

Abstract
Mitochondrial production of reactive oxygen species and oxidation of cardiolipin are key events in initiating apoptosis. We reported that group VIA Ca2+-independent phospholipase A2 (iPLA2β) localizes in and protects β-cell mitochondria from oxidative damage during staurosporine-induced apoptosis. Here, we used iPLA2β-null (iPLA2β−/−) mice to investigate the role of iPLA2β in the repair of mitochondrial membranes. We show that islets isolated from iPLA2β−/− mice are more sensitive to staurosporine-induced apoptosis than those from wild-type littermates and that 2 wk of daily ip administration of staurosporine to iPLA2β−/− mice impairs both the animals’ glucose tolerance and glucose-stimulated insulin secretion by their pancreatic islets. Moreover, the iPLA2β inhibitor bromoenol lactone caused mitochondrial membrane peroxidation and cytochrome c release, and these effects were reversed by N-acetyl cysteine. The mitochondrial antioxidant N-t-butyl hydroxylamine blocked staurosporine-induced cytochrome c release and caspase-3 activation in iPLA2β−/− islets. Furthermore, the collapse of mitochondrial membrane potential in INS-1 insulinoma cells caused by high glucose and fatty acid levels was attenuated by overexpressing iPLA2β. Interestingly, iPLA2β was expressed only at low levels in islet β-cells from obesity- and diabetes-prone db/db mice. These findings support the hypothesis that iPLA2β is important in repairing oxidized mitochondrial membrane components (e.g. cardiolipin) and that this prevents cytochrome c release in response to stimuli that otherwise induce apoptosis. The low iPLA2β expression level in db/db mouse β-cells may render them vulnerable to injury by reactive oxygen species.
PLA2β prevents ROS-induced apoptosis by repair of oxidative modification of mitochondrial membrane phospholipids and relative deficiency of independent PLA2β may contribute to β-cell failure.
Diabetes is one of the most prevalent human metabolic diseases and is characterized by high blood glucose levels and insufficient insulin secretion due to the dysfunction and/or loss of β-cells in pancreatic islets. Type 1 diabetes (T1D) is an autoimmune disease caused by the specific destruction of insulin-producing β-cells (1), whereas type 2 diabetes (T2D) results from insulin resistance and gradual loss of β-cell function and mass to overcome the insulin resistance (2,3). In both T1D and T2D, apoptosis is thought to be a primary mechanism of β-cell death, which results in insufficient insulin production (1,2).
Mitochondrial reactive oxygen species (ROS) generation and the resultant oxidation of the phospholipid cardiolipin in mitochondrial membranes are key steps in the release of cytochrome c and other proapoptotic proteins that trigger caspase activation and apoptosis (4,5). Cytochrome c release is a two-step process that is initiated by its dissociation from cardiolipin, which normally anchors cardiolipin in the inner mitochondrial membrane (6). Oxidation of cardiolipin during apoptosis reduces cytochrome c binding and increases the amount of free cytochrome c in the intermembrane space (5,6). Therefore, oxidative stress controls the fate of cells by affecting cardiolipin oxidation (7,8).
Mitochondrial cardiolipin is a structurally unique dimeric phospholipid rich in polyunsaturated fatty acid substituents, and linoleate (18:2) is the predominant substituent in most mammalian tissues (9). For example, linoleate comprises 70% of the fatty acid substituents in rat pancreatic islet mitochondria (10). It is thought that the enrichment of linoleic acid (18:2) in cardiolipin results from a remodeling process in which a mitochondrial phospholipase A2 (PLA2) deacylates saturated fatty acid substituents from cardiolipin produced by de novo synthesis, and a mitochondrial phospholipid acyltransferase then reacylates the resultant lysocardiolipin with linoleic acid. Defective reacylation resulting from defects in the putative mitochondrial phospholipid acyltransferase Tafazzin (11) alters the composition of cardiolipin molecular species and leads to Barth syndrome (12).
PLA2 is a superfamily of enzymes that catalyze the hydrolysis of the sn-2 fatty acid substituent of glycerophospholipids to yield a free fatty acid and a 2-lysophospholipid (13). Among the intracellular PLA2, group VI Ca2+-independent PLA2 (iPLA2 or iPLA2β) has been found to participate in ongoing membrane phospholipid remodeling via a cycle of deacylation-reacylation (14). It was recently reported that iPLA2β plays an important role in cardiolipin deacylation and monolysocardiolipin accumulation in Barth syndrome (15). We found that iPLA2β localizes to and protects β-cell mitochondria during staurosporine-induced apoptosis (16), which involves mitochondrial ROS generation (17) and cardiolipin oxidation (5). These findings suggest that iPLA2β-mediated deacylation of cardiolipin might play an important role in the repair of mitochondrial phospholipids (e.g. cardiolipin) by removing peroxidized fatty acid substituents.
Interestingly, islets from mice with genetic ablation of iPLA2β gene (iPLA2β−/−) exhibit reduced insulin secretory responses (18), and iPLA2β−/− mice experience more severe deterioration of islet function than wild-type (WT) mice in response to stressors, such as administration of multiple low doses of the β-cell toxin streptozotocin or prolonged consumption of a Western diet with a high-fat content (18), suggesting that iPLA2β−/− β-cells are sensitive to oxidative stress. Here, we further characterize the role of iPLA2β-mediated deacylation in mitochondrial function of islet β-cells in vivo and ex vivo using iPLA2β−/− mice. Our findings presented in this study support our hypothesis that iPLA2β plays an important role in repairing oxidized mitochondrial membrane lipids (e.g. cardiolipin), which prevents cytochrome c release and apoptosis.
Materials and Methods
Materials
Bromoenol lactone (BEL) was obtained from Cayman Chemical (Ann Arbor, MI). Mouse Insulin ELISA kit was purchased from Crystal Chem, Inc. (Downers Grove, IL). DC Protein Assay kit was from Bio-Rad Laboratory (Hercules, CA). Cytochrome c Assay kit was obtained from Invitrogen Corp. (Camarillo, CA). JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzamidazolocarbocyanin iodide) Mitochondrial Membrane Potential Detection kit was from Cell Technology, Inc. (Mountain View, CA). N-t-butyl hydroxylamine (NtBHA) was purchased from Vitaspace (Central Islip, NY). Colorimetric Caspase-3 Assay kit, N-acetyl cysteine (NAC), and other chemicals were from Sigma Chemicals, Inc. (St. Louis, MO) unless otherwise noted.
Animals
We generated iPLA2β-null mice (iPLA2β−/−) by inserting the neomycin resistance gene into exon 9 of the mouse PLA2G6 by homologous recombination (18,19). iPLA2β−/− and WT mice were obtained by mating heterozygote animals and genotyping for the knockout allele by PCR assay. Six- to 10-wk-old db/db (C57BL/KsJ-Leprdb) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed and cared for in animal facilities administered through the Mount Sinai School of Medicine. All animal procedures were performed according to protocols approved by the Mount Sinai School of Medicine Animal Studies Committee.
