

Abstract
The integrity of DNA is constantly challenged throughout the life of a cell by both endogenous and exogenous stresses. A well-organized rapid damage response and proficient DNA repair, therefore, becomes critically important for maintaining genomic stability and cell survival. When DNA is damaged, the DNA damage response (DDR) can be initiated by alterations in chromosomal structure and histone modifications, such as the phosphorylation of the histone H2AX (the phosphorylated form is referred to as γ-H2AX). γ-H2AX plays a crucial role in recruiting DDR factors to damage sites for accurate DNA repair. Upon repair completion,γ-H2AX must then be reverted to H2AX by dephosphorylation for attenuation of the DDR. Here, we report that Wip1 phosphatase, which is often over-expressed in a variety of tumors, effectively dephosphorylates γ-H2AX in vitro and in vivo. Ectopic expression of Wip1 significantly reduces the level of γ-H2AX after ionizing as well as ultraviolet radiation. Forced premature dephosphorylation of γ-H2AX by Wip1 disrupts recruitment of important DNA repair factors to damaged sites, and delays DNA damage repair. Additionally, deletion of Wip1 enhances γ-H2AX levels in cells undergoing constitutive oncogenic stress. Taken together our studies demonstrate that Wip1 is an important mammalian phosphatase for γ-H2AX and demonstrates an additional mechanism for Wip1 in the tumor surveillance network.
Keywords: Wip1, PPM1D, H2AX, gamma-H2AX, ionizing radiation, ultraviolet radiation, DNA damage, DNA repair
Introduction
H2AX is a histone variant that plays a major role in the DNA damage response (DDR) and maintenance of genomic stability. A well-established function of H2AX is the recognition and repair of double-strand breaks (DSBs) created by genotoxic stress, and monitoring the removal of H2AX phosphorylation has been shown to be a useful tool in detecting the completion of DNA repair (1, 2). However, agents that induce other types of DNA damage, such as ultraviolet radiation (UVR), also induce the phosphorylation of H2AX (3). Furthermore, an early action of some oncogenes is thought to be to trigger DNA damage by inducing stalling and collapse of DNA replication forks, which triggers the DDR and leads to phosphorylation of H2AX (4-7). Thus, H2AX phosphorylation may have a variety of roles in genomic stability.
H2AX phosphorylation is a critical event during the DDR [reviewed in (1, 8)]. Briefly, when a DSB is formed by ionizing radiation (IR) exposure, for example, the Mre11-Rad50-NBS1 (MRN) complex recognizes the free DNA ends at the break and recruits the PI3K-like kinases, such as ataxia telangiectasia mutated (ATM), which phosphorylate H2AX on serine 139. This phosphorylated form of H2AX (γ-H2AX) then serves as a platform and facilitates the recruitment of further DNA repair factors necessary to mend the lesion. First, mediator of DNA damage checkpoint protein 1 (MDC1) binds to γ-H2AX and coordinates with the MRN complex to activate ATM. ATM then phosphorylates H2AX, resulting in a cyclical process that leads to as large as a two megabase pair spreading of γ-H2AX molecules from the DSB site. The γ-H2AX molecules surrounding the damage site then act as a scaffold for DNA repair factors that repair the damage, and other DDR response proteins that arrest the cell cycle until the DNA is repaired (1, 8). However, the mechanism of γ-H2AX formation is different depending on the type of genotoxic stress. For example, where as H2AX is primarily phosphorylated by ATM at DSBs after IR exposure, the mechanism of UVR-induced γ-H2AX formation is different and depends on the cell cycle and can occur in G1-phase cells presumably in the absence of DSBs (3, 9-11).
Once DNA damage is repaired, the cell must have a means of terminating DDR signaling, and protein modifications like γ-H2AX that occurred during the DDR need to be removed. γ-H2AX could be removed by dephosphorylation or by histone exchange, and currently there is evidence that both occur. The phosphatases PP2A and PP4C have been shown to dephosphorylate γ-H2AX, which is required for checkpoint recovery (12-14). Additionally, several groups have shown that replacement of γ-H2AX with unmodified H2AX is induced by nucleosomal conformational changes (15), can occur after IR exposure (16), and occurs in the yeast Saccharomyces cerevisiae (17-19).
Due to the presence of γ-H2AX in pre-cancerous lesions and its role in the DDR (1, 4), it is not surprising that H2AX has tumor suppressor properties. The gene encoding H2AX, H2AFX, has a chromosomal location of 11q23, which is a region that is often altered in many types of cancers (20). Furthermore, deletion of the H2AFX gene leads to genomic instability in mice (21), and H2AX has been shown to maintain genomic stability by inhibiting the production of chromosomal aberrations by DNA breaks after IR exposure and DNA replication stress (22-24). Although the H2AFX-/- mice do not develop cancers in higher frequency than wild type mice, even a heterozygous deletion of this gene in a p53-null background renders mice susceptible to the accelerated development of cancerous lesions (20, 25). Additionally, H2AX has been shown to inhibit lymphomagenesis by synchronizing DNA repair with the cell cycle and proliferation (26), which indicates that H2AX can function as a tumor suppressor in certain cellular environments.
