

Abstract
Machine learning methods pool diverse information to perform computer-assisted diagnosis and predict future clinical decline. Here we introduce a machine learning method to boost power in clinical trials. We created a Support Vector Machine algorithm that combines brain imaging and other biomarkers to classify 737 ADNI subjects as AD, MCI, or normal controls. We trained our classifiers based on example data including: MRI measures of hippocampal, ventricular, and temporal lobe volumes, a PET-FDG numerical summary, CSF biomarkers (t-tau, p-tau and aβ42), ApoE genotype, age, sex, and body mass index. MRI measures contributed most to AD classification; PET-FDG and CSF biomarkers, particularly aβ42, contributed more to MCI classification. Using all biomarkers jointly, we used our classifier to select the one-third of the subjects most likely to decline. In this sub-sample, fewer than 40 AD and MCI subjects would be needed to detect a 25% slowing in temporal lobe atrophy rates with 80% power - a substantial boosting of power relative to standard imaging measures.
Keywords: Clinical Trial Enrichment, Alzheimer’s Disease, Mild Cognitive Impairment, Magnetic Resonance Imaging, Neuroimaging, Biomarkers, Classification, Support Vector Machines
1. Introduction
Alzheimer’s Disease (AD), the most common form of dementia, affects approximately 5.3 million people in the United States alone, and its prevalence continues to rise (Alzheimer’s Association, 2009). Research and therapeutic efforts also focus on subjects with Mild Cognitive Impairment (MCI) – an intermediate condition between healthy aging and AD – as they convert to AD at a heightened rate of 10-15% per year (Petersen et al., 1999). Multiple imaging biomarkers have been used for quantifying disease progression and measuring various aspects of AD pathology, such as amyloid and tau deposition, measured by Positron Emission Tomography (PET) and radiotracers that bind to the plaques and tangles in the brain (Klunk et al., 2004; Protas et al., 2010), metabolic decline or perfusion deficits assessed by fluoro-deoxyglucose PET (PET-FDG), brain atrophy on MRI, and risk factors that influence these measures (e.g. ApoE, cardiovascular risks, etc.) (Frisoni et al., 2010; Jack et al., 2010; Petersen, 2010).
Although the disease can be tracked in many ways, methods are also needed to integrate these multiple measures to achieve greater power in diagnosis and prognosis. Machine learning algorithms such as linear discriminant analysis, support vector machines, and boosting have recently been proposed to combine multiple AD features derived from brain imaging and other biomarkers, for AD and MCI classification. Several studies have performed diagnostic classification based on MRI scans, using measures such as whole-brain patterns of atrophy (Davatzikos et al., 2009; Mesrob et al., 2008), tissue densities from voxel-based morphometry (Vemuri et al., 2008) and cortical thickness (Lerch et al., 2008). Vemuri et al. (2008) assigned overall “scores” for each subject’s MRI – called the Structural Abnormality Index (STAND) - based on gray and white matter voxels that best differentiated AD patients from controls. In related work, Davatzikos et al. (2009) assigned “scores” to each subject’s MRI scan based on a minimal set of brain regions that best discriminated AD from normal controls in a training sample; their approached is termed Spatial Pattern of Abnormality for Recognition of Early Alzheimer’s disease, or SPARE-ED.
Researchers have also explored adding other predictors to improve the accuracy of MRI for computer-assisted diagnosis of AD and MCI, and for predicting whether a person will convert from MCI to AD in the near future. PET, for example, offers metabolic or perfusion-based information that complements measures of structural atrophy on MRI (Fan et al., 2008; Hinrichs et al., 2009). Vemuri et al. (2009) adjusted their STAND scores by incorporating demographic variables such as age, sex, and ApoE genotype, and this improved their classification accuracy. Additionally, MRI-based STAND scores were shown to improve the accuracy of CSF biomarkers for predicting cognitive decline, including total tau (t-tau), phosphorylated tau (p-tau) and the beta-amyloid isoform, aβ42 (Vemuri et al., 2009).
It is worth noting that MRI-based machine learning has been used widely for classification not only for AD, but also for predicting changes in patients with brain tumors (Lukas et al., 2004), aphasia (Wilson et al., 2009), autism (Ecker et al., 2010), psychosis (Koutsouleris et al., 2009) and even for classifying patterns of brain activation in functional MRI (Mourão-Miranda et al., 2005). Similar algorithms have been implemented to distinguish AD from other types of dementia such as frontotemporal dementia (Davatzikos et al., 2008; Klöppel et al., 2008). Support vector machines (SVMs) are one of the most widely-used and effective tools for classification of AD and other neurological disorders, and are used in many of the reports listed above. We therefore set out to test how well SVMs would perform for classifying patients as having AD and MCI based on multiple imaging and biological measures in ADNI, as well as for predicting imminent cognitive decline.
A second goal of this paper was to make a conceptual connection between sample size requirements for clinical trials and the power of classifiers to predict future decline. By using our classifiers to predict those most likely to decline, we tested the hypothesis that this subset might experience atrophic rates with greater effect sizes. This concept is termed clinical trial enrichment, as it seeks out a sub-sample of subjects who might be better candidates for demonstrating therapeutic effects, at least from a statistical standpoint (see Discussion for assumptions of this approach).
We recently found that regional numerical summaries derived from tensor-based morphometry of longitudinal MRI (over a 1-year interval) can reduce the estimated sample size requirements to 48 AD and 88 MCI subjects per arm of a hypothetical clinical trial (treatment versus placebo), for detecting a 25% reduction in the mean annual temporal lobe atrophy rate with 80% power (Hua et al., 2009). Power was similar when 3 Tesla or 1.5 Tesla MRI scans were used (Ho et al., 2009); still higher power was possible for trials with longer follow-up intervals (Hua et al., 2010b). Other groups report comparable power for measures based on hippocampal volumes (Schuff et al., 2009). Through the use of multi-modality classifiers, these and other similar sample size estimates can presumably be reduced still further.
In this report, our goals were: (1) to statistically combine baseline MRI measures of hippocampal, temporal and ventricular volumes with age, sex, ApoE genotype and body mass index (BMI) for AD and MCI classification, (2) to examine how the best-performing predictors would be further enhanced by using information on CSF biomarkers and PET-FDG; (3) to evaluate this multi-modality approach for predicting cognitive decline in MCI, and, most importantly, (4) to assess whether we could expect to reduce clinical trial sample size estimates by using our classifiers to target those most likely to decline. Numerous structural MRI-based measures, including hippocampal and ventricular volumes, as well as other temporal lobe summaries, have already been validated as indicators of AD progression, particularly after the MCI stage (Frisoni et al., 2010). We hypothesized that using multiple MRI summaries (rather than choosing one) might offer complementary information to classify patients into the correct diagnostic categories and predict cognitive decline, thereby providing a new way to boost the power of clinical trials.
2. Methods
2.1. Subjects
Baseline neuroimaging and biomarker data were downloaded from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) public database (http://www.loni.ucla.edu/ADNI/Data/) on or before November 20, 2009 and reflect the status of the database at that point; as data collection is ongoing. ADNI is a large five-year study launched in 2004 with the primary goal of testing whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessments at multiple sites (as in a typical clinical trial), can replicate results from smaller single site studies measuring the progression of MCI and early AD. More sensitive and specific markers of early AD progression will help monitor the effectiveness of new treatments, and lessen the time and cost of clinical trials.
Available Data for Baseline Subjects
In what follows, sample sizes for analyses using different predictors are slightly different, as the study is ongoing and not all measures could be collected from all ADNI subjects. For our classification study based on baseline MRI numerical summaries, ApoE, age, sex and BMI, data were available from 737 ADNI subjects (158 AD: 75.4 ± 7.4 years of age, 366 MCI: 74.8 ± 7.3 years of age, and 213 controls: 76.0 ± 5.1 years of age). To equalize the sex distribution, we reduced the MCI subject set to a group of 264 sex-matched subjects. As there were 102 more men than women in the MCI group, we ranked the MCI males based on numbers assigned to them via a computerized random number generator and removed the first 102 to ensure that the elimination process was random and unbiased. For our next classification study, we were limited by the availability of PET-FDG and CSF data, so our studies included subsets of the subjects considered above (328 subjects after adding only CSF, 364 subjects after adding only PET-FDG, and 166 subjects after adding both CSF and PET-FDG). For the first part of the cognitive decline prediction study, we considered 64 sex-matched MCI subjects, of whom 12 converted to AD in 12 months. 64 subjects remained after selecting MCI subjects who had all biomarker information and equalizing the distribution of sex. The fraction of converters here (18.75%) is a slightly higher than the previously estimated rate of conversion in ADNI (13% according to Petersen et al., 2010); the rate is marginally higher as a subgroup of male non-converters was excluded to allow sex matching. A larger sample of 129 sex matched MCI subjects with a reduced number of biomarkers was considered for the second part of the same study, 22 of whom (17.05%) converted to AD within 12 months. Sex matching was performed through a random elimination process as described above. The subjects and biomarkers included in each study are summarized in Table 1.
