

Substoichiometric enantioinduction in the venerable Claisen rearrangement has remained a formidable challenge to the increasingly diverse strategies for catalysis of organic reactions.1 Chiral Lewis acids2,3 and Jacobsen’s ureas4 can induce excellent levels of enantioselectivity in a variety of Claisen rearrangements, but suffer from slow catalyst turnover. In seeking to devise a catalytic Claisen reaction that overcomes this limitation, we considered the possibility of an enantioselective variant of the Coates-Claisen reaction5,6 of enols and acetals of unsaturated aldehydes that would give lactone products as a means of catalyst turnover. In this communication, we document the use of chiral N-heterocyclic carbenes as catalysts for highly enantioselective Claisen rearrangments via the intermediacy of catalytically generated α,β-unsaturated acyl azoliums (Scheme 1).
Scheme 1.

Acyl azoliums are fascinating reactive intermediates with chemistry quite distinct from that of other activated carboxylic acid derivates.7 Our recent work,8 and that of many other groups,9 has focused on their catalytic generation by internal redox reactions of α-functionalized aldehydes, but these species have long been studied for their unusual reactivity and role in biochemical pathways.7,10 Unlike other acylating agents, acyl azoliums display a high preference for ester formation or hydrolysis rather than amide formation.11 This is attributed to the rapid formation of kinetically important hydrates or hemiacetals that undergo general base catalyzed C–C bond cleavage in the acid or ester forming step.7 Therefore, trapping acyl azolium I with a suitable enol should lead to meta-stable hemiacetal II poised for Claisen rearrangement followed by lactonization to effect catalyst turnover (Scheme 1). Importantly, Zeitler has invoked the intermediacy of acyl azolium I in the NHC-catalyzed redox esterification of ynals to give (E)-α,β-unsaturated esters.9b
We selected catalytic, enantioselective Claisen rearrangements of kojic acid derivatives as a starting point for our studies. Wender and others hve established the synthetic utility of the Claisen rearrangement in elaborating readily available kojic acid into platforms for complex molecule synthesis,12 but also noted the failure of chiral Lewis acid approaches,13 such as those documented by Hiersemann,3 to suitably control the absolute stereochemistry. Elegant work on chiral transition metal-catalyzed Claisen rearrangements, including that of Kozlowski and Mikami,14 do not appear to be applicable to this substrate class.
The combination of kojic acids, ynals, and chiral N-mesityl substituted precatalyst 1 led to the formation of somewhat unstable dihydropyranones. Upon completion of the reaction, simply stirring these products in MeOH for 6 hours led to ring opening, a procedure we adopted for product isolation (Table 1). A standard course of optimization identified the conditions shown in Table 1. The addition of a catalytic base (NEt3, N-Me-morpholine) increased the reaction rates, but led to side products and a decrease in enantiopurity. In the absence of a triazolium salt, no reaction occurred regardless of whether or not base was present. Within our focus on reactions of kojic acid derivatives, the reaction proceeded with a broad range of ynals including both aromatic and aliphatic derivatives, all in good yield and with exceptional enantioselectivity.
Table 1.
Catalytic, enantioselective couplings with kojic acids.a
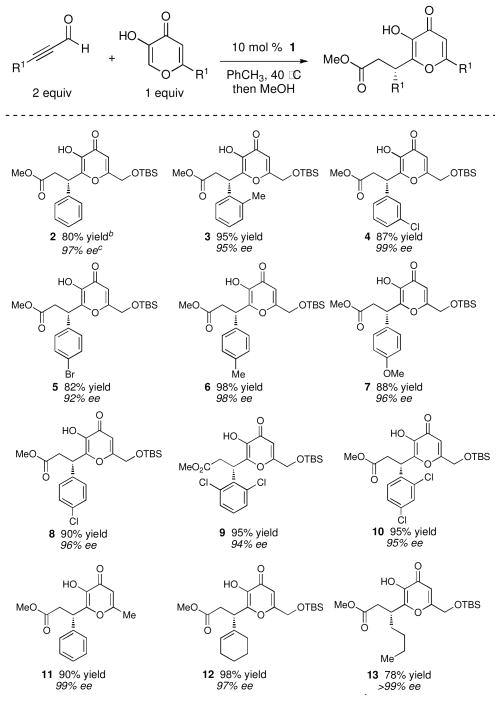 |
Reaction conditions: 0.1 M PhCH3, 24 h.
