


Abstract
The binding of the transcriptional regulator XylR to its cognate upstream activating sequences (UAS) of the σ54-dependent promoter Pu of Pseudomonas putida has been examined in vivo in single copy gene dose and stoichiometry. To this end, we have employed a novel in vivo genomic footprinting procedure that uses short exposures of bacterial cells to diffuse high intensity UV light that causes formation of TT or TC dimers. In contrast to simpler models for activation of σ54-dependent promoters, our results clearly indicate that the XylR protein is not permanently bound in vivo to its target sites in Pu. On the contrary, the UAS appear to be mostly unoccupied at all growth stages. This is in contrast to the integration host factor (IHF), which binds Pu strongly in vivo at stationary phase, as also revealed by UV footprinting. Only overexpression of XylR altered the photoreactivity of the corresponding DNA region to report stable binding of the regulator to the UAS. However, the presence of aromatic XylR inducers reversed the forced occupation caused by increased levels of the activator. These results are compatible with the notion that XylR interacts very transiently with the UAS and detaches from the promoter during transcriptional activation of Pu.
INTRODUCTION
The σ54-dependent Pu promoter regulates transcription of the upper operon of Pseudomonas putida mt-2 TOL plasmid pWW0, which encodes the first enzymes for toluene and m/p-xylene degradation (1). The RNA polymerase holoenzyme containing σ54 (2) can form a closed promoter complex, but is not able to proceed to open complex formation, and thus transcription, in the absence of an activator protein (3–5). XylR is a toluene-responsive activator protein that binds to cognate upstream activating sequences (UAS) of Pu (6,7). DNA looping of the region between the UAS and the –12/–24 motifs for the σ54-RNAP seems to facilitate the interaction between the activator protein and the holoenzyme (8–10). In Pu, this event is assisted by the integration host factor (IHF), which binds to a specific site in the intervening region (Fig. 1), bringing about a strong DNA bend (11–14). In addition, IHF helps the recruitment of the σ54-RNAP to Pu when cells reach stationary phase (15,16).
Figure 1.

Schematic representation of the Pu-lacZ fusion used in this study. The –12/–24 motif for σ54-RNA polymerase recognition, the IHF binding site and the UAS, where the XylR activator binds, are indicated. UAS-D and UAS-P stand, respectively, for the distal and the proximal UAS. The detailed sequence for relevant sites is expanded. Shaded letters indicate adjacent pyrimidine bases that may form dimers upon UV light irradiation. The relative intensity of these bands is a descriptor of specific DNA–protein interactions.
XylR belongs to the NtrC family of prokaryotic enhancer binding proteins (EBPs), named after the most studied member of the group (17). These regulators exhibit a common mechanism of activation of their cognate promoters. Once exposed to a specific environmental condition (i.e. contact with toluene in the case of XylR), the signal-receiving N-terminal domain of the protein initiates a number of conformational changes leading ultimately to formation of a multimer competent to hydrolyze ATP and drive σ54- dependent transcription initiation (18). With little variation, this seems to be true for virtually all prokaryotic enhancer-binding proteins where the issue has been examined (19–24). Under this view, each of the factors that participate in the activation of a given σ54 promoter (EBP, IHF, σ54-RNAP) has an intrinsic ability of interacting with distinct sequences at the corresponding DNA, which in the case of the EBPs may or may not be increased under activation conditions (23,25,26). In either case, the major regulatory step is predicted to be the switching on of the activator already bound to DNA. This simple model is based exclusively on in vitro experiments (21–26), where the concentration of every protein can be varied at will. An additional complexity of σ54 promoters is the assembly/disassembly of the EBP multimer during transcription. At least in the case of XylR, it seems that the multimeric complex dissociates at every round of transcription. This notion is supported by DNase I footprinting and XylR cross-linking experiments, as well as by the behavior of XylR variants locked in either a multimeric or a non-multimeric state (18). Furthermore, potential intermediates of the cyclic process have been observed in vitro by transmission electron microscopy, suggesting that the multimer becomes disassembled every time ATP is hydrolysed (27). Again, all this is based on in vitro evidence with a purified and constitutively active form of XylR. But does all this happen in vivo?
In this work, we employ a non-disruptive technique (high intensity UV footprinting) for monitoring in vivo the occupation of the UAS of Pu by XylR under various physiological and induction conditions, but without affecting the single copy gene dosage of the regulatory elements and its natural stoichiometry in P.putida. The data below indicate that, unlike IHF (which binds Pu stably in stationary phase), the interaction of XylR with its cognate UAS in Pu is surprisingly transient. Moreover, inducer addition (i.e. XylR activation) further decreases such already weak protein–DNA contacts. These results reinforce the view that the transcriptionally competent complex assembles and disassembles at every initiation round.
MATERIALS AND METHODS
Strains and plasmids
Pseudomonas putida MAD1 is a derivative of the reference P.putida strain KT2442 which bears a hybrid mini-Tn5 transposon encoding the sequence of XylR and a transcriptional Pu-lacZ fusion as well (28). This design ensures that all regulatory elements controlling expression from Pu are placed in one copy per chromosome. Pseudomonas putida MAD2 is identical to P.putida MAD1, except that the mini-Tn5 transposon bears a XylR variant (XylRΔA) which is fully constitutive and requires no inducer for promoter activation. Pseudomonas putida SF05 is a strain inserted with the same Pu-lacZ fusion as P.putida MAD2, but without the xylR gene (29). The plasmid used as the template for UV footprinting in vitro (pEZ9) has been described before (12). pEZ9 bears an insert spanning coordinates –208 to +93 of the Pu sequence. pTK19 plasmid is a derivative of the broad host range plasmid pKT230 harboring a 2.4 kb HpaI fragment from the pWW0 plasmid containing the xylR gene expressed under its own promoter (30).
Proteins and enzyme assays
Purified IHF protein of P.putida was the kind gift of F. Bartels (GBF, Braunschweig). Purification of the XylRΔA protein fused to a poly-His metallo-affinity tag has been described before (31). As in vivo with the xylRΔA allele, the XylRΔA protein is a constitutive variant which can fully activate transcription from Pu in the absence of any aromatic inducer (31). β-Galactosidase assays were carried out on cells grown in LB medium until the desired optical density (OD600), permeabilized with chloroform and sodium dodecyl sulphate as described by Miller (56). Induced cultures were exposed to saturating vapors of the XylR effector m-xylene.