Table 1.
ADNI subjects and biomarkers included in each study. Here we outline the subject samples analyzed for different classification tests. Subjects are split into independent training and testing samples approximately in a 3:1 ratio, except for the smaller studies, to ensure correct evaluation of classifier performance. The 3:1 ratio is used in several machine learning studies such as Vemuri et al. (2008a). MRI denotes that a 1.5T MRI scan was available; BMI denotes body mass index. CSF denotes that CSF-derived biomarkers were available
| Number of Subjects (Training + Testing) | ||||
|---|---|---|---|---|
| Study | Biomarkers | AD | MCI | CN |
| 1 | MRI, Age, ApoE, Sex, BMI | 158 (118 + 40) | 264 (184 + 80) | 213 (163 + 50) |
| 2a | MRI, Age, ApoE, CSF | 77 (57 + 20) | 158 (118 + 40) | 93 (68 + 25) |
| 2b | MRI, Age, ApoE, PET-FDG | 79 (59 + 20) | 191 (146 + 45) | 94 (74 + 20) |
| 2c | MRI, Age, ApoE, CSF, PET-FDG | 40 (20 + 20) | 83 (43 + 40) | 43 (23 + 20) |
| 3a | MRI, Age, ApoE, CSF, PET-FDG | - | 64 (41 + 23) | - |
| 3b | MRI, ApoE, PET-FDG | - | 129 (67 + 62) | - |
2.2. Biomarkers
For each subject, the biomarkers we considered included three MRI-derived numerical summaries, a PET-FDG numerical summary, and three CSF biomarkers (t-tau, p-tau and aβ42). In addition to MRI, PET-FDG and CSF can provide important functional and pathological information on AD progression (Jack et al., 2010). We also considered ApoE genotype (coded as 0, 1 or 2 for the number of E4 alleles), age, sex and BMI, as each can influence AD risk (Corder et al., 1993, Lindsay et al., 2002, Azad et al., 2007, Buchman et al., 2005). BMI was included as a number of recent studies found that higher BMI is associated with greater brain atrophy in normal elderly subjects (Raji et al., 2009), and in MCI and AD (Ho et al., 2010b). This effect still holds true after accounting for the effects of hypertension, diabetes, and the level of white matter hyperintensities (Ho et al., 2010b) on the brain. In addition, a commonly carried risk gene for obesity, FTO, was recently reported to be associated with the level of brain atrophy in the ADNI cohort (Ho et al., 2010a), so we included BMI as it is a cardiovascular risk factor associated with brain atrophy. Clinical biomarkers that were used in ADNI to determine diagnosis, such as the sum of boxes Clinical Dementia Rating (sobCDR) and other similar measures are used by physicians for making diagnoses and were therefore not used as features for classification to avoid circular inference. In fact, using sobCDR alone for classification led to almost perfect classification accuracy, as accuracy here is judged in terms of agreement with clinical diagnosis, the best available proxy when post mortem neuropathological data is not yet available. Instead, the annual rate of change in sobCDR was used as an outcome measure of cognitive decline to help define conversion from MCI to AD.
The MRI features included numerical summaries from the hippocampus, lateral ventricles and a TBM-derived measure of atrophy in the temporal lobes. The hippocampal summaries were volumes generated from an automatic segmentation method that we developed based on machine learning; we recently validated this method against manual gold standards (Morra et al., 2008; Morra et al., 2009; Morra et al., 2010). The ventricular summaries were volumes acquired from a semi-automated, multi-atlas segmentation technique that we developed (multi-atlas fluid image alignment or MAFIA; (Chou et al., 2008)). The temporal lobe summaries were obtained from an anatomically defined region-of-interest (ROI) on three-dimensional atrophy maps generated with tensor-based morphometry (Hua et al., 2008a; Hua et al., 2008b). PET-FDG numerical summaries were based on a pre-defined temporal lobe ROI (Landau et al., 2009). All imaging summaries were averaged for the lobes in the left and right brain hemispheres.
CSF samples were obtained through lumbar puncture, after an overnight fast. Samples from various sites were transferred, on dry ice, to the ADNI Biomarker Core Laboratory at the University of Pennsylvania Medical Center, where the levels of t-tau, p-tau and aβ42 are measured with a multiplex immunoassay platform under the direction of Drs. Leslie Shaw and John Trojanowski. ApoE genotyping was performed on DNA samples from subjects’ blood. Genomic DNA samples were analyzed using the Human610-Quad BeadChip (Illumina, Inc. San Diego, CA) at the University of Pennsylvania. Demographic data were obtained from https://www.loni.ucla.edu/ADNI/Data/. It should be emphasized that only baseline values of the biomarkers were used for prediction.
2.3. Support Vector Machines
SVMs are a type of machine learning or pattern recognition method that can be used to classify a dataset into different groups, based on multiple features, or measures, available for each subject (see e.g., Morra et al., 2009b). As with linear discriminant analysis, a number of observations about a subject (here the imaging and other measures) may be assembled into a vector, with as many components as there are measures. Then a mathematical function is estimated (or “learned”) that best combines these features to give an output that indicates, as accurately as possible, which group the individual belongs to. For an introduction to SVM - comparing it to simpler methods such as linear discriminant analysis (LDA) - please see our tutorial (Morra et al., 2009b). As mentioned in the introduction, SVM was chosen as a machine learning algorithm for this report due to its successful performance in the previous AD literature (Davatzikos et al., 2009, Fan et al., 2008, Mesrob et al., 2008, Vemuri et al., 2008), and for other neurobiological applications (Ecker et al., 2010, Koutsouleris et al., 2009, Wilson et al., 2009). SVMs may be considered as generalizations of linear regression, which use a supervised learning method to fit a classification function to the data in a training set of labeled observations. Other types of classifiers, such as adaptive boosting (Freund and Schapire, 1999; Morra et al., 2010), may also be useful for subject classification based on multiple biomarker measures, as they optimally combine predictors that perform weakly individually, but strongly in combination.
SVM is formulated as an optimization problem. Given a set of training data with corresponding class labels, a hyperplane is sought that maximizes the margin (a measure of the ability to differentiate) between different classes. This hyperplane, computed from a training set of example data, can then be utilized to classify newly presented (independent) testing data sets. Data consist of a set of vectors (x1, …, xn) where each vector contains a number of features and the class labels are scalars (y1, …, yn) where yi is either 1 or -1 in a 2-class problem. The optimization problem for a linear SVM is written as subject to yi (xi · w + b≥1, where w and b represent the normal vector to and the intercept of the hyperplane respectively. For cases where a linear surface (hyperplane) cannot effectively separate the data, nonlinear kernels, such as radial basis functions (RBFs), are incorporated into the optimization problem. Additionally, “slack variables” may be introduced with a tunable parameter, C, to allow for a balance between misclassifications and the width of the margin. With this modification, the optimization problem may be restated as subject to yi(xi · w + b)≥1−ξi, where ξi is the slack variable for each i (Vapnik, 1998, Burges, 1998). SVMs may also be utilized for regression, where instead of a binary output, it would predict a continuous output for each subject’s input vector, x. We performed our experiments using the LS-SVM package for classification and regression (Suykens and Vandewalle, 1999) in Matlab (MathWorks, Natick, MA).
2.4. Training and Testing
We divided AD, MCI and control subjects randomly into training and testing sets as shown in Table 1. The training sets were used for parameter optimization (regularization parameter C for a linear kernel; C and kernel-specific parameter, σ, for an RBF kernel) and for leave-one-out cross-validation. The SVM models were tested on independent testing sets to ensure generalizability. Receiver operating characteristic (ROC) curves were obtained to demonstrate the trade-off between sensitivity and specificity. ROC curves were compared, to evaluate different classifiers, using a statistical method developed for ROC analysis (Hanley and McNeil, 1983) in the MedCalc Statistical Software (MedCalc, Mariakerke, Belgium). When SVM was implemented for prediction instead of classification, mean squared errors were used for comparison, instead of misclassification errors.
2.5. Power Analysis
A power analysis was defined by the ADNI Biostatistics Core to estimate the sample size required to detect a 25% reduction in the mean annual rate of atrophy, using a two-sided test and standard significance level (α=0.05) for a hypothetical two-arm study (treatment versus placebo), with 80% power (this number is referred to as n80, and smaller numbers are better). The formula is , where σ and β refer to the mean and standard deviation in the atrophic rates respectively, β is set to be 0.05, and the desired power is 80%. Atrophic rates were determined based on a statistically-defined ROI by training on 22 AD subjects, as described more fully in (Hua et al., 2009). Brain atrophy rates measured by MRI correlate with the progression of Alzheimer’s disease, and offer baseline and transitional predictive power for diagnosis, making them clinically relevant endpoints for power analysis (Duara et al., 2008, Fox et al., 2000, Jack et al., 2004).
