figure 3). This damage includes mismatches,74,75 oxidative lesions,76 bending by DNA-binding proteins,77,78 and abasic sites. Although at equilibrium most mismatches have a similar structure to well-matched DNA, more unstacked configurations are dynamically sampled. The extent of this destacking is mirrored by the extent that each individual base mismatch attenuates CT.75,79 The stable, well-stacked GG mismatch is poorly discriminated by CT,80 although a similarly stable GT wobble base pair gives significant attenuation. Similarly, proteins that induce destacking, such as TATA-binding protein, attenuate CT.77,78,81 In a particularly illustrative example, the methylase M.HhaI, which extrudes a cytidine from the duplex and replaces it with glutamine, sharply suppresses the current through DNA.78 Upon incubation with M.HhaI Q237W, which substitutes the aromatic, well-stacking base tryptophan for the intercalating glutamine, the current is restored to nearly that found through the unperturbed DNA, demonstrating that the stacking interactions determine the CT competence. The restriction enzyme R.BamHI contains a guanidinium on R155 which forms a hydrogen bond with guanine in the cognate site. This protein inhibits CT through its binding site without bending the DNA, presumably due to modulation of the potential of the guanine.82
Figure 3.
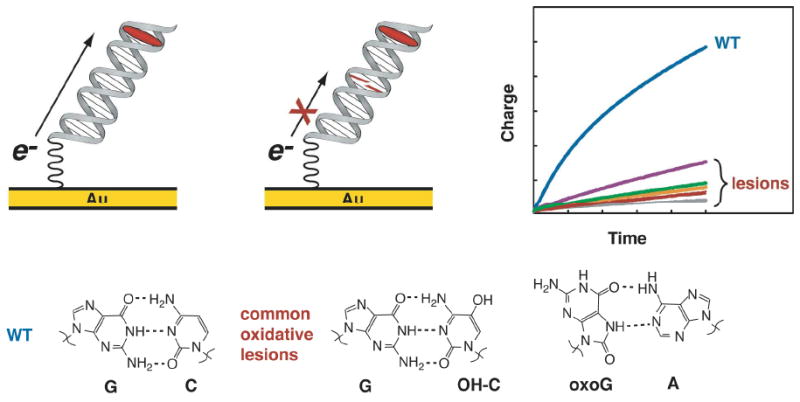
DNA-mediated CT is sharply attenuated by the presence of a mismatch or other lesions that affect stacking. The accumulated current through the DNA is far less when oxidative lesions are present, including the physiologically relevant 8-oxoguanosine base-paired with adenosine and 5-hydroxycytidine base-paired with guanine.
In addition to affecting CT through the DNA, some DNA-binding proteins can also participate in reactions with the oxidized DNA. One class consists of the proteins that are redox-active themselves (vide infra). DNA-binding proteins containing iron-sulfur clusters that we have studied, including EndoIII, MutY, UDG, and SoxR, are readily oxidized at DNA films that allow direct access to the iron sulfur cluster, demonstrating that an appropriate electron transfer path exists from the DNA to the bound proteins.83-86 Without DNA to mediate CT, protein electrochemistry is more challenging.85, 87
Proteins and peptides can also react with DNA, oxidized from a distance, to form protein-DNA crosslinks. The guanine radical that is generated upon DNA photooxidation degrades to 8-oxo-7,8-dihydroguanine (oxoG), among other products, following nucleophilic attack by water at the C8 carbon.26,88 After it was demonstrated that the tripeptides KYK and KWK are oxidized by DNA with the radical localized on the tyrosine and tryptophan respectively (Figure 4),89-91 several groups explored reactions of DNA-binding peptides that lack a stable radical acceptor but have nucleophilic groups that might attack the guanine radical.92-94 Consistent with the oxidative decomposition products of covalent guanosine-lysine conjugates,95 trilysine for ms crosslinks with oxidized DNA. Cytochrome c, which is not natively a DNA-binding protein but has extensive lysine content, also undergoes crosslinking with DNA, demonstrating the generality of this decomposition. The rate of crosslinking is slower than 104 s-1,94 comparable to that which has been reported for degradation of guanine radical in the presence of in situ generated superoxide. In the absence of protein, superoxide or other diffusing reductants, guanine radical survives for seconds.96 Importantly, further oxidation events can lead to more extensive damage products, both from the initial oxoG intermediate97-99 and from the later protein-DNA adduct.100 Ultimately, this pathway of decomposition both competes and cooperates with oxoG and other guanine decomposition products. In fact, one can consider that the timescale of guanine radical degradation through secondary reactions serves as the in vivo clock that competes with the diffusion of charge through the genome.
Figure 4.
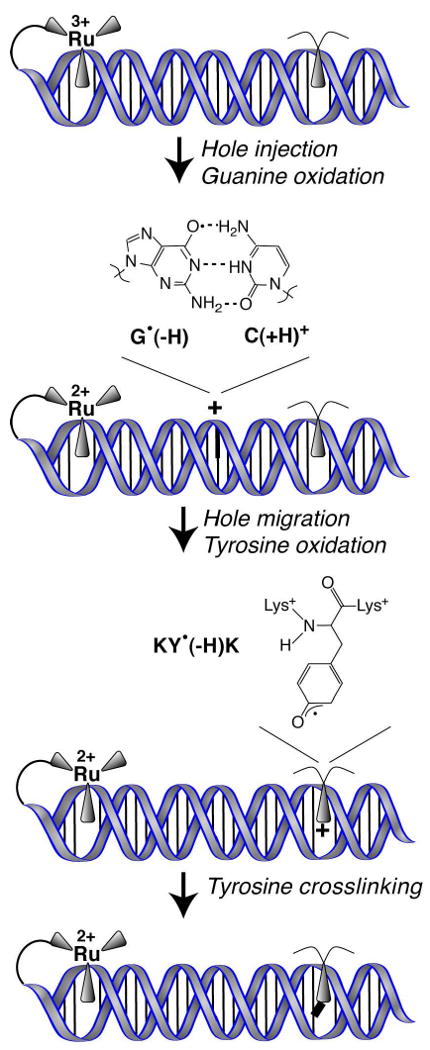
DNA-mediated CT from a strongly oxidizing intercalated ruthenium complex to an intercalated lysine-tyrosine-lysine tripeptide (KYK) proceeds through a guanine radical intermediate. Oxidation of KYK leads to DNA-tyrosine cross-linking, while KWK is oxidized but does not form a covalent product with the DNA. This serves as a model system for the oxidation of DNA-bound proteins by radical species in DNA.
Since strong coupling is required for facile CT into and out of DNA, it is not surprising that the chemical nature of the oxidants plays a strong role in determining oxidation of DNA. Small molecules can readily access the bases through either groove. Pulse radiolysis studies employing SeO4 and SO4 have found that the mechanism of radical injection varied with the oxidant; oxidants of different energy abstract different hydrogens from the bases.70,101 Intercalated photooxidants typically oxidize DNA directly by single electron abstraction, either coincidentally with or followed by proton transfer to generate a neutral radical.2,26,102,103 The nature of the chemical pathway of oxidation can have profound effects on the role that DNA-mediated CT has in determining the damage products.104,105 More biologically relevant, superoxide and hydrogen peroxide are powerful oxidants and are associated with devastating DNA damage in the absence of cellular defenses.106-108 Despite this, they do not facilely oxidize DNA on their own.9,109 Rather, superoxide oxidatively induces the release of Fe2+ from exposed iron-sulfur clusters, which in turn reductively cleaves hydrogen peroxide to hydroxyl anion and hydroxyl radical.110 It is the latter species that readily oxidizes all four nucleobases by radical attack111 or hydrogen abstraction,112 as well as the backbone sugars by hydrogen abstraction.113 The base oxidative products, in turn, can degrade to many products, including oxoG, imidazolone, spiroiminodihyantoin, and oxazolone, with guanine neutral radical as an intermediate following condensation.88 This general mechanism for peroxide toxicity has profound implications for the role of cellular defenses against ROS and helps explain the effectiveness of the cellular machinery in responding to superoxide, i.e. that this molecule is several steps upstream of the initial damage event, and requires involvement of free metal ions.
Thus, it is clear that it is not appropriate to discuss oxidative stress of DNA as monolithic; individual oxidants will vary both in their ability to chemically oxidize the bases, and in the products that they generate.105,114,115 Different oxidants can produce the same products by different pathways, depending on the availability of nucleophilic and radical species, and some intermediates are reactive to a broad spectrum of available species. Despite this, any process that generates neutral or cationic guanine radical will allow CT to equilibrate it over hundreds of base pairs prior to decomposition or crosslinking of this intermediate.11
It is important to note that these DNA radical quenching mechanisms have mostly been studied in the absence of the variety of proteins that are normally bound. The histones that package DNA in chromosomes provide a competing target for oxidation. Yet once holes are injected into the DNA, seemingly protected by the nucleosome, efficient DNA CT can still occur. There is, moreover, a growing compendium of redox-active proteins that bind and process DNA (vide infra). Many of these DNA-bound proteins may be facilely reduced or oxidized through DNA-mediated CT.4,83 In fact, in some bacteria, a variety of stress conditions also lead to the expression of the ferritin analogue DNA-binding protein from starved cells (Dps), which protects DNA from oxidative damage116-118 by sequestration of oxidative equivalents and free Fe2.119-120 Whether oxidizing equivalents are similarly funneled into DNA-bound Dps has not yet been determined.
III. Shuttling of Oxidative Damage
Since pathways also exist for chemically irreversible decomposition of guanine neutral and cationic radicals, guanine damage is an eventual signature of DNA oxidation. Guanine damage can lead to mutations during transcription, depending on both the oxidation product and the particular polymerase involved.115 A slight mutation rate is an adaptive advantage for a population, but all organisms have a strong incentive to limit mutations to their genomes and the consequent risk of lethality or cancer.
Mutation rates are variable within genomes, with some regions particularly “hot” for even silent genomic change.121,122 Hot regions tend to be correlated with genes that are less vital for cellular survival.123 One proposal is that the variability in mutation rate might be due to chromosomal structures that allow damage agents preferable access to certain regions.124,125 Another explanation involves DNA-mediated CT.