Staurosporine treatment of mice
Six-month-old iPLA2β−/− mice (C57BL6 background) were randomly divided into control (n = 10) and staurosporine (n = 10) groups. Control mice were ip injected with 0.5% sodium carboxymethyl cellulose at a volume of 1 ml/kg, and staurosporine group was ip injected with staurosporine (0.4 mg/kg) daily. Staurosporine was dissolved in 0.5% sodium carboxymethyl cellulose at a volume of 1 ml/kg. Age-matched normal C57BL6 mice (WT) were also divided into control (n = 10) and staurosporine (n = 10) groups and treated identically to the iPLA2β−/− mice. After 2 wk of treatment, the mice of each group were subjected to metabolic analysis, and their islets were isolated for further study.
Glucose tolerance tests (GTT)
Intraperitoneal GTT (IPGTT) were performed after 2 wk of staurosporine treatment (20). After fasting overnight, mice were injected ip with 10% glucose (2 mg/g body weight) and glucose levels were determined after 0, 30, 60, 90, and 120 min by a Glucometer Elite (Bayer Corp., Elkhart, IN) (21).
Insulin tolerance test (ITT)
ITT were performed as described (20). Briefly, after 6-h fasting, human regular insulin (0.75 U/kg) (Sigma Chemicals, Inc.) was ip administrated to mice that freely accessed to water and chow. Blood samples were collected from tails before and after insulin administration every 15 min for 120 min, and serum glucose levels were determined.
Islet isolation and cell culture
Islets were isolated from pancreases removed from iPLA2β−/− mice and their WT littermates by Liberase (Roche Diagnostics, Indianapolis, IN) digestion, followed by discontinuous Ficoll gradient separation and manual stereomicroscopic selection to exclude contaminating tissues (20). The isolated islets were counted and cultured in islet culture medium (composed of RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 1% sodium pyruvate, 50 μm β-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin).
Rat insulinoma INS-1 cells and iPLA2-INS cells were cultured as described previously (16,22). iPLA2-INS is an iPLA2β-stable expression INS-1 cell line that was previously established using the retroviral vector pMSCVneo (CLONTECH, Palo Alto, CA) containing iPLA2β cDNA downstream of a 5′ long terminal repeat from the PCMV virus as described (22).
Determination of static insulin secretion
Isolated islets were incubated for 2 h in glass vials containing 500 μl Krebs-Ringer bicarbonate buffer supplemented with 10 mm HEPES and 2 mg/ml of BSA in the presence of 1.7 mm in humidified air with 5% CO2 at 37 C, and then further incubated for 60 min at 37 C in the presence of 1.7 or 16.7 mm glucose. At the end of the incubation, the medium was removed and stored at −20 C for insulin assay. Islets were washed briefly in Hanks’ solution and homogenized by sonication for total protein measurement. Insulin concentrations in the media were determined using the Ultra Sensitive Mouse Insulin ELISA kit (Crystal Chem, Inc.) and normalized to the total protein amounts.
Detection of apoptosis by Annexin V-FLUOS staining
For detection of phosphatidylserine externalization, an early characteristic of early-stage apoptosis, an Annexin V-FLUOS staining kit (Roche Molecular Biochemicals, Indianapolis, IN) was used to stain cells with fluorescent isothiocyanate-conjugated annexin V and propidium iodide (PI) according to the manufacturer’s protocol. The late-stage apoptotic and necrotic cells were stained with PI. Briefly, about 106 cells were harvested, washed with PBS by centrifugation at 200 × g for 5 min, and resuspended in 100 μl of Annexin-V-FLUOS labeling solution containing annexin-V-PLURO. Cells were incubated 10–15 min at 15–25 C and immediately analyzed by flow cytometry on a Becton Dickinson FACSCalibur (BD Biosciences, San Jose, California) (23,24). Cells were considered to be early apoptotic when they were annexin V-positive and PI-negative and late apoptotic when they were both annexin V- and PI-positive.
Measurement of cytochrome c concentration
After attachment, apoptosis was induced in isolated islets or in INS-1 β-cells in the presence or absence of NAC or NtBHA. After 0, 4, 8, and 12 h, islets were collected, and mitochondria and cytosolic fractions were extracted using a Mitochondria/Cytosol Fractionation kit (BioVsion Research Products, Mountain View, CA). The protein concentration of the supernatant was determined by Bradford assay using BSA as the standard. Cytochrome c levels in both fractions were measured using Cytochrome c Assay kit (Invitrogen Corp.). Data are presented as ng/mg proteins.
Western blot analysis
The cytosol and mitochondrial fractions of cells were obtained, and the protein concentration of the supernatant was determined as described above. Aliquots (containing 60 μg proteins for cytoplasmic fractions or 15 μg for mitochondrial fractions) were separated by SDS-PAGE, transferred to nitrocellulose, blotted with corresponding antibodies, and detected by enhanced chemiluminescence Western blotting detection system (Amersham Biosciences, Piscataway, NJ). Anticytochrome c and antiactin antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used for protein detection.
Measurement of caspase-3 activity
After attachment, apoptosis was induced in isolated islets or in INS-1 β-cells in the presence or absence of NAC or NtBHA. After 0, 4, 8, and 12 h, islets were collected, proteins extracted, and caspase-3 activity measured with a Colorimetric Caspase-3 Assay kit (Sigma Chemicals, Inc.) according to the manufacturer’s instructions. The concentration of p-nitroaniline released from the substrate was calculated from the absorbance values at 405 nm and is presented as nmol p-nitroaniline/min·μg proteins.
Detection of mitochondrial membrane potential
Mitochondrial membrane potentials were determined by the JC-1 Mitochondrial Membrane Potential Detection kit according to the manufacturer’s protocol (Cell Technology, Inc.). JC-1 is a lipophilic, cationic dye that selectively enters mitochondria and reversibly changes color from green to red as the membrane potential increases. In healthy cells with high mitochondrial potential, JC-1 aggregates spontaneously with an intense red fluorescence. Conversely, in apoptotic or unhealthy cells with low mitochondrial potential, JC-1 remains in the monomeric, green fluorescing form. Briefly, after induction of apoptosis, cells were washed with PBS, labeled with the JC-1 reagent for 15 min, and washed again with PBS. For live cell analysis, JC-1 stained cells were analyzed using LiveCell Microscope (magnification, ×20). For quantification, JC-1 staining was analyzed by a Becton Dickinson FACSCalibur (BD Biosciences) flow cytometer. Mitochondria with red JC-1 aggregates in healthy cells (upper) and green JC-1 monomers in apoptotic cells (lower) were separated in the FL2 channel. The loss of mitochondrial membrane potential was calculated as the percentage of green cells in the total.
Measurement of membrane phospholipid peroxidation
Decomposition of unstable peroxides derived from polyunsaturated fatty acids results in the formation of malondialdehyde, which reacts with thiobarbituric acid. Therefore, the thiobarbituric acid reactive substances (TBARS) assay is commonly used to detect lipid peroxidation (25). The sensitivity of TBARS Assay kit (ZeptoMetrix Corp., Buffalo, NY) increases dramatically (1 nmol/ml) when a fluorometer is used. After apoptotic induction, the cells (4 × 107) were collected, and mitochondria cytosol fractions were extracted using a Mitochondria/Cytosol Fractionation kit (BioVsion Research Products) according to the manufacturer’s instructions. Mitochondria or cytosol samples were subjected to Bradford and TBARS assays with excitation set at 530 nm and emission at 550nm. The amount of TBARS formed was normalized to total protein amounts.