3. Results
3.1. AD and MCI Classification based on MRI markers, ApoE genotype and demographic information
We first used the 3 MRI-derived summaries, ApoE genotype and demographic variables (age, sex and BMI) for AD and MCI classification with 635 ADNI subjects. SVM training was performed with all seven features using a linear kernel with C = 1, and the contributions of the different biomarkers were put into a rank order (best to worst) based on their SVM weights, assessed by wi2 in the notation of SVM described in the methods. The rank orders are shown in Table 2.
Table 2.
Rank order list with relative SVM weights for MRI, ApoE, Age, Sex and BMI in AD and MCI classification. Hippocampal volumes were the most influential feature for differentiating AD from controls, closely followed by ApoE genotype, which outperformed all the other MRI-derived markers. For classifying subjects as either MCI or controls, the exact same features were useful, in the same order of priority. This is somewhat in line with expectation, as hippocampal volume is so widely used and is perhaps the most well-validated MRI measure in AD studies. This rank order refers to a situation in which all measures are used jointly for classification. Also, the gray highlighted measures are the ones that, when used jointly, gave the best classification accuracy in our independent test datasets (see Figure 1 for ROC curves)
| Biomarker |
||
|---|---|---|
| Rank | AD vs. control (weight / wi2) |
MCI vs. control (weight / wi2) |
| 1 | MRI Hipa 0.1664 |
MRI Hip 0.1045 |
| 2 | ApoE 0.1063 |
ApoE 0.0938 |
| 3 | Age 0.0369 |
Age 0.0188 |
| 4 | MRI Ventb 0.0349 |
MRI Vent 0.0103 |
| 5 | MRI Tempc 0.0210 |
MRI Temp 0.0045 |
| 6 | BMI 0.0147 |
BMI 0.0019 |
| 7 | Sex 0.0013 |
Sex 0.0009 |
Hippocampal volume summary
Ventricular volume summary
Temporal lobe summary from tensor-based morphometry (TBM)
Groups of biomarkers yielding the highest leave-one-out accuracy are highlighted.
We then aimed to find the top N (N ranging from 1 to 7) features that yielded the highest leave-one-out accuracy in the training set, using an RBF kernel with parameter optimization. Both linear and RBF kernels identified the same set of top features, but the RBF kernel gave better performance, so we only present those results here. For AD vs. control, the best combination included the top 4 features (baseline hippocampal and ventricular volumes, as well as ApoE and age); this joint classifier yielded a leave-one-out accuracy of 82.21% correct classification, with a corresponding area under the ROC curve (AUC) of 0.945, which is relatively high. For classifying MCI vs. control, the best feature combination consisted of the top 3 (baseline hippocampal volume, ApoE and age), which gave 70.89% accurate classification, with a corresponding area under the ROC curve of 0.860. As expected, MCI classification accuracy was slightly poorer than AD classification, as there is substantial overlap on all known measures, between MCI and normal aging. The best biomarker sets for each classification are highlighted in Table 2. Figure 1 shows the ROC curves. In Table 2, only a subset of features was actually used: the best classifiers did not include BMI, sex, and the TBM-derived numeric summary. Also in Table 2, it is interesting that ventricular volume was helpful for the AD classification problem but not for distinguishing MCI from controls. This is reasonable given past findings by ourselves and others that ventricular expansion in MCI is relatively mild; there is also substantial cross-subject variation in ventricular volume, even in healthy subjects (Chou et al., 2009b), and this may throw off a classifier’s accuracy unless the disease effect outweighs this natural variation (Chou et al., 2008; Chou et al., 2009a; Chou et al., 2009b).
Figure 1.
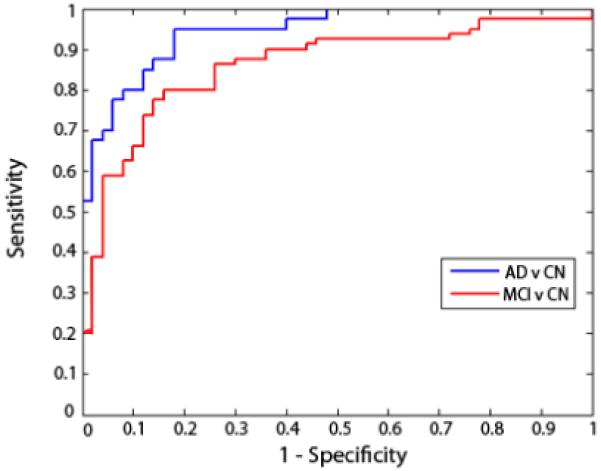
ROC curves for AD and MCI classification. These curves show the trade-off between specificity and sensitivity for classifiers that best distinguished MCI from controls (red curve) and AD from controls (blue curve). The AD classifier used 4 measures and the MCI classifier only used 3. These evaluations are based on finding the top set of features that yielded the highest leave-one-out accuracies on the training set. The curves gradually rise, meaning that there is a natural trade-off: the parameters of the classifier’s decision boundary can be adjusted to be stricter or more lenient. For stricter classification settings, false positive classifications will decrease but so will the rate of true positives. Curves are slightly jagged and not perfectly smooth as they are based on a finite set of test data; with more data, they would be smoother.
3.2. Adding PET-FDG and CSF for multi-modality classification
In this study, our goal was to compare the predictive power from the best combination of features obtained above, which included MRI, ApoE, and age, to that obtained when also including the PET-FDG temporal summary and CSF biomarkers. This may seem like an artificial distinction between two lists of biomarkers, but, from a practical point of view, the first classifier could be applied to a study that only used MRI, while the extended classifier would also need PET scans and lumbar puncture to be performed. Although using more features is almost certainly better statistically, we wanted to assess how much difference it made, given the added expense, logistics, and possible attrition effects of performing multiple assessments.
Here, we considered three subsets of the ADNI subjects (N=328 when adding CSF alone, N=364 when adding PET-FDG alone, and N=166 when adding both CSF and PET-FDG) of the ADNI subjects, for whom the data from these additional diagnostic modalities were available. We applied the same ranking algorithm based on SVM weights and obtained rank orders for the biomarkers, with CSF and PET-FDG taken into account. We found the top set of biomarkers yielding the highest leave-one-out accuracies on the training set for each classification. The rank orders and best sets of biomarkers are displayed in Table 3. CSF t-tau and aβ42 were included in the best set of biomarkers for both AD and MCI classification. PET-FDG also contributed substantially to AD and MCI classification. The remaining top biomarkers were essentially the same as the ones identified in the above study.
Table 3.
Rank order list with relative SVM weights for MRI, ApoE, Age, Sex, BMI and either (a) CSF or (b) PET-FDG, for AD and MCI classification. Biomarkers are ranked according to their relative weights (contributions) in an SVM classifier that includes them all. A secondary question is which subset of these gives best classification accuracy, and this sublist is shown in gray. In these sublists, some features are omitted as adding them does not improve classification accuracy. Of the CSF markers, p-tau is relatively unhelpful but both t-tau and aβ42 provide independent predictive value. PET-FDG is a useful feature; whether it ranks above MRI hippocampal measures or not depends on whether the task is MCI or AD classification (hippocampal volume is slightly more useful than PET for MCI). PET measures are also somewhat correlated with MRI measures, so that when they are both included, each absorbs some of the variance; this may explain why ApoE genotype rises to the top of the predictors in terms of its independent contribution when MRI and PET are both included (last two columns)
| Biomarker | ||||
|---|---|---|---|---|
| a. MRI + CSF | b. MRI + PET-FDG | |||
| Rank | AD vs. control (weight / wi2) |
MCI vs. control (weight / wi2) |
AD vs. control (weight / wi2) |
MCI vs. control (weight / wi2) |
| 1 | MRI Hipa 0.0794 |
MRI Hip 0.0519 |
ApoE 0.1529 |
ApoE 0.0929 |
| 2 | CSF t-tau 0.0614 |
CSF aβ42 0.0313 |
PET-FDG 0.1022 |
MRI Hip 0.0354 |
| 3 | CSF aβ42 0.0505 |
Age 0.0308 |
MRI Hip 0.0846 |
PET-FDG 0.0289 |
| 4 | ApoE 0.0268 |
ApoE 0.0292 |
MRI Vent 0.0181 |
Age 0.0161 |
| 5 | MRI Ventb 0.0238 |
CSF t-tau 0.0231 |
Age 0.0080 |
MRI Temp 0.0075 |
| 6 | Age 0.0210 |
Sex 0.0157 |
MRI Temp 0.0057 |
Sex 0.0036 |
| 7 | MRI Tempc 0.0163 |
MRI Temp 0.0085 |
BMI 0.0010 |
MRI Vent 0.0021 |
| 8 | BMI 0.0077 |
BMI 0.0017 |
Sex 0.0004 |
BMI 0.0020 |
| 9 | CSF p-tau 0.0003 |
CSF p-tau 0.0014 |
||
| 10 | Sex 0.0001 |
MRI Vent 0.0013 |
||
Hippocampal volume summary
Ventricular volume summary
Temporal lobe summary from tensor-based morphometry (TBM)
Sets of biomarkers yielding the highest leave-one-out accuracy are highlighted.