It has been proposed that long-range hole migration in DNA might serve a protective role, by controlling the distribution of mutation rates.126,127 Oxidative damage accumulates most readily in polypurine regions, particularly at multiple G sites. If holes accumulate at these sites, then they will be depleted from other regions of DNA, and damage will occur selectively in the polypurine regions. Interestingly, polypurine regions have been shown to be statistically enriched in promoter regions versus transcribed or intergenic sequences of DNA.128,129 Furthermore, oxidative damage to polypurine regions within promoter regions might modulate transcription factor binding; there is evidence for this in the binding sites for NF-kB, AP-1 and HIF-1.130-134 Polypurine regions are also critical to quadruplex formation in telomeres, though in this case it appears that the specific conformation of the quadruplex determines whether DNA CT funnels damage to the guanines inside the structure.135,136
An obvious challenge to this mechanism is the presence of DNA sequence motifs, such as ATAT, that do not mediate CT well. In one notable experiment, GG radical decomposition was measured with and without a nearby oxoG, which serves as a low-potential radical trap.137 With some sequences, the presence of the oxoG protected the GG from oxidative damage. With intervening ATTA, however, the oxoG had no effect on GG damage. Similarly, a TTTT bridge was found to prevent transport between GG and oxoG.138 Of course, as discussed above, the superoxide generated in these experiments is associated with an increase in the guanine radical decomposition rate of about two orders of magnitude.96,103 [Rh(phi)2(bpy)]2+, the reduced state generated from oxidation of guanine by photoexcited [Rh(phi)2(bpy)]3+, cannot generate superoxide. In this case, guanine radical equilibrates over 200 Å prior to chemical decomposition.11 Furthermore, the equilibration of damage is not affected by nucleosome formation when Rh is used as the photooxidant.139 A contrary result was found for another sequence in which nucleosome formation protects a single GG in the binding domain from damage when anthraquinone is used as the photooxidant, with evidence for DNA-protein crosslink formation near the photooxidant;140,141 it was not established whether superoxide generation plays a role in the damage distribution, although presumably since the protected guanine site is accessible to hydroxyl radical in the presence and absence of the nucleosome, it is also accessible to superoxide in both cases.
Two series of experiments have assayed for whether this damage funneling occurs inside the organelles of living cells. In the first, the distribution of oxidative DNA damage induced by photoexcited [Rh(phi)2(bpy)]3+ was determined inside isolated HeLa nuclei; in the second, HeLa mitochondrial DNA was studied.12,142 The type of damage induced by [Rh(phi)2(bpy)]3+ is dictated by the excited state of the metal complex. Excitation at 365 nm generates the interligand CT state which is a powerful oxidant with E(Rh*3+/2+) of 2.0 V vs NHE, competent for direct oxidation of guanine in DNA to produce guanine radical.29,143 At higher energy excitation (308 nm), a ligand-centered state abstracts a sugar hydrogen to promote strand scission at the binding site of the metal complex.144 By comparing the sites of guanine damage induced by photoexcitation at these two wavelengths, the binding profile of the metal complex can be directly compared to the oxidative damage profile. In the absence of DNA-mediated CT, these two profiles should be identical.
For both isolated nuclei and isolated mitochondria, the damage sites are distinct from the binding sites. Damage occurs preferentially at the 5′ guanine of multiple guanine sites, indicating CT as the damage mechanism. The presence of DNA-bound protein affects neither the formation of the formamidopyridine damage product in nuclei,12 nor the formation of alkali-sensitive damage products in mitochondrial DNA.142 The damage profiles in the hypervariable region of the mitochondrial genome were identical both for photooxidation inside the isolated mitochondria and for photooxidation on the mitochondrial DNA in isolation (Figure 5).142,145 Damage in a noncoding region, termed the hypervariable region, localizes specifically to three regions; one is a flexible sequence, while the other two are long polypurine tracts of presumably lower potential. One of these is in conserved sequence block II (CSBII), which is the site of replication initiation for the mitochondrial genome.146 Mitochondria are subject to extensive oxidative damage, due to the leakage of ROS from the respiratory pathway.147,148 Simultaneously, it is essential to protect the integrity of mitochondrial genome, as the mitochondria are important actors in the apoptotic cascade; mutation of the mitochondrial genome in the presence of oxidative damage directly affects the ability of the cell to respond to an excess of genomic damage. Hence, it is not surprising that mitochondrial replication is strictly regulated. Presumably, damage to CSBII serves to inhibit binding of a ribonuclease, MRP, which is required for subsequent replication, and hence this damage prevents the propagation of mitochondrial genomes that are under severe oxidative stress. As further evidence for the importance of this site to mitochondrial genomic maintenance, CSBII is a hot spot for mutation in breast and neck cancer.149-151 Hot spots for oxidatively induced mutation that are outside of CSBII also correlate with the hot spots for mutation found in cancer.151,152
Figure 5.
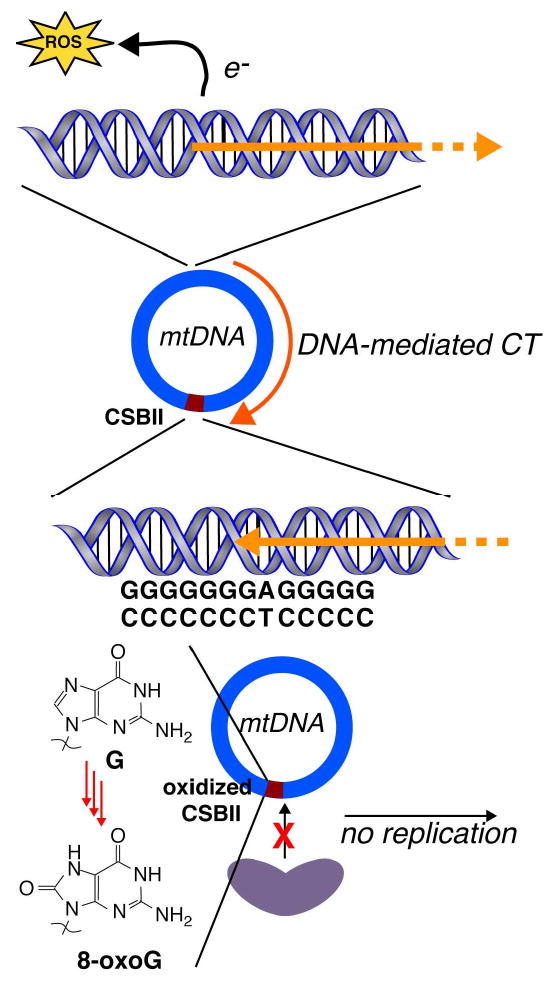
The conserved sequence block II (CSBII) serves as a checkpoint for mitochondrial DNA replication. Reactive oxygen species (ROS) introduce oxidative damage to the DNA, which we have shown to be funneled to the long polypurine region in CSBII, leading to oxidative decomposition of guanine to 8-oxoguanine at this site. This damage might interrupt specific CSBII-RNAse interactions that are required for replication.
What are the consequences of DNA CT on the profile of damage? First, oxidative damage will be preferentially localized to specific regions of the genome. Secondly, although oxoG is a major biomarker of oxidative stress oxoG can also absorb oxidative radicals in the DNA and undergo decomposition to further products, and a substantial amount of oxidative damage will instead lead to protein crosslinks. Finally, redox-active proteins can serve to protect DNA from oxidative damage by absorbing the holes;153 we have directly observed this behavior with both SoxR and MutY.13,154 In fact, oxidation of the DNA-bound protein can not only repair the damage but also activate a response.
IV. DNA-mediated Redox Signaling and Sensing
The general way that the cell responds to oxidative stress involves the activation of transcription factors that are sensitive to the overall redox state of the cell.155 Sensing of cellular redox transformation by these DNA-bound proteins elicits a specific response that is propagated to the transcriptional machinery which then alters expression of genes that repair or mitigate oxidative damage. These ROS-sensitive transcriptional regulators are present throughout the three domains of life and their diversity illustrates the breadth of oxidative reactions experienced by different organisms. In general, prokaryotic and other single-celled organisms experience acute exposure to ROS and the corresponding stress response is highly dynamic to maximize individual cell survival.156 Multicellular eukaryotic organisms experience more chronic oxidative stress. In these organisms, regulatory responses are often decoupled from ROS sensing. The regulatory consequences are focused on long-term, constitutive protection and are intimately linked to regulation of cell cycle progression, apoptosis, and senescence. In all organisms, the chemical mechanisms governing activation of redox sensitive transcription factors are not well understood even though these processes lie at the crux of physiologically relevant processes such as cancer and aging in eukaryotes and antibiotic resistance and virulence in prokaryotes.
a. Activation of SoxR by DNA-mediated CT
SoxR is a well-characterized example of a bacterial redox sensitive transcriptional regulator.157 While most studied in enteric bacteria (such as E. coli), SoxR homologs are widely represented throughout bacteria though they are not conserved in higher organisms.158 SoxR is an interesting target for study, since general mechanisms of activation may be conserved in other transcriptional regulators. Furthermore, SoxR in Pseudomonas aeruginosa has been linked to the metabolism of phenazine derivatives,158,159 small molecules implicated in virulence in pathogenic strains of P. aeruginosa.160 Activation of E. coli SoxR turns on the soxS response, which induces transcription of ∼50 genes involved in antioxidant production and oxidative damage repair as well as genes involved in multidrug resistance and heavy metal detoxification.161 SoxR homologues beyond the enterics do not trigger expression of soxS homologs, but instead induce expression of genes putatively involved in export and transformation of redox-active small molecules.158,159 These molecules are implicated in a variety of biological processes within the cell including modulation of the NAD+/NADH pool, quorum sensing, and defense against competitor bacteria.
SoxR is related to the MerR-like family of transcriptional sensors, all of which share a common scaffold.162 These transcription factors adopt a homodimer configuration where each monomer consists of a DNA binding domain, a coiled-coil dimerization domain, and a functionally unique sensor domain. The sensor domain in SoxR contains a [2Fe2S] cluster, and the sensing capacity of this domain resides in the redox activity of its iron-sulfur cluster.163 Oxidation of the [2Fe2S]1+ cluster in SoxR activates soxS transcription up to 100-fold.
SoxR binds to a symmetric sequence flanked by the -10 and -35 elements of the soxS promoter.162 These elements are separated by a 19 bp spacer which hinders RNA polymerase recruitment in the absence of SoxR activation. Upon activation, the SoxR-bound DNA undergoes significant distortion and underwinding to bring the soxS promoter elements into the optimal position. A recent 2.8 Å X-ray crystal structure of oxidized SoxR in complex with a 20 bp fragment of the soxS promoter sequence confirms these DNA conformational changes.164
The mechanism of SoxR activation at the sensor domain is not well understood, however. Diverse superoxide-generating agents such as paraquat are known to induce E. coli SoxR activation in vitro and in vivo.163,165 SoxR activation has also been demonstrated upon exposure to macrophage-generated nitric oxide, resulting in SoxR-bound dinitrosyl iron complexes.166 In either case, activation is rapid, with maximal SoxR oxidation and soxS induction occurring within minutes of cellular exposure to redox cycling agents.165 It is not known, however, if ROS react directly with the sensor moiety in SoxR or if activation occurs by alternate mechanisms. The possibility of the latter case is supported by the high reactivity of ROS with many other cellular components, including DNA.9,111,167
Furthermore, in bacterial species outside the enterics, SoxR activation may involve redox-active antibiotics.158,159 In these bacteria, SoxR induction can occur in the absence of oxygen. Interestingly, soxR genes from these disparate species can complement each other,168 indicating that activation of SoxR throughout bacteria may occur by common pathways or that SoxR has inherent flexibility in its activation mechanism. In sum, it is unlikely that direct superoxide reaction with the sensing domain of SoxR is the primary option for activation of these proteins.
