

Abstract
Multiple sclerosis (MS) is an autoimmune disease characterized by peripheral activation of CD4+ T cells that migrate into the central nervous system (CNS) and mount an autoimmune neuroinflammatory attack on myelin and oligodendrocytes. Secondary to these events, however equally destructive, is the generation of inflammatory-mediated reactive oxygen and nitrogen species generated by persistently activated microglia and astrocytes. Nuclear factor-erythroid 2–related factor 2 (Nrf2) is a basic leucine zipper transcription factor that regulates genetic expression of many protective antioxidant and detoxication enzymes. Here we describe the Nrf2 modulation of innate and adaptive immune responses in an acute autoimmune model of MS, experimental autoimmune encephalomyelitis (EAE). Wild-type (WT) mice and Nrf2 knockout mice were immunized with myelin oligodendrocyte glycoprotein (MOG 35-55) and monitored daily for clinical scores of disease. Disruption of Nrf2 resulted in a more severe clinical course, a more rapid onset, and a greater percentage of mice with the disease. Furthermore, increased immune cell infiltration and glial cell activation in spine was observed. In conjunction, we observed increased inflammatory enzyme (iNOS, phox-47, gp91-phox, and phox-67), cytokine (IFN-gamma, IL1-b, TNF-alpha, and IL-12), and chemokine (BLC and MIG) gene expression levels in the Nrf2-deficient mice compared to the WT mice, supporting the notion that Nrf2 can modulate an autoimmune neuroinflammatory response. Our results show that the absence of Nrf2 exacerbates the development of EAE and thus suggests that activation of Nrf2 may then attenuate pathogenesis of autoimmune diseases such as MS as well as other neurodegenerative diseases that present with neuroinflammation.
Keywords: Neuroinflammation, Multiple Sclerosis, Nrf2, Antioxidant Response Element
The National Multiple Sclerosis Society estimates that over 2 million persons worldwide are affected with the autoimmune demyelinating disease known as multiple sclerosis (MS). MS is regarded as a heterogenic disease and presents with variable symptoms and severity ranging from loss of vision to debilitating ataxia (Noseworthy et al., 2000). In most cases, MS is considered to be a chronic inflammatory disease of the central nervous system (CNS) with intervals of remission followed by relapse. However, acute progressive cases are documented. Onset of this disease initiates outside of the CNS through activation of CD4+ T cells by myelin-like antigenic peptides. These cells then migrate across the blood brain barrier (BBB) initiating focal inflammation. In addition, activation of infiltrating macrophages, CD8+ T cells, B cells, as well as resident microglia and astrocytes have all been implicated in the pathology of MS. Sclerotic lesions associated with this disease result from an attack on the myelin sheathing surrounding the neuronal axons and on the oligodendrocytes, resulting in axonal retraction and subsequent astrogliosis. Current therapies for this disease primarily focus on prevention of the penetration of immune cells across the BBB (Dhib-Jalbut, 2007; Noseworthy et al., 2000). However, oxidative stress is also documented in the pathogenesis of MS. Here, activation of macrophages, microglia, and astrocytes produce reactive oxygen species (ROS) such as super oxide radicals and reactive nitrogen species (RNS), which are injurious to cells and also contribute to the cellular and tissue damage observed in the MS lesion (Liu et al., 2001; van Horssen et al., 2008; Zeis et al., 2008).
Many studies clearly demonstrate that the pro-antioxidant transcription factor, nuclear factor-erythroid 2–related factor 2 (Nrf2), promotes cell survival or tumor prevention via disruption of the Keap1-Nrf2 cytosolic complex, an event mediated by electrophilic or free radical molecules (Chan et al., 2001; Durchdewald et al., 2007; Kang et al., 2004; Lee et al., 2005; Li et al., 2005; Liu et al., 2008). The actin-bound Keap1 sequesters Nrf2 in the cytosol and serves as an E3 ligase, shuttling the continuously ubiquinated Nrf2 to the proteasome for degradation (Zhang and Hannink, 2003). The turnover of Nrf2 is thus rapid, and its short half-life in cell lines and macrophages is estimated to be less than 20 min. Hence, basal levels of Nrf2-driven genes can be quite low. Upon cytosolic activation of Nrf2, the transcription factor releases from Keap1, translocates to the cell nucleus, and binds to the antioxidant response element (ARE; Itoh et al., 2003, 1997). The ARE is a cis-acting DNA-responsive element located in the promoter region of a battery of genes whose functions are to promote cell survival. Clusters transcribed include genes that (1) metabolize and conjugate xenobiotics or electrophiles, such as the phase II enzymes NAD(P)H-quinone oxidoreductase (NQO1), glutathione S-transferases (GSTs), and UDP-glycosyltransferase 1A6 (UGT), and (2) boost a cells antioxidant potential that include enzymes and proteins, such as glutamate-cysteine ligase (glutathione [GSH] production), heme oxygenase-1, thioredoxin reductase-1 (TXNRD1), thioredoxin, and ferritin (Kraft et al., 2004; Lee et al., 2003; Li et al., 2002; Shih et al., 2003). Nrf2 is thus considered an important mediator of cellular oxidative stress particularly in diseases that present with neuroinflammation such as Parkinson's disease, Alzheimer's disease, and MS.