Assessing mitochondrial cardiolipin oxidation
It is known that mitochondrial generation of ROS and resultant oxidation of cardiolipin cause release cytochrome c during induction of apoptosis (5). Simultaneous measurements of mitochondrial membrane peroxidation and cytochrome c release can be used to assess the extent of cardiolipin oxidation. Briefly, after inducing cells (2–4 × 107) to undergo apoptosis, mitochondrial and cytosolic fractions were isolated using a Mitochondria/Cytosol Fractionation Kit (BioVsion Research Products). Isolated mitochondria were sonicated in 200 μl of PBS on ice and subjected to protein, cytochrome c, and TBARS assays. The amount of TBARS formed was normalized to total mitochondrial proteins or mitochondrial DNA (Supplemental Fig. 1 published on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org), and the amount of free cytochrome c was calculated (total − remained cytochrome c/total cytochrome c). Increased levels of TBARS and of free cytochrome c reflect cardiolipin oxidation (5).
Immunohistochemistry
Isolated mouse pancreases were fixed overnight in 10% formalin at 4 C. Four-micrometer-thick sections were cut using ThermoShandon Finesse Microtome (Thermo Shandon, Inc., Pittsburgh, PA). Slides were incubated in 4% H2O2 to block the endogenous peroxidase and Powerblock universal blocking reagent (10×; Biogenex Corp., San Ramon, CA) was used to eliminate the background. iPLA2 were stained with goat anti-iPLA2 antibodies (Santa Cruz Biotechnology, Inc.). Biotinylated rabbit antigoat IgG (Vector Corp., Burliname, CA) was used as the second antibody followed by a peroxidase-conjugated streptavidin (Biogenex Corp.) and Dako liquid 3,3′ diaminobenzidine substrate-chromogen reagent (Dako, Carpinteria, CA). Slides were counterstained with hematoxylyn to localize cell nuclei. Finally, the slides were mounted with cytoseal xylene-based mounting medium (Stephens Scientific, Kalamazoo, MI) for visualization. Image scale bars were elucidated using ImageJ.
iPLA2β activity assay
iPLA2β activity was determined using a kit originally designed for cPLA2 (cPLA2 Assay kit; Cayman Chemicals) that had been modified as described (26). Briefly, after specific treatments, tissues from mice or cultured cells were collected and homogenized in buffer [50 mm HEPES (pH 7.4) and 1 mm EDTA] followed by centrifugation at 14,000 × g for 20 min at 4 C. The supernatant was removed, and the concentration of proteins was determined. iPLA2β activity was assayed by incubating the samples with arachidonoyl thio-phosphatidylcholine for 1 h at 25 C in a Ca2+-free buffer [4 mm EGTA, 160 mm HEPES (pH 7.4), 300 mm NaCl, 8 mm Triton X-100, 60% glycerol, and 2 mg/ml BSA]. The reaction was stopped by addition of 5,5′-dithiobis (nitrobenzoic acid) for 5 min, and the absorbance was determined at 414 nm using a μQuant microplate reader (BioTek Instruments, Inc., Winooski, VT). The specific activity of iPLA2β was calculated and expressed in nmol/min·mg of total proteins. The background basal lipase activity, which was determined by inhibiting all specific iPLA2β activity in control samples with BEL, was subtracted from all readings as described (26).
Calculations and statistical analysis
The incremental areas under the curve (IAUCs) of GTT were calculated for each group by using PRISM (version 3.0; GraphPad Software, Inc., San Diego, CA). All AUCs below the baseline (fasting glucose levels of WT control) were excluded from the calculations. Data were expressed as means ± sem. Significant differences among groups were evaluated by one-way ANOVA and Tukey’s multiple comparisons test or by unpaired two-tailed Student’s t test by using PRISM. Significance levels are described in individual figure legends.
Results
Staurosporine induced more extensive apoptosis in islets isolated from iPLA2β−/− mice than from their WT littermates
To examine genetically the role of iPLA2β-mediated deacylation in protection of β-cells from apoptosis, islets were isolated from 6-month-old iPLA2β−/− mice and their WT littermates and then incubated with 1 μm staurosporine. Apoptosis was assessed by measuring the time course of cytochrome c release and the increase in caspase-3 activity. As shown in Fig. 1, staurosporine induced significantly higher levels of cytochrome c release and caspase-3 activation in iPLA2β−/− islets than from WT islets. Overexpressing iPLA2β in INS-1 cells significantly blunted staurosporine-induced cytochrome c release (Fig. 1C) and annexin-V staining (Fig. 1D), the latter is a marker of phosphatidylserine externalization during apoptosis. These genetic and molecular observations support the hypothesis that iPLA2β acts to reduce the extent of apoptosis induced by staurosporine.
Figure 1.
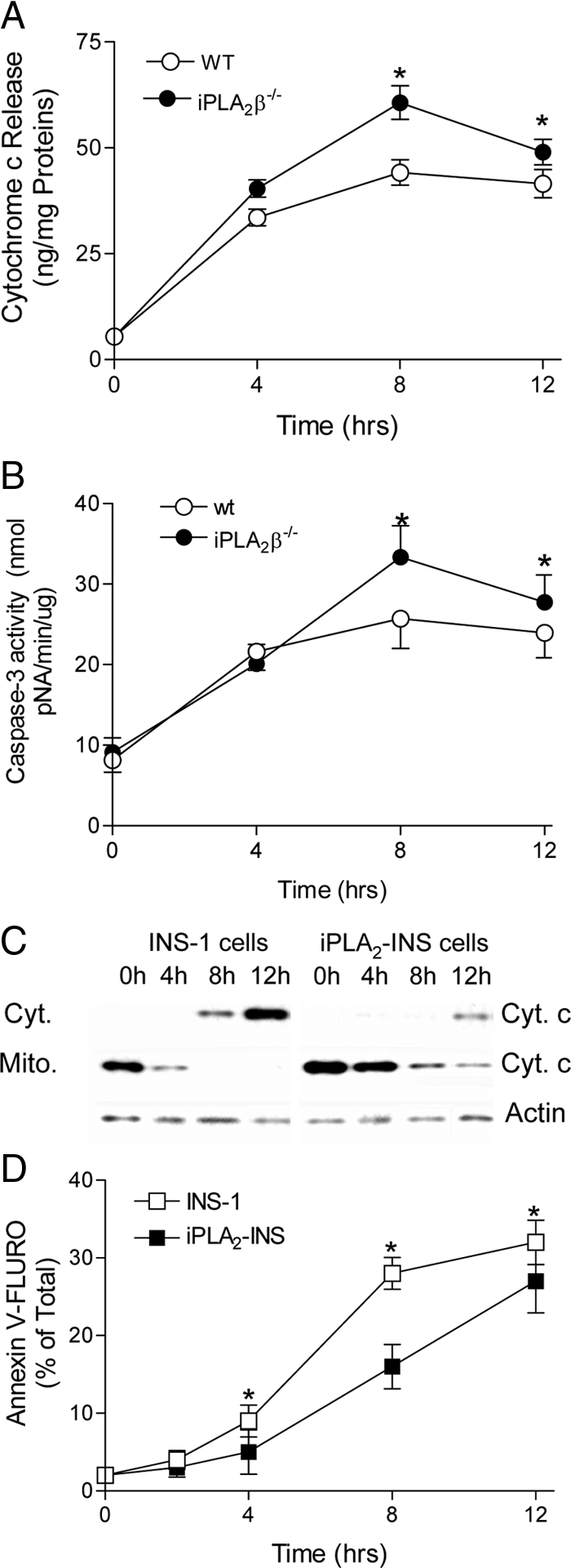
Effects of staurosporine on iPLA2β−/− islets. Islets isolated from 6-month-old iPLA2β−/− mice, and their normal littermates were treated with 1 μm staurosporine for up to 12 h. After extraction of mitochondria (Mito.) and cytosol (Cyt.) fractions, the released cytochrome c (A) and caspase-3 activities (B) were measured. Data are presented as the mean ± sem of three independent experiments. *, P < 0.05. INS-1 cells and iPLA2β-overexpression INS-1 cells (iPLA2-INS) were treated with 1 μm staurosporine over time. Then the cell lysates were prepared for analysis of cytochrome (Cyt. c) by the Western blot analysis (C), and the cells were stained with annexin-V and immediately analyzed by flow cytometry (D). Data are presented as the mean ± sem of six independent experiments. *, P < 0.05.