In understanding the possible activation mechanisms of SoxR, it is important to consider the redox potential of the [2Fe2S] cluster. The potentials of E. coli and P. aeruginosa SoxR in the absence of DNA have been determined by redox titration as -290 mV vs. NHE.163,169 This relatively low value, in light of the overall cytosolic potential within these cells (largely governed by the NADPH/NADP+ potential of -340 mV vs. NHE),170 indicates that free SoxR could be significantly oxidized under ambient conditions. Independent experimental evidence, however, predicts that SoxR is mostly reduced during normal aerobic growth.
Within the cell, however, SoxR is likely bound to the soxS promoter site most of the time, and interaction with DNA may change the environment of the iron-sulfur cluster and its redox potential. Indeed, measurements of SoxR bound to DNA-modified electrodes reveal that the midpoint potentials of P. aeruginosa and E. coli SoxR are both ∼ +200 mV vs. NHE.85 These higher values are reasonable given the reduced resting state of SoxR in these organisms, and, significantly, they indicate that only strong oxidants may induce DNA-bound SoxR activation. The nearly 500 mV shift in the redox potential that occurs upon DNA association also raises interesting questions about the origins of this phenomenon. SoxR does not display differential binding affinity for the soxS promoter site in the oxidized and reduced forms.171 While a comparison of the X-ray crystal structures of oxidized SoxR with and without DNA indicates no significant structural changes in SoxR in the presence of DNA, these structures show dramatic changes in the DNA structure in the activated SoxR:DNA complex.166 Thus, the potential shift may provide the driving force for conformational changes induced in the protein or DNA upon SoxR oxidation. (Figure 6)
Figure 6.

SoxR is able to exploit DNA as an antenna for sensing oxidative stress conditions. Oxidative damage generates guanine radical intermediates, which migrate over long distances in DNA. These species oxidize SoxR, activating it to promote the transcription of soxS, inducing the oxidative stress response in E. coli.
DNA-bound SoxR has a high redox potential, a low copy number (and thus likely remains bound to its promoter site the majority of the time within the cell), and a fast response time upon exposure to environmental oxidants. In this context, it is interesting to consider the possibility that SoxR may be activated through the DNA via DNA-mediated CT chemistry. This possibility has already been examined in DNA-mediated electrochemical and photooxidation experiments.13,85 SoxR is readily accessible to oxidation and reduction via the DNA base pair stack. Importantly, harnessing this chemistry may allow SoxR to rapidly sense oxidative damage events in DNA from a distance. In this model for SoxR activation, ROS and other oxidants extract electrons from the DNA, leading to the formation of DNA base radicals.111 Since guanine-rich sites have the lowest oxidation potential within the genome, the resulting holes will migrate via DNA CT to these sites.11 These guanine radicals can then either react with solution molecules to form stable damage products or they can continue to migrate to DNA-bound proteins such as SoxR. Recall that the timescale for DNA CT is nanoseconds or faster versus guanine reaction with water, which is on the millisecond timescale. Activation of SoxR from a distance in this fashion would allow these proteins to expand the range of sensing beyond the physical space occupied by the protein within the cell. Interestingly, the soxS promoter site in E. coli contains two GGGG sites within 100 bp, and these sites are conserved in many other enterics containing soxRS regulons.
This model is consistent with what is known about SoxR and provides a rationale for rapid and sensitive activation of SoxR as observed in vivo. In keeping with this model, when guanine radicals are selectively generated in DNA by photoexcited, [Rh(phi)2(bpy)]2+ tethered 80 bp (270 Å) from the SoxR binding site, SoxR is activated and transcription is initiated, as determined by a reconstituted abortive transcription assay.13 In vivo, treatment of live E. coli with [Rh(phi)2(bpy)]2+ also induces generation of the soxS transcript in response to illumination.
It is interesting that DNA-mediated oxidation of SoxR is so facile, given the 20 Å separation between the cluster and the DNA in the crystal structure of the oxidized species. It is possible that the cluster is closer to the DNA in the reduced state. Time-resolved transient absorption measurements154 and computational study86 would both be useful in characterizing CT across the SoxR-DNA interface.
b. Redox Signaling between BER Enzymes
While DNA repair proteins are not typically viewed as participants in redox signaling pathways, a class of base-excision repair proteins, largely involved in repair of oxidative DNA damage, binds an iron-sulfur cluster cofactor much like SoxR.172 It is intriguing to consider whether this cofactor might confer upon these DNA repair proteins additional redox regulatory and functional properties.
MutY and Endonuclease III (EndoIII) are the most well conserved enzymes in this class, with homologs present in a wide variety of prokaryotic and eukaryotic organisms.172 In base excision repair, glycosylase enzymes, often present in low copy number, must locate and recognize specific isolated damaged bases in the genome and then excise these lesions.173 The first step of this process, the initial search for lesions in the genome, is not well understood. The presence of a redox-active iron-sulfur cluster in these proteins may facilitate DNA-mediated redox signaling as a particularly efficient damage detection mechanism in this repair pathway.
MutY and EndoIII repair oxidatively damaged bases in the genome.172,173 MutY removes adenines mispaired with 8-oxo-guanine while EndoIII excises a variety of oxidized pyrimidine bases from DNA. Both enzymes are present within the cell in extremely low copy number (< 30/cell for MutY, 500/cell for EndoIII).174 Despite their low abundance, MutY and EndoIII are extremely effective in vivo.175 Thus, how these enzymes efficiently locate their substrates is a major question, as is the function of the iron-sulfur cluster in these proteins.
A primary focus of study with these enzymes has been exploring the mechanism, properties, and specificity of the excision reaction both in vitro and in vivo.172,173 In addition, X-ray crystal structures have been solved for E. coli MutY and EndoIII as well as Geobacillus stereothermophilus MutY and EndoIII crosslinked to substrate analogs in a short DNA helix.176,177 Much is now understood regarding the structural basis for substrate recognition in these proteins and the nature of the enzymatic reaction. Both enzymes extrude their substrate bases from the helix and bind them in a specific active site pocket. Recognition is achieved based on specific interactions between protein sites and functional groups on the lesion base. Scission of the glycosidic bond occurs by different mechanisms in these enzymes and these are also well characterized.172,173 However, repair assays in E. coli cells indicate that, in vivo, the rate-limiting step in base excision repair by MutY is the initial detection of lesions rather than the base excision reaction.178 These studies have revealed very little, however, about the explicit role of the iron-sulfur cluster. DNA-bound crystal structures reveal that the iron-sulfur cluster, though close to the DNA backbone, is located far from the enzyme active site and substrate recognition pocket and does not play a distinct part in the excision reaction pathway.176,177 The cofactor is required for non-specific DNA binding and, thus, is essential for overall activity.
Our laboratory has further investigated the role of the iron-sulfur cluster in these proteins. MutY and EndoIII each contain a [4Fe4S]2+ cluster ligated by four cysteine residues.176,177,179 In the absence of DNA, the iron-sulfur cluster is resistant to oxidation and reduction; oxidation by solution-bourne oxidants results in loss of an iron atom to form [3Fe4S]1+ while reduction could only be accomplished using mediators with potentials < -600 mV vs. NHE.179 When examined at DNA-modified electrode surfaces, MutY and EndoIII both display markedly different properties to those seen in solution without DNA.83,84,87 Robust and quasi-reversible signals are observed with midpoint potentials (E1/2) between +50 and +100 mV vs. NHE. Observation of these electrochemical signals requires an intact base pair π-stack; the presence of an abasic site dramatically diminishes the intensity of the electrochemical features. Consistent with mediator experiments, electrochemical examination of EndoIII in the absence of DNA reveals features at both higher (> +250 mV vs. NHE) and lower potentials (< -400 mV).87 These peaks are far less reversible and distinct than those present at DNA-modified surfaces. These features at high and low potential are interpreted as the [4Fe4S]2+/3+ and [4Fe4S]2+/1+ redox couples, respectively. When bound to DNA, the [4Fe4S]2+/3+ couple shifts by − 200 mV vs. NHE and redox activity is much more robust. The DNA-associated forms of MutY and EndoIII are both more easily oxidized than the free forms of the proteins, and more stable in the oxidized state. Furthermore, a distinct requirement exists that iron-sulfur cluster oxidation and reduction be mediated by the DNA base-pair stack.
The − 200 mV shift in redox potential upon DNA binding, as with SoxR, is indicative of some form of energetic change upon DNA binding by MutY and EndoIII, such as a change in binding affinity or protein/DNA conformation. In these proteins, unlike SoxR, it is known that the structure of the protein does not change significantly upon DNA binding nor do they induce DNA distortion when non-specifically bound.176,177,180,181 Moreover, for the repair proteins, DNA-binding causes a negative shift in redox potential, as one might expect upon binding to a polyanion, versus the large positive shift in potential associated with binding of SoxR to DNA. Thus, it is proposed that the redox potential shift for the repair proteins translates to a differential DNA-binding affinity for the oxidized and reduced forms of the protein. The 200 mV shift estimated in electrochemical studies must correspond to an increased DNA-binding affinity for the [4Fe4S]3+ form by > 3 orders of magnitude.
A redox-active iron-sulfur cluster could allow MutY and EndoIII to participate in redox reactions when bound to DNA. The possibility that these reactions can occur via the DNA π-stack may also permit these proteins to harness the exquisite sensitivity of DNA-mediated CT reactions towards myriad damaged and mismatched bases (including the substrates for MutY and EndoIII).76,79 Our laboratory has described a model illustrating how MutY and EndoIII might exploit DNA CT to accomplish the first step of base excision repair, which is the initial scan of the genome for lesions (Figure 7).182 BER glycosylases initially bind to DNA in a nonspecific fashion. In this first step, the iron-sulfur clusters in the BER enzymes (e.g. MutY) are in the [4Fe4S]2+ state. Upon binding to DNA, MutY is now more accessible to oxidation, and upon transformation to the [4Fe4S]3+ state by myriad cellular oxidants, including guanine radicals, now binds DNA much more tightly. If the surrounding genome is free of damaged and mismatched bases, this MutY molecule can be reduced, via the DNA π-stack, by distally bound redox-active proteins of similar or greater reduction potential. Instead, if a damaged site is present, oxidized repair proteins will be less accessible for reduction via the DNA base-pair stack and are more likely to remain tightly associated to the DNA near the lesion site. This model provides not only a rationale for the presence of a redox-active iron-sulfur cluster in BER enzymes, but it also provides an explanation for fast and efficient lesion detection by these enzymes in a genomic context. It has been shown by simulation that allowing CT between individual copies of MutY, for certain parameter ranges, can lead to protein accumulation in the vicinity of a lesion.183,184
Figure 7.