Modulation of the innate immune response by Nrf2 has been observed in mice with lung inflammation or acute lung injury (ALI). Previous studies showed that if lungs of mice were instilled with LPS or carrageenan, there was an increased presence and persistence of inflammatory cells in Nrf2-KO mice when compared to WT controls. This corresponded to increased activation of nuclear factor KB in the Nrf2-KO mice. (Itoh et al., 2004; Thimmulappa et al., 2006a). Expression of genes associated with inflammation, such as tumor necrosis factor-alpha (TNF-alpha), IL-1beta, and IL-6, were also dramatically increased. Sepsis-induced mortality in these same mice was greater than in a wild-type (WT) cohort. This response correlated to the lack of Nrf2, since pretreatment with an activator of Nrf2, the triterpenoid analogue CDDO-imidazolide (CDDO-Im), failed to attenuate the innate inflammatory response and mortality in the Nrf2-knockout (KO) mice. However, administration of a cellular antioxidant, N-acetyl-cysteine, was able to partially protect (Thimmulappa et al., 2006b, 2007).
Previous studies in our laboratory have demonstrated the protective effects of Nrf2 in mouse cortical cultures treated with H2O2 as well as in mouse models of neurodegeneration, including Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis. Moreover, an increased occurrence of activated microglia and astrocytes was evident in the brains or spines from Nrf2-KO mice with these diseases compared with WT controls (Calkins et al., 2005; Chen et al., 2009; Jakel et al., 2007; Kraft et al., 2004, 2006; Vargas et al., 2008). These observations further underline the significance of Nrf2 in the modulation of neuroinflammation as well as the protection from oxidative stress.
The present study attempts to understand the role of Nrf2 in an autoimmune inflammatory model of MS. Nrf2-KO and WT mice were immunized with MOG 35-55 to induce experimental autoimmune encephalomyelitis (EAE). Clinical scores of disease and disease onset were monitored, and cellular and molecular mechanisms of the disease in Nrf2-KO and WT mice were analyzed.
MATERIALS AND METHODS
Mice.
All experiments were performed using Biozzi ABH mice backcrossed onto Nrf2-KO mice for a minimum of seven generations producing congenic strains of mice containing over 99% of the recipient genome. The Biozzi ABH mice were graciously provided by Dr Ian Duncan, School of Veterinary Medicine, University of Wisconsin, Madison (Note: these mice were originated by Dr David Baker, Institute of Neurology, University College London). ABH mice are widely used in studying diseases of autoimmunity. These mice have a unique major histocompatibility complex haplotype designated H-2dq1, yielding them highly susceptible to antigenic agents (Amor et al., 2005). Nrf2-KO mice were generated by displacement of the basic leucine zipper/DNA-binding domain of Nrf2 with a B-galactosidase cassette (Chan et al., 1996) and were kindly provided by Dr Yuet Wai Kan, Howard Hughes Medical Institute, University of California, San Francisco. For sake of simplicity, the ABH × Nrf2-KO mice are hereafter referred to as Nrf2-KO.
Induction of EAE and clinical scoring.
EAE was induced in 8- to 16-week-old male and female Nrf2-KO mice and WT littermate controls by sc injecting 100 μg of MOG 35-55 peptide (AnaSpec, Inc., San Jose, CA) emulsified in complete Freund's adjuvant (CFA) including Mycobacterium tuberculosis (SIGMA, St Louis, MO) into the scapular region. This was concomitant with ip injections of 200 ng of pertussis toxin (SIGMA) on days 0 and 2. EAE was monitored daily for clinical scores of disease until completion of the experiment using the following scoring system—1, complete tail atony; 2, hind limb weakness; 3, hind limb paralysis; 4, complete hind limb paralysis and front limb weakness; and 5, moribund. If mice with EAE were deemed to be within two scorings, a 0.5 value was added to the lower of the clinical score. Total body weights were also monitored daily. It is noted that all animals were treated and cared for in accordance with and approval of the University of Wisconsin's Institutional Animal Care and Use Committee.
LFB staining and measurement of demyelination.
At 14–16 days after EAE induction, mice were transcardially perfused with 0.1M PBS. Spinal cords were removed from the spinal column by liquid expulsion, fixed in 10% formalin, and embedded in paraffin. Spinal cords were cut, and middle lumbar and lower sacro-lumbar regions were included on each slide. Sections (10 μm) were cut and stained with Luxol Fast Blue (LFB) to detect myelin. Four intact cross-sections from each mouse (four to five mice per group) were photographed on an Olympus BX40 microscope (Olympus America Inc., Melville, NY) using a CMOS Pro 1000 series digital camera (Sound Vision Inc., Wayland, MA). Adobe Photoshop images were imported into the ImageJ image processing and analysis program (http://rsb.info.nih.gov/ij/index.html). Normally myelinated areas and areas of demyelination in the dorsal region were measured on each section. Areas from each animal were totaled, and the percent demyelination was calculated. GraphPad Prism 4 (GraphPad Software) statistical software was used to plot data and to calculate unpaired t test–generated p-values for comparison of demyelination between groups of mice (p < 0.05 was considered significant).
Cell infiltration.
Paraffin sections of spine from the above experiment were also stained with hematoxylin and eosin (H&E) for analysis of total cell infiltration in perivascular and subpial regions. High-resolution images of sections were captured at ×200 magnification using the Mosaic Scanning System (Zeiss Microscopy, Germany). Paneled sections were then stitched, and the total cell infiltration was blindly assessed and scored by adding the total number of areas of perivascular cuffing to the arbitrary scoring system for subpial cell infiltration that consisted of the following: 0, no infiltration; 1, minor infiltration; 2, numerous infiltrating cells; 3, most of the pia involved; and 4, all of the pia involved. Statistical analysis was performed as described above.
Immunohistochemistry.