In vitro high intensity UV footprinting
Fifty nanograms of supercoiled pEZ9 plasmid were incubated in a volume of 40 µl for 15 min at 30°C in binding buffer (20 mM Tris–HCl pH 8.0, 2 mM MgCl2, 40 mM KCl, 0.1 mM EDTA, 100 µg/ml bovine serum albumin) in the absence or presence of 400 nM XylRΔA or 100 nM IHF. Following incubation, 20 µl of each mixture were transferred to 0.5 ml Eppendorf tubes, and irradiated for 15–20 s with the UV beam from a 500 W Hg/Xe arc lamp (Oriel Instruments, CT). This instrument produces emissions throughout the UV spectrum, including major peaks at 235, 246, 265, 280, 289, 296, 302 and 315 nm, as well as several emission lines in the infrared (IR) range (820, 880 and 1000 nm). The resulting radiation is thus passed through a water filter (Oriel Instruments, ref. 61955) to eliminate IR. The beam is then collimated in a condenser and carried through a fused silica fiber optics bundle (Oriel Instruments, ref. 77575), the outlet of which is placed at ∼15 mm from the treated samples. In this way, the whole UV output of the lamp is concentrated on the DNA–protein sample. After irradiation, the samples were either processed immediately or stored at –20°C. The primer used for extension of the DNA samples was Pu14 (5′-CCCGCTTTGAGGATATACATGGCGAAAGC-3). Pu14 is complementary to positions +63/+91 on the coding strand of the Pu promoter. In the presence of Taq polymerase and dNTPs, Pu14 primes an extension reaction in vitro which covers entirely the UAS and the IHF site of Pu. This primer was end-labeled with [γ32-P]ATP and T4 polynucleotide kinase (32). For detection of pyrimidine dimers in the DNA, 10 µl of the samples with the irradiated plasmid were added to 6 µl of an amplification mix containing 10 pmol of each of the dNTPs, 1 pmol of the end-labeled primer and 2 U of Taq polymerase in 3× GeneAmp PCR buffer with MgCl2 (Perkin Elmer). Samples were then denatured for 3 min at 94°C and subjected to 10 cycles of extension (1 min 94°C, 1 min 61°C, 1 min 72°C) plus a final incubation for 10 min at 72°C. Ten microliters of formamide loading buffer were then added to each sample. Then 2 µl samples were electrophoresed in 6% denaturing polyacrylamide sequencing gels (32). The resulting pattern of radioactive bands was registered in a Molecular Imager System β-sensitive screen and visualized with Quantity One software (Bio-Rad). The different lanes were scanned with the same system and the line graphs were transferred to Microsoft Excel for quantitative analysis. Intensity normalization was performed with respect to the band appearing at position –104, which lies outside both the UASs and the IHF binding site and is therefore unaffected by protein binding.
In vivo UV footprinting
Cultures of P.putida MAD1 or P.putida MAD2, bearing plasmids or not as indicated, were grown at 30°C in LB liquid medium in the presence or absence of m-xylene (see above) until the desired optical densities at 600 (OD600) were reached. Ten 50-µl aliquots of each of the cultures were then dispensed into white Costar™ 96-well plates (Corning Incorporated, Corning, NY) and each well was irradiated for 25–30 s under the same conditions as for the in vitro assay above. Replicated samples were immediately collected after treatment, mixed to make 500 µl volumes and chilled at once. The genomic DNA was extracted using the miniprep protocol (32). Ten micrograms of such DNA were used for the primer extension reactions with 5′ labeled oligo Pu14 described above. These were run in 100 µl volumes, which contained 25 pmol of each dNTP, 5 pmol of labeled primers and 5 U of Taq polymerase in a PCR buffer with 2.5 mM MgCl2 as above. Unlike the in vitro extensions, the DNA samples from intact cells were submitted to 45 extension cycles (1 min 94°C, 1 min 59°C, 1 min 72°C) and a final extension for 10 min at 72°C. DNA was then precipitated by addition of 10 µl of 3 M sodium acetate, pH 7.5 and 220 µl of ethanol. DNA pellets were rinsed with 70% ethanol, dried and resuspended in 5 µl of TE. After addition of 10 µl of formamide loading buffer (32), 6–7 µl of each reaction were run on a denaturing polyacrylamide gel and analysed as described above. Experiments were repeated at least three times and similar relative band intensities were seen.
RESULTS AND DISCUSSION
Rationale of UV footprinting for detecting occupation of Pu by regulatory proteins
The technique employed in this work for addressing XylR binding to Pu under various conditions in vivo is based on formation of TT and TC dimers in DNA irradiated with UV light in the range of 230–300 nm (33–36). Since the efficiency of the photochemical reaction is related to the local DNA topology, DNA-binding proteins influence the efficiency of formation of such dimers (37). These changes are revealed by primer extension, since they interfere with DNA polymerization (34). The pattern of such an extension can thus be instrumental for describing the occupation of a given site by a cognate protein. Different variants of this concept have been employed for studying DNA–protein interaction in vitro and in vivo. For instance, UV-laser footprinting captures the pattern of DNA–protein interactions by subjecting samples to short pulses (∼6 ns) of high-energy, coherent 266 nm emissions (38). On the other hand, some features of DNA–protein contacts have been addressed in a few cases by exposing binding mixtures to much longer times (>10 min) of diffuse UV emissions, such as those produced by a commercial transilluminator (39). In our case, we strived to combine the imaging of interactions with a high off rate (as was predicted for XylR-Pu, see below) with a relatively short-irradiation period. To this end, we adapted a method in which small sample volumes were irradiated with a non-coherent but high intensity UV light for a few seconds (33), for visualizing in vivo XylR binding to the UAS in the chromosome. The technique is operatively referred to as footprinting, although, in reality, what is reflected after UV irradiation is not protection to DNA damage due to bound protein, but the conformation of a DNA segment and its immediate environment.
Prior to examining occupation of Pu by XylR, we validated the performance of the UV footprinting procedure. Using UV laser footprinting, we had previously described that binding of IHF to its target site causes a deformation of the DNA helix that is readily converted into increased formation of TT dimers at positions –57 and –81/–77 as well as into hyporeactivity of the –65 CT dimer (16). We compared the pattern of TT/TC bands raised by the binding of IHF in vitro to its target site in Pu following either a 6 ns UV laser pulse (16) or a 15 s exposure to non-coherent UV beam. The band patterns obtained in the IHF binding site were transformed into intensity plots. The normalized scans of each lane for this region are plotted in Figure 2. Perusal of the results indicates that the intrinsic descriptors of the occupation of the Pu promoter by IHF were reproduced at a comparable resolution. We thus assumed that the information revealed by either technique was virtually the same within the scope of this work.
Figure 2.