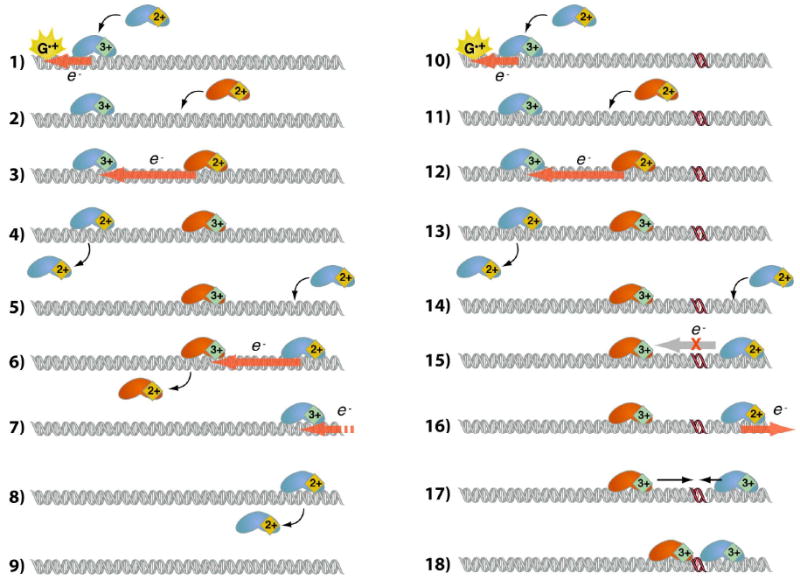
Redox-active base excision repair (BER) enzymes can exploit DNA-mediated CT to rapidly assay regions of DNA for damage, such as oxidative lesions. The presence of a lesion serves as a barrier for DNA CT, leading to accumulation of BER proteins near the lesion. Importantly, BER enzymes of similar potential can assist each other in searching for damage. Here, the orange and blue proteins represent two different FeS cluster containing proteins of similar potential. In this model, guanine cation radical or another oxidative intermediate oxidizes a nearby BER FeS cluster to the tight-binding 3+ state (1). Binding of reduced protein nearby (2) allows self-exchange (3), following which the first protein, now in the 2+ state, has decreased affinity for DNA and can diffuse away (4). Steps 2-4 correspond to a net translation of 3+ protein on the DNA. This process is repeated when another reduced protein binds the DNA on the other side (5,7), followed by self-exchange and dissociation of the newly reduced protein (6,8). The DNA is left empty of the 3+, and hence strongly bound, protein (9). The directionality portrayed in (1-9) is illustrative, as the CT-mediated migration of 3+ protein is diffusive. When a lesion is present, steps 10-14 mirror steps 1-5. Reduced protein distal to the lesion, however, is unable to reduce the oxidized protein through DNA-mediated CT (15), increasing the residence of that particular 3+ protein near the lesion. CT-mediated migration of oxidized protein, as shown in the previous steps, can oxidize reduced protein on the distal side of the lesion (16), and 3+ protein accumulated near the lesion can slide along the DNA (17) and repair the lesion upon direct contact (18).
The first step of this damage detection pathway requires oxidation of DNA-bound [4Fe4S] cluster repair proteins. As proposed with SoxR, this could also occur via DNA CT through formation of guanine radical cations that occur as a consequence of oxidative DNA damage.86,111 This could be especially significant for DNA repair, recruiting BER enzymes to local genomic sites actively undergoing oxidative stress. We have established that this chemistry can occur in vitro using a Ru-DNA assembly to generate guanine radicals via flash-quench reactions.154 Guanine radicals are monitored directly via EPR and transient absorption spectroscopies and indirectly via trapping to form permanent oxidative damage products, visualized by gel electrophoresis. Flash-quench of these assemblies in the presence of MutY shows quenching of the guanine radical and formation of spectroscopic features typical of oxidized iron-sulfur clusters. These results are consistent with electron transfer from MutY to fill the hole present at oxidized guanine sites, and suggest that this activation pathway is feasible within the cell.
The ultimate outcome of the proposed model is the clustering of enzymes near damaged sites. This idea has been tested by AFM imaging of EndoIII bound to long DNAs containing a site-specific mismatch (C:A) known to attenuate DNA CT.182 In these experiments, a mixture of long strands containing a single C:A mismatch and short matched strands are incubated with EndoIII. Consistent with the CT model, a greater proportion of proteins (1.6:1) are found bound to the long strand containing the mismatch. When all strands are matched, the long/short ratio is 0.9:1. Note that a C:A mismatch is not a substrate for EndoIII, so the preference for the mismatched strand does not reflect a direct affinity for the mismatched site. The long/short ratio increases with increasing concentrations of external oxidants such as hydrogen peroxide, indicating a relationship between the accumulation of EndoIII near the mismatch site and the oxidation state of the iron-sulfur cluster. These experiments corroborate the prediction from the model that enzymes using DNA CT for genome scanning will redistribute onto regions near damaged sites.
Another important feature of the DNA CT damage detection model is its ability to accommodate cooperative lesion detection by disparate DNA repair proteins. Electrons may transfer to any redox active DNA-binding protein, and in doing so, will transmit information about the integrity of the surrounding DNA. This may provide a further explanation for the low abundance yet high in vivo effectiveness of BER enzymes. We have tested this putative cooperativity using established assays for MutY activity in E. coli cells. If MutY and EndoIII, both present in E. coli, search for lesions cooperatively via DNA CT, then genetic inactivation of EndoIII should hinder damage detection by MutY within the cell. EndoIII knockouts (nth-) indeed cause a small decrease in MutY activity inside the cell. We have examined the origin of this defect using site-directed mutagenesis. Introduction of excision deficient EndoIII mutant (D138A) into the MutY reporter nth- strain restores activity, while a mutant that is deficient in protein/DNA electron transfer (Y82A EndoIII) remains inactive. These results support the idea that MutY and EndoIII engage in a cooperative functional relationship inside the cell that requires a fully redox-active iron-sulfur cluster in EndoIII.
Many other models have been proposed for target location by these DNA-binding proteins. All of these rely upon physical interaction with genome sites to verify target or non-target status. These strategies may allow for efficient repair over a single cell cycle for DNA repair enzymes in high copy number, but for low copy number enzymes, such as MutY, they are insufficient for effective substrate location. We have calculated the improvement in genome scanning time for MutY in E. coli that would be achieved using the DNA CT scanning model. In these calculations, the interprotein charge transport distance and the proportion of protein initially in the oxidized state are taken as variables and cooperative CT is assumed between MutY and EndoIII only. A baseline genome scanning time in the absence of DNA CT is determined for MutY, assuming only facilitated diffusion of the protein with instantaneous interrogation of potential targets, and is too long to account for effective repair within the cell cycle. Allowing CT over 200-500 bp with 10-20% oxidized protein, modeled with the diffusion of reduced protein to within CT distance of a DNA-bound oxidized protein as the rate limiting step, gives genome scanning times of at least an order of magnitude faster, comfortably within the cell cycle. Therefore, DNA CT provides an explanation for efficient DNA repair by MutY in vivo, whereas other models fail in this respect. Interestingly, these calculations indicate switch-like behavior in the dependence of genome scanning time on the fraction of oxidized protein at low levels of oxidation. This could allow for an additional level of redox regulation in DNA repair by MutY.
c. Activation of p53 by DNA-mediated CT
The protein p53 is a well known example of a eukaryotic transcription factor regulated by redox processes.185 p53 is also of high clinical relevance as it is found mutated in over 50% of cancers.186 As a transcription factor, it activates many different genes involved in apoptosis, cell cycle regulation, oxidative stress response, repair, and autophagy.187 This transcriptional regulation occurs in response to cellular stress that includes DNA damage, oncogene activation, telomere elimination, and hypoxia. p53 can respond to these events directly or in a protein-mediated fashion. While in many cases, the relationship between p53 and a particular stress response is known, the chemical mechanisms governing p53 activation are often not well understood. Understanding these relationships is important since p53 plays a key role in cancer prevention and aging.
The p53 gene product binds DNA as a tetramer and consists of three individual domains (N-terminal transactivation, DNA-binding, and C-terminal tetramerization) as well as several regions that are relatively unstructured.188 The DNA-binding domain is where the majority of p53 mutations occur that are associated with cancer. p53 binds DNA across two palindromic half-sites of the form 5′-A/G A/G A/G C A/T A/T G C/T C/T C/T-3′ separated by 0–13 bp. The DNA binding domain forms a β sandwich structure with distinct faces that interact with its binding site over the major and minor groove. This domain also binds a zinc ion important for stability and DNA affinity. Many contacts between protein side chains and DNA functional groups are apparent that likely dictate sequence recognition and specificity.
The protein p53 is one of many eukaryotic transcription factors whose DNA affinity is modulated by thiol-disulfide redox chemistry.185 In vitro DNA binding by p53 requires a reducing environment. Similarly, reagents that specifically oxidize thiol groups prevent p53/DNA association. p53 contains seven conserved cysteines in its DNA-binding domain.188 Three of these are involved in binding zinc (C176, C238, C242) while two are proposed to form a disulfide bond based on structural proximity (C275, C277). The remaining cysteines are C135 and C141 are in close proximity to the DNA backbone. While the redox modulation of p53 is established in vitro, the mechanism and functional consequences of this chemistry within the cell is not well understood.
Redox modulation of p53 from a distance via DNA CT has been examined using a pendant photooxidant assembly consisting of a DNA sequence bearing a p53-specific promoter target functionalized with a tethered anthraquinone (AQ).189 Irradiation of AQ promotes DNA-mediated oxidation reactions. The ability of this assembly to oxidize thiols from a distance has been established using oligonucleotides containing thiol modifiers in the DNA backbone.190 Photoinitiated oxidation of p53 bound to a consensus promoter sequence yields dissociation of the protein. Mass spectrometry confirms chemical changes in the protein consistent with oxidation of conserved cysteines in the DNA binding domain. Experiments with p53 promoters for genes involved in apoptosis (p21), negative feedback regulation (mdm2), and DNA repair (GADD45) reveal sequence-specific effects on DNA-mediated p53 oxidation. In particular, upon photooxidation from a distance, p53 dissociates from the GADD45 and mdm2 promoters. p53 is, however, not released from the p21 promoter as a consequence of DNA-mediated oxidation. These results indicate that p53 can also be oxidized from a distance when bound to natural promoter sites and that redox regulation may be sequence-specific (Figure 8). It is interesting to consider the functional consequences of the latter idea; DNA-mediated oxidation from a distance may signal extreme stress such that, under these circumstances, p53 will still induce apoptosis but does not initiate DNA repair or engage in negative feedback.
Figure 8.
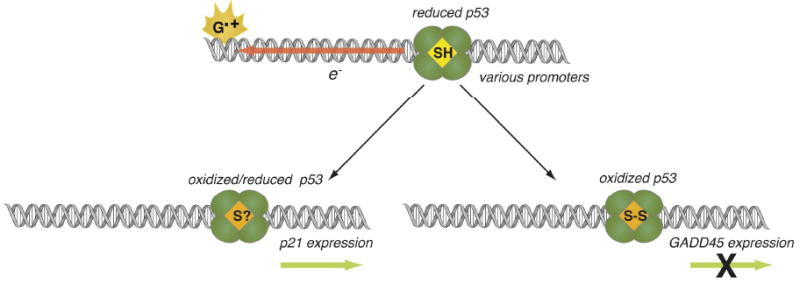
The delivery of radical holes induces dissociation of p53 from the promoter for GADD45, but not that for p21. DNA-mediated CT serves as a mechanism for efficient delivery of oxidation to p53, allowing a rapid redistribution from pro-repair to pro-apoptotic promoter sites.