Paraffin sections of spine from the above experiment were cut into 10-μm sections, rehydrated, and immunohistochemically stained using rabbit anti-ionized calcium-binding adaptor molecule 1 (Iba1) antibody (1:250; Wako Pure Chemical Industries, Osaka, Japan) or rabbit anti-Glial fibrillary acidic protein (GFAP) antibody (1:2000; Dako). Antigen retrieval was performed prior to staining for MOG by boiling sections for 20 min in 0.01M sodium citrate, pH 6.0. All sections were then blocked with buffer containing 0.4% Triton X-100, 10% goat serum or horse serum, and 0.5% bovine serum albumin in PBS. Sections were incubated with primary antibodies diluted in blocking solution overnight at 4°C. Secondary staining of sections was performed using the appropriate antibodies conjugated to Alexafluor® 488 and 647 (Molecular Probes, Inc., Eugene, OR) that were diluted in blocking solution (1:250) and incubated for 1 h at room temperature. Hoescht 33258 (5 μg/ml, SIGMA) was added to the secondary antibody solutions to stain cell nuclei. Sections were washed with PBS and mounted with Fluoro-Gel (EMS). Control staining was performed using appropriate IgG antibodies in place of the primary antibody. Sections were visualized using a Zeiss epifluorescent microscope (Carl Zeiss, Germany) and photographic images of immunofluorescence captured using AxioVision 4 software (Carl Zeiss).
qPCR.
In some experiments, total RNA was extracted from whole spinal cords at day 14–16 after induction of EAE. Spines were expelled from spinal column and stored in RNAlater® (Ambion, Austin, TX) for at least 24 h at 4°C until further processing. Spinal cords were then homogenized in Trizol reagent (Invitrogen, Inc.), and total RNA was extracted following the manufacturer's instructions. Integrity and concentrations of RNA were determined using the RNA 6000 Nano chip and analyzed by the Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA). Only samples that carried an RNA Integrity Number of 8.0 or greater were used for further analysis. RNA (1 μg) was reverse transcribed using an Oligo-dT 15 primer in accordance with the Reverse Transcription System (Promega Corporation, Madison, WI). PCR amplification and quantification (qPCR) of resulting cDNA was performed in real time using the LightCycler® 480 System (Roche Applied Science), using Cycler480 SYBR Green I Master (Roche Applied Science), and following manufacturer's instructions. All PCR product quantification was subject to relative standard curves that yielded amplification efficiencies greater than 1.79 and less than 2.19 and with error less than 0.2. Primers used for the qPCR analysis are listed in the Supplementary table 1.
RESULTS
Nrf2-KO Mice with EAE Have Higher Clinical Scores of Disease and Incidence
To understand the significance of Nrf2 in modulating inflammation and demyelination, Nrf2-KO and WT mice were immunized with MOG 35-55 emulsified in CFA or CFA alone. Clinical scores of the disease were assessed starting at day 13 after the initial immunizations, and the onset and severity of EAE were monitored. Mice lacking Nrf2 showed a more severe course of disease than the WT mice (Fig. 1A). When mice were categorized as having a mild (clinical score ≥ 0 and < 2) or severe (clinical score ≥ 2) course of disease, the percentage of Nrf2-KO mouse had greater incidence in both mild and severe EAE at all time points after disease onset (Fig. 1B). Further, the Nrf2-KO mice had an earlier onset of EAE and had a greater percentage of the mice responding to the immunization of MOG 35-55 (100% of Nrf2-KO mice responding vs. 64% of WT) (Fig. 1C). It is noted that male and female mice were randomly and evenly distributed among the experimental treatment groups, and no significant gender differences in clinical scores, percent incidence, or severity were observed.
FIG. 1.
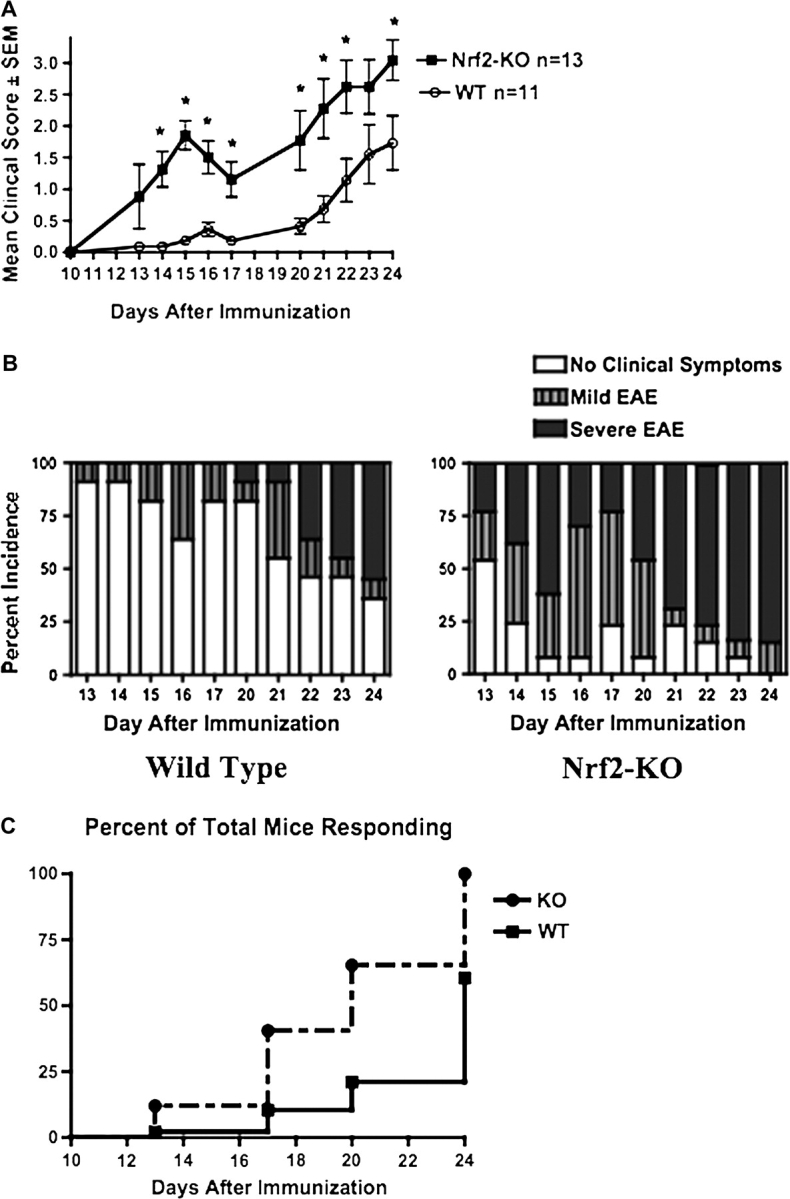
Mice lacking the transcription factor Nrf2 show a more severe response to EAE. (A) Clinical scores of disease as described in the Materials and Methods were monitored daily after injection of 100 μg of MOG-35-55 emulsified in CFA. (B) Percent incidence of the severity of disease was documented as follows—0, no clinical score; 0.5–1.5, mild EAE; and 2.0 or above, severe EAE. (C) Onset of EAE was tracked, and the percent of total numbers of mice responding to immunization with MOG 35-55 is represented for WT and Nrf2-KO mice. Onset curves are significantly different, p < 0.003 (χ2 = 8.853). Data in (A) are presented as mean ± SEM. *Significantly different than WT control (p < 0.05).