Wild-type p53-induced phosphatase 1 (Wip1) is a nuclear, oncogenic type 2C protein phosphatase (PP2C). Wip1 has been found to be overexpressed and in some cases amplified in many types of human cancers such as breast cancer, ovarian clear cell carcinoma, and medulloblastomas (27-33). Further work using various cancer mouse models has validated the oncogenic properties of Wip1. Specifically, Wip1 has been shown to act as an oncogene by inhibiting tumor suppressors, such as p53, and complementing other oncogenes, such as H-Ras1 (34). The involvement of Wip1 in tumorigenesis makes it an attractive drug target for the treatment of cancers, and recently this notion was demonstrated in ovarian clear cell carcinoma with Wip1 pharmacological inhibitor (35). Therefore, unraveling the molecular functions of Wip1 is necessary.
Wip1 was discovered as a protein induced after ionizing radiation (IR) in a p53-dependent manner (36). Investigation of Wip1 molecular functions indicates that it facilitates in returning the cell to homeostasis after stress, and the literature to date focuses on the role of Wip1 after genotoxic stress such as IR and UVR. Once it is induced after DNA damage, Wip1 is then responsible for terminating stress-induced signaling pathways by dephosphorylating a number of proteins involved in these pathways such as p38, p53, ATM and MDM2 (34). The functional consequences of these dephosphorylation events all support the survival and potential onset of genomic instability and tumorigenesis if Wip1 is overexpressed by reversing cell cycle arrest, inhibiting DNA repair and inhibiting apoptotic signaling (34).
In this study, we identified Wip1 as a novel phosphatase for γ-H2AX. Phosphorylation of H2AX, caused by IR and UVR, is significantly reduced by ectopic expression of Wip1 in vivo. Furthermore, deletion of Wip1 in oncogene-transformed mouse embryonic fibroblasts (MEF) results in heightened basal and IR-induced γ-H2AX levels. Wip1 constitutively associates with H2AX and dephosphorylates γ-H2AX in an in vitro phosphatase assay. We also show that premature dephosphorylation of γ-H2AX after IR by Wip1 expression results in failure to recruit various DNA repair molecules such as the MRN components and MDC1 to damaged foci, which slows DNA repair and is consistent with the H2AXF-/- phenotype (21). The present study extends our understanding of γ-H2AX regulation and of Wip1 as a modulator of the DDR and an important regulator of the tumor surveillance network.
Materials and Methods
Cell Culture
MCF7, HCT116, H1299, and transformed mouse embryonic fibroblasts (MEF) were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, California) supplemented with 10% FBS and penicillin (100 U/ml)/streptomycin (100 μg/ml) at 37°C in 5% CO2. A tet-on inducible Wip1 H1299 cell line was generated in the same manner as the previously described Saos2 cell line, and pooled clones were used after selection (37).
Plasmids, siRNA and Transfections
The Flag-tagged Wip1 was a gift from Ettore Appella (37). The Flag-tagged Wip1 314DA was constructed by using the QuikChange® site-directed mutagenesis kit as the manufacture recommended (Stratagene, CA, Cat # 200518). siRNA from Dharmacon was used for targeting either Wip1 (Cat #: D-004554-01) or GFP (Cat #: D-001940-01). Lipofectamine 2000 (Invitrogen, CA) was used for the transfection of plasmids and siRNA into cells per the manufacturer’s protocols, and experiments were performed 48 hours after transfection.
Antibodies
Antibodies obtained from Cell Signaling Technology (Danver, MA) include those recognizing phosphoS139-H2AX (Cat#: 2577), H2AX (Cat#: 2595), phosphoS1981-ATM (Cat#4526), and 53BP1 (Cat#: 4937). Antibodies for Rad50 (Cat#: GTX70228), NBS (Cat#: GTX70224), Mdc1 (Cat#: GTX11170) and Mre11 (Cat#: GTX70212) were purchased from GeneTex, Inc (San Antonio, TX). Wip1 antibodies were obtained from R&D systems, (Minneapolis, MN, Cat#: 2380-MC-100) and Bethyl Laboratories, Inc. (Montgomery, TX, Cat#: A300-664A). Antibodies obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) include those recognizing tubulin (B-7, Cat#: SC5286), ERK2 (C-14, Cat#: SC-154), β-actin (C-4, Cat#: SC47778) and p38 (C-20, Cat#: SC-535).