It may seem paradoxical that when we list the biomarkers in order of priority (Table 3) some of them are listed even though they are not ultimately used in the best-performing classifier (only the lists of features in gray are used in the best classifier). The reason this occurs is that when all features are included, some features are given non-zero weights, which means that they are useful for classifying the training set. Even so, these features may give no detectable improvement in classifying the test set, so they were dropped from the final classifier. This does not mean that they are not useful predictors under any circumstances; it just means that in this sample, they did not improve classification accuracy on the independent evaluation data.
We then compared the performance of AD and MCI classifiers trained with the top biomarkers from the previous (N=635) study to those trained with the top biomarkers that included either CSF or the PET-FDG temporal summary or with both combined. Comparison of leave-one-out accuracies on the training set improved classification, implying that PET-FDG and CSF provide complementary information to MRI, ApoE and age. Leave-one-out accuracies for AD vs. control improved by 6.4%, 3.8%, and 11.6% by adding CSF alone, PET alone and both CSF and PET respectively. The corresponding improvements for MCI vs. control were 2.3%, 2.7%, and 4.6%.
When we compared the ROC curve AUCs, however, the improvement obtained by adding CSF, PET-FDG or both measures to the MRI measures was not statistically significant (p values > 0.05; Table 4). This lack of statistical significance may be due to the small size of the testing sets. If, however, this lack of significance is verified in even larger studies, it could have considerable implications for clinical trials in terms of total cost, efficiency and adverse effects.
Table 4.
Comparison of AD and MCI classification accuracy and false positive/false negative trade-offs (ROC analyses) for classifiers that use different types of information: MRI, MRI+CSF, MRI+PET-FDG, and MRI+CSF+PET-FDG. Information for the top MRI classifier is listed twice, because MRI data were available for all ADNI subjects, but CSF and PET data were available only for a subset of those who had MRI. So it is only fair to report the classification accuracy on the full sample of MRIs, as well as on the subsamples in which head-to-head comparisons could be made with classifiers that also included the available CSF and PET data. The classifiers include ApoE and age, but not sex or BMI as the latter two did not contribute to the classification accuracies
| AD versus control | MCI versus control | |||||
|---|---|---|---|---|---|---|
| Biomarkers | LOOCV Accuracy |
ROC AUC ± SE |
Δ AUCa (p value) |
LOOCV Accuracy |
ROC AUC ± SE |
Δ AUC (p value) |
| Top MRIb | 0.8160 | 0.8940 ± 0.0499 | - | 0.8421 | 0.8350 ± 0.0632 | - |
| Top MRI+CSFc | 0.8800 | 0.9560 ± 0.0273 | 0.0620 (0.191) | 0.8647 | 0.8125 ± 0.0672 | 0.0225 (0.722) |
|
| ||||||
| Top MRIb | 0.7634 | 0.7760 ± 0.0585 | - | 0.7227 | 0.7067 ± 0.0696 | - |
| Top MRI+PET−FDGd | 0.8011 | 0.7820 ± 0.0580 | 0.0060 (0.906) | 0.7500 | 0.7444 ± 0.0672 | 0.0377 (0.382) |
|
| ||||||
| Top MRIb | 0.7907 | 0.8850 ± 0.0501 | - | 0.7121 | 0.7488 ± 0.0649 | - |
| Top MRI+CSF+PET−FDGe | 0.9070 | 0.9175 ± 0.0413 | 0.0325 (0.357) | 0.7576 | 0.7688 ± 0.0669 | 0.0200 (0.709) |
AUC difference relative to using the top MRI-based classifier only
Top biomarkers identified in the N=635 study with MRI
Top biomarkers identified in the N=328 study with MRI and CSF
Top biomarkers identified in the N=364 study with MRI and PET-FDG
Top biomarkers identified in the N=166 study with MRI, CSF and PET-FDG
LOOCV: leave-one-out cross-validatino
3.3. Boosting Power for Clinical Trials
A novel use of classifiers is to identify subjects who are more likely to decline. Under some reasonable assumptions (see Discussion), this can lead to larger effect sizes for detecting changes in biomarkers over time; this may also be useful for reducing sample size requirements for clinical trials of potential disease-modifying therapies. In the past, several authors have suggested that people in the lowest 50% (or some other quantile) of hippocampal volume are more likely to show future decline, both clinically (e.g., conversion from MCI to AD) and on imaging (see, e.g., Frisoni et al, 2010). Of course, this idea could be generalized to defining a sample based on the k% of subjects that a classifier declares as most likely to decline clinically in the future. Such a classifier could include not just MRI but any biomarker relevant for improving prediction.
As such, we computed minimum sample size estimates (n80) for the top k percent of subjects (for different values of k noted below) classified as most likely to have AD with our best AD classifier, using MRI hippocampal and ventricular summaries, ApoE and age as features. This k% of people are subjects in the independent test datasets (not used to train the classifier) who are assigned by the classifier to the AD class; they are those classified as AD who are farthest from the “SVM classifier decision boundary”. We did not include PET-FDG and CSF biomarkers here, since adding these covariates limited our sample size and, as shown above, did not significantly improve classification in our tests. The subjects were ranked based on the SVM classifier output, the arithmetic sign of which determines the class assigned to each subject. A few AD subjects were excluded from the training and testing sets to avoid any overlap with the training set used in our prior report (Hua et al., 2009) for creating the statistical ROIs. The results are shown in Figure 2a. When k is less than about 33%, the power estimates for AD subjects are improved compared to the minimal sample size of 48 AD subjects reported by Hua et al. (2009). There is a drop in the sample sizes needed to show a specific slowing effect, as the more AD-like subjects are selected. This has to be weighed against other factors (see Discussion), but it is interesting that the changes in these subjects have a greater effect size. It is also by no means obvious in advance that these subjects would give greater effect sizes. For the effect size to be greater, the changes have to be large and their variance has to be small; restricting the sample did not lead to an increase in the variability of the change measures sufficient to deplete effect sizes.
Figure 2.
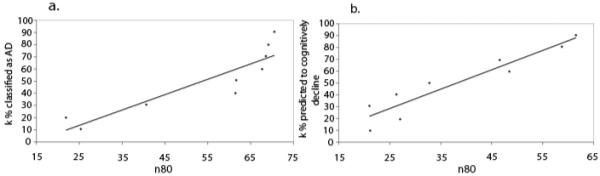
n80 estimates (i.e., sample sizes required to detect a 25% slowing of the rate of atrophy with 80% power) as a function of restricting the sample to likely decliners. (a) Samples are based on the top k% classified, based on all biomarkers, as most likely to have AD (lower k gives smaller samples). (b) Here samples are based on the top k% of MCI subjects predicted by the classifier as most likely to decline (again lower values of k give dramatically lower samples). If only one-third of the most likely decliners were kept, in a sub-analysis based on the classifier’s predictions, then the sample size needed (n80) for an MCI trial would only be around 30 subjects per arm (see Discussion for caveats of this approach).
We could use the classifiers in many different ways to define a subsample – the diagnostic classifiers single out those who are most likely, based on all their imaging measures, to fall into a specific diagnostic category (e.g., AD). We also tested the benefit of defining a subsample of subjects with a classifier trained to identify likely decliners, based on all their imaging measures and other biomarkers, all at baseline.
To obtain similar n80 estimates for MCI subjects predicted to undergo cognitive decline, we considered 64 MCI subjects using MRI measures (3 features), PET-FDG, CSF biomarkers (3 features), ApoE and age. Here, the output of the SVM algorithm was set to be the 12-month rate of change in sobCDR, instead of a binary output for the classification approach used in the studies above. Training with all possible 29-1 feature combinations using a linear kernel (parameter C = 1) revealed PET-FDG, MRI ventricular and temporal summaries, and ApoE as the best set of features, with the lowest mean squared error on the testing set.
To increase our sample size for evaluating this classifier, we considered a larger group of 129 MCI subjects with only the four features identified above and trained a model that predicted the rate of sobCDR change in a novel testing set. We ranked the testing MCI subjects in order of predicted cognitive decline and computed n80 estimates for the top k% percent (for different values of k) of MCI subjects who the classifier predicted to be most likely to decline within a year (Figure 2b). The n80 values were even lower than the 88 MCI individuals we reported before as the minimal sample size for MCI (Hua et al., 2009). In addition to sample size estimates reported by Hua et al. (2009), similar estimates have also been made by other investigators such as Fox et al. (2000), Jack et al. (2004) and Schuff et al. (2009), and we were able to improve upon these too with our approach.