Histopathological Analysis for Demyelination and Cell Infiltration in Nrf-KO and WT Mice with EAE
A hallmark of tissue damage in EAE is loss of myelin in white matter regions of spinal cord. Lumbar spine regions from day 14 after MOG 35-55 immunizations were assessed for demyelination by staining with LFB. Sections from lumbar spinal cord from both Nrf2-KO and WT mice with EAE showed loss of LFB stain in multifocal areas of the lumbar spine. The demyelination was then measured and analyzed using ImageJ (Fig. 2A and 2B). Demyelination, shown by the loss of LFB stain in multifocal areas of the lumbar spine, was then measured and analyzed using ImageJ. Here, Nrf2-KO mice showed a 5.4% loss of myelin, which was significantly greater than the 1.7% loss seen in WT mice (Fig. 2B). Quantitative measurement of the gene expression levels for MOG was determined by qPCR from whole spine total RNA. Although not significant, a trend showing greater reductions in MOG expression in Nrf2-KO mice at 35% compared with WT mice at 26% as compared with their respective CFA-treated control mice (data not shown).
FIG. 2.

Nrf2-KO mice have greater demyelination than WT mice with EAE. (A and B) LFB staining of representative paraffin-embedded sections from lumbar spine of nondiseased (left panel) and diseased (MOG-induced EAE, right panel) mice. Bolded black arrows point to areas exemplifying demyelination. (C) Quantification of demyelination of sections described in (A and B) using ImageJ processing software and represented as mean ± SEM (WT, n = 4; Nrf2-KO, n = 5). *Significantly different than WT control (p < 0.05).
Based on the previous data showing a greater amount of demyelination in the Nrf2-KO mouse with EAE, we wanted to determine the cellular composition of infiltrating immune cells and to what extent these cells were involved in the pathological process. At day 14–16 after immunization, a time point corresponding to the early stages of disease onset, Nrf2-KO and WT mice from all experimental groups were sacrificed for histopathological and molecular examination. Representative sections of lumbar spinal cord stained with H&E from mice with and without EAE (MOG 35-55 treated) are shown (Fig. 3A). Cell infiltration was observed in all mice with EAE as measured by the number of perivascular cuffings and the amount of subpial infiltration; however, there was a significant increase in the inflammatory index of Nrf2-KO compared with WT (Fig. 3B). Total RNA from whole spinal cord was also harvested and measured to identify what cell types and to what extent the cells had breached and infiltrated the BBB. By measuring gene expression using qPCR, we were able to identify and quantify CD4+ T cell, CD8a+ T cell, and CD19 B cell lymphocytes. All mice with EAE disease had significantly augmented expression levels of these genes compared with the nondiseased CFA-immunized mice (Fig. 3C). Further, gene expression levels for CD4+ T cell, CD19, a cellular marker for B cells, as well as myeloperoxidase, a neutrophil cell marker, were significantly greater in the Nrf2-KO mice compared with WT, both with EAE (Fig. 3C).
FIG. 3.

Nrf2-KO mice with EAE show a greater total cell infiltration. (A) Representative paraffin-embedded spinal cord lumbar sections (20 μm) stained with H&E from CFA and MOG 35-55–immunized mice (×50). The solid and stippled black arrows display the perivascular and subpial cell infiltration, respectively. (B) Inflammatory index reflecting quantification of total cell infiltration in lumbar spinal regions from Nrf2-KO (n = 6) and WT (n = 6). *Significantly different from corresponding WT mice (p < 0.05). (C) Quantitative PCR analysis of whole spine extracts for mRNA levels of CD4, CD8 CD19, and myeloperoxidase (MPO). All data were standardized to GAPDH mRNA and presented as the mean fold change ± SEM when compared with the CFA-immunized mice (WT-CFA, n = 5; WT-MOG, n = 6; KO-CFA, n = 5; KO-MOG, n = 5). GraphPad Prism 4 (GraphPad Software) statistical software was used to analyze and plot two-way ANOVA followed by Bonferroni post hoc tests. *Significantly different than CFA-immunized mice within respective genotype cohort (p < 0.05). **Significantly different than MOG 35-55–immunized WT mice (p < 0.05).