Yield refers to isolated yield after chromatography.
Absolute configuration determined by conversion to (S)-phenylsuccinic acid.
Although we have not exhaustively explored the scope of this reaction, we have found that many enolic compounds couple with ynals to give the expected products. We were pleased to find that pyruvic esters are excellent substrates, provided that a weak amine base is added to promote enol formation. Phenolic compounds, such as naphthols, give Claisen products but with diminished levels of selectivity. The key α,β-unsaturated acyl triazolium I could be generated from other substrates, including α,β-unsaturated aldehdyes via in situ oxidation.15
In all prior reports on the use of azolium salts as catalysts or precatalysts, including our own work, the addition of a base was required, presumably to generate the N-heterocyclic carbene, which is thought to be the active nucleophilic catalyst. This annulation proceeds smoothly in absence of added base, but is slower and counterion dependent; in contrast, NHC-catalyzed reactions in the presence of base show no counterion effects.16 In the absence of base, 1H NMR investigations reveal no detectable loss of the azolium C-2 proton during the course of the reaction.17 While we cannot rule out alternative mechanisms at this time,18 our current understanding is that the chloride ion plays the role of base in generating a trace amount of the nucleophilic carbene, which quickly attacks the aldehyde to initiate the catalytic cycle. This is supported by comparing the reactivity of precatalysts bearing different counterions (Table 2). The acetate precatalyst is much more reactive than the chloride, but led to product epimerization. Precatalysts with less basic counterions, such as SbF6− or ClO4− are unreactive. The addition of a catalytic amount of a weak amine base restores the activity to levels similar to that observed with the chloride counterion.
Table 2.
Effect of azolium counterion on conversion.a
 | |||
|---|---|---|---|
| X = | % conv. 1 h | % conv. 2.5 h | % conv. 12 h |
| Cl− | 10 | 16 | 70 |
| OAc− | 100 | na | na |
| CF3CO2− | 7 | 10 | 20 |
| ClO4− | 0 | 0 | trace |
| SbF6− | 0 | 0 | 0 |
| SbF6− + NMM(30%) | 30 | 35 | 86 |
Determined by 1H NMR analysis against an internal standard.
During the preparation of this manuscript, Lupton reported a racemic dihydropyranone-forming annulation from enolates via α,β-unsaturated acyl azolium species, promoted by imidazolium-derived NHCs under basic conditions,19 Although the starting materials and reaction conditions are different from our process, the postulated intermediates are identical and the reactions are likely to be mechanistically related. Lupton proposes a direct Michael addition of an enolate to I but our observations (vide supra) to date are more consistent with the Claisen mechanism shown in Figure 1.20
Figure 1.

Possible mechanistic pathways for NHC-catalyzed annulation.