DNA-mediated oxidation of p53 has also been demonstrated within the cell.189 Incubation of HeLa cells with a rhodium photooxidant ([Rh(phi)2(bpy)]3+) yields formation of oxidized p53 as determined by western blotting. Similar results are also observed upon cellular exposure to hydrogen peroxide. Thus, p53 can be oxidized via DNA-mediated CT in an in vivo setting as well.
These experiments establish that thiol-disulfide redox chemistry is accessible via DNA-mediated charge transport. Furthermore, they describe a novel chemical pathway for redox modulation of p53. Additional exploration of the sequence specificity of DNA-mediated p53 oxidation may provide new insight into both the structural requirements for efficient protein-DNA charge transport while illuminating new aspects of p53 regulation of possible clinical importance.
Conclusions and Outlook
Signaling and sensing processes, as forms of communication, are inherently coupled to location and distance. By offering a medium for chemistry at a distance, the ability of DNA to mediate CT provides a natural mechanism for redox sensing and signaling in the genome. Here, we have illustrated how charge migration in DNA may be employed to solve a series of challenges to both prokaryotes and eukaryotes. Charge migration through DNA allows oxidative stress to be funneled either to a sensing protein, such as SoxR or p53, or to damage hotspots that then affect protein recognition. Electron exchange between proteins allows redox signaling to redistribute repair proteins in the vicinity of lesions. Given the power of this chemistry, how else might it be exploited by nature?
Table I lists a sampling of DNA-binding proteins, from a variety of organisms, that contain moieties that are traditionally redox-active under physiological conditions. We do not mean to suggest that each of these proteins will be found to exploit DNA-mediated CT in a physiological context. Indeed, redox-potentials for most of these have not yet been measured, and even fewer measurements have been taken in the presence of DNA, though such efforts are now ongoing in our laboratory. Rather, we find it enticing that so many DNA-binding proteins have been recently identified that have the potential to participate in redox reactions. For many of these proteins, particularly for those containing an iron-sulfur cluster, the redox-active moiety serves no apparent catalytic role yet is highly conserved. The examples in humans are particularly intriguing. FancJ is required for repair of double strand breaks and of inter-strand crosslinks, while XPD is required for other forms of nucleotide excision repair. The p58 component of human DNA primase is required for replication and some repair processes. Increasingly, proteins with iron-sulfur clusters are being identified as essential to early steps in each of the various types of DNA processing. Eukaryotic DNA processing typically involves macromolecular assemblies containing a diversity of protein components, many of which are now unknown. Perhaps a common component of these assemblies is generally available for redox-sensing. Certainly as new components of these macromolecular machines are revealed and characterized, the list of proteins involved that contain iron-sulfur clusters will continue to grow. As new biological roles for the iron-sulfur clusters in Table I are determined, new insight will be gained into how redox reactions can be chemically exploited to enable the functions of DNA-binding proteins.
Table 1.
| Potential (V vs. NHE) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein | Organism | Function | Redox-Active Moiety | +DNA | -DNA | Redox Function | Phylogenetic Distribution | Clinical Relevance | References |
| MutY | E. coli | BER | [4Fe-4S] | +0.06 | N. D. | Lesion Detection | P E | Y | 83, 84 |
| Endolll | E. coli | BER | [4Fe-4S] | +0.06 | +0.35 | Lesion Detection | P A E | N | 83, 87 |
| UDG | A. fulgidus | BER | [4Fe-4S] | +0.06 | N. D. | Lesion Detection | A | N | 83 |
| SoxR | E. coli | oxidative stress response TF | [2Fe-2S] | +0.20 | -0.30 | ROS Detection | P | Y | 85 |
| FNR | E. coli | oxygen sensing TF | [4Fe-4S] | N. D. | N. D. | ROS Detection | P | N | 191 |
| IscR | E. coli | TF regulating FeS cluster synthesis | [2Fe-2S] | N. D. | N. D. | Unknown | P | N | 192, 193 |
| SP lyase | B. subtilis | spore photoproduct thymine dimer repair | [4Fe-4S] | N. D. | N. D. | Catalytic | P | N | 194, 195 |
| XPD | S. acidocaldarius | NER helicase | [4Fe-4S] | N. D. | N. D. | Unknown | A E | Y | 196-199 |
| FancJ/BACH1/BRIP1 | human | NER, HR helicase | [4Fe-4S] | N. D. | N. D. | Unknown | A E | Y | 196 |
| Dna2 | human | NER helicase | [4Fe-4S] | N. D. | N. D. | Unknown | A E | N | 196 |
| DinG | E. coli | NER | [4Fe-4S] | N. D. | -0.39 | NO detection? | P | N | 200 |
| primase p58 | human | primase | [4Fe-4S] | N. D. | N. D. | Unknown | A E | N | 201, 202 |
| AddB | E. coli | DSB helicase/nuclease | [4Fe-4S] | N. D. | N. D. | Unknown | P | N | 203 |
| RNA polymerase | S. solfataricus | DSB helicase/nuclease | [4Fe-4S] | N. D. | N. D. | Unknown | A | N | 204 |
| Elp3 | yeast | histone acetyltransferase | [4Fe-4S] | N. D. | N. D. | Non-catalytic | A E | N | 205, 206 |
| p53 | human | tumor suppressor TF | thiol/disulfide | N. D. | N. D. | ROS detection | E | Y | 189 |
| MexR | P. aeruginosa | multidrug efflux TF | thiol/disulfide | -0.16 | N. D. | ROS detection (2° signal, antibiotic) | P | Y | 207 |
| OhrR | B. subtilis | peroxide response TF | thiol/sulfenic acid/disulfide | N. D. | N. D. | peroxide detection | P | N | 208, 209 |
| OxyR | E. coli | peroxide response TF | thiol/disulfide | -0.19 | N. D. | peroxide detection | P | N | 7 |
| CrtJ | R. capsulatus | photosystem regulating TF | thiol/disulfide | -0.19 | N. D. | oxygen detection | P | N | 210 |
| Ape1/Ref-1 | human | AP-1, NF-κB activation abasic site excision | thiol/sulfenic acid | N. D. | N. D. | Trx signalling | E | Y | 211, 212 |
| RsrA | gram positive actinomycetes | σR repressor ROS response | Zn-2S/disulfide | N. D. | N. D. | ROS detection | P | N | 155 |
| Bach1 | human | Fe metabolism and ROS response TF | Fe heme | N. D. | N. D. | unknown | E | N? | 213 |
| Dps | E. coli | Fe storage DNA Protection | Ferritin | N. D. | N. D. | unknown | P | N | 216, 119 |
| AidB | E. coli | alkylation response | FAD | N. D. | N. D. | detoxification? | P | N | 214 |
| Photolyase | E. coli | cyclobutane thymine dimer repair | FAD | N. D. | +0.04 | photoexcitation for catalytic redox repair | P | N | 215 |
| Rex | S. coelicor | TF regulating NADH dehydrogenase | NAD(H) | N. D. | N. D. | detection of global NAD/NADH balance | P | N | 155, 216 |
| Hap1 | yeast | heme synthase and ferrochelatase TF | heme | N. D. | N. D. | heme detection | E | N | 217, 218 |
| Notl | N. otitidis-caviarum | rare-cutting restriction enzyme | FeS4 | N. D. | N. D. | non-catalytic | P | N | 219 |
Abbreviations: N.D., not determined; E, euakaryota; P, prokaryota; A, archaea.; FAD, flavin adenine dinucleotide The list of thiol/disulfide sensing proteins is illustrative rather than exhaustive.
But an iron-sulfur cluster is not essential for this signaling. It has been well established that many transcription factors are regulated by the redox state of cysteine. We have demonstrated that one of the most significant of these, p53, can be regulated in a site-selective manner by DNA-mediated CT. Detailed chemical mechanisms of reduction and oxidation have not been worked out for the majority of these factors, and it is unknown how many are sufficiently well-coupled to the DNA π stack to be regulated by redox reaction through DNA. Using the entire genome as an antenna for oxidative stress is an attractive alternative to relying on diffusion of ROS to the sensing protein.
We have demonstrated a few ways in which a chemical reaction, DNA-mediated CT, is exploited for biological roles. The chemistry of DNA-mediated CT to and between proteins has only begun to be characterized. As new understanding of this process emerges and new participants are identified, undoubtedly, critical chemical mechanisms inside living cells will be revealed.
Supplementary Material
Acknowledgments
We thank the NIH for their financial support of the research described here. We also thank our coworkers and collaborators for their hard work, fortitude, courage and creativity in elucidating this chemistry.
Footnotes
The full citation for reference 125 is provided.
References
- 1.Delaney S, Barton JK. J Org Chem. 2003;68:6475. doi: 10.1021/jo030095y. [DOI] [PubMed] [Google Scholar]
- 2.O'Neill MA, Barton JK. In: Topics in Current Chemistry. Schuster GB, editor. Vol. 236. Springer; Berlin: 2004. p. 67. [Google Scholar]
- 3.Genereux JC, Barton JK. Chem Rev. 2009 doi: 10.1021/cr900228f. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorodetsky AA, Buzzeo MC, Barton JK. Bioconug Chem. 2008;19:2285. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DNA-mediated CT is also facile through DNA/RNA hybrids. See Sartor V, Henderson PT, Schuster GB. J Am Chem Soc. 1999;121:11027.O'Neill MA, Barton JK. J Am Chem Soc. 2002;124:13053. doi: 10.1021/ja0208198.