Immuno-assays
Immunoblot analysis and immunofluorescence were performed as previously described (38). Co-immunoprecipitations were done as follows. To analyze Mdc1 and NBS1 interaction, cell lysate (1μg/μl) was incubated with 1 μg of primary Mdc1 antibody at 4°C for 4 hours, followed by 20 μl protein A slurry (Amersham Biosciences, Piscataway, NJ) for another 16 hours. Alternatively, to detect the interaction between Wip1 and H2AX, Flag-tagged Wip1 protein was immunoprecipitated with M2 anti-Flag antibody conjugated beads (Sigma-Aldrich, St Louis, MO) per the manufacturer’s protocol. Protein/bead complexes were washed with phosphate-buffered saline three times. Precipitated proteins were then resuspended in 40 μl of SDS-PAGE sample buffer (50 mM Tris pH 6.8, 2% SDS, 10% glycerol, and 2% β-mercaptoethanol) and were analyzed by immunoblot analysis.
In vitro phosphatase assay
Purified histone γ-H2AX protein (Axxora, San Diego, CA) was incubated with purified bacterial recombinant Wip1 protein (39) in 20 μl of phosphatase buffer containing 20 mM MOPS, pH 7.0 and 20 mM MgCl2 for 30 min at 30 °C. The reaction was terminated by adding 20 μl of SDS-PAGE sample buffer as described previously (40) and then analyzed by immunoblot analysis. A chemical inhibitor for Wip1 [“Compound M,” (40)] was used to inhibit the phosphatase activity of the recombinant Wip1 protein as described in the text.
Radiation
Cells were exposed to x-rays using a model Philips MGC40 x-irradiator (dose rate 0.86 Gy/minute). Unless otherwise indicated, cells were analyzed 4 hours after a 5 Gy dose. For ultraviolet radiation (UVR), cells were exposed to 20 Jm-2 of UV light (254 nm) and harvested at the indicated time points.
Comet Assay
DNA double-stranded breaks were analyzed using a commercial comet assay (Trevigen, Inc., Gaithersburg, M.D.) following the manufacturer’s protocol. For quantification, comet positive cells were scored in random fields of cells. Over 200 cells from each sample were scored.
Results
Wip1 decreases IR-induced γ-H2AX
To study the effects of Wip1 on genotoxic stress-induced γ-H2AX levels, lung carcinoma H1299 cells were modified to express Wip1 by incubation with doxycycline using the tet-on system (Clonetech, CA). Wip1 was clearly expressed in the vast majority of cells in the presence of doxycycline as seen with immunofluorescence and immunoblot analysis of Wip1 (“+ Dox”, Fig. 1A, 1B). γ-H2AX foci in cells that express Wip1 were compared to control cells after IR. A dramatic increase in the amount of γ-H2AX foci was seen in the absence of Wip1 expression (“-Dox”), whereas γ-H2AX foci were similar to basal levels in cells expressing Wip1 at the same time point (“+Dox,” Fig. 1A). A minority of the tet-on Wip1 inducible H1299 cells failed to express Wip1 after doxycycline treatment. Since these cells can serve as internal negative controls, select fields including these infrequent doxycycline-unresponsive cells were chosen. As seen in Figure 1A (lower panel), a white arrowhead designates a cell that did not express Wip1 and, therefore, had a high level of γ-H2AX foci compared to the other Wip1-expressing cells in the field after radiation exposure. Immunoblot analysis (Fig. 1B) demonstrated similar findings – γ-H2AX levels were reduced in H1299 cells that express Wip1 after IR.
Figure 1. Wip1 reduced IR-induced γ-H2AX signal.

A, immunocytochemistry of either sham-irradiated (No IR) or IR-exposed H1299 cells induced to express Wip1 showed reduced γ-H2AX foci [+Dox, Wip1 induction; −Dox, no Wip1 induction; green, Wip1; red, γ-H2AX; blue, 4′,6-diamidino-2-phenylindole (DAPI)]. The white arrowhead points to a single non–Wip1-expressing cell that showed elevated γ-H2AX levels. B, immunoblot analysis showed decreased IR-induced γ-H2AX levels in H1299 cells expressing Wip1 (+Dox). p38 immunoblot is used as a loading control. C, immunocytochemistry of IR-exposed HCT116 cells that overexpress Wip1 showed reduced γ-H2AX foci (white arrows). D, immunoblotting of HCT116 cells showed that depletion of Wip1 by siRNA (Wip1i) or overexpression of Wip1 (Wip1) by transient transfection of a Flag-tagged Wip1 construct resulted in increased or decreased levels of γ-H2AX, respectively, basally after IR exposure compared with the control (GFPi). Tubulin is used as a loading control.