In this report, we have considered AD and MCI classification as well as prediction of MCI conversion. Classifiers can also be trained to distinguish MCI converters from diagnostic groups other than MCI. For instance, when we performed classification with a small group of 12 MCI converters versus 12 healthy controls using all features in our study, we obtained a reasonably promising 71% accuracy, as this discrimination is more challenging than separating AD patients from controls.
In general, however, we do not want to discriminate MCI decliners from groups other than MCI for the prediction of later decline. We assumed here that MCI diagnosis was given, and we aimed to predict who would decline within that group. Predicting decline in a mixed group of controls and MCIs is a little easier, as the knowledge that a person is MCI is already fairly good evidence that future decline is likely. Because of that, we wanted to assess the specific additive value of neuroimaging markers once a person is diagnosed as MCI (and it is reasonably helpful).
4. Discussion
We explored the power of several baseline biomarkers for AD and MCI, used jointly for diagnostic classification and for predicting future (1-year) cognitive decline in MCI. We also showed how to apply the multi-modality classifiers to choose sub-samples of subjects for boosting power in clinical trials. We determined combinations of regional MRI numerical summaries with demographic variables and ApoE that best classified AD vs. control and MCI vs. control. The top set of complementary biomarkers for AD classification (when used together) were the MRI hippocampal volume summary (measured with the method of Morra et al., 2008), ApoE genotype, age and the MRI ventricular summary (measured with the method of Chou et al., 2009) in that order, resulting in an 82.21% accuracy, and an ROC AUC of 0.945, which is quite strong. Biologically, hippocampal atrophy and ventricular enlargement are established manifestations of AD pathology, and the two structures are routinely monitored via MRI for AD clinical trials (Frisoni et al., 2010). ApoE and advancing age are also well-known risk factors for AD (Carlsson et al., 2009), and age is associated with atrophic rates in ADNI (Hua et al., 2010a). The best set of features identified agrees with the AD literature. The one exception is the MRI temporal lobe summary, which did not improve classification power. This is not entirely surprising as it is quite highly correlated with the other two measures of atrophy (hippocampal and ventricular volume), so it may not add very much independent information for diagnostic classification. As expected, MCI classification was less accurate, and ventricular summaries were not as helpful; the best MCI diagnostic classifier only used hippocampal volume, ApoE genotype and age (Frisoni et al., 2010; Petersen, 2010).
When compared to accuracy results reported by groups such as Vemuri et al. (2008), Klöppel et al. (2008) and Fan et al. (2008), our accuracies may seem a bit low. Perhaps, the main reason the accuracy values are not so high is that we are using numerical summary measures (single values for each imaging modality) as opposed to voxel-wise maps (which are implemented in papers that report higher accuracies). Even so, it is difficult to compare the results across papers as different subject samples are used. For example, ADNI considers only AD patients with relatively mild AD, and classification of AD is clearly easier in cohorts with a greater proportion of more severely affected patients. Nonetheless, if some future classifier performs better, it could also be used to boost power using the same subpopulation selection method shown here.
By separately adding CSF biomarkers and PET-FDG as covariates for classification, where available, we obtained new rank order lists. These demonstrated how much the additional diagnostic measures contributed to AD and MCI classification, at least with this type of classifier. Different classes of AD biomarkers have dynamic trajectories that are thought to be temporally ordered with respect to the progression of the disease; in general, markers of amyloid deposition are thought to rise earlier than markers of neurodegeneration detectable on MRI, and these in turn become abnormal before tests of clinical function (Braskie et al., 2008; Jack et al., 2010; Petersen, 2010; Protas et al., 2010).
It is therefore plausible to expect classifiers to perform best with biomarkers that are maximally dynamic during the stages of disease being considered; measurement reproducibility and precision are important. The top feature lists are generally consistent with this hypothesis, as MRI contributes more strongly to AD classification, whereas PET-FDG and CSF biomarkers, particularly aβ42, play more important roles in MCI classification. The observation that CSF tau levels were more important for AD classification, and CSF aβ42 more contributory to MCI classification is also consistent with Jack et al.’s model, in which the dynamic range of aβ42 precedes that of tau in the progression of AD. ApoE is consistently included among the best biomarkers for both AD and MCI classification, which agrees with another component of the Jack et al. (2010) hypothesis, stating that carrying E4 alleles may shift the sequence of biomarker activities to earlier time points relative to the onset of overtly detectable clinical symptoms.
Predicting future decline in MCI subjects is more challenging than AD and MCI classification, as differences among MCI subjects are subtle. Instead of approaching this problem with a binary classifier, we adapted the algorithm to predict a continuous cognitive outcome, which is the 12-month change in sobCDR. The baseline PET-FDG temporal summary, MRI temporal and ventricular summaries, and ApoE, were the best predictors of future cognitive decline in MCI (assessed over a 1-year follow-up interval). The combination of PET-FDG and ApoE genotype has been previously shown to provide good accuracy for predicting MCI conversion (Mosconi et al., 2004). MRI-based temporal and ventricular volumes have also been reported for their predictive power in MCI subjects (Fleisher et al., 2008; Korf et al., 2004). It is mechanistically reasonable for this combination of structural, functional and genetic information to supply complementary predictive power. By using a multi-modality regression approach to predicting cognitive decline in ADNI subjects, a very recent study found that a linear combination of MRI and PET-FDG was a better predictor of cognitive decline than CSF biomarkers (Walhovd et al., 2010), consistent with our best set of biomarkers. Unexpectedly, however, the MRI hippocampal summaries were not incorporated into our predictive model, which is surprising as hippocampal volume can be useful for prediction of MCI progression to AD (Apostolova et al., 2006a; Apostolova et al., 2006b; Apostolova et al., 2007; Frisoni et al., 2010). The presence of detectable extra-hippocampal atrophy (e.g. in the ventricles and white matter) may also be good predictors of whether an MCI patient is deteriorating.
Our choice of brain regions and imaging measures to analyze was based on discussions among the ADNI Clinical, MRI and PET Cores. We chose imaging measures that had been used successfully in the past for disease classification or to monitor disease progression, preferring those measures that could be derived efficiently from a large dataset, without substantial manual interaction with the images. Clinical ratings were based on those widely used in clinical trials – CDR and mini-mental status examination (MMSE) – and the CSF biomarker measures were those found to be most promising in pilot studies (Shaw et al., 2009). Needless to say, more brain regions or alternative cognitive tests could be proposed, and could be added to those analyzed here to boost performance even further. Specifically, in conference abstracts, Alexander et al. (2008) and Zhang et al. (2008) have advocated a multivariate network analysis in which a very large number of regional brain volumes are jointly used as predictors, in an SVM model. Other groups have parcellated the brain into a large number of subregions, but found that temporal lobe regions showed the greatest disease-related changes and significantly outperformed any of the clinical or cognitive measures examined for both AD and MCI (Holland et al., 2009). To single out brain regions that are most promising for analysis of disease-related brain change, we also focused on pre-selecting voxels in maps of brain change that show greatest effect sizes in independent samples. We and others have found that a classifier can be given and entire brain image, and from it can derive the voxels whose signals are most promising for group classification (Sun et al., 2009). By comparing different imaging measures (voxel-based, ROI-based, or surface-based; Gutman et al., 2008), and different classifiers (SVM versus others), future studies may be able to gauge which aspects of the classifier (its mathematical design or the features used) are most relevant for boosting performance.
In addition to scanning all the subjects with MRI at 1.5 T field strength, one quarter of ADNI’s subjects also received 3 T scans. In prior work (Ho et al., 2009), we studied 110 ADNI subjects scanned longitudinally at both 3 and 1.5 T, across a one-year interval. Our power analyses found that 37 AD and 108 MCI subjects would be needed at 1.5 T versus 49 AD and 166 MCI subjects at 3 T, to detect a 25% slowing of atrophy with 80% power, but these estimates did not differ significantly with field strengths. At both field strengths, temporal lobe atrophy rates were highly correlated with interval decline in Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog), MMSE and sobCDR scores. To avoid modeling the effects of scanner field strength as a confound, here we used the 1.5T ADNI data only. Some additional work may be needed to show that 3T scanners perform equally well for all biomarkers assessed here. The few ADNI studies that have assessed the field strength effect (Ho et al., 2009; Kruggel et al., 2010) suggest that 1.5 and 3 T scanners did not significantly differ in their power to detect neurodegenerative changes over a year.