- 6.Storz G, Tartaglia LA, Ames BN. Science. 1990;248:189. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 7.Zheng M, Åslund F, Storz G. Science. 1998;279:1718. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 8.Stone JR, Yang S. Antioxidants Redox Signaling. 2006;8:243. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 9.Imlay JA. Ann Rev Biochem. 2008;77:755. doi: 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thannickal VJ, Fanburg B. In: Signal Transduction by Reactive Oxygen and Nitrogen Species. Forman HJ, Fukuto J, Torres M, editors. Kluwer Academic; Norwell, MA: 2003. p. 291. [Google Scholar]
- 11.Núñez ME, Hall DB, Barton JK. Chem Biol. 1999;6:85. doi: 10.1016/S1074-5521(99)80005-2. [DOI] [PubMed] [Google Scholar]
- 12.Núñez ME, Holmquist GP, Barton JK. Biochemistry. 2001;40:12465. doi: 10.1021/bi011560t. [DOI] [PubMed] [Google Scholar]
- 13.Lee PE, Demple B, Barton JK. Proc Natl Acad Sci USA. 2009;106:13164. doi: 10.1073/pnas.0906429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Osakada Y, Kawai K, Fujitsuka M, Majima T. Proc Natl Acad Sci USA. 2006;103:18072. doi: 10.1073/pnas.0607148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takada T, Kawai K, Fujitsuka M, Majima T. Proc Natl Acad Sci USA. 2004;101:14002. doi: 10.1073/pnas.0402756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu CS, Hernandez R, Schuster GB. J Am Chem Soc. 2004;126:2877. doi: 10.1021/ja0378254. [DOI] [PubMed] [Google Scholar]
- 17.Odom DT, Dill EA, Barton JK. Chem Biol. 2000;7:475. doi: 10.1016/s1074-5521(00)00133-2. [DOI] [PubMed] [Google Scholar]
- 18.Delaney S, Pascaly M, Bhattacharya PK, Han K, Barton JK. Inorg Chem. 2002;41:1966. doi: 10.1021/ic0111738. [DOI] [PubMed] [Google Scholar]
- 19.Boon EM, Jackson NM, Wrightman MD, Kelley SO, Hill MG, Barton JK. J Phys Chem B. 2003;107:11805. [Google Scholar]
- 20.Breslin DT, Coury JE, Anderson JR, McFail-Isom L, Kan Y, Williams LD, Bottomley LA, Schuster GB. J Am Chem Soc. 1997;119:5043. [Google Scholar]
- 21.Yavin E, Stemp EDA, O'Shea VL, David SS, Barton JK. Proc Natl Acad Sci USA. 2006;103:3610. doi: 10.1073/pnas.0600239103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda R, Chiba J, Inouye M. e-J Surf Sci Nanotechnol. 2005;3:393. [Google Scholar]
- 23.Gorodetsky AA, Green O, Yavin E, Barton JK. Bioconj Chem. 2007;18:1434. doi: 10.1021/bc0700483. [DOI] [PubMed] [Google Scholar]
- 24.Murphy CJ, Arkin MR, Jenkins Y, Ghatlia ND, Bossmann SH, Turro NJ, Barton JK. Science. 1993;262:1025. doi: 10.1126/science.7802858. [DOI] [PubMed] [Google Scholar]
- 25.Murphy CJ, Arkin MR, Ghatlia ND, Bossman SH, Turro NJ, Barton JK. Proc Natl Acad Sci USA. 1994;91:5315. doi: 10.1073/pnas.91.12.5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasai H, Yaamaizumi Z, Berger M, Cadet J. J Am Chem Soc. 1992;114:9692. [Google Scholar]
- 27.Armitage B, Yu C, Devadoss C, Schuster GB. J Am Chem Soc. 1994;116:9847. [Google Scholar]
- 28.Saito I, Takayama M, Sugiyama H, Nakatani K. J Am Chem Soc. 1995;117:6406. [Google Scholar]
- 29.Hall DB, Holmlin RE, Barton JK. Nature. 1996;382:731. doi: 10.1038/382731a0. [DOI] [PubMed] [Google Scholar]
- 30.Marcus RA, Sutin N. Biochim Biophys Acta. 1985;811:265. [Google Scholar]
- 31.Giese B. Annu Rev Biochem. 2002;71:51. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 32.Wan C, Fiebig T, Kelley SO, Treadway CR, Barton JK, Zewail AH. Proc Natl Acad Sci USA. 1999;96:6014. doi: 10.1073/pnas.96.11.6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valis L, Wang Q, Raytchev M, Buchvarov I, Wagenknecht HA, Fiebig T. Proc Natl Avad Sci USA. 2006;103:10192. doi: 10.1073/pnas.0600957103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis FD, Wu T, Zhang Y, Letsinger RL, Greenfield SR, Wasielewski MR. Science. 1997;277:673. doi: 10.1126/science.277.5326.673. [DOI] [PubMed] [Google Scholar]
- 35.Lewis FD, Kalgutkar RS, Wu Y, Liu X, Liu J, Hayes RT, Miller SE, Wasielewski MR. J Am Chem Soc. 2000;122:12346. [Google Scholar]
- 36.Lewis FD, Liu J, Weigel W, Rettig W, Kurnikov IV, Beratan DN. Proc Natl Acad Sci USA. 2002;99:12536. doi: 10.1073/pnas.192432899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LeBard DN, Lilichenko M, Matyushov DV, Berlin YA, Ratner MA. J Phys Chem B. 2003;107:14509. [Google Scholar]
- 38.Jortner J, Bixon M, Langenbacher T, Michel-Beyerle ME. Proc Natl Acad Sci USA. 1998;95:12759. doi: 10.1073/pnas.95.22.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jortner J, Bixon M, Voityuk AA, Rösch N. J Phys Chem A. 2002;106:7599. [Google Scholar]
- 40.Berlin YA, Burin AL, Ratner MA. J Am Chem Soc. 2001;123:260. doi: 10.1021/ja001496n. [DOI] [PubMed] [Google Scholar]
- 41.Berlin YA, Ratner MA. In: Charge Migration in DNA. Chakraborty T, editor. Springer; Berlin: 2008. p. 45. [Google Scholar]
- 42.Takada T, Kawai K, Cai X, Sugimoto A, Fujitsuka M, Majima T. J Am Chem Soc. 2004;126:1125. doi: 10.1021/ja035730w. [DOI] [PubMed] [Google Scholar]
- 43.Kodera H, Osakada Y, Kawai K, Majima T. Nat Chem. 2009;1:156. doi: 10.1038/nchem.171. [DOI] [PubMed] [Google Scholar]
- 44.Meggers E, Michel-Beyerle ME, Giese B. J Am Chem Soc. 1998;120:12950. [Google Scholar]
- 45.Giese B, Spichty M. ChemPhysChem. 2000;1:195. doi: 10.1002/1439-7641(20001215)1:4<195::AID-CPHC195>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 46.Lewis FD, Zhu H, Daublain P, Cohen B, Wasielewski MR. Angew Chem Int Ed. 2006;45:7982. doi: 10.1002/anie.200603455. [DOI] [PubMed] [Google Scholar]
- 47.Behrens C, Cichon MK, Grolle F, Hennecke U, Carell T. In: Topics in Current Chemistry. Schuster GB, editor. Vol. 236. Springer; Berlin: 2004. p. 187. [Google Scholar]
- 48.Cordes M, Giese B. Chem Soc Rev. 2009;38:892. doi: 10.1039/b805743p. [DOI] [PubMed] [Google Scholar]
- 49.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 50.Shih C, Museth AK, Abrahamsson M, Blanco-Rodriguez AM, Di Billio AJ, Sudhamsu J, Crane BR, Ronayne KL, Towrie M, Vicek A, Richards JH, Winkler JR, Gray HB. Science. 2008;320:1760. doi: 10.1126/science.1158241. [DOI] [PubMed] [Google Scholar]
- 51.Gray HB, Winkler JR. Q Rev Biophys. 2003;36:341. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 52.Grozema FC, Tonzani S, Berlin YA, Schatz GC, Siebbeles LDA, Ratner MA. J Am Chem Soc. 2008;130:5157. doi: 10.1021/ja078162j. [DOI] [PubMed] [Google Scholar]
- 53.Grozema FC, Tonzani S, Berlin YA, Schatz GC, Siebbeles LDA, Ratner MA. J Am Chem Soc. 2009;131:14204. doi: 10.1021/ja906863k. [DOI] [PubMed] [Google Scholar]
- 54.Giese B, Amaudrut, Köhler AK, Sporman M, Wessely S. Nature. 2001;412:318. doi: 10.1038/35085542. [DOI] [PubMed] [Google Scholar]
- 55.Renger T, Marcus RA. J Phys Chem A. 2003;107:8404. [Google Scholar]
- 56.O'Neill MA, Barton JK. J Am Chem Soc. 2004;126:11471. doi: 10.1021/ja048956n. [DOI] [PubMed] [Google Scholar]
- 57.Barnett RN, Cleveland CL, Joy A, Landman U, Schuster GB. Science. 2001;294:567. doi: 10.1126/science.1062864. [DOI] [PubMed] [Google Scholar]
- 58.Conwell EM, Park JH, Choi HY. J Phys Chem B. 2005;109:9760. doi: 10.1021/jp044485f. [DOI] [PubMed] [Google Scholar]
- 59.Basko DM, Conwell EM. Phys Rev Lett. 2002;88:098102. doi: 10.1103/PhysRevLett.88.098102. [DOI] [PubMed] [Google Scholar]
- 60.Genereux JC, Augustyn KE, Davis ML, Shao F, Barton JK. J Am Chem Soc. 2008;130:15150. doi: 10.1021/ja8052738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeidan TA, Carmieli R, Kelley RF, Wilson TM, Lewis FD, Wasielewski MR. J Am Chem Soc. 2008;130:13945. doi: 10.1021/ja803765r. [DOI] [PubMed] [Google Scholar]
- 62.O'Neill MA, Barton JK. J Am Chem Soc. 2004;126:13234. doi: 10.1021/ja0455897. [DOI] [PubMed] [Google Scholar]
- 63.O'Neill MA, Becker HC, Wan C, Barton JK, Zewail AH. Angew Chem Int Ed. 2003;42:5896. doi: 10.1002/anie.200352831. [DOI] [PubMed] [Google Scholar]
- 64.Troisi A, Orlandi G. J Phys Chem B. 2002;106:2093. [Google Scholar]
- 65.Kelley SO, Jackson NM, Hill MG, Barton JK. Angew Chem, Int Ed. 1999;38:941. doi: 10.1002/(SICI)1521-3773(19990401)38:7<941::AID-ANIE941>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 66.Wong ES, Gooding JJ. J Am Chem Soc. 2007;129:8950. doi: 10.1021/ja0723075. [DOI] [PubMed] [Google Scholar]
- 67.Drummond TG, Hill MG, Barton JK. J Am Chem Soc. 2004;126:15010. doi: 10.1021/ja044910i. [DOI] [PubMed] [Google Scholar]
- 68.Crespo-Hernández CE, Close DM, Gorb L, Leszczynski J. J Phys Chem B. 2007;111:5386. doi: 10.1021/jp0684224. [DOI] [PubMed] [Google Scholar]
- 69.Seidel CAM, Shulz A, Sauer MHM. J Phys Chem. 1996;100:5541. [Google Scholar]
- 70.Shinde SS, Maroz A, Hay MP, Anderson RF. J Am Chem Soc. 2009;131:5203. doi: 10.1021/ja8087339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kubař T, Elstner M. J Phys Chem B. 2008;112:8788. doi: 10.1021/jp803661f. [DOI] [PubMed] [Google Scholar]
- 72.Sugiyama H, Saito I. J Am Chem Soc. 1996;118:7063. [Google Scholar]
- 73.Sistare MF, Codden SJ, Heimlich G, Thorp HH. J Am Chem Soc. 2000;122:4742. [Google Scholar]
- 74.Osakada Y, Kawai K, Fujitsuka M, Majima T. Nuc Acids Res. 2008;36:5562. doi: 10.1093/nar/gkn505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bhattacharya PK, Barton JK. J Am Chem Soc. 2001;123:8649. doi: 10.1021/ja010996t. [DOI] [PubMed] [Google Scholar]
- 76.Boal AK, Barton JK. Bioconj Chem. 2005;16:312. doi: 10.1021/bc0497362. [DOI] [PubMed] [Google Scholar]
- 77.Rajski SR, Barton JK. Biochem. 2001;40:5556. doi: 10.1021/bi002684t. [DOI] [PubMed] [Google Scholar]