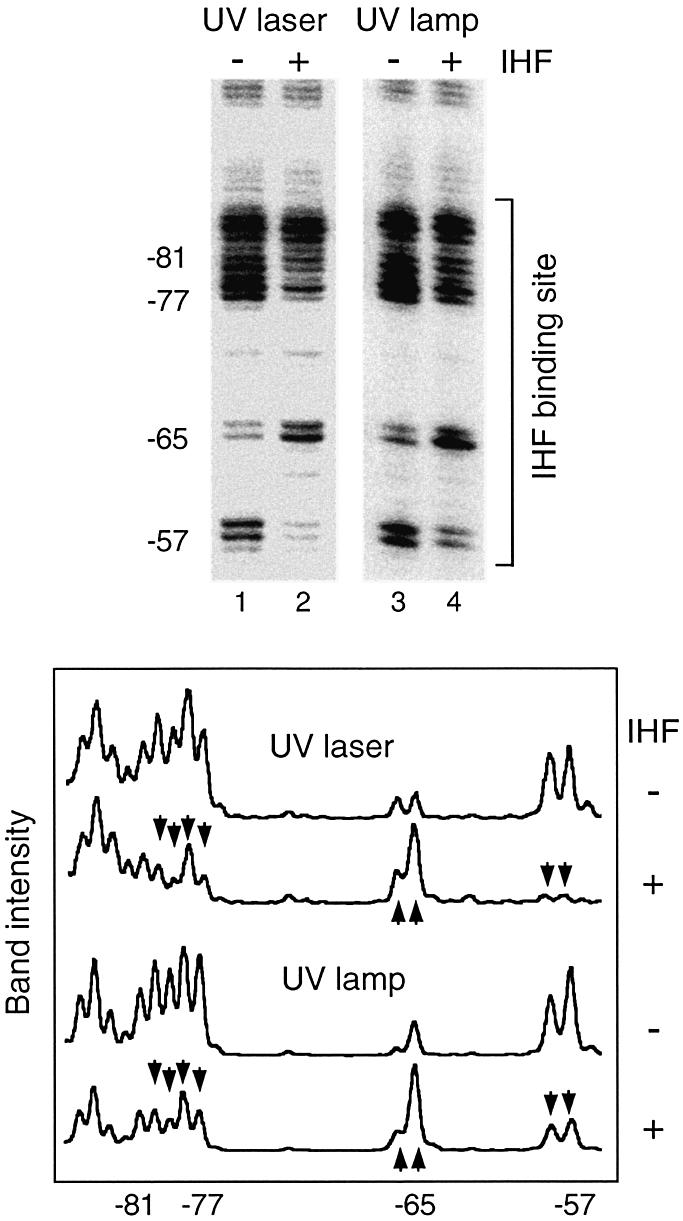
Comparison of UV laser versus non-coherent high intensity UV for DNA footprinting. The gel shows the band patterns obtained after primer extension of the Pu promoter irradiated in vitro with either a 6 ns UV laser pulse or 15 s of non-coherent UV beam in the presence or absence of the purified IHF protein (see Materials and Methods). The observed photoreactions correspond to formation of dimers between adjacent pyrimidines in the sequence shown in Figure 1. Protein binding alters the physicochemical environment and distorts DNA, affecting such reactivity. This can be visualized after primer extension by changes in band intensities. The gel was scanned and each lane was transformed into a normalized intensity plot shown at the bottom of the figure. It is apparent that formation of pyrimidine dimers occurs in both conditions to a similar extent, and is equally affected by protein binding, resulting in comparable footprints. Compare (lanes 2 and 4) the decreased photoreactivity of positions –77/–81 and –57 and the increase in –65 band intensity upon IHF binding detected by both procedures.
Setting the descriptors of XylR binding to Pu
In order to assign the changes in photoreactivity (i.e. band intensity) of Pu due to XylR binding, the effect of UV was first examined in vitro. Supercoiled pEZ9 plasmid was UV-irradiated in the presence or absence of purified XylRΔA, a constitutively active regulator variant (31). The concentrations of XylRΔA employed in this experiment (400 nM) were those known to produce in vitro a detectable DNase I footprint, to form complexes that were visible with electron microscopy and to trigger transcription in an in vitro assay (27,31). Following UV exposure, samples were then extended with Taq polymerase and products separated in a DNA sequencing gel. The resulting band patterns reporting the physical interaction of XylR with its binding sites are shown in Figure 3. As expected, the changes caused by XylRΔA correspond to termination of primer extension in the vicinity of putative pyrimidine dimers on the template strand (positions –129, –134 in the proximal UAS and –162, –171 in the distal UAS). It is noteworthy that the reactive bases are not identical in each of the UAS, believed to bind XylR dimers separately. This suggests that even unbound DNA has significant local differences in the base–base wedges and their stacking along the DNA helix.
Figure 3.
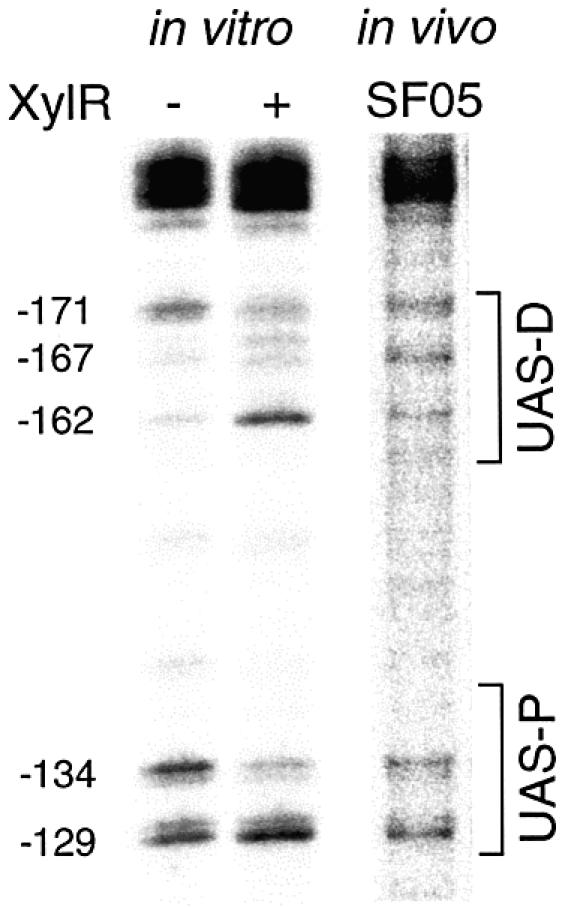
In vitro effect of XylR binding to Pu and in vivo reactivity of the naked promoter. The UV footprinting procedure described in this work was used to monitor the effect of XylR binding on DNA photoreactivity. A supercoiled plasmid bearing Pu was UV-irradiated in the presence or absence of the purified XylRΔA protein (a constitutively active variant of the XylR activator). Alternatively, the Pu-lacZ but xylR-minus strain P.putida SF05 was irradiated with UV and its DNA extracted as described in Materials and Methods. In either case, the DNA was extended with a specific primer and the resulting products run in a denaturing polyacrylamide gel. A blow up of the gel zone corresponding to the Pu upstream region is shown. Informative positions are indicated on the left. Note the distinct band pattern produced by XylRΔA binding (lane 2), with hyper-reactive pyrimidine dimers at –162 and –129 and a hypo-reactivity in –171 and –134. All these positions fall within the well-defined UAS where the XylR regulator binds to bring about transcription activation.
Informative changes in the pattern of pyrimidine dimers did occur upon XylRΔA addition. Inspection of the resulting extension products revealed that the addition of XylRΔA caused a hyper-reactivity of the TC dimer at –162 concurrent with hypo-reactivity of the TT pair at –171 at the distal UAS sequence. Similarly, bands at positions –129 and –134 of the proximal UAS region were, respectively, enhanced or weakened upon XylRΔA binding. To obtain a more reliable reference for XylRΔA binding, the bands were scanned, the intensity of the signals normalized with respect to a band that does not appear to vary under any of the conditions tested (–104), and finally plotted as shown in Figure 4. Although changes in the pattern of photoreactivity induced by XylRΔA in Pu cannot be interpreted in structural terms, such variations should reflect changes in the wedge angle between bases following XylRΔA binding. Inspection of the scan shown in Figure 4 reveals that bands corresponding to positions –171, –162, –134 and –129, are intrinsic descriptors of the occupation of the Pu promoter by XylRΔA, since their relative intensity increases or decreases depending on XylRΔA binding. That XylRΔA caused a sharp reduction in bands –171 and –134, along with a clear augmentation of bands –129 and –162 gave us a useful reference to judge the occupation of the Pu promoter in vivo as explained below.
Figure 4.