Staurosporine administration impairs glucose tolerance via effects on insulin secretion
Because islets isolated from iPLA2β−/− mice are more sensitive than those from WT mice to staurosporine-induced apoptosis (Fig. 1, A and B), we examined whether staurosporine affects islet β-cell function in vivo by ip injection of staurosporine. Six-month-old iPLA2β−/− mice (on a C57BL/6J genetic background) and their age-matched littermates were treated with daily ip injections of staurosporine for 2 wk, and IPGTT was conducted. As shown in Fig. 2A, the fasting glucose levels (116.0 ± 5.6 mg/dl) in staurosporine-treated iPLA2β−/− mice were significantly higher than all of other groups (84.0 ± 5.8 mg/dl in iPLA2β−/− control, P < 0.01; 95.0 ± 5.4 mg/dl in staurosporine-treated WT group, P < 0.05; 77.0 ± 7.5 mg/dl in WT control, P < 0.001). After glucose loading, staurosporine-treated iPLA2β−/− mice exhibited significantly higher glucose levels than all other groups at 30, 60, and 90 min in the GTT. We then calculated the IAUC during the GTT to quantitate glucose intolerance (27). The IAUCs in the staurosporine-treated iPLA2β−/− mice were significantly higher than that in all of other groups, and the IAUCs in the untreated iPLA2β−/− and staurosporine-treated WT littermates were also significantly higher than that in the WT control (Fig. 2B). However, no significant differences among the four groups were observed in ITT (Fig. 2C). These results indicate that the staurosporine-induced deterioration in glucose tolerance reflects impaired β-cell function, which is more severe with iPLA2β−/− islets than with WT islets in vivo. We next isolated islets from staurosporine-treated or untreated mice and examined their ability to secrete insulin ex vivo. As shown in Fig. 2D, islets from the staurosporine-treated iPLA2β−/− mice secreted significantly less insulin in response to 16.7 mm glucose than did islets from the other three groups.
Figure 2.
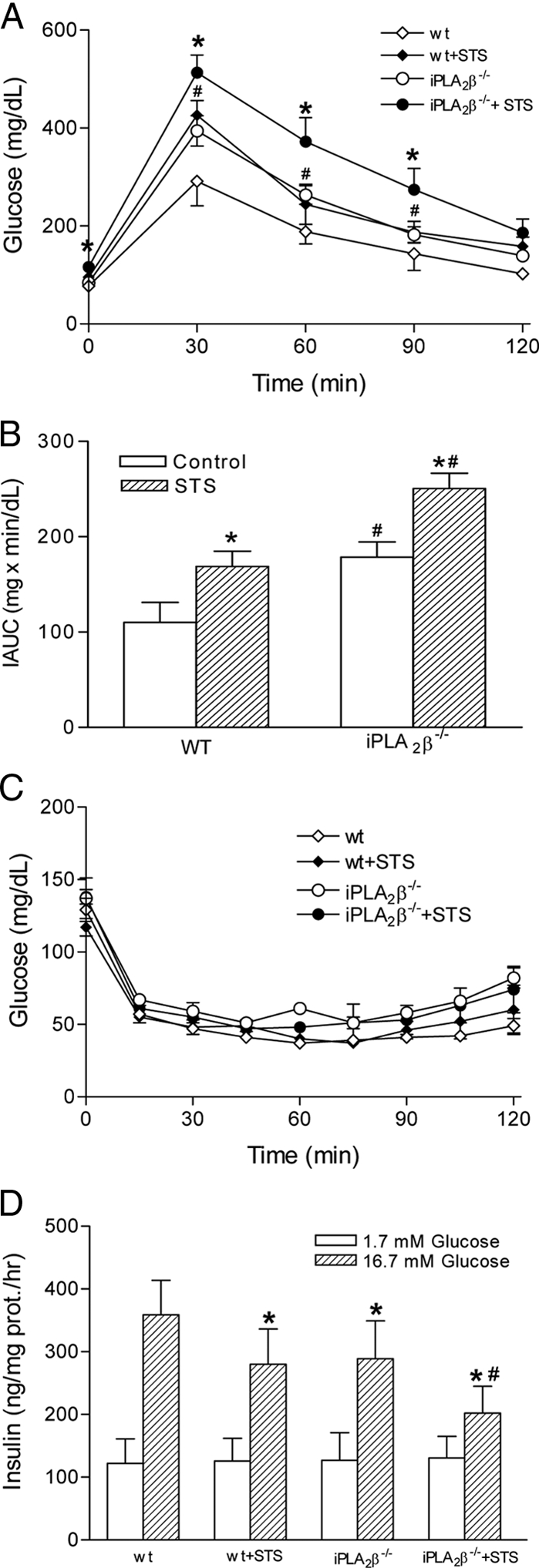
Effects of staurosporine (STS) on the metabolism of iPLA2β−/− mice. Six-month-old iPLA2β−/− (C57BL6 background) mice and their normal littermates were randomly divided into control (n = 10) and staurosporine (n = 10) groups. Control mice were injected ip with 0.5% sodium carboxymethyl cellulose at a volume of 1 ml/kg, and staurosporine groups were injected ip with staurosporine (0.4 mg/kg) daily for 2 wk. A, IPGTT (2 g/kg body weight). Data are presented as mean ± sem (n = 10); *, P < 0.05, iPLA2β−/− + STS vs. all other groups; #, P < 0.05, iPLA2β−/− control or WT + STS vs. the WT control. B, IAUCs of the GTTs. Data are presented as mean ± sem (n = 10); *, P < 0.001, STS-treated groups vs. their controls; #, P < 0.001, iPLA2β−/− groups vs. corresponding WT groups. C, ITT. Data are presented as mean ± sem; P > 0.05. D, Glucose-stimulated insulin secretion by isolated islets ex vivo. The islets were isolated from the mice described above for insulin secretion assays. Insulin secretion of each group in 16.7 mm glucose is significantly higher than that in 1.7 mm glucose (P < 0.01). *, P < 0.05, compared with WT 16.7 mm glucose; #, P < 0.05, iPLA2β−/− + STS vs. all of other groups.