- 78.Boon EM, Salas JE, Barton JK. Nat Biotech. 2002;20:282. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 79.Boon EM, Ceres DM, Drummond TG, Hill MG, Barton JK. Nat Biotech. 2000;18:1096. doi: 10.1038/80301. [DOI] [PubMed] [Google Scholar]
- 80.Bhattacharya PK, Cha J, Barton JK. Nuc Acids Res. 2002;30:4740. doi: 10.1093/nar/gkf601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gorodetsky AA, Ebrahim A, Barton JK. J Am Chem Soc. 2008;130:2924. doi: 10.1021/ja7106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakatani K, Dohno C, Ogawa A, Saito I. Chem Biol. 2002;9:361. doi: 10.1016/s1074-5521(02)00119-9. [DOI] [PubMed] [Google Scholar]
- 83.Boal AK, Yavin E, Lukianova OA, O'Shea VL, David SS, Barton JK. Biochemistry. 2005;44:8397. doi: 10.1021/bi047494n. [DOI] [PubMed] [Google Scholar]
- 84.Boon EM, Livingston AL, Chmiel NH, David SS, Barton JK. Proc Natl Acad Sci USA. 2003;100:12543. doi: 10.1073/pnas.2035257100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gorodetsky AA, Dietrich LEP, Lee PE, Demple B, Newman DK, Barton JK. Proc Natl Acad Sci USA. 2008;105:3684. doi: 10.1073/pnas.0800093105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin JC, Singh RR, Cox DL. Biophys J. 2008;95:3259. doi: 10.1529/biophysj.108.132183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gorodetsky AA, Boal AK, Barton JK. J Am Chem Soc. 2006;128:12082. doi: 10.1021/ja064784d. [DOI] [PubMed] [Google Scholar]
- 88.Cadet J, Douki T, Ravanat JL. Acc Chem Res. 2008;41:1075. doi: 10.1021/ar700245e. [DOI] [PubMed] [Google Scholar]
- 89.Wagenknecht HA, Stemp EDA, Barton JK. J Am Chem Soc. 2000;122:1. [Google Scholar]
- 90.Wagenknecht HA, Stemp EDA, Barton JK. Biochemistry. 2000;39:5483. doi: 10.1021/bi992897m. [DOI] [PubMed] [Google Scholar]
- 91.Wagenknecht HA, Rajski SR, Pascaly M, Stemp EDA, Barton JK. J Am Chem Soc. 2001;123:4400. doi: 10.1021/ja003986l. [DOI] [PubMed] [Google Scholar]
- 92.Copeland KD, Lueras AMK, Stemp EDA, Barton JK. Biochemistry. 2002;41:12785. doi: 10.1021/bi020407b. [DOI] [PubMed] [Google Scholar]
- 93.Kurbanyan K, Nguyen KL, To P, Rivas EV, Lueras AMK, Kosinski C, Steryo M, Gonzalez A, Mah DA, Stemp EDA. Biochemistry. 2003;42:10269. doi: 10.1021/bi020713p. [DOI] [PubMed] [Google Scholar]
- 94.Nguyen KL, Steryo M, Kurbanyan K, Nowitzki KM, Butterfield SM, Ward SR, Stemp EDA. J Am Chem Soc. 2000;122:3585. [Google Scholar]
- 95.Morin B, Cadet J. J Am Chem Soc. 1995;117:12408. [Google Scholar]
- 96.Misiaszek R, Joffe J, Geacintov NE, Shafirovich V. J Biol Chem. 2004;279:32106. doi: 10.1074/jbc.M313904200. [DOI] [PubMed] [Google Scholar]
- 97.Ravanat JL, Saint-Pierre C, Cadet J. 2003;125:2030. doi: 10.1021/ja028608q. [DOI] [PubMed] [Google Scholar]
- 98.Xu X, Fleming AM, Muller JG, Burrows CJ. J Am Chem Soc. 2008;130:10080. doi: 10.1021/ja803896d. [DOI] [PubMed] [Google Scholar]
- 99.Johansen ME, Muller JG, Xu X, Burrows CJ. Biochemistry. 2005;44:5660. doi: 10.1021/bi047580n. [DOI] [PubMed] [Google Scholar]
- 100.Perrier S, Hau J, Gasparutto D, Cadet J, Favier A, Ravanat JL. J Am Chem Soc. 2006;128:5703. doi: 10.1021/ja057656i. [DOI] [PubMed] [Google Scholar]
- 101.Kobayashi K, Yamagami R, Tagawa S. J Phys Chem B. 2008;112:10752. doi: 10.1021/jp804005t. [DOI] [PubMed] [Google Scholar]
- 102.Stemp EDA, Arkin M, Barton JK. J Am Chem Soc. 1997;119:2921. [Google Scholar]
- 103.Shafirovich V, Dourandin A, Huang W, Geacintov NE. J Biol Chem. 2001;276:24621. doi: 10.1074/jbc.M101131200. [DOI] [PubMed] [Google Scholar]
- 104.Lee YA, Yun BH, Kim SK, Margolin Y, Dedon PC, Geacintov NE, Shafirovich V. Chem Eur J. 2007;13:4571. doi: 10.1002/chem.200601434. [DOI] [PubMed] [Google Scholar]
- 105.Lee YA, Durandin A, Dedon PC, Geacintov NE, Shafirovich V. J Phys Chem B. 2008;112:1834. doi: 10.1021/jp076777x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ananthaswamy HN, Eisenstark A. J Bacteriology. 1977;130:187. doi: 10.1128/jb.130.1.187-191.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Demple B, Halbrook J, Linn S. J Bacteriology. 1983;153:1079. doi: 10.1128/jb.153.2.1079-1082.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park S, You X, Imlay JA. Proc Natl Acad Sci USA. 2005;102:9317. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park S, Imlay JA. J Bacteriology. 2003;185:1942. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fenton HJH. J Chem Soc, Trans. 1894;65:899. [Google Scholar]
- 111.Burrows CJ, Muller JG. Chem Rev. 1998;98:1109. doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 112.Chatgilialoglu C, D'Angelantonio M, Guerra M, Kaloudis P, Mulazzani QG. Angew Chem Int Ed. 2009;48:2214. doi: 10.1002/anie.200805372. [DOI] [PubMed] [Google Scholar]
- 113.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 114.Gimisis T, Cismas C. Eur J Org Chem. 2006:1351. [Google Scholar]
- 115.Neeley JL, Essigman JM. Chem Res Toxicology. 2006;19:491. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 116.Hong Y, Wang G, Meier RJ. Free Radical Res. 2006;40:597. doi: 10.1080/10715760600618882. [DOI] [PubMed] [Google Scholar]
- 117.Martinez A, Kolter R. J Bacteriology. 1997;179:5188. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park M, Yun ST, Hwang SY, Chun CI, Ahn TI. J Bacteriology. 2006;188:7572. doi: 10.1128/JB.00576-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ishikawa T, Mizunoe Y, Kawabata S, Takade A, Harada M, Wai SN, Yoshida S. J Bacteriology. 2003;185:1010. doi: 10.1128/JB.185.3.1010-1017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pulliainen AT, Haataja S, Kahkonen S, Finne J. J Biol Chem. 2003;278:7996. doi: 10.1074/jbc.M210174200. [DOI] [PubMed] [Google Scholar]
- 121.Wolfe KH, Sharp PM, Li WH. Nature. 1989;337:283. doi: 10.1038/337283a0. [DOI] [PubMed] [Google Scholar]
- 122.Arnheim N, Calabrese P. Nat Rev Genetics. 2009;10:478. doi: 10.1038/nrg2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chuang JH, Li H. PLoS Biol. 2004;2:0253. doi: 10.1371/journal.pbio.0020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pendergrast JGD, Campbell H, Gilbert N, Dunlop MG, Bickmore WA, Semple CAM. BMC Evol Biol. 2007;7:72. doi: 10.1186/1471-2148-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sasaki S, et al. Science. 2009;323:5912. [Google Scholar]
- 126.Friedman KA, Heller A. J Phys Chem B. 2001;105:11859. [Google Scholar]
- 127.Heller A. Farad Disc. 2000;116:1. [Google Scholar]
- 128.Ramon Goñi J, de la Cruz X, Orozco M. Nuc Acids Res. 2004;32:354. doi: 10.1093/nar/gkh188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wu Q, Gaddis SS, MacLeod MC, Walborg EF, Thames HD, DiGiovanni J, Vasquez KM. Mol Carcinogen. 2007;46:15. doi: 10.1002/mc.20261. [DOI] [PubMed] [Google Scholar]
- 130.Ghosh R, Mitchell DL. Nuc Acids Res. 1999;27:3213. doi: 10.1093/nar/27.15.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramon O, Wong HK, Joyeux M, Riondel J, Halimi S, Ravanat JL, Favier A, Cadet J, Faure P. Free Rad Biol Med. 2001;30:107. doi: 10.1016/s0891-5849(00)00451-2. [DOI] [PubMed] [Google Scholar]
- 132.Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Biochemistry. 2003;42:9761. doi: 10.1021/bi034546k. [DOI] [PubMed] [Google Scholar]
- 133.Ziel KA, Grishko V, Campbell CC, Breit JF, Wilson GL, Gillespie MN. FASEB J. 2005;19:387. doi: 10.1096/fj.04-2805com. [DOI] [PubMed] [Google Scholar]
- 134.Mitchell D, Ghosh R. In: Oxidative Damage to Nucleic Acids. Evans MD, Cooke MS, editors. Landes Bioscience; Georgetown, TX: 2007. pp. 91–99. [Google Scholar]
- 135.Delaney S, Barton JK. Biochemistry. 2003;42:14159. doi: 10.1021/bi0351965. [DOI] [PubMed] [Google Scholar]
- 136.Huang YC, Cheng AKH, Yu HZ, Sen D. Biochemistry. 2009;48:6794. doi: 10.1021/bi9007484. [DOI] [PubMed] [Google Scholar]
- 137.Kanvah S, Schuster GB. Nuc Acids Res. 2005;33:5133. doi: 10.1093/nar/gki801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Joseph J, Schuster GB. J Am Chem Soc. 2006;128:6070. doi: 10.1021/ja060655l. [DOI] [PubMed] [Google Scholar]
- 139.Núñez ME, Noyes KT, Barton JK. Chem Biol. 2002;9:403. doi: 10.1016/s1074-5521(02)00121-7. [DOI] [PubMed] [Google Scholar]
- 140.Bjorklund CC, Davis WB. Nuc Acids Res. 2006;34:1836. doi: 10.1093/nar/gkl030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bjorklund CC, Davis WB. Biochemistry. 2007;46:10745. doi: 10.1021/bi700475b. [DOI] [PubMed] [Google Scholar]
- 142.Merino EJ, Barton JK. Biochemistry. 2008;47:1511. doi: 10.1021/bi701775s. [DOI] [PubMed] [Google Scholar]
- 143.Turro C, Evenzahav A, Bossman SH, Barton JK, Turro NJ. Inorg Chim Acta. 1996;243:101. [Google Scholar]
- 144.Turro C, Hall DB, Chen W, Zuilhof H, Barton JK, Turro NJ. J Phys Chem A. 1998;102:5708. [Google Scholar]
- 145.Merino EJ, Barton JK. Biochemistry. 2007;46:2805. doi: 10.1021/bi062024+. [DOI] [PubMed] [Google Scholar]
- 146.Clayton DA. Annu Rev Cell Biol. 1991;7:453. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 147.Richter C, Park JW, Ames BN. Proc Natl Acad Sci USA. 1988;85:6465. doi: 10.1073/pnas.85.17.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Andreyev Yu A, Kushnareva Yu E, Starkov AA. Biochemistry (Moscow) 2005;70:200. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 149.Tan DJ, Bal RK, Wong LJC. Cancer Res. 2002;62:972. [PubMed] [Google Scholar]
- 150.Lièvre A, Blons H, Houllier AM, Laccourreye O, Brasnu S, Beaune P, Laurent-Puig P. Br J Cancer. 2006;94:692. doi: 10.1038/sj.bjc.6602993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yu M, Shi Y, Zhang F, Zhou Y, Yang Y, Wei X, Zhang L, Niu R. J Biomed Sci. 2008;15:535. doi: 10.1007/s11373-007-9229-4. [DOI] [PubMed] [Google Scholar]