Some clinical measures, such as sobCDR, were not used as features for classification to avoid circular inference. Since these measures are used in making a diagnosis, it would be circular to incorporate them into our diagnostic classifiers and then test their empirical accuracy relative to the diagnosis given by physicians in the clinic. Even so, if used in practice to assist diagnosis, a classifier could use more cognitive measures – including those conventionally used for diagnosis and any other relevant information. However, the diagnostic accuracy of such a classifier could not then be “independently” validated in the same way as we did here. Doing so would require some other form of independent diagnostic ground truth, not used by the classifier, such as autopsy confirmation of characteristic signs of AD neuropathology. This could in principle be done, but neuropathology is not available in large numbers for the ADNI cohort.
A major clinical application of disease classifiers is for boosting power for clinical trials by reducing sample size estimates required to observe therapeutic effects. The idea of targeting a subgroup for analysis of treatment effects is not new (Frisoni et al., 2010). In fact, a drug trial for prodromal AD is currently recruiting subjects, with an inclusion criterion based on CSF aβ42 and t-tau (http://clinicaltrials.gov/ct2/show/NCT00890890?term=bms+alzheimer%27s&rank=2). It appears new, however, to base the selection on a machine learning-based classifier that combines numerous biomarkers, which include neuroimaging measures. Combinations of disease markers are more likely to achieve sample size reductions than using single measures, such as subpopulation selection based on hippocampal volume only (of course statistical power must be traded off against the logistical complexity and cost of collecting and analyzing multiple biomarker assessments). When we considered the subset of subjects classified as most likely to have AD by our multi-feature AD classifier, and the most likely decliners in MCI, we were able to reduce the n80 estimates to fewer than 40 subjects for both AD and MCI, improving on those estimates we reported before (Ho et al., 2009; Hua et al., 2009; Hua et al., 2010b). This result supports the concept of clinical trial enrichment, which has been previously advocated (Cummings et al., 2007; Frisoni et al., 2010; Hampel and Broich, 2009). Our enrichment strategy works because the subpopulation of subjects who are more likely to decline are selected based on disease classifiers and outcome predictors that integrate information from a number of complementary biomarkers.
We chose to compute sample sizes needed to detect a 25% slowing of atrophy with 80% power. While 25% is a reasonable target for a treatment that aims to slow atrophy, the exact number chosen is arbitrary. It is simple to compute sample size estimates for other percentage reductions in the atrophic rate, such as 5% or 50%, for example. As we noted in Hua et al. (2010b), treatments may slow atrophy to different degrees, which may be denoted by k%, for different k. The sample size estimates required to detect a k% slowing of atrophy may be easily derived by multiplying the sample size estimates (n80) in this paper by (25/k)2, as the numbers follow an inverse-square law. For example, 4 times as many subjects would be needed to detect a 12.5% slowing of atrophy (half of 25%), versus a 25% slowing of atrophy (Ho et al., 2009). The quadratic relationship between the sample size estimates and the percentage atrophic rate is illustrated in (Hua et al., 2010b). Similarly, the results of this paper can be easily translated to studies aiming to detect a different level of treatment effect, and our findings remain unaffected as multiplying the variables by a constant (25/k)2 does not alter the ranking of the effect sizes in the statistical tests (it is a monotone transformation, i.e., it preserves the rank order).
As a caveat, the n80 “minimal sample size” measure is practical but has limitations: first, it is based on changes in the patient groups only, and not their difference from controls; second, it assumes that a treatment would slow atrophy in the same places as it normally occurs, with the clinical outcome as observing an untreated sample with less atrophy. Finally, any treatment effects in a sub-analysis might only apply to people who fit the selection criteria for that sub-analysis; even so, evidence of an effect in a sub-analysis might suffice to initiate a broader study.
The approach and results reported here are relevant to future work in the neuroimaging of AD in several ways. First, several authors advocate “enrichment” in clinical trials by trying to select those most likely to decline, based on clinical criteria, or occasionally based on imaging criteria. This can be done by applying thresholds or cut-offs to volumetric measures on MRI scans, such as hippocampal volume, but here we advocate using the full armory of imaging and CSF measures to classify subjects first, and then use the classifier’s output to select subpopulations for later statistical testing.
Although this may seem like basing the statistical approach in part on the data collected, rather than specifying it all in advance of the study, this approach would identify subjects whose imaging data made them most likely to show treatment effects, regardless of the treatment. A similar approach to boost the power of imaging biomarkers is voxel-set pre-selection, which substantially boosts power to detect the slowing of atrophy (Hua et al., 2010; Chen et al., 2010).
For these statistically-guided measures to be widely adopted as outcome measures in clinical trials, there needs to be some flexibility on the part of regulatory bodies that some features of the data collected may play a role in establishing which measures or subjects are evaluated. The analysis strategy can then adapt to the incoming data, and can exploit the power of Bayesian statistics and machine learning to obtain more powerful measures. It is quite defensible - and even advisable - for these machine learning approaches to be used, so long as the independence of statistical training and test samples is rigorously maintained.
A limitation of our study is that sample sizes become small when multiple imaging modalities and biomarkers are considered. In longitudinal studies especially, assessments of many kinds bring added costs, complexity, logistical difficulty, subject burden, and subject attrition (although in ADNI, attrition rates are only around 7% per year). Larger cohorts of subjects with available data from multiple biomarkers would allow more powerful classifiers and predictors to be developed, incorporating the best combinations of available diagnostic tools. More accurate ranking of biomarkers for verifying the details of Jack et al.’s temporal sequence hypothesis would become feasible. In addition, future studies will include additional diagnostic modalities such as Pittsburgh compound B (PiB), diffusion tensor imaging (DTI), arterial spin labeling (ASL) and resting state functional MRI for disease classification. PiB has been collected in a small subsample of ADNI subjects, but we did not evaluate it here as requiring all biomarkers would have further limited our sample sizes. Another future direction would be to employ machine learning algorithms other than SVM (e.g., boosting; Morra et al., 2009b), or classifiers based on features in voxel-based maps (Sun et al., 2009), to improve classification and prediction accuracy. More powerful classifiers may then be implemented to improve upon our clinical trial boosting results. Furthermore, machine learning can perhaps be used to discover genetic (Stein et al., 2009; Stein et al., 2010), epidemiological and physiological factors that influence the progression of AD.
Acknowledgements
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., and Wyeth, as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org <http://www.fnih.org> ). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129, K01 AG030514, and the Dana Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement for authors: The authors have no potential financial or personal conflicts of interest including relationships with other people or organization within three years of beginning the work submitted that could inappropriately influence their work.