iPLA2β inhibition enhances mitochondrial membrane peroxidation and cytochrome c release in response to glucose
If iPLA2β acts to remove oxidized fatty acid substituents from mitochondrial phospholipids, so that it can be reacylated with unoxidized substituents, this could confer relative resistance to stimuli that induce apoptosis. Conversely, reducing iPLA2β activity could increase susceptibility to apoptosis. To examine this possibility, we treated INS-1 cells with increasing concentrations of BEL in the presence of 11 mm glucose, which is at a moderately high level. As shown in Fig. 3A, BEL induced apoptosis in INS-1 cells in a concentration-dependent manner, and protection from this effect was achieved by including the antioxidant NAC in the incubation medium (Fig. 3B). BEL also induced the peroxidation of mitochondrial membranes, as reflected by TBARS formation, but this was not observed with nonmitochondrial membranes (Fig. 3C). In contrast, treating the cells with H2O2 resulted in peroxidation of both mitochondrial and nonmitochondrial membrane lipids (Fig. 3C), which indicates that there is a specific effect of BEL to enhance mitochondrial membrane susceptibility to oxidation. Moreover, BEL induced release of cytochrome c (Fig. 3D), and both this effect and mitochondrial membrane peroxidation were prevented by including NAC in the incubation medium (Fig. 3, C and D). These observations indicate that inhibition of iPLA2β in cells incubated with 11 mm glucose may impair the repair of oxidized mitochondrial membrane lipids, such as cardiolipin, and this could increase their susceptibility to apoptosis.
Figure 3.
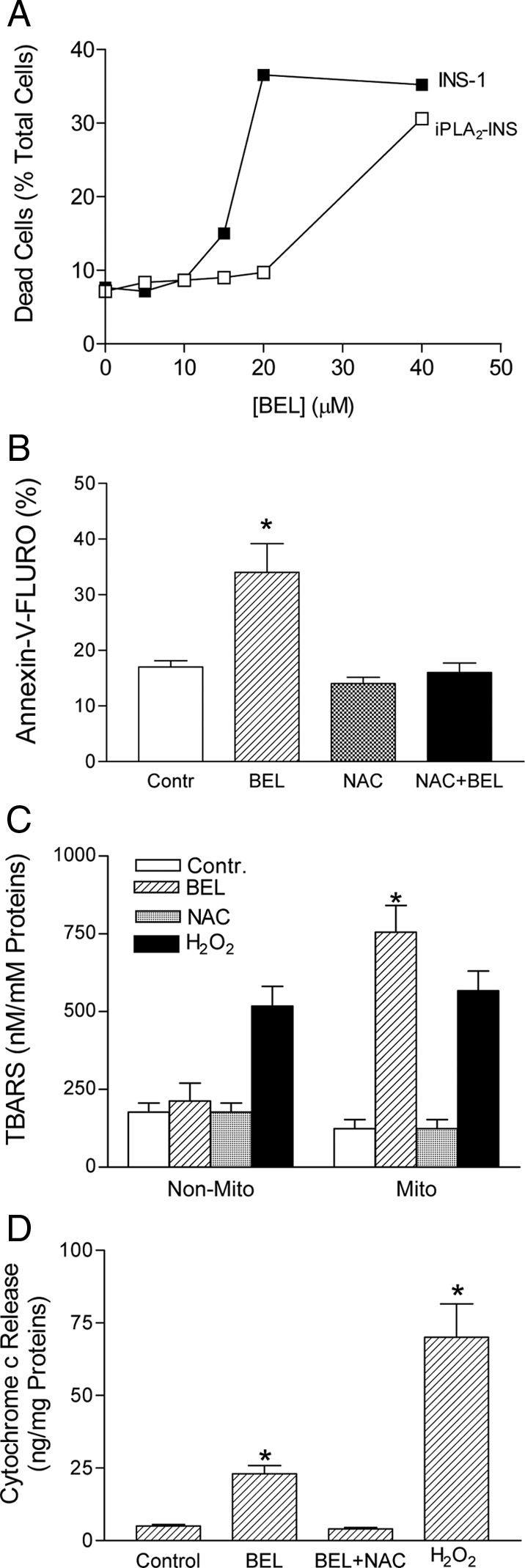
Inhibition of iPLA2β induced the peroxidation of mitochondrial membranes. A, BEL-induced apoptosis at high concentrations. INS-1 and iPLA2-INS cells were incubated with increasing concentrations of BEL in the presence of 11 mm glucose for 48 h. The cells were then stained with annexin-V and analyzed by flow cytometry. B, BEL-induced apoptosis was blocked by NAC. INS-1 cells were treated with 25 μm of BEL in the presence of 11 mm of glucose for 48 h. Apoptosis was analyzed by Annexin V-staining and fluorescence-activated cell sorter (*, compared with control). C, TBARS assay of mitochondrial membrane peroxidation in INS-1 cells. INS-1 cells were incubated with 25 μm BEL in the presence of 11 mm glucose for 48 h. Mitochondria (Mito) were isolated, and the Mito and non-Mito fractions were subjected to TBARS assay (*, compared with non-Mito). D, Cytochrome c release assay. P < 0.05 (n = 4); *, compared with control. Data are presented as the mean ± sem of three independent experiments.
Blocking mitochondrial membrane peroxidation inhibits staurosporine-induced apoptosis in iPLA2β−/− islets
To further examine whether selective peroxidation of mitochondrial membranes contributes to apoptosis in iPLA2β−/− β-cells, pancreatic islets isolated from iPLA2β−/− mice were incubated with staurosporine in the presence or absence of NtBHA, an antioxidant that accumulates in mitochondria and blocks peroxidation of their phospholipids. As shown in Fig. 4, staurosporine induced both cytochrome c release and caspase-3 activation in iPLA2β−/− islets, and both effects were essentially blocked when NtBHA was included in the incubation medium.
Figure 4.
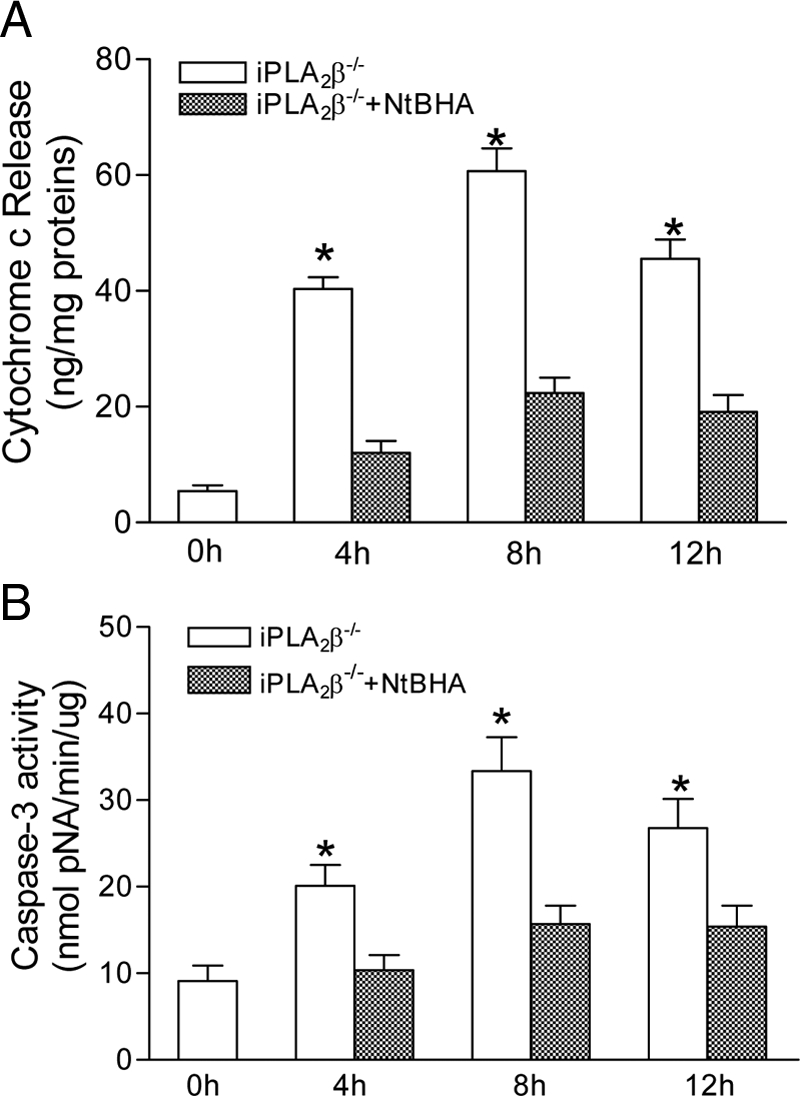
Effect on apoptosis of blocking the peroxidation of mitochondrial membranes. A, Isolated islets from iPLA2β−/− mice were treated with staurosporine in the presence or absence of NtBHA over time. Cytochrome c release (A) and caspase-3 activity (B) were measured. Data are presented as the mean ± sem of three independent experiments. *, P < 0.05 compared with the control.