- 152.Merino EJ, Barton JK. Biochemistry. 2009;48:660. doi: 10.1021/bi801570j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ly A, Bullick S, Won JH, Milligan JR. Intl J Rad Biol. 2006;82:421. doi: 10.1080/09553000600771531. [DOI] [PubMed] [Google Scholar]
- 154.Yavin E, Boal AK, Stemp EDA, Boon EM, Livingston AL, O'Shea VL, David SS, Barton JK. Proc Natl Acad Sci USA. 2005;102:3546. doi: 10.1073/pnas.0409410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Green J, Paget M. Nat Rev Microbiol. 2004;2:954. doi: 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- 156.D'Autreaux B, Toldeano MB. Nat Rev Mol Cell Biol. 2007;8:813. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 157.Pomposiello PJ, Demple B. Trends Biotechnol. 2001;19:109. doi: 10.1016/s0167-7799(00)01542-0. [DOI] [PubMed] [Google Scholar]
- 158.Dietrich LEP, Teal TK, Price-Whelan A, Newman DK. Science. 2008;321:1203. doi: 10.1126/science.1160619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Price-Whelan A, Dietrich LEP, Newman DK. J Bacteriol. 2007;189:6372. doi: 10.1128/JB.00505-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kerr JR. Infect Dis Rev. 2000;2:184. [Google Scholar]
- 161.Pomposiello PJ, Bennik MH, Demple B. J Bacteriol. 2001;183:3890. doi: 10.1128/JB.183.13.3890-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hobman JL, Wilkie J, Brown NL. BioMetals. 2005;18:429. doi: 10.1007/s10534-005-3717-7. [DOI] [PubMed] [Google Scholar]
- 163.Ding H, Hidalgo E, Demple B. J Biol Chem. 1996;271:33173. doi: 10.1074/jbc.271.52.33173. [DOI] [PubMed] [Google Scholar]
- 164.Watanabe S, Kita A, Kobayashi K, Miki K. Proc Natl Acad Sci USA. 2008;105:4121. doi: 10.1073/pnas.0709188105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ding H, Demple B. Proc Natl Acad Sci USA. 1997;94:8445. doi: 10.1073/pnas.94.16.8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ding H, Demple B. Proc Natl Acad Sci USA. 2000;97:5146. doi: 10.1073/pnas.97.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gort AS, Imlay JA. J Bacteriol. 1998;180:1402. doi: 10.1128/jb.180.6.1402-1410.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dietrich LEP, Price-Whelan A, Petersen A, Whiteley M, Newman DK. Mol Microbiol. 2006;61:1308. doi: 10.1111/j.1365-2958.2006.05306.x. [DOI] [PubMed] [Google Scholar]
- 169.Kobayashi K, Tagawa S. J Biochem. 2004;136:607. doi: 10.1093/jb/mvh168. [DOI] [PubMed] [Google Scholar]
- 170.Zheng M, Storz G. Biochem Pharmacol. 2000;59:1. doi: 10.1016/s0006-2952(99)00289-0. [DOI] [PubMed] [Google Scholar]
- 171.Hidalgo E, Demple B. EMBO J. 1994;13:138. doi: 10.1002/j.1460-2075.1994.tb06243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.David SS, Williams SD. Chem Rev. 1998;98:1221. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 173.David SS, O'Shea VL, Kundu S. Nature. 2007;447:941. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Demple B, Harrison L. Annu Rev Biochem. 1994;63:915. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 175.Cupples CG, Miller JH. Proc Natl Acad Sci USA. 1989;86:5345. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Nature. 2004;427:652. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 177.Fromme JC, Verdine GL. EMBO J. 2003;22:3461. doi: 10.1093/emboj/cdg311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Livingston AL, O'Shea VL, Kim T, Kool ET, David SS. Nat Chem Biol. 2008;4:51. doi: 10.1038/nchembio.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Biochemistry. 1989;28:4450. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- 180.Thayer MM, Ahern H, Xing D, Cunningham RP, Tainer JA. EMBO J. 1995;14:4108. doi: 10.1002/j.1460-2075.1995.tb00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd S, Tainer JA. Nat Struct Biol. 1998;5:1058. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 182.Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DA, Barton JK. Proc Natl Acad Sci USA. 2009;106:15237. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fok PW, Guo CL, Chou T. J Chem Phys. 2008;129:235101. doi: 10.1063/1.3026735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fok PW, Chou T. Biophys J. 2009;96:2949. doi: 10.1016/j.bpj.2009.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Meplan C, Richard MJ, Hainaut P. Biochem Biopharmacol. 2000;59:25. doi: 10.1016/s0006-2952(99)00297-x. [DOI] [PubMed] [Google Scholar]
- 186.Whibley C, Pharoah PD, Hollstein M. Nat Rev Cancer. 2009;9:95. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- 187.Vousden KH, Prives C. Cell. 2009;137:413. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 188.Joerger AC, Fersht AR. Annu Rev Biochem. 2008;77:557. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 189.Augustyn KE, Merino EJ, Barton JK. Proc Natl Acad Sci USA. 2007;104:18907. doi: 10.1073/pnas.0709326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Takada T, Barton JK. J Am Chem Soc. 2005;127:12204. doi: 10.1021/ja054306n. [DOI] [PubMed] [Google Scholar]
- 191.Kiley PJ, Beinert H. FEMS Microbiol Rev. 1998;22:341. doi: 10.1111/j.1574-6976.1998.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 192.Nesbit AD, Giel JL, Rose JC, Kiley PJ. J Mol Biol. 2009;387:28. doi: 10.1016/j.jmb.2009.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Schwartz CJ, Giel JL, Patschkowski T, Luther C, Ruzicka FJ, Beinert H, Kiley PJ. Proc Natl Acad Sci USA. 2001;98:14895. doi: 10.1073/pnas.251550898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Buis JM, Cheek J, Kalliri E, Broderick JB. J Biol Chem. 2006;281:25994. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- 195.Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-De-Choudens S, Fontecaye M, Atta M. J Biol Chem. 2008;283:36361. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. Mol Cell. 2006;23:801. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 197.Wolski SC, Kuper J, Hanzelmann P, Truglio JJ, Croteau DL, Van Houten B, Kisker C. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. Cell. 2008;133:789. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu H, Rudolf J, Johnson KA, McMahon SA, Oke M, Carter L, McRobbie AM, Brown SE, Naismith JH, White MF. Cell. 2008;133:801. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ren B, Duan XW, Ding HG. J Biol Chem. 2009;284:4829. doi: 10.1074/jbc.M807943200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Klinge S, Hirst J, Maman JD, Krude T, Pellegrini L. Nat Struct Mol Biol. 2007;14:875. doi: 10.1038/nsmb1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Weiner BE, Huang H, Dattilo BM, Bilges MJ, Fanning E, Chazin WJ. J Biol Chem. 2007;282:33444. doi: 10.1074/jbc.M705826200. [DOI] [PubMed] [Google Scholar]
- 203.Yeeles JTP, Cammack R, Dillingham MS. J Biol Chem. 2009;284:7746. doi: 10.1074/jbc.M808526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hirata A, Klein BJ, Murakami KS. Nature. 2008;451:851. doi: 10.1038/nature06530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Paraskevopoulou C, Fairhurst SA, Brick P, Onesti S. Mol Microbiol. 2006;59:795. doi: 10.1111/j.1365-2958.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 206.Greenwood C, Selth LA, Dirac-Svejstrup AB, Svejstrup JQ. J Biol Chem. 2009;284:141. doi: 10.1074/jbc.M805312200. [DOI] [PubMed] [Google Scholar]
- 207.Chen H, Hu J, Chen PR, Lan LF, Li ZL, Hicks LM, Dinner AR, He C. Proc Natl Acad Sci USA. 2008;36:13586. doi: 10.1073/pnas.0803391105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Panmanee W, Vatanaviboon P, Poole LB, Mongkolsuk S. J Bacteriol. 2006;188:1389. doi: 10.1128/JB.188.4.1389-1395.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Eiamphugporn W, Soonsanga S, Lee JW, Helmann JD. Nuc Acids Res. 2009;37:1174. doi: 10.1093/nar/gkn1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Masuda S, Dong C, Swem D, Setterdahl AT, Knaff DB, Bauer CE. Proc Natl Acad Sci USA. 2002;99:7078. doi: 10.1073/pnas.102013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hiroto K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. Proc Natl Acad Sci USA. 1997;94:3633. doi: 10.1073/pnas.94.8.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S, Laszlo A, Spitz DR, Goswami PC, Yodoi J, Gius D. Cancer Res. 2000;60:6688. [PubMed] [Google Scholar]
- 213.Outten FW, Theil EC. Antioxid Redox Signal. 2009;11:1029. doi: 10.1089/ars.2008.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Bowles T, Metz AH, O'Quin J, Wawrzak Z, Eichman BF. Proc Natl Acad Sci USA. 2008;105:15299. doi: 10.1073/pnas.0806521105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.DeRosa MC, Sancar A, Barton JK. Proc Natl Acad Sci USA. 2005;102:10788. doi: 10.1073/pnas.0503527102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. J Bacteriol. 2006;188:7062. doi: 10.1128/JB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hon T, Dodd A, Dirmeier R, Gorman N, Sinclair PR, Zhang L, Poyton RO. J Biol Chem. 2003;278:50771. doi: 10.1074/jbc.M303677200. [DOI] [PubMed] [Google Scholar]
- 218.Hickman MJ, Winston F. Mol Cell Biol. 2007;27:7414. doi: 10.1128/MCB.00887-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lambert AR, Sussman D, Shen B, Maunus R, Nix J, Samuelson J, Xu SY, Stoddard BL. Structure. 2008;16:558. doi: 10.1016/j.str.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Takeda K, Noguchi T, Naguro I, Ichijo H. Annu Rev Pharmacol Toxicol. 2008;48:199. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- 221.Ando K, Hirao S, Kabe Y, Ogura O, Sato I, Yamaguchi Y, Wada T, Handa H. Nuc Acids Res. 2008;36:4327. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.