Normalized representation of in vivo versus in vitro footprints corresponding to the UAS region. The intensity of each signal is represented in arbitrary units. Bands informative of protein binding are identified at the bottom of the scans with respect to the transcription start site. The photoreactivity profiles obtained in vitro for the naked DNA (–XylR), and after addition of XylRΔA are shown at the top. An in vivo control sample to visualize the reactivity pattern of naked DNA is indicated as Sample 1, and corresponds to P.putida SF05 (Pu-lacZ but lacking xylR) grown to stationary phase. Some discrepancies in the band pattern observed in the non-occupied UAS in vivo versus in vitro are apparent at the distal UAS (UAS-D), in positions –167 and –162, as discussed in the text. The three scans at the top correspond to the lanes in the gel of Figure 3. The in vivo samples 2–5 correspond to those of the experiment in Figure 5, as follows: Sample 2: P.putida MAD1 (pTK19) irradiated during exponential growth (exp.), without inducer. Sample 3: P.putida MAD1 (pKT19) irradiated at stationary phase, non-induced. Sample 4: Plasmidless P.putida MAD1 cells (single copy Pu-lacZ and xylR), irradiated at stationary phase. Sample 5: P.putida MAD1 (pKT19) irradiated at stationary phase after induction with m-xylene (ind.). Observe the distinct alteration of band intensity (arrows) upon XylRΔA addition in vitro, and the comparable profile obtained in vivo only in stationary cells with increased levels of XylR. Note the lack of XylR binding signals in P.putida MAD1 (pTK19) cells induced with m-xylene.
Pu reactivity to UV in vivo
Once a reference for XylRΔA binding to Pu was produced in vitro, we set out to determine whether and under what conditions the regulator was bound to the promoter. Since the target DNA represents a very small fraction of the whole chromosome we adapted the protocol developed for single-copy UV-laser footprint (16) to the new irradiation procedure. With the method described in Materials and Methods, we were able to produce a single-base resolution and signal-to-noise ratio compatible with the results in vitro.
To address UAS occupation by XylR in vivo, we first subjected cells of P.putida SF05 grown in LB to UV irradiation. This strain bears a single-copy insertion of Pu but lacks the xylR gene. Therefore, the pattern of pyrimidine dimer formation should reflect the XylR-free state of the DNA region. The result of such a condition is shown in lane 3 of the gel in Figure 3. Comparison of the band pattern in vivo versus that in vitro allowed an easy match between the major bands found in each case. A positive control is provided by the IHF site, which is occupied by this factor following a well-defined and virtually identical banding arrangement in vivo and in vitro (see above). As for the UAS, the degree of similarity varies between the proximal and distal sequences. The distinct bands at –134 and –129 were well equivalent in vitro and in vivo both in intensity and relative ratio. But the status with the distal UAS was different. Although the band at –171 which is predominant in vitro appears also in vivo, live cells produce an extra band at –167 which is poorly visible in vitro (Figs 3 and 4). Furthermore, the extension product at –162, which is produced only weakly in vitro in the absence of XylR is clearly apparent in vivo.
While the similarity in vivo and in vitro between the footprints of the proximal UAS with UV is expected, the discrepancies in the distal UAS are not. These differences, which reflect genuine variations in DNA topology in vivo and in vitro, are the only significant ones throughout the whole promoter region (see below). They are thus unlikely to be caused by variations in local supercoiling or any other reason inherent to DNA alone. Instead, the in vivo versus in vitro disparity of the UV footprints of distal UAS calls for the presence in vivo of an additional factor different from XylR. This factor would bind the sequence and modify the local conformation of DNA, producing the otherwise unlikely CT dimer at –167 and somewhat enhancing the formation of the one at –162. Although we do not know yet the identity of such a putative factor(s), gel retardation assays of radiolabeled UAS with protein extracts of P.putida KT2442 indicate the existence of one or more proteins which bind the region in the absence of XylR (Bertoni,G. et al., in preparation). Whether or not the two observations are connected (unusual UV footprints in distal UAS in the absence of XylR, factors in P.putida other than XylR binding the region) deserves further studies. In any case, the data above reveal the formation in vivo of the pyrimidine dimers, which are informative of XylR binding to the UAS. With the cautions triggered by the appearance of an extra band at –167, the evolution of such bands under various growth conditions was then followed and interpreted as explained below.
The UAS of Pu stay predominantly clear of bound XylR at any growth stage
We next set out to visualize XylR binding in vivo to the Pu UAS under conditions ensuring the binding of the factor to target DNA. This is not trivial, since the number of XylR molecules per cell is very low [in the range of 5–30 hexamers/cell (40)] and the resulting intracellular levels are in the 0.5 nM range at its maximum during stationary phase. This figure is several orders of magnitude below the XylRΔA concentrations that produce clear footprints in vitro. Furthermore, given the proposed cyclic fashion of Pu activation by XylR (18), we expected XylR–UAS interactions to be more stable under non-induced conditions. The reference in vivo conditions to examine occupation of UAS by XylR were thus cells of P.putida MAD1 (xylR+, Pu-lacZ) transformed with a multicopy xylR+ plasmid (pTK19) in the absence of aromatic inducers. Although, due to its pedigree, pTK19 also bears XylR binding sites (12), this plasmid increases the intracellular concentration of XylR at least 10 times (41) and should thus result in a more favorable occupation of the target sequences. To examine this issue, we grew the P.putida MAD1 (pTK19) in LB medium in conditions similar to those used for P.putida SF05 above. Samples were collected at the exponential and the stationary phases of growth and irradiated with UV. After the standard processing, reactive pyrimidines were detected as described above. Figure 5 shows the results of this experiment that can be better visualized in the plots of Figure 4. The positive control for the performance of the procedure, and for checking the physiological status of the cell was provided by the IHF site in Pu. As expected, the very specific band pattern indicative of IHF binding did appear in DNA from samples taken at stationary phase (Fig. 5, lanes 2–4), but not in those from exponential growth (Fig. 5, lane 1). As for the UAS region, we detected changes descriptive of XylR binding at both growth stages, in particular the relative ratios of bands –171 versus –162 and –134 versus –129 (Figs 4 and 5). These changes were more visible in samples from stationary cells, perhaps reflecting a further excess in intracellular XylR concentration brought about by the multicopy state of xylR under its own promoter (40). Interestingly, the extra band at –167 in distal UAS was present under all conditions tested and reinforced in the sample from the strain overproducing XylR. This suggested not only that the corresponding DNA distortion involved continues along various growth stages, but also that it is somehow exacerbated by XylR binding.
Figure 5.