References
- Alexander GE, Hanson KD, Chen K, Reiman EM, Bernstein MA, Kornak J, Schuff NW, Fox NC, Thompson PM, Weiner MW, Jack CR., Jr. Six month MRI gray matter declines in Alzheimer’s dementia evaluated by voxel-based morphometry with multivariate network analysis: Preliminary findings from the ADNI study; Presented at The International Conference on Alzheimer’s Disease; Chicago. 2008.Jul 26-31, [Google Scholar]
- Alzheimer’s Association Alzheimer’s disease facts and figures. Alzheimer’s and Dementia. 2009;5(3):234–70. doi: 10.1016/j.jalz.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Dinov ID, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. 3D comparison of hippocampal atrophy in amnestic mild cognitive impairment and Alzheimer’s disease. Brain. 2006a;129(11):2867–73. doi: 10.1093/brain/awl274. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Archives of Neurology. 2006b;63(5):693–9. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Steiner CA, Akopyan GG, Dutton RA, Hayashi KM, Toga AW, Cummings JL, Thompson PM. Three-dimensional gray matter atrophy mapping in mild cognitive impairment and mild Alzheimer disease. Archives of Neurology. 2007;64(10):1489–95. doi: 10.1001/archneur.64.10.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad NA, Al Bugami M, Loy-English I. Gender differences in dementia risk factors. Gender Medicine. 2007;4(2):121–129. doi: 10.1016/s1550-8579(07)80026-x. [DOI] [PubMed] [Google Scholar]
- Braskie MN, Klunder AD, Hayashi KM, Protas H, Kepe V, Miller KJ, Huang SC, Barrio JR, Ercoli LM, Siddarth P, Satyamurthy N, Liu J, Toga AW, Bookheimer SY, Small GW, Thompson PM. Plaque and tangle imaging and cognition in normal aging and Alzheimer’s disease. Neurobiology of Aging. 2008 doi: 10.1016/j.neurobiolaging.2008.09.012. doi:10.1016/j.neurobiolaging.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman AS, Wilson RS, Bienias JL, Shah RC, Evans DA, Bennett DA. Change in body mass index and risk of incident Alzheimer disease. Neurology. 2005;65:892–897. doi: 10.1212/01.wnl.0000176061.33817.90. [DOI] [PubMed] [Google Scholar]
- Burges CJC. A Tutorial on Support Vector Machines for Pattern Recognition. Data Mining and Knowledge Discovery. 1998;2:121–67. [Google Scholar]
- Carlsson CM, Gleason CE, Puglielli L, Asthana S. Chapter 65. Dementia Including Alzheimer’s Disease. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, editors. Hazzard’s Geriatric Medicine and Gerontology. The McGraw-Hill Companies; 2009. p. 6e. URL: http://www.accessmedicine.com/content.aspx?aID=5122625. [Google Scholar]
- Chen K, Langbaum JB, Fleisher AS, Ayutyanont N, Reschke C, Lee W, Liu X, Bandy D, Alexander GE, Thompson PM, Foster NL, Harvey DJ, de Leon MJ, Koeppe RA, Jagust WJ, Weiner MW, Reiman EM. Twelve-Month Metabolic Declines in Probable Alzheimer’s Disease and Amnestic Mild Cognitive Impairment Assessed Using an Empirically Pre-Defined Statistical Region-of-Interest: Findings from the Alzheimer’s Disease Neuroimaging Initiative. NeuroImage. 2010 doi: 10.1016/j.neuroimage.2010.02.064. doi:10.1016/j.neuroimage.2010.02.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YY, Lepore N, Avedissian C, Madsen SK, Parikshak N, Hua X, Shaw LM, Trojanowski JQ, Weiner MW, Toga AW, Thompson PM. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer’s disease, mild cognitive impairment and elderly controls. NeuroImage. 2009a;46(2):394–410. doi: 10.1016/j.neuroimage.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YY, Lepore N, Chiang MC, Avedissian C, Barysheva M, McMahon KL, de Zubicaray GI, Meredith M, Wright MJ, Toga AW, Thompson PM. Mapping genetic influences on ventricular structure in twins. NeuroImage. 2009b;44(4):1312–23. doi: 10.1016/j.neuroimage.2008.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YY, Lepore N, de Zubicaray GI, Carmichael OT, Becker JT, Toga AW, Thompson PM. Automated ventricular mapping with multi-atlas fluid image alignment reveals genetic effects in Alzheimer’s disease. NeuroImage. 2008;40(2):615–30. doi: 10.1016/j.neuroimage.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Doody R, Clark C. Disease-modifying therapies for Alzheimer disease: challenges to early intervention. Neurology. 2007;69(16):1622–34. doi: 10.1212/01.wnl.0000295996.54210.69. [DOI] [PubMed] [Google Scholar]
- Davatzikos C, Resnick SM, Wu X, Parmpi P, Clark CM. Individual patient diagnosis of AD and FTD via high-dimensional pattern classification of MRI. NeuroImage. 2008;41(4):1220–7. doi: 10.1016/j.neuroimage.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davatzikos C, Xu F, Yang A, Yong F, Resnick SM. Longitudinal progression of Alzheimer’s-like patterns of atrophy in normal older adults: the SPARE-AD index. Brain. 2009;132(8):2026–35. doi: 10.1093/brain/awp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duara R, Loewenstein DA, Potter E, Appel J, Greig MT, Urs R, Shen Q, Raj A, Small B, Barker W, Schofield E, Wu Y, Potter H. Medial temporal lobe atrophy on MRI scans and the diagnosis of Alzheimer disease. Neurology. 2008;71:1986–1992. doi: 10.1212/01.wnl.0000336925.79704.9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecker C, Rocha-Rego V, Johnston P, Mourao-Miranda J, Marquand A, Daly EM, Brammer MJ, Murphy C, Murphy DG. Investigating the predictive value of whole-brain structural MR scans in autism: A pattern classification approach. NeuroImage. 2010;49(1):44–56. doi: 10.1016/j.neuroimage.2009.08.024. [DOI] [PubMed] [Google Scholar]
- Fan Y, Resnick SM, Wu X, Davatzikos C. Structural and functional biomarkers of prodromal Alzheimer’s disease: a high-dimensional pattern classification study. NeuroImage. 2008;41(2):277–85. doi: 10.1016/j.neuroimage.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Sun S, Taylor C, Ward CP, Gamst AC, Petersen RC, Jack CR, Jr., Aisen PS, Thal LJ. Volumetric MRI vs clinical predictors of Alzheimer disease in mild cognitive impairment. Neurology. 2008;70(3):191–9. doi: 10.1212/01.wnl.0000287091.57376.65. [DOI] [PubMed] [Google Scholar]
- Fox NC, Cousens S, Scahill R, Harvey RJ, Rossor MN. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease. Arch Neurol. 2000;57:339–344. doi: 10.1001/archneur.57.3.339. [DOI] [PubMed] [Google Scholar]
- Freund Y, Schapire RE. A Short Introduction to Boosting. Journal of Japanese Society for Artificial Intelligence. 1999;14(5):771–780. [Google Scholar]
- Frisoni GB, Fox NC, Jack CR, Jr, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nature Reviews | Neurology. 2010;6:1–11. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutman B, Wang YL, Morra JH, Tu Z, Jack CR, Weiner MW, Toga AW, Thompson PM. Disease Classification with Hippocampal Surface Invariants; MICCAI Workshop on Hippocampal Mapping; March 2008.2008. [Google Scholar]
- Hampel H, Broich K. Enrichment of MCI and early Alzheimer’s disease treatment trials using neurochemical & imaging candidate biomarkers. The Journal of Nutrition, Health and Aging. 2009;13(4):373–5. doi: 10.1007/s12603-009-0048-3. [DOI] [PubMed] [Google Scholar]
- Hanley JA, McNeil BJ. A Method of Comparing the Areas under Receiver Operating Characteristic Curves Derived from the Same Cases. Radiology. 1983;148(3):839–43. doi: 10.1148/radiology.148.3.6878708. [DOI] [PubMed] [Google Scholar]
- Hinrichs C, Singh V, Mukherjee L, Xu G, Chung MK, Johnson SC. Spatially augmented LPboosting for AD classification with evaluations on the ADNI dataset. NeuroImage. 2009;48(1):138–49. doi: 10.1016/j.neuroimage.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, Dinov ID, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen A, Corneveaux J, Stephan DA, Webster J, DeChairo BM, Potkin SG, Jack CR, Weiner MW, Raji CA, Lopez OL, Becker JT, Thompson PM. Commonly carried allele within FTO, an obesity-associated gene, relates to accelerated brain degeneration in the elderly. Proc Natl Acad Sci. 2010a doi: 10.1073/pnas.0910878107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AJ, Raji CA, Becker JT, Lopez OL, Kuller LH, Hua X, Lee S, Hibar D, Dinov ID, Stein JL, Jack CR, Weiner MW, Toga AW, Thompson PM. Obesity and brain structure in 700 MCI and AD patients. Neurobiology of Aging. 2010b doi: 10.1016/j.neurobiolaging.2010.04.006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AJ, Hua X, Lee S, Yanovsky I, Leow AD, Gutman B, Dinov ID, Toga AW, Jack CR, Jr., Bernstein MA, Reiman EM, Harvey D, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Comparing 3T and 1.5T MRI for tracking AD progression with tensor-based morphometry. Human Brain Mapping. 2009 doi: 10.1002/hbm.20882. doi:10.1002/hbm.20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Brewer JB, Hagler DJ, Fenema-Notestine C, Dale AM. Subregional neuroanatomical change as a biomarker for Alzheimer’s disease. Proc Natl Acad Sci. 2009;106(49):20954–20959. doi: 10.1073/pnas.0906053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Hibar DP, Lee S, Toga AW, Jack CR, Jr., Weiner MW, Thompson PM. Sex and age differences in atrophic rates: an ADNI study with N=1368 MRI scans. Neurobiology of Aging. 2010a doi: 10.1016/j.neurobiolaging.2010.04.033. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Lee S, Hibar DP, Yanovsky I, Leow AD, Toga AW, Jack CR, Jr, Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Mapping Alzheimer’s disease progression in 1309 MRI scans: power estimates for different inter-scan intervals. NeuroImage. 2010b doi: 10.1016/j.neuroimage.2010.01.104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Lee S, Yanovsky I, Leow AD, Chou YY, Ho AJ, Gutman B, Toga AW, Jack CR, Jr., Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM. Optimizing power to track brain degeneration in Alzheimer’s disease and mild cognitive impairment with tensor-based morphometry: an ADNI study of 515 subjects. NeuroImage. 