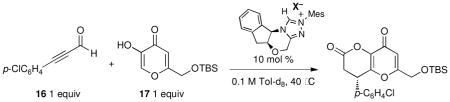
We considered five possible rate-determining steps for the overall catalytic reaction, listed in red in Figure 1: 1) the initial redox reaction/protonation to form I; 2) the 1,2 addition of kojic acid to give hemiacetal II; 3) the conjugate addition of kojic acid to III; 4) the Claisen rearrangement of II to give III; 5) the lactonization of III to give IV and effect catalyst turnover. These mechanistic possibilities can be differentiated by analysis of the reaction kinetics. Kinetic studies of azolium-catalyzed processes such as the benzoin and Stetter reactions are often plagued by the presence of multiple rate determining steps and catalyst inhibition;21 however, this system proved somewhat more tractable to analysis. We determined the empirical rate law for the reaction of kojic acid 17 with 2,4-dichlorophenyl substituted ynal 18 to be:
| (1) |
From this experimental rate law, we also determined the activation parameters from the rate constant via an Eyring plot, which revealed an activation enthalpy of 15.3 kcal/mol and activation entropy of −25.5 cal/K·mol. The lactonization is excluded as the rate-limiting step by the activation parameters. The redox reaction–protonation step is excluded by isotopic labeling experiments, which show no kinetic isotope effect for the protonation.22 The 1,2 addition of kojic acid to I, as well as the 1,4-addition pathway, are excluded by the observed negative order of the reaction in kojic acid. These experimental results are consistent with a derived rate law (eq 2) for the Claisen rearrangement that takes into consideration generation of the NHC from the azolium, a steady-state approximation of the initial aldehyde–NHC adduct, and rapid formation of hemiacetal II.23
| (2) |
This rate law rationalizes the partial positive order of the reaction in aldehyde and the negative order of kojic acid, which inhibits generation of the N-heterocyclic carbene by acting as a general acid (HX in eq 2). It explains the observation that the reaction rate increases in the presence of added base by generating more of the NHC and provides a role for the counterion of the azolium salt as a genral base to generate the active catalyst (X− in eq 2).
Other observations are also most consistent with the Claisen pathway. All attempts to effect conjugate additions with nucleophiles that cannot undergo Claisen rearrangement, such as indoles or hydroxamic acids,24 have failed; in most cases 1,2-adducts are obtained instead. The well-studied chemistry of acyl azoliums documents the rapid formation of hydrates and hemiacetals in their acylation reactions.7,25 These hydrates are stable under acidic conditions and require general base catalysis to give hydrolysis products. This may explain the cleaner and higher yielding outcome of this reaction under our acidic conditions. The reaction is relatively insensitive to sterics; an ortho,ortho-dichloro derivative is competitive with its ortho,para-isomer, which would not be expected in the conjugate addition pathway (see Supporting Information).26 Finally, the excellent enantioselectivity is best rationalized by a reversible, but stereochemically determining, 1,2-addition adjacent to the chiral triazolium to give the Claisen precursor. In contrast, a C–C bond forming conjugate addition would be an irreversible stereochemically determining step.
Although we cannot completely rule out a short-lived enolate-acyl azolium ion pair, our results, including the activation entropy, are consistent with the detailed studies of Coates and Curran on the related Claisen-rearrangement of 2-alkoxy-substituted vinyl ethers. More importantly, these studies provide a new mode of substrate activation unique to chiral N-heterocyclic carbenes and, via their ability to access multiple catalytically generated reactive species in a tandem fashion,27 a pathway to induce high levels of enantioselectivity and provide solution to catalyst turnover in a catalytic Claisen manifold.
Supplementary Material
Scheme 2.

Azolium-catalyzed annulations of ynals via Claisen rearrangements.