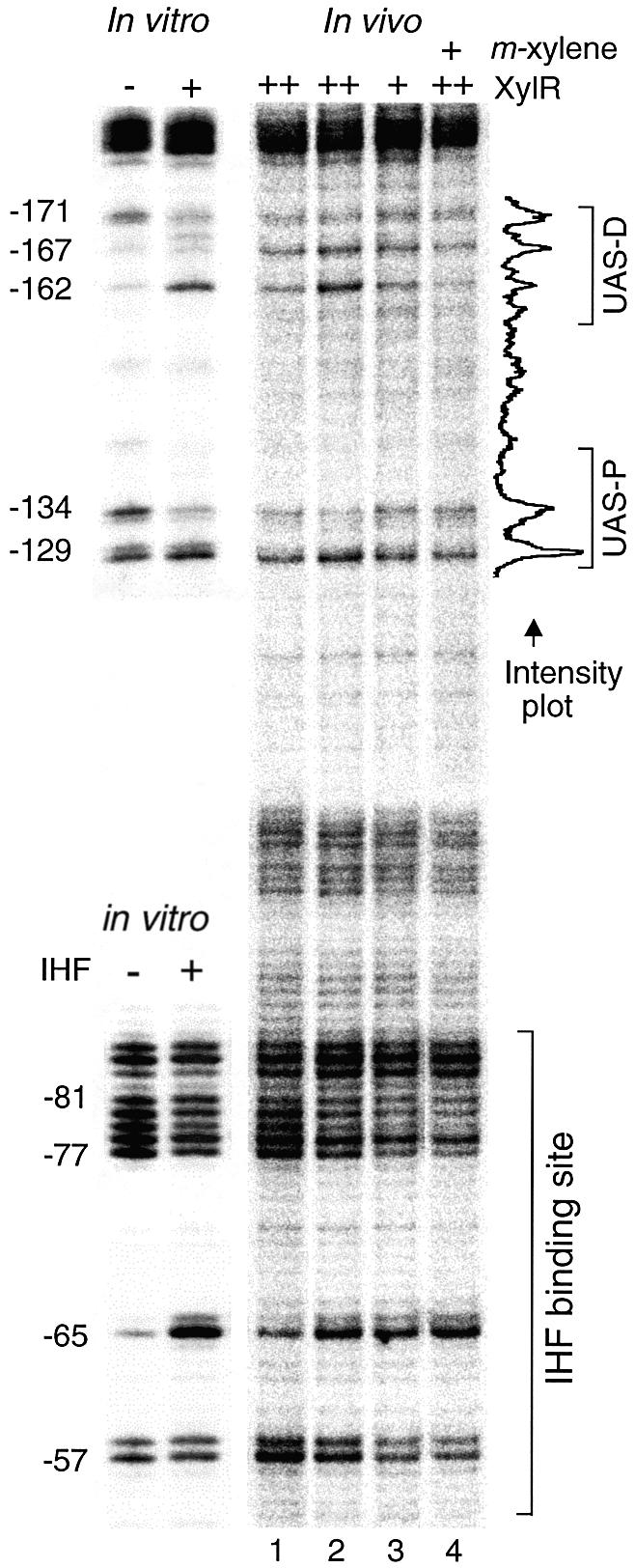
In vivo UV footprinting of the Pu promoter. Different P.putida strains were grown in LB medium as indicated below and exposed to non-coherent UV irradiation. Lane 1: Exponentially growing P.putida MAD1 strain (single copy Pu-lacZ and xylR) transformed with xylR+ plasmid pTK19 for increased (++) intracellular levels of XylR protein. Lane 2: P.putida MAD1 (pTK19) cells irradiated at stationary phase. Lane 3: stationary P.putida MAD1 cells devoid of plasmids and thus containing native intracellular levels (+) of XylR. Lane 4: stationary P.putida MAD1 (pTK19) cells induced with m-xylene. For reference, in vitro footprintings of the UAS and IHF sequences in the absence (–) or presence (+) of the corresponding proteins are shown to the left. Bands informative of protein binding are identified to the left of the gels in respect of the transcription start site. An example of band intensity profile resulting from the scan of lane 4 through the UAS is shown to the right (see full analysis in Fig. 4).
To examine whether XylR binding to Pu could also be detected in P.putida cells bearing both Pu and xylR in single copy (i.e. in its native stoichiometry), we repeated the procedure with P.putida MAD1 strain devoid of any extra plasmid. As shown in Figure 4, the difference between the P.putida strains SF05 (xylR–) and MAD1 (xylR+) through the UAS region was minimal if existing at all, indicating that the corresponding DNA was basically clear of bound XylR. This was in contrast to the distinct attachment of IHF to its downstream target site (Fig. 4). These results are somewhat unexpected, as they show that, under non-induced, native conditions the binding of XylR to Pu is very poor and altogether insufficient to occupy the UAS permanently. Whether this is due to the low intracellular concentration of the activator, to a poor affinity for the UAS or to competition by already bound factors, only overexpression of XylR gave evidence of binding to Pu in vivo.
Transient binding of XylR to Pu during promoter activation
That XylR must necessarily bind the UAS in vivo for activating transcription was demonstrated because a Pu variant deleted of such a region is completely inactive (7,29). On this basis, we inspected the UAS region under conditions where Pu is active. This was achieved by either exposing xylR+ cells to aromatic inducers such as m-xylene or by replacing the wild-type xylR gene by its xylRΔA counterpart, which encodes the constitutively active XylRΔA protein. Since the bands descriptive of XylR binding in vitro and in vivo had been identified (see above), we ran a separate experiment to compare the pyrimidine dimer formation patterns of the UAS in cells actively transcribing Pu. Such transcriptional activity was followed by virtue of the single copy Pu-lacZ fusion built in P.putida MAD1 (xylR+) and MAD2 (xylRΔA+) strains. Lanes 1, 2 and 3 of the gel shown in Figure 6 compare equivalent regions of P.putida MAD1 induced or not with m-xylene along with the same picture for P.putida MAD2. Despite the fact that the Pu promoter is being actively transcribed in cells corresponding to samples 2 and 3 (Fig. 6), we could not find any significant difference in the UV footprint of their UAS in respect to those of the inactive Pu of sample 1 (at least within the resolution of this technique). This observation rules out the view that inducer addition brings about a real increase in the affinity of XylR for DNA and reinforces the notion that, under native stoichiometry XylR-DNA contacts are as transient as to be elusive to detection with the UV footprinting procedure.
Figure 6.
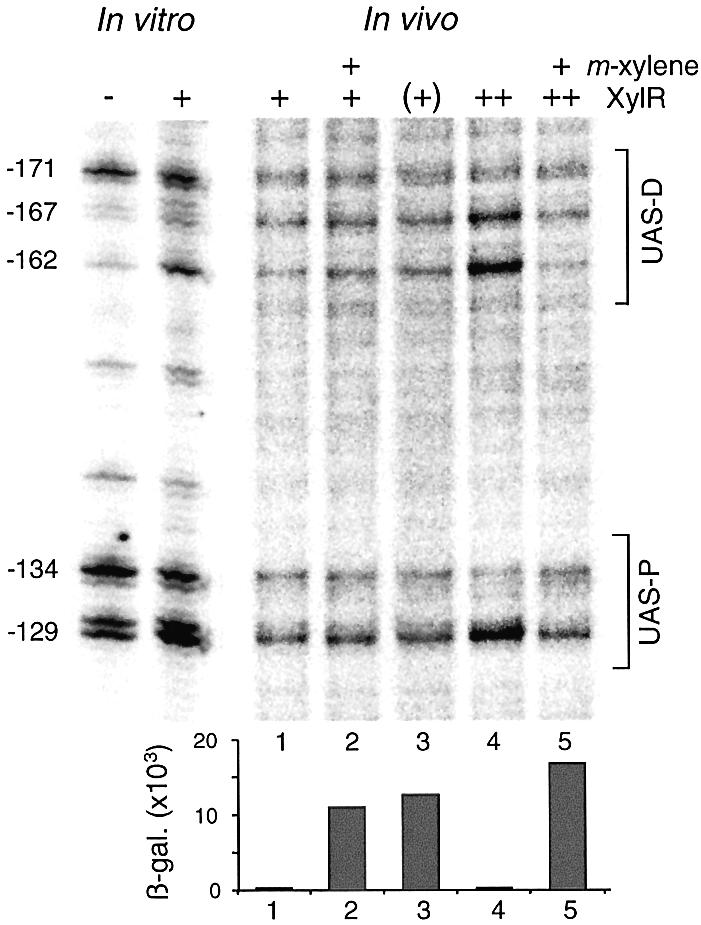
In vivo UV footprinting of the UAS of Pu promoter in induced versus uninduced P.putida cells. For reference, the in vitro footprintings of the same region, with indications of relevant nucleotides are shown to the left. All cultures were irradiated at stationary phase. Sample 1: P.putida MAD1 non-induced. Sample 2: P.putida MAD1, induced with m-xylene. Sample 3: P.putida MAD2, bearing the constitutive xylR allele named xylRΔA and thus indicated as (+). Sample 4: P.putida MAD1 (pTK19) non-induced. Sample 5: P.putida MAD1 (pTK19), induced with m-xylene. Note that inducer addition to P.putida MAD1 (pTK19) reverses the band changes that describe XylR binding to the UAS (notice bands –167, –162, –134 and –129 in lane 5 versus those of lane 4). The levels of β-galactosidase (Miller units) accumulated by each of the Pu-lacZ strains at the moment of irradiation under the conditions specified are represented at the bottom of the gel.