Evidence of Astrogliosis in Lumbar Spines of Nrf2-KO and WT Mice with EAE
Activation of astrocytes and microglia is associated with degeneration of neurons and oligodendrocytes, directly or indirectly or both. Lumbar spinal cord from WT and Nrf2-KO mice with EAE were stained for the microglial cell marker Iba1 and the astrocytic cell marker GFAP. Compared with their nondiseased (CFA alone) littermate controls, both WT and Nrf2-KO mice showed increased activation of both cell types (Fig. 4A; data not shown for CFA alone). Staining for Iba1, however, indicated more robust microglial activation in the Nrf2-KO mice than WT (Fig. 4A). In fact, gene expression levels for Iba1 were significantly higher in the diseased Nrf2-KO mice when compared with both nondiseased Nrf2-KO and diseased WT mice (Fig. 4B). Staining and gene expression levels for GFAP showed increased and significant activation in both Nrf2-KO and WT mice (Fig. 4A and 4B).
FIG. 4.
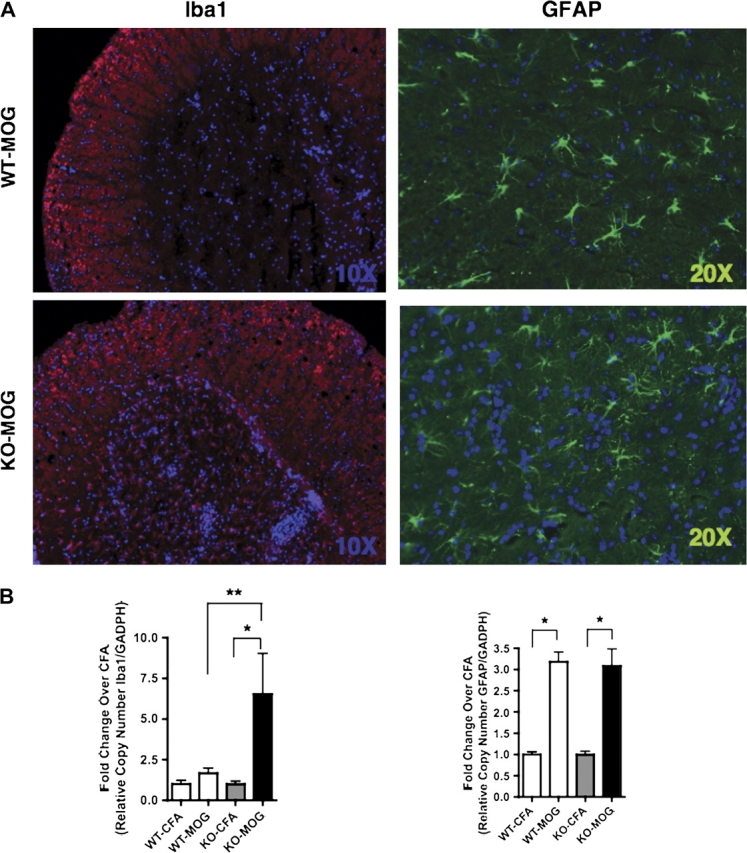
Evidence of astrogliosis and microgliosis in mice with EAE. (A and B) Representative immunofluorescent staining of lumbar spine for the cellular markers GFAP (astrocytes) and Iba1 (microglia) from mice immunized with CFA only or CFA and MOG 35-55. (B) Quantitative PCR analysis of whole spine extracts for mRNA levels of GFAP and Iba1. All data were standardized to GAPDH mRNA and presented as the mean fold change ± SEM when compared with the CFA-immunized mice (WT-CFA, n = 5; WT-MOG, n = 6; KO-CFA, n = 5; KO-MOG, n = 5). GraphPad Prism 4 (GraphPad Software) statistical software was used to analyze and plot two-way ANOVA followed by Bonferroni post hoc tests. *Significantly different than CFA-immunized mice within respective genotype cohort (p < 0.05). **Significantly different than MOG 35-55–immunized WT mice (p < 0.05).
Altered Expression Levels of Proinflammatory, Cytokine, and Chemokine Genes in Nrf2-KO and WT Mice with EAE
To better understand the biological and molecular mechanisms responsible for the differences in the clinical scores as well as the more robust presence of microglia in Nrf2-KO mice with EAE, the genetic profiles of proinflammatory enzymes and canonical cytokines and chemokines were assessed. Whole spine extracts of RNA from diseased and nondiseased mice in both Nrf2-KO and WT were analyzed by qPCR. Results showed that the proinflammatory enzyme, inducible nitric oxide synthase (iNOS), was significantly increased by 23- and 96-folds in WT and Nrf2-KO with EAE, respectively, compared with the littermate controls (CFA only) (Fig. 5A). The 96-fold induction of iNOS in the Nrf2-KO mice was also significantly greater than in WT mice. Subunits of the multimeric enzyme, NADPH oxidase, a significant contributor of ROS (O2·−), were also analyzed. Gene expression levels for the subunits, phox-47, phox-67, and gp91-phox, were all increased in the diseased mice; gp91-phox showed the greatest induction in Nrf2-KO mice with EAE at 156-fold. Furthermore, all subunits were also significantly increased in Nrf2-KO versus WT mice with EAE (Fig. 5A). Gene expression levels for all cytokines, chemokines, and proinflammatory enzymes were similar and not significantly different between CFA-treated WT and Nrf2-KO (data not shown).
FIG. 5.