iPLA2 prevents the collapse of mitochondrial membrane potential in response to high concentrations of glucose or palmitic acid
We next examined the role of iPLA2β in protecting β-cell mitochondrial membranes in response to high concentrations of glucose or the fatty acid palmitate, because both obesity and chronic hyperglycemia are known to increase mitochondrial superoxide production (28). Parental INS-1 insulinoma cells or INS-1 cells that had been stably transfected to overexpress iPLA2β (iPLA2-INS) were incubated in culture medium containing 5.6 or 22 mm glucose for 72 h. The cells were then stained with JC-1 and analyzed by fluorescence-activated cell sorter to assess loss of mitochondrial membrane potential as previously described (16). As shown in Fig. 5A, iPLA2-INS cells were found to be more resistant than parental INS-1 cells to the collapse of mitochondrial membrane potential induced by incubation in medium containing 22 mm glucose.
Figure 5.

iPLA2β prevented the loss of mitochondrial membrane potential in INS-1 cells in response to glucose or fatty acids. A, Flow cytometry analysis of glucose-induced loss of mitochondrial membrane potential. INS-1 cells and iPLA2-INS were treated with or without (control) 22 mm glucose for 72 h. The cells were then stained with JC-1 and analyzed by flow cytometry. B, Microscopic analysis of living cells treated with palmitate. INS-1 cells and iPLA2-INS were treated with or without 0.5 mm palmitate for 48 h. The cells were then stained with JC-1, analyzed by fluorescent microscopy, and quantified by flow cytometry. Living cells are stained both red and green, apoptotic cells are stained with green only. Data are presented as the mean ± sem of nine independent experiments. *, P < 0.05, treated groups vs. the controls; #, P < 0.05, the treated iPLA2-INS vs. the treated INS-1 cells. Pal, Palmitate; Glc, glucose.
Similarly, the cells were also cultured with and without 0.5 mm palmitic acid for 48 h, stained with JC-1, and analyzed by fluorescence microscopy. As shown in Fig. 5B, palmitate treatment caused a significant collapse of mitochondrial membrane potential in the parental INS-1 cells (visualized as green only), but many iPLA2-INS cells retained functional mitochondria under these conditions (visualized here as orange).
Islets from db/db mice express only low levels of iPLA2β
The findings described above suggest that iPLA2β deficiency increases β-cell susceptibility to mitochondrial injury by ROS, and this might predispose mice to β-cell loss or dysfunction resulting in development of diabetes. The db/db mouse model is diabetes prone, and this might be attributable in part to relative deficiency of iPLA2β in db/db mouse islets. To evaluate this possibility, pancreata were removed from age-matched (6–10 wk old) C57/BL6J and male db/db mice (C57BL/KsJ-Leprdb), and sections were stained for iPLA2β expression, which was observed uniformly in islet cells from C57/BL6J mice but was barely detectable in exocrine acinar cells of these mice (Fig. 6A, a).
Figure 6.

Immunohistochemical analysis of iPLA2β expression in the pancreases of db/db mice. A, Six- to 10-wk-old normal (a) and db/db (C57BL/KsJ-Leprdb) mice (b–d) were killed, and the pancreases were fixed and sliced for iPLA2 staining with goat anti-iPLA2β antibodies. B, iPLA2β activity analysis. The islets were isolated from normal and db/db mice at the ages of 6–10 wk, and cell lysates were prepared for iPLA2β analysis (n = 4); *, P < 0.05, compared with control (Contr.) in the absence of BEL. Contr, Control.
In contrast, iPLA2β expression in β-cells in pancreatic islets from db/db mice was dramatically reduced compared with that in age-matched controls (Fig. 6A, b–d). Most db/db islets exhibited only patchy expression of iPLA2β (Fig. 6A, b). Some islets exhibited low expression overall (Fig. 6A, c), whereas in others, the uniformity of iPLA2β expression was lost (Fig. 6A, d). In every case, iPLA2β expression in β-cells was substantially lower than that observed in the control mice. Consistent with these observations, BEL-sensitive iPLA2 activity was much lower in db/db islets than in the control islets (Fig. 6B). It is thus possible that the low level of iPLA2β in islets of db/db mice may contribute to the enhanced susceptibility of these animals to obesity-associated β-cell failure and diabetes.
Discussion
Mitochondrial failure in β-cells has emerged as an important step in the pathogenesis of T2D (29,30). The generation of ROS in response to multiple stresses may cause mitochondrial dysfunction and trigger β-cells apoptosis (3). We previously reported that iPLA2β localizes in and protects the mitochondria of pancreatic β-cells during apoptotic induction by staurosporine (16). Here, we further determine the role of iPLA2β in protection of mitochondrial membranes in pancreatic β-cells in a mouse model with genetic ablated iPLA2β (iPLA2β−/−). Key findings include: 1) islets isolated from iPLA2β−/− mice are more prone to be apoptotic in the presence of staurosporine; 2) iPLA2β−/− mice exhibit increased fasting glucose levels, impaired glucose tolerance, and reduced insulin secretion by their islets; 3) specific inhibition of iPLA2β by BEL enhances peroxidation of mitochondrial membranes and release of cytopchrome c in response to glucose or palmitate in INS-1 cells; 4) the antioxidant NtBHA, which accumulates in mitochondria and prevents peroxidation of their phospholipids, blunts staurosporine-induced apoptosis in islets from iPLA2β−/− mice; and 5) expression of iPLA2β is significantly decreased in the pancreatic β-cells of db/db mice. These findings support the hypothesis that iPLA2β confers protection against ROS-induced apoptosis by attenuating or reversing the effects of oxidative modification of mitochondrial membrane phospholipids, such as cardiolipin, and suggest that relative deficiency of iPLA2β may contribute to β-cell failure during the development of T2D in db/db mice.