Since occupation of Pu under induction could not be revealed in vivo with the indigenous concentration of XylR, we resorted to re-examining the DNA in strain P.putida MAD1 (pTK19), which bears higher intracellular levels of XylR (see above). Samples were taken from m-xylene-induced and non-induced samples, and the UAS region of each of them processed as usual. The samples were loaded in lanes 4 and 5 of the gel of Figure 6. The most outstanding feature of their comparison was that the increase in the bands which were indicative of Pu occupation (–162, –129) reversed their intensity upon exposure to m-xylene (a higher resolution picture of the same effect can be seen in lane 4 of the gel of Fig. 5). This suggested that during Pu functioning, the already weak binding of XylR to the promoter is destabilized rather than increased. In any case, these observations revealed a new view of the functioning of Pu in vivo that diverges from the simpler view of most σ54 promoters, in which the EBP is mostly bound to the UAS irrespective of the level of transcription (5). In the well-studied case of NtrC (42), there is evidence for stable binding of this archetypical EBP to the glnAp1 and glnL promoters under physiological conditions. Yet, this may not be a general feature of every σ54 promoter: our observations open the possibility that such EBP-DNA binding can itself be regulated by additional factors and environmental conditions.
CONCLUSION
We have shown previously that the intracellular concentration of both IHF (16) and XylR (40) in P.putida varies during growth. In the case of IHF, we demonstrated that the levels of the factor during exponential growth are insufficient to saturate the Pu promoter (16). This is seen not only in the loss of optimal promoter geometry (13,43), but also in the lack of recruitment of σ54-RNAP to the promoter (15). As a consequence, the transcriptional capacity of Pu is very low during exponential growth but extraordinarily high in the stationary phase. In addition to IHF, the level of XylR itself is subject to some degree of growth-phase control, as the number of XylR molecules (monomers) increases from 30 to 120. If [as believed for other EBPs (24,44,45)], the active form of XylR is a hexamer or a larger oligomer, then the number of active XylR species is extremely limited and changes in concentration with growth could have a regulatory effect. As described above, we set out to examine Pu occupation by XylR under various conditions adapting the least disruptive in vivo footprinting procedure available, based on short-time UV irradiation of cell cultures.
The most salient conclusion of the experiments above is the lack of stable binding of XylR to its target sites at the Pu promoter. Only when XylR was overexpressed could the pyrimidine dimer formation pattern through the UAS in vivo be matched with that raised by the purified XylRΔA protein on Pu DNA in vitro. We believe that this result is not due to a lack of sensitivity of the technique employed, but it reflects the bona fide state of the promoter under physiological conditions. More intricate explanations (e.g. formation of a different type of protein–DNA complex which may not be detected with UV footprinting) cannot be ruled out. But on the basis of the data available we must favor the simpler view. Parallel experiments run with much shorter pulses of high-energy UV-laser on the same samples gave basically the same results (not shown). The lack of occupation of Pu by XylR contrasts with the evident interaction of IHF with its target in cells at stationary phase, as revealed both with UV-laser footprinting (16,46) and high intensity UV irradiation (this work). The results described in this work add to the notion that proteins present in small number in the cell may exhibit a distinct behavior, in which the DNA–protein interactions are hardly explained by classical thermodynamic equations (47). The low concentration of XylR in the cell, and its weak interactions with the UAS make it plausible that XylR multimerization—an event that is crucial for Pu activation (18)—could occur in solution before contacting Pu. It could even happen that the activator multimer interacts with σ54 or the RNA holoenzyme prior to DNA binding. Such a pre-recruitment type of mechanism has recently been described for the SoxR regulator in E.coli (48) and could also be present in other cases.
It is revealing to compare our results with those reported in 1991 by Abril et al. using dimethyl sulphate as the footprinting agent in vivo (11). In this case, the authors employed E.coli as the host to probe XylR–Pu interactions in a multicopy set-up in which the xylR gene was present in large excess. Changes in the UAS region were detected in the presence or absence of XylR, as well as in induced versus non-induced conditions. Yet, such an assay system did not maintain either the native host of the system or the stoichiometry of the regulatory elements, so it failed to detect the clearance of Pu under most circumstances. In our hands, only the artificial overproduction of XylR allowed visualization of protein binding to Pu in vivo, as compared to the singular conditions pondered by Abril et al. (11).
Despite these limitations, we could still picture the effect of inducer addition in the binding of XylR to Pu by forcing a longer term occupation of the promoter by an increased intracellular concentration of the regulator (Figs 4 and 6). The unanticipated result of these experiments was that inducer addition (and the predicted activation or XylR to a transcription-competent form) decreased, rather than increased the binding of XylR to its target DNA. This may be due to the fact that intracellular concentrations of XylR appear to decrease upon inducer addition, conceivably due to a higher protein turnover (40). In addition, the binding can be further reduced by the disassembly of the upstream nucleoprotein complex at every transcription round, as has been proposed not only for Pu (18,27) but also for the glnAp2 promoter of E.coli (49). In any case, it does appear that XylR is not strongly bound to Pu under any circumstance thereby allowing extra regulatory checks that increase or further decrease such an interaction. In this respect, the possibility of an extra factor binding the distal UAS (as hinted at by the TC dimer at –167 in vivo, see above) deserves future studies.
The main paradox between the functioning of σ54 promoters in vivo and in vitro is that factor binding to the corresponding DNA sequence is not granted in live bacteria. In fact, that the whole initiation complex is re-assembled at every transcription round might be a general trait of σ54 promoters (49). In our case, there is only an invariable number of ∼80 U of σ54 per P.putida cell (40), which may appear insufficient for saturation of the ∼50 σ54-promoters in the genome (50) and for efficient sigma factor competition for available core RNA polymerase (51). Only at stationary stage does IHF reach intracellular levels high enough to saturate Pu, and perhaps many other IHF-dependent σ54-promoters. The amount of XylR (and perhaps other EBPs as well) also varies in a growth-dependent fashion and is only transiently bound to the UAS. It is possible that each of these variables are connected to specific physiological conditions, so that promoter output can be finely tuned to the degree of transcription required in the diversity of environments where P.putida thrives (52,53). In this respect, we advocate that the organization and functioning of σ54-promoters is particularly well suited for interweaving a large number of physiological signals in the final transcriptional output (53–55).
Acknowledgments
ACKNOWLEDGEMENTS
The authors are indebted to V. Shingler and T. Galvao for critical reading of the manuscript and to F. Velázquez and M. Mencía for inspiring discussions. This work was supported in part by contracts QLK3-CT-2002-01933, QLK3-CT-2002-01923, and INCO-CT-2002-1001, by grant BIO2001-2274 of the Spanish Comisión Interministerial de Ciencia y Tecnología (CICYT) and by the Strategic Research Groups Program of the Autonomous Community of Madrid. M.V. is a beneficiary of the I3P Programme of the CSIC.