Gene expression levels for proinflammatory enzymes, cytokines, and chemokines are greater in Nrf2-KO than WT with EAE. (A and B) At days 14–16, total RNA from whole spine was extracted from mice immunized with CFA or CFA and MOG 35-55. RNA was reverse transcribed and the cDNA product was subject to quantitative PCR analysis for mRNA expression levels of the following gene clusters—proinflammatory enzymes: iNOS, phox-47, gp91-phox, and phox-67; cytokines: IFN-gamma, IL1-b, TNF-alpha, and IL-12; and chemokines: BLC and MIG. All data were standardized to GAPDH mRNA and presented as the mean fold change ± SEM when compared with the CFA-immunized mice (WT-CFA, n = 5; WT-MOG, n = 6; KO-CFA, n = 5; KO-MOG, n = 5). GraphPad Prism 4 (GraphPad Software) statistical software was used to analyze and plot two-way ANOVA followed by Bonferroni post hoc tests. *Significantly different than CFA-immunized mice within respective genotype cohort (p < 0.05). **Significantly different than MOG 35-55–immunized WT mice (p < 0.05).
Levels for cytokines and chemokines were also monitored in these same spine extracts. The canonical cytokines, INF-gamma, IL1-b, TNF-alpha, and IL-12, were all significantly increased in both Nrf2-KO and WT-diseased mice. All but TNF-alpha were significant in the Nrf2-KO compared with WT mice with EAE (Fig. 5B). The chemokines, monokine induced by interferon-gamma (MIG) and B cell attractant chemokine (BLC) known to attract monocytes/macrophages and lymphocytes to sites of injury, respectively, were also analyzed. All genes analyzed from diseased mice showed significance compared with nondiseased mice. Similar to the cytokines analyzed, gene expression levels for MIP1-gamma and BLC with EAE were significantly increased in the Nrf2-KO mice when compared with WT (Fig. 5B). In particular, BLC and MIG were nearly an order of magnitude greater in the Nrf2-KO mice with EAE.
DISCUSSION
Inflammation is an essential defense mechanism against a wide selection of pathogens as well as tumor formation. However, when the inflammatory process fails to subside and becomes dysregulated, such as in sepsis or during neurodegeneration, or if immune cells start to attack self, then destructive and irreversible tissue damage can occur (Innamorato et al., 2008, 2009; Johnson et al., 2008). The data presented herein demonstrate that the transcription factor Nrf2 can modulate innate and adaptive immune responses in an acute autoimmune model of MS, EAE. Disruption of Nrf2 resulted in a more severe clinical course, a more rapid onset, and a greater percentage of mice with the disease. Furthermore, increased immune cell infiltration and glial cell activation in spine was observed, indicative of a greater pathological response in the Nrf2-KO mice with EAE. In conjunction, we observed increased inflammatory, cytokine, and chemokine gene expression levels in the Nrf2-deficient mice compared with the WT mice, supporting the notion that Nrf2 can modulate an inflammatory response.
Our data are consistent with previous studies researching the role of Nrf2 in modulation of inflammation in models outside the CNS. When LPS was injected ip or after cecal ligation/puncture, a more dramatic rate of mortality in Nrf2-KO versus WT mice was observed. Septic Nrf2-KO mice had serum TNF-alpha levels that were significantly greater than in WT, suggesting that mice lacking Nrf2 had a more severe innate immune response to the bacteria or endotoxin (Thimmulappa et al., 2006a). In nonlethal inflammatory studies, lungs from Nrf2-KO or WT mice were instilled with LPS or carrageenan, or mice had ip injections of LPS or TNF-alpha. In all cases, Nrf2-KO mice had a greater influx of inflammatory cells from bronchoaleveolor lavage fluids (Itoh et al., 2004; Thimmulappa et al., 2006a). Carrageenan, a potent activator of COX-2, also showed increased ALI in Nrf2-KO mice. The latter data are interesting since resolution of inflammation is also thought to occur through the COX-2 generation of cyclopentanone 15d-PGJ2, an endogenous electrophile and activator of Nrf2. Previous reports had shown that 15d-PGJ2 increased expression of Nrf2-driven genes in peritoneal macrophages. In fact, 15d-PGJ2 has been shown to covalently bind to Keap1, whereby Nrf2 is then activated and expression of ARE-driven genes again is increased. Enzymatic inhibition of 15d-PGJ2 production using NS-398, a selective COX-2 inhibitor, exacerbated ALI in WT mice to that observed in Nrf2-KO. Moreover, exogenous administration of 15d-PGJ2 reversed ALI in WT but not in Nrf2-KO mice, supporting the notion that Nrf2 does indeed modulate inflammatory response (Itoh et al., 2004).
Microarray analysis of lung tissue from LPS-injected Nrf2-KO mice showed greater gene expression levels of several cytokines, chemokines, and adhesion molecules, at 30 min and 1 h. We also observed increased gene expression levels for cytokines and chemokines of which IL-12, BLC, and MIG were significantly increased in Nrf2-KO mice and were common to the LPS study described above (Thimmulappa et al., 2006a). In contrast, our gene expression studies were performed on whole spine samples taken at 14–16 days after initial injection of MOG 35-55. We chose this time point because onset of EAE begins at or near day 14 and inflammatory responses are continuously occurring. Perhaps, analysis of cytokine and chemokine profiles at earlier time points is warranted, as it appears from the LPS studies that gene expression levels are dramatically changed within minutes after injection. Interestingly, TNF-alpha levels, although significantly increased in both WT and Nrf2-KO with EAE, were consistently increased in both cohorts. Studies done in mice lacking TNF-alpha showed delays in remyelination. It was suggested that TNF-alpha may actually play role in the regeneration and repair of oligodendrocytes (Arnett et al., 2001). Although we would expect significant increases in the Nrf2-KO mice with EAE, perhaps the TNF-alpha levels, in this respect, are actually protective.