Acknowledgments
We are grateful to NIGMS (National Institutes of Health, GM–079339), the National Science Foundation (CHE-0449587) and Bristol Myers Squibb for support of this research. J.K. is a graduate fellow of the Royal Thai Government. We thank Marisa Kozlowski (UPenn), Franziska Schönebeck (ETH-Zürich) and John A. Kowalski (GlaxoSmithKline) for helpful discussions.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Ito H, Taguchi T. Chem Soc Rev. 1999;28:43–50. [Google Scholar]; (b) Hiersemann M, Abraham L. Eur J Org Chem. 2002;9:1461–1471. [Google Scholar]; (c) Hiersemann M, Nubbemeyer U, editors. The Claisen Rearrangment. Wiley-VCH; Weinheim, Germany: 2007. [Google Scholar]
- 2.(a) Maruoka K, Banno H, Yamamoto H. J Am Chem Soc. 1990;112:7791–7793. [Google Scholar]; (b) Yoon TP, MacMillan DWC. J Am Chem Soc. 2001;123:2911–2912. doi: 10.1021/ja015612d. [DOI] [PubMed] [Google Scholar]
- 3.Abraham L, Czerwonka R, Hiersemann M. Angew Chem, Int Ed. 2001;40:4700–4703. doi: 10.1002/1521-3773(20011217)40:24<4700::aid-anie4700>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Uyeda C, Jacobsen EN. J Am Chem Soc. 2008;130:9228–9229. doi: 10.1021/ja803370x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Coates RM, Shah SK, Mason RW. J Am Chem Soc. 1982;104:2198–2208. [Google Scholar]; (b) Coates RM, Hobbes SJ. J Org Chem. 1984;49:140–152. [Google Scholar]; (c) Coates RM, Rogers BD, Hobbs SJ, Peck DR, Curran DP. J Am Chem Soc. 1987;109:1160–1170. [Google Scholar]
- 6.For related reactions thought to proceed by Claisen rearrangement, see: Dolby LJ, Elliger CA, Esfandri S, Marshall KS. J Org Chem. 1968;33:4508–4511.Clarke DG, Crombie L, Whiting DA. J Chem Soc Perkin Trans 1. 1974:1007–1015.
- 7.(a) Bruice TC, Kundu NG. J Am Chem Soc. 1966;88:4097–4098. [Google Scholar]; (b) Lienhard G. J Am Chem Soc. 1966;88:5642–5649. doi: 10.1021/ja00969a017. [DOI] [PubMed] [Google Scholar]; (c) Owen TC, Richards A. J Am Chem Soc. 1987;109:2520–2521. [Google Scholar]; (d) Owens TC, Harris JN. J Am Chem Soc. 1990;112:6136–6137. [Google Scholar]
- 8.(a) Chow YK, Bode JW. J Am Chem Soc. 2004;126:8126–8127. doi: 10.1021/ja047407e. [DOI] [PubMed] [Google Scholar]; (b) Sohn SS, Bode JW. Org Lett. 2005;7:3873–3876. doi: 10.1021/ol051269w. [DOI] [PubMed] [Google Scholar]
- 9.(a) Reynolds NT, Read de Alaniz J, Rovis T. J Am Chem Soc. 2004;126:9518–9519. doi: 10.1021/ja046991o. [DOI] [PubMed] [Google Scholar]; (b) Zeitler K. Org Lett. 2006;8:637–640. doi: 10.1021/ol052826h. [DOI] [PubMed] [Google Scholar]
- 10.(a) Khaleeli N, Li R, Townsend CA. J Am Chem Soc. 1999;121:9223–9224. [Google Scholar]; (b) Merski M, Townsend CA. J Am Chem Soc. 2007;129:15750–15751. doi: 10.1021/ja076704r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Movassaghi M, Schmidt MA. Org Lett. 2005;7:2453–2456. doi: 10.1021/ol050773y. [DOI] [PubMed] [Google Scholar]; (b) Bode JW, Sohn SS. J Am Chem Soc. 2007;129:13798–13799. doi: 10.1021/ja0768136. [DOI] [PubMed] [Google Scholar]; (c) Vora HU, Rovis T. J Am Chem Soc. 2007;129:13796–13797. doi: 10.1021/ja0764052. [DOI] [PMC free article] [PubMed] [Google Scholar]; De Sarkar S, Grimme S, Studer A. J Am Chem Soc. 2010;132:1190–1191. doi: 10.1021/ja910540j. [DOI] [PubMed] [Google Scholar]