It is known that mitochondrial phospholipid oxidation, particularly cardiolipin oxidation, sets cytochrome c free and release to trigger apoptosis (4,5). The inner mitochondrial membranes contain a high proportion of cardiolipin that is rich at polyunsaturated fatty acid linoleic acid (9) and thus are particularly susceptible to ROS attack. Because the polyunsaturated fatty acids in phospholipids tend to be located in the sn-2 position, it is likely that members of the PLA2 group of enzymes, which can hydrolyze oxidized fatty acids at sn-2 (13,31), are involved in the repair of oxidized membrane phospholipids (32,33,34). iPLA2β localizes in mitochondria (16) and was recently reported to play an important role in deacylation during cardiolipin remodeling in a Tafazzin-deficient Drosophila model of Barth syndrome (15). Even though we did not directly measure the cardiolipin oxidation in this study, it is likely that iPLA2β-mediated deacylation, in addition to its role in cardiolipin remodeling, also serves to cleave oxidized fatty acids from cardiolipin as evidenced by that both peroxidation of mitochondrial phospholipids and cytochrome c release are significantly increased in iPLA2β−/− islets. Loss of such repair mechanism by knocking out iPLA2β may render mice more sensitive to the attack by ROS induced by staurosporine (Fig. 2) or by high-fat diet (18). Indeed, mutations in the iPLA2β gene PLA2G6 are recently found to underlie the syndromes of infantile neuroaxonal dystrophy and neurodegeneration with brain iron accumulation in humans (35).
In this study, we found that mitochondrial phospholipid peroxidation can be induced more readily when iPLA2β activity is inhibited by its suicide substrate BEL. It is possible that oxidation of mitochondrial phospholipids, including cardiolipin, serves to rapidly trap ROS to protect mitochondrial proteins or DNA from oxidative injury or to transduce a ROS signal to respiratory chain proteins associated with it to mitigate ROS generation (36). Repair of oxidized mitochondrial phospholipids by iPLA2β-mediated deacylation followed by acyltransferase-catalyzed reacylation might complete a cycle of oxidation and repair that could modulate ROS levels and effects during stress responses. In circumstances where ROS levels and oxidation of mitochondrial phospholipids, particularly cardiolipin oxidation, exceed the capacity of repair or where the repair system is defective, such as in iPLA2β−/− mice, unrestrained modification of mitochondrial membrane phospholipids, such as cardiolipin, could compromise membrane integrity and result in release of cytochrome c. When this occurs in β-cells, the resultant induction of apoptosis could represent a mechanism of β-cell loss that ultimately leads to diabetes.
The generation of ROS in response to the high concentrations of glucose and free fatty acids may cause mitochondrial dysfunction and trigger β-cells apoptosis (3). Here we demonstrate that iPLA2β overexpression in β-cells can confer protection against the collapse of mitochondrial membrane potential induced by their exposure to the high concentrations of glucose and the free fatty acid palmitate. Interestingly, db/db islet β-cells express lower levels of iPLA2β than islets from the controls, and this could impair cardiolipin remodeling and repair in db/db β-cells and render them more susceptible to oxidative injury and thereby contribute to their propensity to undergo obesity-associated reduction of β-cell mass and the development of T2D. Indeed, iPLA2β-null mice exhibited more severe deterioration in their islet function when they were fed with a high-fat diet (18).
iPLA2β contains several caspase-3 consensus sites, and cleavage at these sites irreversibly activates iPLA2β (13,37,38). Endoplasmic reticulum stress-induced or caspase-3-induced activation of iPLA2β has been reported to facilitate apoptosis (37,39,40,41,42). Because caspase-3 activation marks cells for death, iPLA2β activation by caspase-3 may contribute to the characteristic biologic and morphologic changes of apoptosis. It is known that iPLA2β activation during apoptosis produces the chemoattractant lysophosphatidylcholine to recruit professional phagocytes to engulf the apoptotic cells (38). Effective clearance of apoptotic cells is an important physiologic homeostatic mechanism that is required to prevent secondary necrosis and inflammation (43,44). Interestingly, db/db mice (C57BL/KsJ-Leprdb) exhibit perturbations in cell-mediated immunity (45,46,47,48). Because disruption of the process of apoptotic cell clearance contributes to the development of a number of inflammatory and autoimmune diseases (43,44), it will be interesting to investigate whether low expression of iPLA2β in db/db islets results in the failure of clearance of apoptotic β-cells leading to the development of antiislets immunity in the db/db mice.
In summary, accumulating evidence indicates that iPLA2β plays an important role in cells as elucidated in Fig. 7. We propose that the intrinsic function of iPLA2β is to maintain optimal mitochondrial membrane composition by participating in cardiolipin remodeling and in repairing oxidative modifications caused by mitochondrial ROS production. Once caspase-3 activation makes cell death inevitable, however, activated iPLA2β facilitates phagocytic clearance of apoptotic cells by release of the chemoattractant lysophosphatidylcholine to set up the antiinflammatory environment (38,43).
Figure 7.

Proposed model for the role of iPLA2β in mitochondrial function. (1) Cardiolipin remodeling. Nascent cardiolipin is remodeled to mature cardiolipin through a cycle of deacylation by iPLA2β and reacylation by monolysocardiolipin acyltransferase (MLCLAT). (2) Repair of cardiolipin oxidation. Under low oxidative stress, mitochondrial cardiolipin is oxidized, which may lower ROS levels, and is repaired promptly by iPLA2β and MLCLAT-mediated deacylation and reacylation. Under high oxidative stress, cardiolipin oxidation frees cytochrome c and initiates apoptosis. (3) Facilitation of apoptotic process. The permanent activation of iPLA2β by caspase-3 may facilitate clearance of apoptotic cells by releasing lysophosphatidylcholine (LPC). sFA, Saturated fatty acids; usFA, unsaturated fatty acids. Gray arrows, The biochemical reactions catalyzed by iPLA2β; black arrows, stimulatory pathway; dashed arrow, inhibitory pathway.
Supplementary Material
Footnotes
This work was supported by the National Institutes of Health Grant DK074805 and the Iacocca Family Foundation.
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 12, 2010
Abbreviations: BEL, Bromoenol lactone; cPLA2, cytosolic PLA2; GTT, glucose tolerance tests; IAUC, incremental area under the curve; IPGTT, intraperitoneal GTT; iPLA2 or iPLA2β, group VI Ca2+-independent PLA2; ITT, Insulin tolerance test; JC-1, 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzamidazolocarbocyanin iodide; NAC, N-acetyl cysteine; NtBHA, N-t-butyl hydroxylamine; PI, propidium iodide; PLA2, phospholipase A2; ROS, reactive oxygen species; TBARS, thiobarbituric acid reactive substances; T1D, type 1 diabetes; T2D, type 2 diabetes; WT, wild type.