The immunomodulatory potential of Nrf2 was demonstrated in cultured human blood monocytes and neutrophils as well as mouse peritoneal macrophages treated with various Nrf2-activating compounds, including tert-butyl hydroquinone, diethyl maleate, CDDO-Im, and sulforaphane (Ishii et al., 2000; Liu et al., 2008; Thimmulappa et al., 2007). In some of these studies, activation of Nrf2 attenuated inflammation induced by subsequent treatments with LPS. ROS measured after the LPS treatments, was decreased in Nrf2-activated macrophages and, as expected, when Nrf2 was disrupted in these cultures, loss of the protection against inflammation was observed (Thimmulappa et al., 2007). Further reinforcing evidence that the transcription factor Nrf2 can prevent oxidative stress in inflammation was demonstrated in a study using peritoneal macrophage cultures that were isolated from mice without the gene glutathione peroxidase-1 (GPX). GPX is a selenium-dependent enzyme that catalyzes the reduction of hydrogen perioxides to water and lipid hydroperoxide radicals to alcohols using GSH for donation of electrons in these processes. Nrf2 regulates gene expression levels of GPX. In this study, GPX-KO and WT macrophages were treated with LPS, interferon-gamma (IFN-gamma), and diquat, a super oxide anion-generating molecule. Peritoneal macrophages from GPX-KO mice showed significantly enhanced levels of oxidative stress as detected by levels of NO− and protein carbonyls (Fu et al., 2001). Our studies did indeed see increased gene expression levels of enzymes involved in the generation of ROS and RNS in mice with EAE supporting these concepts. More specifically, iNOS and the multimeric subunits of NADPH oxidase were all significantly increased in Nrf2-KO when compared with WT mice. Our future studies will include direct measurement of oxidative stress in a cell-specific manner.
Implications that Nrf2 modulates autoimmune diseases are found in studies investigating aged Nrf2-KO mice. Systemic lupus erythematous and autoimmune nephritis diseases have been documented in female Nrf2-KO mice that were over 12 months of age (Li et al., 2004; Yoh et al., 2001). Auto-antibodies against double-stranded DNA and Smith antigen were detected in the serums from these mice. In addition, deposition of IgG and IgM was noted in kidney, liver, heart, and brain. Since Nrf2-deficient mice have a decreased capacity to compensate for conditions of oxidative stress, acute and chronic cell damage can occur. Increased damage resulting from lipid peroxidation as well as modifications of DNA and proteins can trigger immunologic reactions leading to dysregulated autoimmunity responses (Li et al., 2004). Of note, the studies described in this manuscript were performed using Nrf2-KO mice that were 2–5 months of age, a period of time that is well before the onset of the endogenous autoimmune disease.
Further evidence of increased neuroinflammation in Nrf2-KO mice was demonstrated by heightened reactive microgliosis in spines from Nrf2-KO mice with EAE. Iba1-positive microglia were clearly visible in immunostained sections of spine as was increased gene expression levels for Iba1. However, GFAP staining for reactive astrocytes was increased similarly in spines from WT and Nrf2-KO with EAE. Interestingly, many of the GFAP-positive cells from Nrf2-KO appeared damaged as if sick or unhealthy. We speculated that these Nrf2-deficient astrocytes, unable to mount an adequate response to the increased oxidative stress generated by reactive microglia, had become diseased themselves. In fact, it was presented in active and chronic active MS lesions that NQO1, an Nrf2-driven antioxidant enzyme, was significantly upregulated in hypertrophic astrocytes and myelin-laden macrophages, underlining the importance of Nrf2 activation as an endogenous response to oxidative stress (van Horssen et al., 2006, 2008). Previous studies from our laboratory have described similar neuroinflammatory responses. Kainate-induced loss of neurons in the hippocampus of Nrf2-KO mice had enhanced microgliosis. It was postulated that this heightened microglia response exacerbated the neuronal damage that was observed (Kraft et al., 2006). Similar to that observed in our EAE studies, the processes of reactive astrocytes from Nrf2-KO mice appeared broken and damaged. In another study, astrocyte-selective activation of Nrf2 was able to confer protection to motor neurons in a mouse model of amyotrophic lateral sclerosis and was followed by a delayed astrocyte and microglia activation (Vargas et al., 2008). A more recent study by Innamorato et al. found that Nrf2-KO mice injected with LPS were hypersensitive and evidence of a greater influx or activation of macrophage/microglia in the hippocampus which was not prevented by pretreatments with an Nrf2-activating compound, sulforaphane (Innamorato et al., 2008). Enhanced adaptive neuroinflammatory response was also observed in spines from Nrf2-KO mice with EAE. Gene expression levels for CD4, CD8, and CD19 (B cell)-positive lymphocytes were all increased in WT and Nrf2-KO mice with EAE. CD4 and CD19 expression levels were greater in the Nrf2-KO and were significantly different. With respect to generation of oxidative stress, these lymphocytic cells contribute by chemotactically attracting microglia/macrophages to sites of disease or damaged tissue. (Of note, the robust changes seen by measuring expression levels of specific genes associated with inflammatory cells allow for a quantitative measure of cell infiltration observed in the spines from mice with EAE that cannot be achieved simply by immunostaining.) In all cases discussed, Nrf2 was implicated in the mitigation of neuroinflammation as well as neuroprotection.