2009;48(4):668–81. doi: 10.1016/j.neuroimage.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Lee S, Klunder AD, Toga AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, Jack CR, Jr., Bernstein MA, Britson PJ, Ward CP, Whitwell JL, Borowski B, Fleisher AS, Fox NC, Boyes RG, Barnes J, Harvey D, Kornak J, Schuff N, Boreta L, Alexander GE, Weiner MW, Thompson PM. 3D characterization of brain atrophy in Alzheimer’s disease and mild cognitive impairment using tensor-based morphometry. NeuroImage. 2008a;41(1):19–34. doi: 10.1016/j.neuroimage.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Parikshak N, Lee S, Chiang MC, Toga AW, Jack CR, Jr., Weiner MW, Thompson PM. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer’s disease: an MRI study of 676 AD, MCI, and normal subjects. NeuroImage. 2008b;43(3):458–69. doi: 10.1016/j.neuroimage.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Knopman DSK, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurology. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr., Shiung MM, Gunter JL, O’Brien PC, Weigand SD, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Cha RH, Tangalos EG, Petersen RC. Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology. 2004;62(4):591–600. doi: 10.1212/01.wnl.0000110315.26026.ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöppel S, Stonnington CM, Chu C, Draganski B, Scahill RI, Rohrer JD, Fox NC, Jack CR, Jr., Ashburner J, Frackowiak RSJ. Automatic classification of MR scans in Alzheimer’s disease. Brain. 2008;131:681–9. doi: 10.1093/brain/awm319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Korf ESC, Wahlund LO, Visser PJ, Scheltens P. Medial temporal lobe atrophy on MRI predicts dementia in patients with mild cognitive impairment. Neurology. 2004;63:94–100. doi: 10.1212/01.wnl.0000133114.92694.93. [DOI] [PubMed] [Google Scholar]
- Koutsouleris N, Meisenzahl EM, Davatzikos C, Bottlender R, Frodl T, Scheuerecker J, Schmitt G, Zetzsche T, Decker P, Reiser M, Möller HJ, Gaser C. Use of Neuroanatomical Pattern Classification to Identify Subjects in At-Risk Mental States of Psychosis and Predict Disease Transition. Archives of General Psychiatry. 2009;66(7):700–12. doi: 10.1001/archgenpsychiatry.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruggel F, Turner J, Muftuler LT. Impact of scanner hardware and imaging protocol on image quality and compartment volume precision in the ADNI cohort. Neuroimage. 2010;49(3):2123–33. doi: 10.1016/j.neuroimage.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, Weiner MW, Jagust WJ. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiology of Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.07.002. doi:10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch JP, Pruessner J, Zijdenbos AP, Collins DL, Teipel SJ, Hampel H, Evans AC. Automated cortical thickness measurements from MRI can accurately separate Alzheimer’s patients from normal elderly controls. Neurobiology of Aging. 2008;29(1):23–30. doi: 10.1016/j.neurobiolaging.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I. Risk Factors for Alzheimer’s Disease: A Prospective Analysis from the Canadian Study of Health and Aging. American Journal of Epidemiology. 2002;156(5):445–53. doi: 10.1093/aje/kwf074. [DOI] [PubMed] [Google Scholar]
- Lukas L, Devos A, Suykens JAK, Vanhamme L, Howe FA, Majós C, Moreno-Torres A, Van Der Graaf M, Tate AR, Arús C, Van Huffel S. Brain tumor classification based on long echo proton MRS signals. Artificial Intelligence in Medicine. 2004;31(1):73–89. doi: 10.1016/j.artmed.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Mesrob L, Magnin B, Colliot O, Sarazin M, Hahn-Barma V, Dubois B, Gallinari P, Lehéricy S, Kinkingnéhun S, Benali H. Identification of Atrophy Patterns in Alzheimer’s Disease Based on SVM Feature Selection and Anatomical Parcellation. Lecture Notes in Computer Science. 2008;5128:124–32. [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr., Weiner MW, Thompson PM. Validation of a fully automated 3D hippocampal segmentation method using subjects with Alzheimer’s disease mild cognitive impairment, and elderly controls. NeuroImage. 2008;43(1):59–68. doi: 10.1016/j.neuroimage.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Toga AW, Jack CR, Jr., Schuff N, Weiner MW, Thompson PM. Automated mapping of hippocampal atrophy in 1-year repeat MRI data from 490 subjects with Alzheimer’s disease, mild cognitive impairment, and elderly controls. NeuroImage. 2009;45(1 Suppl):S3–15. doi: 10.1016/j.neuroimage.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Toga AW, Thompson PM. Comparison of AdaBoost and support vector machines for detecting Alzheimer’s disease through automated hippocampal segmentation. IEEE Transactions on Medical Imaging. 2010;29(1):30–43. doi: 10.1109/TMI.2009.2021941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Perani D, Sorbi S, Herholz K, Nacmias B, Holthoff V, Salmon E, Baron J-C, De Cristofaro MTR, Padovani A, Borroni B, Franceschi M, Bracco L, Pupi A. MCI conversion to dementia and the APOE genotype, A prediction study with FDG-PET. Neurology. 2004;63:2332–40. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- Mourão-Miranda J, Bokde ALW, Born C, Hampel H, Stetter M. Classifying brain states and determining the discriminating activation patterns: Support Vector Machine on functional MRI data. NeuroImage. 2005;28(4):980–95. doi: 10.1016/j.neuroimage.2005.06.070. [DOI] [PubMed] [Google Scholar]
- Petersen RC. Alzheimer’s disease: progress in prediction. Lancet Neurology. 2010;9(1):4–5. doi: 10.1016/S1474-4422(09)70330-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of Neurology. 1999;56(3):303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- Protas HD, Huang SC, Kepe V, Hayashi K, Klunder A, Braskie MN, Ercoli L, Bookheimer S, Thompson PM, Small GW, Barrio JR. FDDNP binding using MR derived cortical surface maps. NeuroImage. 2010;49(1):240–8. doi: 10.1016/j.neuroimage.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raji CA, Ho AJ, Parikshak N, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD, Toga AW, Thompson PM. Brain Structure and Obesity. Human Brain Mapping. 2008 doi: 10.1002/hbm.20870. doi:10.1002/hbm.20870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR, Jr., Weiner MW. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(4):1067–77. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–13. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JL, Hua X, Lee S, Ho AJ, Leow AD, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen A, Corneveaux J, Stephan DA, Webster J, DeChairo BM, Potkin SG, Jack CR, Jr., Weiner MW, Thompson PM. Voxelwise Genome-Wide Association Study (vGWAS) NeuroImage. 2009 doi: 10.1016/j.neuroimage.2010.02.032. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein JL, Hua X, Morra JH, Lee S, Hibar DP, Ho AJ, Leow AD, Toga AW, Sul JH, Kang H, Eskin E, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Stephan DA, Webster J, DeChairo BM, Potkin SG, Jack CR, Jr., Weiner MW, Thompson PM. Genome-wide association study of temporal lobe structure identifies novel quantitative trait loci for neurodegeneration in Alzheimer’s disease. NeuroImage. 2010 doi: 10.1016/j.neuroimage.2010.02.068. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, van Erp TGM, Thompson PM, Bearden CE, Daley M, Kushan L, Hardt ME, Nuechterlein K, Toga AW, Cannon TD. Elucidating an MRI-based biomarker for psychosis: classification using probabilistic brain atlas and machine learning algorithms. Biological Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.07.019. doi:10.1016/j.biopsych.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suykens JAK, Vandewalle J. Least Squares Support Vector Machine Classifiers. Neural Processing Letters. 1999;9:293–300. [Google Scholar]
- Vapnik V. The Nature of Statistical Learning Theory. Springer; New York: 1995. [Google Scholar]
- Vemuri P, Gunter JL, Senjem ML, Whitwell JL, Kantarci K, Knopman DS, Boeve BF, Petersen RC, Jack CR., Jr. Alzheimer’s disease diagnosis in individual subjects using structural MR images: validation studies. NeuroImage. 2008;39(3):1186–97. doi: 10.1016/j.neuroimage.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR., Jr. MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology. 2009;73(4):294–301. doi: 10.1212/WNL.0b013e3181af79fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhovd KB, Fjell AM, Brewer J, McEvoy LK, Fennema-Notestine C, Hagler DJ, Jr., Jennings RG, Karow D, Dale AM. Combining MR Imaging, Positron-Emission Tomography, and CSF Biomarkers in the Diagnosis and Prognosis of Alzheimer Disease. American Journal of Neuroradiology. 2010 doi: 10.3174/ajnr.A1809. doi: 10.3174/ajnr.A1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Ogar JM, Laluz V, Growdon M, Jang J, Glenn S, Miller BL, Weiner MW, Gorno-Tempini ML. Automated MRI-based classification of primary progressive aphasia variants. NeuroImage. 2009;47(4):1558–67. doi: 10.1016/j.neuroimage.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Wu T, Bae M, Reiman EM, Alexander GE, Jack CR, Jr., Thompson PM, Chen K. Use Of The Support Vector Machine And Sensitivity Of an AD-related Region-of-interest Gray Matter Classifier In Identifying Amnestic MCI Subjects Who Convert To AD: Preliminary Findings From The AD Neuroimaging Initiative; Presented at The International Conference on Alzheimer’s Disease; Chicago. 2008.Jul 26-31, [Google Scholar]