References
- Mathis D, Vence L, Benoist C 2001 β-Cell death during progression to diabetes. Nature 414:792–798 [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC 2003 β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110 [DOI] [PubMed] [Google Scholar]
- Prentki M, Nolan CJ 2006 Islet β cell failure in type 2 diabetes. J Clin Invest 116:1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ 1993 Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 75:241–251 [DOI] [PubMed] [Google Scholar]
- Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG 2005 Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1:223–232 [DOI] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S 2002 Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99:1259–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Zhivotovsky B, Orrenius S 2007 Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ 14:1243–1247 [DOI] [PubMed] [Google Scholar]
- Schug ZT, Gottlieb E 2009 Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta 1788:2022–2031 [DOI] [PubMed] [Google Scholar]
- Schlame M, Rua D, Greenberg ML 2000 The biosynthesis and functional role of cardiolipin. Prog Lipid Res 39:257–288 [DOI] [PubMed] [Google Scholar]
- Turk J, Wolf BA, Lefkowith JB, Stump WT, McDaniel ML 1986 Glucose-induced phospholipid hydrolysis in isolated pancreatic islets: quantitative effects on the phospholipid content of arachidonate and other fatty acids. Biochim Biophys Acta 879:399–409 [DOI] [PubMed] [Google Scholar]
- Bione S, D'Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, Toniolo D 1996 A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 12:385–389 [DOI] [PubMed] [Google Scholar]
- Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJA, Barth PG 2000 Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome. Biochem Bioph Res Commun 279:378–382 [DOI] [PubMed] [Google Scholar]
- Ma Z, Turk J 2001 The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acids Res Mol Biol 67:1–33 [DOI] [PubMed] [Google Scholar]
- Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA 1995 Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci USA 92:8527–8531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Edelman-Novemsky I, Xu Y, Plesken H, Ma J, Schlame M, Ren M 2009 Role of calcium-independent phospholipase A2 in the pathogenesis of Barth syndrome. Proc Natl Acad Sci USA 106:2337–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA 2006 Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem 281:22275–22288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Jones DP 1998 Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 273:11401–11404 [DOI] [PubMed] [Google Scholar]
- Bao S, Song H, Wohltmann M, Ramanadham S, Jin W, Bohrer A, Turk J 2006 Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J Biol Chem 281:20958–20973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J 2004 Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem 279:38194–38200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, Ma ZA 2009 Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 150:2569–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Lee YJ, Kim SK, Kim HJ, Shim WS, Ahn CW, Lee HC, Cha BS, Ma ZA 2009 Rosiglitazone and fenofibrate improve insulin sensitivity of pre-diabetic OLETF rats by reducing malonyl-CoA levels in the liver and skeletal muscle. Life Sci 84:688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J 2001 Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids 36:689–700 [DOI] [PubMed] [Google Scholar]
- Zhang XH, Zhao C, Seleznev K, Song K, Manfredi JJ, Ma ZA 2006 Disruption of G1-phase phospholipid turnover by inhibition of Ca2+-independent phospholipase A2 induces a p53-dependent cell-cycle arrest in G1 phase. J Cell Sci 119:1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH, Zhao C, Ma ZA 2007 The increase of cell-membranous phosphatidylcholines containing polyunsaturated fatty acid residues induces phosphorylation of p53 through activation of ATR. J Cell Sci 120:4134–4143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi K 1998 Simple procedure for specific assay of lipid hydroperoxides in serum or plasma. Methods Mol Biol 108:107–110 [DOI] [PubMed] [Google Scholar]
- Song K, Zhang X, Zhao C, Ang NT, Ma ZA 2005 Inhibition of Ca2+-independent phospholipase A2 results in insufficient insulin secretion and impaired glucose tolerance. Mol Endocrinol 19:504–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolever TM, Jenkins DJ, Jenkins AL, Josse RG 1991 The glycemic index: methodology and clinical implications. Am J Clin Nutr 54:846–854 [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, Lowell BB 2003 Superoxide-mediated activation of uncoupling protein 2 causes pancreatic β cell dysfunction. J Clin Invest 112:1831–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB 2001 Mitochondrial function in normal and diabetic β-cells. Nature 414:807–812 [DOI] [PubMed] [Google Scholar]
- Lowell BB, Shulman GI 2005 Mitochondrial dysfunction and type 2 diabetes. Science 307:384–387 [DOI] [PubMed] [Google Scholar]
- Six DA, Dennis EA 2000 The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim Biophys Acta 1488:1–19 [DOI] [PubMed] [Google Scholar]
- Sevanian A 1988 Lipid damage and repair. In: Davies K, ed. Oxidative damage and repair. New York: Pergamon Press; 543–549 [Google Scholar]
- Sevanian A, Hochstein P 1985 Mechanisms and consequences of lipid peroxidation in biological systems. Annu Rev Nutr 5:365–390 [DOI] [PubMed] [Google Scholar]
- Nigam S, Schewe T 2000 Phospholipase A2s and lipid peroxidation. Biochim Biophys Acta 1488:167–181 [DOI] [PubMed] [Google Scholar]
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ 2006 PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38:752–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg M 2009 Cardiolipin and mitochondrial carriers. Biochim Biophys Acta 1788:2048–2058 [DOI] [PubMed] [Google Scholar]
- Atsumi GI, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I 1998 Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem 273:13870–13877 [DOI] [PubMed] [Google Scholar]
- Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S 2003 Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113:717–730 [DOI] [PubMed] [Google Scholar]
- Mancuso DJ, Abendschein DR, Jenkins CM, Han X, Saffitz JE, Schuessler RB, Gross RW 2003 Cardiac ischemia activates calcium-independent phospholipase A2β, precipitating ventricular tachyarrhythmias in transgenic mice: rescue of the lethal electrophysiologic phenotype by mechanism-based inhibition. J Biol Chem 278:22231–22236 [DOI] [PubMed] [Google Scholar]
- Pérez R, Melero R, Balboa MA, Balsinde J 2004 Role of group VIA calcium-independent phospholipase A2 in arachidonic acid release, phospholipid fatty acid incorporation, and apoptosis in U937 cells responding to hydrogen peroxide. J Biol Chem 279:40385–40391 [DOI] [PubMed] [Google Scholar]
- Lei X, Zhang S, Bohrer A, Ramanadham S 2008 Calcium-independent phospholipase A2 (iPLA2 β)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J Biol Chem 283:34819–34832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Zhang S, Bohrer A, Bao S, Song H, Ramanadham S 2007 The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 46:10170–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauber K, Blumenthal SG, Waibel M, Wesselborg S 2004 Clearance of apoptotic cells: getting rid of the corpses. Mol Cell 14:277–287 [DOI] [PubMed] [Google Scholar]
- Michlewska S, McColl A, Rossi AG, Megson IL, Dransfield I 2007 Clearance of dying cells and autoimmunity. Autoimmunity 40:267–273 [DOI] [PubMed] [Google Scholar]
- Fernandes G, Handwerger BS, Yunis EJ, Brown DM 1978 Immune response in the mutant diabetic C57BL/Ks-dt+ mouse. Discrepancies between in vitro and in vivo immunological assays. J Clin Invest 61:243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MA, Mahmoud AA 1978 Impairment of cell-mediated immunity in mutation diabetic mice (db/db). J Immunol 120:1375–1377 [PubMed] [Google Scholar]
- Debray-Sachs M, Dardenne M, Sai P, Savino W, Quiniou MC, Boillot D, Gepts W, Assan R 1983 Anti-islet immunity and thymic dysfunction in the mutant diabetic C57BL/KsJ db/db mouse. Diabetes 32:1048–1054 [DOI] [PubMed] [Google Scholar]
- Debray-Sachs M, Saï P, Boitard C, Assan R, Hamburger J 1983 Anti-pancreatic immunity in genetically diabetic mice. Clin Exp Immunol 51:1–7 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.