- 12.(a) Mascarenas JL, Wender PA. Tetrahedron Lett. 1992;33(16):2115–2118. [Google Scholar]; (b) McDonald FE, Wender PA. J Am Chem Soc. 1990;112:4956–4958. [Google Scholar]; (c) Mascarenas JL, Wender PA. J Org Chem. 1991;56:6267–6269. [Google Scholar]; (d) Xiong X, Pirrung MX. Org Lett. 2008;10:1151–1154. doi: 10.1021/ol800058d. [DOI] [PubMed] [Google Scholar]
- 13.Wender PA, D’Angelo N, Elitzin VI, Ernst M, Jackson-Ugueto EE, Kowalski JA, McKendry S, Rehfeuter M, Sun R, Voigtlaender D. Org Lett. 2007;9:1829–1832. doi: 10.1021/ol0705649. [DOI] [PubMed] [Google Scholar]
- 14.(a) Linton EC, Kozlowski MC. J Am Chem Soc. 2008;130:16162–16163. doi: 10.1021/ja807026z. [DOI] [PubMed] [Google Scholar]; (b) Akiyama K, Mikami K. Tetrahedron Lett. 2004;45:7217–7220. [Google Scholar]
- 15.(a) Maki BE, Chan A, Phillips EM, Scheidt KA. Org Lett. 2007;9:371–374. doi: 10.1021/ol062940f. [DOI] [PubMed] [Google Scholar]; (b) Guin J, De Sarkar S, Grimme S, Studer A. Angew Chem, Int Ed. 2008;47:8727–8730. doi: 10.1002/anie.200802735. [DOI] [PubMed] [Google Scholar]; (c) Inoue H, Higashiura K. J Chem Soc Chem Commun. 1980;12:549–550. [Google Scholar]
- 16.Struble JR, Kaeobamrung J, Bode JW. Org Lett. 2008;10:957–960. doi: 10.1021/ol800006m. [DOI] [PubMed] [Google Scholar]
- 17.Imidazolinium salts substituted at the 2-postion, which cannot form carbenes, have been employed as catalyzed for aza-Diels Alder reactions. In this case the salts are thought to act as Lewis acids: Jurcik V, Wilhelm R. Org Biomol Chem. 2005;3:239–244. doi: 10.1039/b415023f.For a recent report on the use of imidazolium salts for acid catalyzed acetylations, see: Myles L, Gore R, Spulák M, Gathergood N, Connon SJ. Green Chem. 2010 doi: 10.1039/c003301d. in press.
- 18.For an alternative mechanism for the addition of azolium salts to aldehydes, see: Crane EJ, III, Washabaugh MW. Bioorganic Chem. 1991;19:351–368.
- 19.Ryan SJ, Candish L, Lupton DW. J Am Chem Soc. 2009;131:14176–14177. doi: 10.1021/ja905501z. [DOI] [PubMed] [Google Scholar]
- 20.Townsend has postulated a conjugate addition to an α,β-unsaturated acyl azolium in the biosynthesis of clavulanic acid, see ref. 10.
- 21.White MJ, Leeper FJ. J Org Chem. 2001;66:5124–5131. doi: 10.1021/jo010244h. [DOI] [PubMed] [Google Scholar]
- 22.Noyce DS, Pollack RM. J Am Chem Soc. 1969;91:119–214. [Google Scholar]
- 23.See the Supporting Information for the derivation of this rate law.
- 24.Hydroxamic acids are known to be excellent nucleophiles for conjugate additions, see: Ibrahem I, Rios R, Vesely J, Zhao G-L, Córdova Chem Comm. 2007;8:849–851. doi: 10.1039/b613410f.Yamagiwa N, Hongbo Q, Matsunaga S, Shibasaki M. J Am Chem Soc. 2005;127:13419–13427. doi: 10.1021/ja054066b.
- 25.Owen TC. J Heterocyclic Chem. 1990;27:987–990. [Google Scholar]
- 26.Usera AR, Posner GH. J Org Chem. 2007;72:2329–2334. doi: 10.1021/jo062155g. [DOI] [PubMed] [Google Scholar]
- 27.For our work on other enantioselective tandem NHC-catalyzed reactions, see: Chiang PC, Kaeobamrung J, Bode JW. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543.Kaeobamrung J, Bode JW. Org Lett. 2009;11:677–680. doi: 10.1021/ol802739d.He M, Bode JW. J Am Chem Soc. 2008;130:418–419. doi: 10.1021/ja0778592.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.