In conclusion, we postulate that the absence of Nrf2-mediated protective mechanisms against oxidative stress exacerbates EAE-induced neuroinflammation due to ongoing cellular and tissue damage by the inflammatory-mediated generation of ROS and RNS. Damaged tissue in turn reactivates resident microglia and astrocytes, and continuous activation of an adaptive immune response can also result. Alleviation of neuroinflammatory-induced oxidative stress via activation of Nrf2 may thus prove to be an important adjuvant therapy in the treatment of MS as well as other neurodegenerative diseases.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environment Health Sciences grant (NIEHS; R01-ES10042, R01-ES08089).
Supplementary Material
Acknowledgments
We would like to thank Toshi Kinoshita from the Histopathology Core Service, Department of Pathology and Laboratory Medicine-University of Wisconsin, for his expert histology assistance.
References
- Amor S, Smith PA, Hart B, Baker D. Biozzi mice: of mice and human neurological diseases. J. Neuroimmunol. 2005;165:1–10. doi: 10.1016/j.jneuroim.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc. Natl Acad. Sci. USA. 2005;102:244–249. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc. Natl Acad. Sci. USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl Acad. Sci. USA. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Vargas MR, Pani AK, Smeyne RJ, Johnson DA, Kan YW, Johnson JA. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson's disease: critical role for the astrocyte. Proc. Natl Acad. Sci. USA. 2009;106:2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhib-Jalbut S. Pathogenesis of myelin/oligodendrocyte damage in multiple sclerosis. Neurology. 2007;68:S13–21. doi: 10.1212/01.wnl.0000275228.13012.7b. discussion S43–54. [DOI] [PubMed] [Google Scholar]
- Durchdewald M, Beyer TA, Johnson DA, Johnson JA, Werner S, auf dem Keller U. Electrophilic chemicals but not UV irradiation or reactive oxygen species activate Nrf2 in keratinocytes in vitro and in vivo. J. Invest. Dermatol. 2007;127:646–653. doi: 10.1038/sj.jid.5700585. [DOI] [PubMed] [Google Scholar]
- Fu Y, McCormick CC, Roneker C, Lei XG. Lipopolysaccharide and interferon-gamma-induced nitric oxide production and protein oxidation in mouse peritoneal macrophages are affected by glutathione peroxidase-1 gene knockout. Free Radic. Biol. Med. 2001;31:450–459. doi: 10.1016/s0891-5849(01)00607-4. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Lastres-Becker I, Cuadrado A. Role of microglial redox balance in modulation of neuroinflammation. Curr. Opin. Neurol. 2009;22:308–314. doi: 10.1097/WCO.0b013e32832a3225. [DOI] [PubMed] [Google Scholar]
- Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, O'Connor T, Yamamoto M. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2) Mol. Cell. Biol. 2004;24:36–45. doi: 10.1128/MCB.24.1.36-45.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, O'Connor T, Yamamoto M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Townsend JA, Kraft AD, Johnson JA. Nrf2-mediated protection against 6-hydroxydopamine. Brain Res. 2007;1144:192–201. doi: 10.1016/j.brainres.2007.01.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, Chen PC. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann. N Y Acad. Sci. 2008;1147:61–69. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl Acad. Sci. USA. 2004;101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J. Neurochem. 2006;98:1852–1865. doi: 10.1111/j.1471-4159.2006.04019.x. [DOI] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- Lee JM, Li J, Johnson DA, Kan YW, Johnson JA. Nrf2, a multi-organ protector? FASEB J. 2005;19:1061–1066. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J. Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol. Sci. 2005;83:313–328. doi: 10.1093/toxsci/kfi027. [DOI] [PubMed] [Google Scholar]
- Li J, Lee JM, Johnson JA. Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J. Biol. Chem. 2002;277:388–394. doi: 10.1074/jbc.M109380200. [DOI] [PubMed] [Google Scholar]
- Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genomics. 2004;18:261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- Liu H, Dinkova-Kostova AT, Talalay P. Coordinate regulation of enzyme markers for inflammation and for protection against oxidants and electrophiles. Proc. Natl Acad. Sci. USA. 2008;105:15926–15931. doi: 10.1073/pnas.0808346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JS, Zhao ML, Brosnan CF, Lee SC. Expression of inducible nitric oxide synthase and nitrotyrosine in multiple sclerosis lesions. Am. J. Pathol. 2001;158:2057–2066. doi: 10.1016/S0002-9440(10)64677-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N. Engl. J. Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Fuchs RJ, Malhotra D, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Preclinical evaluation of targeting the Nrf2 pathway by triterpenoids (CDDO-Im and CDDO-Me) for protection from LPS-induced inflammatory response and reactive oxygen species in human peripheral blood mononuclear cells and neutrophils. Antioxid. Redox Signal. 2007;9:1963–1970. doi: 10.1089/ars.2007.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Lee H, Rangasamy T, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006a;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Scollick C, Traore K, Scollick C, Traore K, Bream HJ, Trush MA, Liby KT, Sporn MB, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006b;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Horssen J, Schreibelt G, Bo L, Montagne L, Drukarch B, van Muiswinkel FL, de Vries HE. NAD(P)H:quinone oxidoreductase 1 expression in multiple sclerosis lesions. Free Radic. Biol. Med. 2006;41:311–317. doi: 10.1016/j.freeradbiomed.2006.04.013. [DOI] [PubMed] [Google Scholar]
- van Horssen J, Schreibelt G, Drexhage J, Hazes T, Dijkstra CD, van der Valk P, de Vries HE. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008;45:1729–1737. doi: 10.1016/j.freeradbiomed.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008;28:13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60:1343–1353. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- Zeis T, Probst A, Steck AJ, Stadelmann C, Bruck W, Schaeren-Wiemers N. Molecular changes in white matter adjacent to an active demyelinating lesion in early multiple sclerosis. Brain Pathol. 2008;19:459–466. doi: 10.1111/j.1750-3639.2008.00231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.