NOTE ADDED IN PROOF
Lee et al. (57) have recently reported that the active form of an enhancer-binding protein is a heptamer.
REFERENCES
- 1.Ramos J.L. and Marqués,S. (1997) Transcriptional control of the Pseudomonas TOL plasmid catabolic operons is achieved through an interplay of host factors and plasmid-encoded regulators. Annu. Rev. Microbiol., 51, 341–373. [DOI] [PubMed] [Google Scholar]
- 2.Merrick M. (1993) In a class of its own—the RNA polymerase sigma factor σ54 (σN). Mol. Microbiol., 10, 903–909. [DOI] [PubMed] [Google Scholar]
- 3.Buck M., Gallegos,M.T., Studholme,D.J., Guo,Y. and Gralla,J.D. (2000) The bacterial enhancer-dependent σ54 (σN) transcription factor. J. Bacteriol., 182, 4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Popham D.L., Szeto,D., Keener,J. and Kustu,S. (1989) Function of a bacterial activator protein that binds to transcriptional enhancers. Science, 243, 629–635. [DOI] [PubMed] [Google Scholar]
- 5.Sasse-Dwight S. and Gralla,J.D. (1988) Probing the Escherichia coli glnALG upstream activation mechanism in vivo. Proc. Natl Acad. Sci. USA, 85, 8934–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abril M.A. and Ramos,J.L. (1993) Physical organization of the upper pathway operon promoter of the Pseudomonas TOL plasmid. Sequence and positional requirements for XylR-dependent activation of transcription. Mol. Gen. Genet., 239, 281–288. [DOI] [PubMed] [Google Scholar]
- 7.Pérez-Martí J. and de Lorenzo,V. (1996) Physical and functional analysis of the prokaryotic enhancer of the σ54-promoters of the TOL plasmid of Pseudomonas putida. J. Mol. Biol., 258, 562–574. [DOI] [PubMed] [Google Scholar]
- 8.Wedel A., Weiss,D.S., Popham,D., Dröge,P. and Kustu,S. (1990) A bacterial enhancer functions to tether a transcriptional activator near a promoter. Science, 248, 486–490. [DOI] [PubMed] [Google Scholar]
- 9.Carmona M., Claverie-Martín,F. and Magasanik,B. (1997) DNA bending and the initiation of transcription at σ54-dependent bacterial promoters. Proc. Natl Acad. Sci. USA, 94, 9568–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu H. and Hoover,T.R. (2001) Transcriptional regulation at a distance in bacteria. Curr. Opin. Microbiol., 4, 138–144. [DOI] [PubMed] [Google Scholar]
- 11.Abril M.A., Buck,M. and Ramos,J.L. (1991) Activation of the Pseudomonas TOL plasmid upper pathway operon. J. Biol. Chem., 266, 15832–15838. [PubMed] [Google Scholar]
- 12.deLorenzo V., Herrero,M., Metzke,M. and Timmis,K.N. (1991) An upstream XylR- and IHF-induced nucleoprotein complex regulates the sigma 54-dependent Pu promoter of TOL plasmid. EMBO J., 10, 1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pérez-Martín J., Timmis,K.N. and de Lorenzo,V. (1994) Co-regulation by bent DNA. Functional substitutions of the integration host factor site at σ54-dependent promoter Pu of the upper-TOL operon by intrinsically curved sequences. J. Biol. Chem., 269, 22657–22662. [PubMed] [Google Scholar]
- 14.Seong G.H., Kobatake,E., Miura,K., Nakazawa,A. and Aizawa,M. (2002) Direct atomic force microscopy visualization of integration host factor-induced DNA bending structure of the promoter regulatory region on the Pseudomonas TOL plasmid. Biochem. Biophys. Res. Commun., 291, 361–366. [DOI] [PubMed] [Google Scholar]
- 15.Bertoni G., Fujita,N., Ishihama,A. and de Lorenzo,V. (1998) Active recruitment of sigma54-RNA polymerase to the Pu promoter of Pseudomonas putida: role of IHF and αCTD. EMBO J., 17, 5120–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valls M., Buckle,M. and de Lorenzo,V. (2002) In vivo UV laser footprinting of the Pseudomonas putida sigma 54 Pu promoter reveals that integration host factor couples transcriptional activity to growth phase. J. Biol. Chem., 277, 2169–2175. [DOI] [PubMed] [Google Scholar]
- 17.Morett E. and Segovia,L. (1993) The σ54 bacterial enhancer-binding protein family: mechanism of action and phylogenetic relationship of their functional domains. J. Bacteriol., 175, 6067–6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Martin J. and de Lorenzo,V. (1996) ATP binding to the sigma 54-dependent activator XylR triggers a protein multimerization cycle catalyzed by UAS DNA. Cell, 86, 331–339. [DOI] [PubMed] [Google Scholar]
- 19.Hwang I., Thorgeirsson,T., Lee,J., Kustu,S. and Shin,Y.K. (1999) Physical evidence for a phosphorylation-dependent conformational change in the enhancer-binding protein NtrC. Proc. Natl Acad. Sci. USA, 96, 4880–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y.K., Lee,J.H., Brewer,J.M. and Hoover,T.R. (1997) A conserved region in the σ54-dependent activator DctD is involved in both binding to RNA polymerase and coupling ATP hydrolysis to activation. Mol. Microbiol., 26, 373–386. [DOI] [PubMed] [Google Scholar]
- 21.Lee H.S., Berger,D.K. and Kustu,S. (1993) Activity of purified NIFA, a transcriptional activator of nitrogen fixation genes. Proc. Natl Acad. Sci. USA, 90, 2266–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bordes P., Wigneshweraraj,S.R., Schumacher,J., Zhang,X., Chaney,M. and Buck,M. (2003) The ATP hydrolyzing transcription activator phage shock protein F of Escherichia coli: Identifying a surface that binds sigma 54. Proc. Natl Acad. Sci. USA, 100, 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tropel D. and van der Meer,J.R. (2002) Identification and physical characterization of the HbpR binding sites of the hbpC and hbpD promoters. J. Bacteriol., 184, 2914–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wikstrom P., O’Neill,E., Ng,L.C. and Shingler,V. (2001) The regulatory N-terminal region of the aromatic-responsive transcriptional activator DmpR constrains nucleotide-triggered multimerisation. J. Mol. Biol., 314, 971–984. [DOI] [PubMed] [Google Scholar]
- 25.Wang X.Y., Kolb,A., Cannon,W. and Buck,M. (1997) Nucleoprotein complex formation by the enhancer binding protein NifA. Nucleic Acids Res., 25, 3478–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyman C., Rombel,I., North,A.K., Bustamante,C. and Kustu,S. (1997) Unusual oligomerization required for activity of NtrC, a bacterial enhancer-binding protein. Science, 275, 1658–1661. [DOI] [PubMed] [Google Scholar]
- 27.Garmendia J. and de Lorenzo,V. (2000) Visualization of DNA–protein intermediates during activation of the Pu promoter of the TOL plasmid of Pseudomonas putida. Microbiology, 146, 2555–2563. [DOI] [PubMed] [Google Scholar]
- 28.Fernandez S., de Lorenzo,V. and Perez-Martin,J. (1995) Activation of the transcriptional regulator XylR of Pseudomonas putida by release of repression between functional domains. Mol. Microbiol., 16, 205–213. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez S., Shingler,V. and De Lorenzo,V. (1994) Cross-regulation by XylR and DmpR activators of Pseudomonas putida suggests that transcriptional control of biodegradative operons evolves independently of catabolic genes. J. Bacteriol., 176, 5052–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.deLorenzo V., Fernandez,S., Herrero,M., Jakubzik,U. and Timmis,K.N. (1993) Engineering of alkyl- and haloaromatic-responsive gene expression with mini-transposons containing regulated promoters of biodegradative pathways of Pseudomonas. Gene, 130, 41–46. [DOI] [PubMed] [Google Scholar]
- 31.Pérez-Martín J. and de Lorenzo,V. (1996) In vitro activities of an N-terminal truncated form of XylR, a σ54-dependent transcriptional activator of Pseudomonas putida. J. Mol. Biol., 258, 575–587. [DOI] [PubMed] [Google Scholar]
- 32.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1994) Current Protocols in Molecular Biology. John Wiley and Sons, New York. [Google Scholar]
- 33.Becker M.M. and Wang,J.C. (1984) Use of light for footprinting DNA in vivo. Nature, 309, 682–687. [DOI] [PubMed] [Google Scholar]
- 34.Buckle M., Geiselmann,J., Kolb,A. and Buc,H. (1991) Protein–DNA cross-linking at the lac promoter. Nucleic Acids Res., 19, 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuluncsics Z., Perdiz,D., Brulay,E., Muel,B. and Sage,E. (1999) Wavelength dependence of ultraviolet-induced DNA damage distribution: involvement of direct or indirect mechanisms and possible artefacts. J. Photochem. Photobiol. B Biol., 49, 71–80. [DOI] [PubMed] [Google Scholar]
- 36.Gurzadyan G.G., Gorner,H. and Schulte-Frohlinde,D. (1995) Ultraviolet (193, 216 and 254 nm) photoinactivation of Escherichia coli strains with different repair deficiencies. Radiat. Res., 141, 244–251. [PubMed] [Google Scholar]
- 37.Engelhorn M., Boccard,F., Murtin,C., Prentki,P. and Geiselmann,J. (1995) In vivo interaction of the Escherichia coli integration host factor with its specific binding sites. Nucleic Acids Res., 23, 2959–2965. [PubMed] [Google Scholar]
- 38.Hockensmith J.W., Kubasek,W.L., Vorachek,W.R. and von Hippel,P.H. (1993) Laser cross-linking of proteins to nucleic acids. I. Examining physical parameters of protein-nucleic acid complexes. J. Biol. Chem., 268, 15712–15720. [PubMed] [Google Scholar]
- 39.Buck M. and Cannon,W. (1994) A simple procedure for visualising protein-nucleic acid complexes by photochemical crosslinking. Nucleic Acids Res., 22, 1119–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraile S., Roncal,F., Fernandez,L.A. and de Lorenzo,V. (2001) Monitoring intracellular levels of XylR in Pseudomonas putida with a single-chain antibody specific for aromatic-responsive enhancer-binding proteins. J. Bacteriol., 183, 5571–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garmendia J. and de Lorenzo,V. (2000) The role of the interdomain B linker in the activation of the XylR protein of Pseudomonas putida. Mol. Microbiol., 38, 401–410. [DOI] [PubMed] [Google Scholar]
- 42.Shiau S.P., Schneider,B.L., Gu,W. and Reitzer,L.J. (1992) Role of nitrogen regulator I (NtrC), the transcriptional activator of glnA in enteric bacteria, in reducing expression of glnA during nitrogen-limited growth. J. Bacteriol., 174, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pérez-Martín J. and de Lorenzo,V. (1995) Integration host factor suppresses promiscuous activation of the σ54-dependent promoter Pu of Pseudomonas putida. Proc. Natl Acad. Sci. USA, 92, 7278–7281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Farez-Vidal M.E., Wilson,T.J., Davidson,B.E., Howlett,G.J., Austin,S. and Dixon,R.A. (1996) Effector-induced self-association and conformational changes in the enhancer-binding protein NtrC. Mol. Microbiol., 22, 779–788. [DOI] [PubMed] [Google Scholar]
- 45.Rippe K., Guthold,M., Hippel,P.H. and Bustamante,C. (1997) Transcriptional activation via DNA-looping: visualization of intermediates in the activation pathway of E.coli RNA polymerase σ54 holoenzyme by scanning force microscopy. J. Mol. Biol., 270, 125–138. [DOI] [PubMed] [Google Scholar]
- 46.Murtin C., Engelhorn,M., Geiselmann,J. and Boccard,F. (1998) A quantitative UV laser footprinting analysis of the interaction of IHF with specific binding sites: re-evaluation of the effective concentration of IHF in the cell. J. Mol. Biol., 284, 949–961. [DOI] [PubMed] [Google Scholar]
- 47.Halling P.J. (1989) Do the laws of chemistry apply to living cells? Trends Biochem. Sci., 14, 317–318. [DOI] [PubMed] [Google Scholar]
- 48.Griffith K.L., Shah,I.M., Myers,T.E., O’Neill,M.C. and Wolf,R.E. (2002) Evidence for ‘pre-recruitment’ as a new mechanism of transcription activation in Escherichia coli: the large excess of SoxS binding sites per cell relative to the number of SoxS molecules per cell. Biochem. Biophys. Res. Commun., 291, 979–986. [DOI] [PubMed] [Google Scholar]
- 49.Bondarenko V., Liu,Y., Ninfa,A. and Studitsky,V.M. (2002) Function of prokaryotic enhancer over a distance does not require continued presence of promoter-bound sigma54 subunit. Nucleic Acids Res., 30, 636–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cases I. and de Lorenzo,V. (2003) The sigma 54 regulon (sigmulon) of Pseudomonas putida. Microbiology, in press. [DOI] [PubMed] [Google Scholar]
- 51.Laurie A.D., Bernardo,L.M., Sze,C.C., Skarfstad,E., Szalewska-Palasz,A., Nystrom,T. and Shingler,V. (2003) The role of the alarmone (p)ppGpp in sigma N competition for core RNA polymerase. J. Biol. Chem., 278, 1494–1503. [DOI] [PubMed] [Google Scholar]
- 52.Cases I. and de Lorenzo,V. (2001) The limits to genomic predictions: role of σN in environmental stress survival of Pseudomonas putida. FEMS Microbiol. Ecol., 35, 217–221. [DOI] [PubMed] [Google Scholar]
- 53.Shingler V. (2003) Integrated regulation in response to aromatic compounds: from signal sensing to attractive behaviour. Environ. Microbiol., in press. [DOI] [PubMed] [Google Scholar]
- 54.Cases I. and de Lorenzo,V. (2001) The black cat/white cat principle of signal integration in bacterial promoters. EMBO J., 20, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cases I., Lopez,J.A., Albar,J.P. and de Lorenzo,V. (2001) Evidence of multiple regulatory functions for the PtsN (IIA(Ntr)) protein of Pseudomonas putida. J. Bacteriol., 183, 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 57.Lee S.Y., De La Torre,A., Yan,D., Kustu,S., Nixon,B.T. and Wemmer,D.E. (2003) Regulation of the transcriptional activator NtrC1: structural studies of the regulatory and AAA+ ATPase domains. Genes Dev., 17, 2552–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]