In order to confirm that the negative effect of Wip1 on γ-H2AX is not H1299 specific, the colon carcinoma cell line HCT116 was also used. HCT116 cells were transiently transfected with a Flag-tagged Wip1 expression vector and were harvested for immunocytochemistry after IR exposure as depicted in Figure 1C. White arrows point to transfected cells that over-express Wip1 and had a much lower level of γ-H2AX foci than the neighboring untransfected cells. Since over-expression of Wip1 decreased γ-H2AX levels, inhibiting endogenous Wip1 expression should enhance γ-H2AX levels. To test this possibility, HCT116 cells were transfected with either control siRNA directed towards GFP (GFPi), Wip1-directed siRNA (Wip1i) to deplete endogenous Wip1, or the Flag-tagged Wip1 expression vector (Wip1) to over-express Wip1 (Fig. 1D). The transfected cells were either unexposed (“control”) or x-irradiated, and then harvested for immunoblot analysis as shown in Figure 1D. γ-H2AX levels increased in the control (GFPi) irradiated cells as expected; however, γ-H2AX levels were enhanced in cells that have depleted Wip1 and reduced in cells over-expressing Wip1, which is consistent with our previous results. Similar results with siRNA suppression of Wip1 were seen in MCF-7 cells (Supplementary Figure 7a). Taken together, these results showed that Wip1 functions to reduce IR-induced γ-H2AX levels.
Wip1 does not block the formation of γ-H2AX
The mechanism by which Wip1 reduces cellular γ-H2AX levels could be by either 1) inhibiting initial H2AX phosphorylation or 2) reversing H2AX phosphorylation. The latter seems the most likely possibility, since Wip1 is a phosphatase. To test this possibility, γ-H2AX levels at various times after exposure to 5 Gy were measured in the tet-on Wip1 inducible H1299 system. With immunofluorescence and immunoblot analysis, γ-H2AX is seen as early as 10 minutes after 5 Gy in all cells, regardless of Wip1 expression level (Fig. 2A and 2B; similar panels of Fig. 2A are seen in Supplementary Figure 1a). On the other hand, there is a drastic decrease in γ-H2AX levels in cells expressing Wip1 at 2 and 4 hours after 5 Gy (Fig. 2A and 2B), indicating that Wip1 does not affect γ-H2AX formation and instead reverses γ-H2AX at a later time point.
Figure 2. Wip1 did not block phosphorylation of H2AX after stress.

A) Immunocytochemistry of H1299 cells induced to express Wip1 either sham irradiated (“No IR”) or harvested at the indicated time points after IR exposure showed reduced γ̃H2AX foci at later time points. Arrows designate cells that do not express Wip1. B) Immunoblot analysis of the cell lysates from A) showed decreased IR-induced γ̃H2AX levels at later time points in H1299 cells expressing Wip1 (“+dox”) compared to the control (“-dox”). C) Immunocytochemistry showed reduced γ̃H2AX foci in Wip1 expressing H1299 cells compared to control cells (white arrows) after UVR exposure. (Wip1, green; γ̃H2AX, red; DAPI, blue) D) Immunoblot analysis of the cell lysates from C) showed similar results. The p38 immunoblot is used as a loading control.
To further validate that Wip1 does not block H2AX phosphorylation, UVR-induced γ-H2AX levels were measured in the tet-on Wip1 inducible H1299 cells. The rationale is that H2AX is not phosphorylated after UVR exposure by known Wip1 targets, such as ATM, and is instead phosphorylated by other kinases such as ATR and JNK (11, 41) or is dependent on nucleotide excision repair (NER) repair factors (3). Therefore, if Wip1 expression reduces UVR-induced γ-H2AX levels, then the mechanism is unlikely through inhibition of an H2AX kinase. As depicted in Figure 2C, immunocytochemistry reveals an expected increase in γ-H2AX pan nuclear staining in cells that do not express Wip1 4 hours after UVR exposure, whereas cells that express Wip1 (white arrow) have no detectable γ-H2AX staining. Similar results are seen with immunoblot analysis (Fig. 2D) – doxycycline treated cells (“+dox”) that express Wip1 have reduced UVR-induced γ-H2AX levels compared to the non-expressing Wip1 cells (“-dox”).
Since Wip1 is highly expressed before IR exposure in the over-expression studies in Fig. 2, Wip1 may have effects on γ-H2AX prior to irradiation. To eliminate this possibility, we induced Wip1 expression in the tet-on Wip1 inducible H1299 cells after IR exposure and measured IR-induced γ-H2AX levels. The cells were irradiated, treated with doxycycline 30 minutes after irradiation, and then harvested for immunoblot analysis and immunofluorescence at several time points post-IR (see Fig. 3A for a schematic of the experimental design). As shown in Fig. 3B, immunoblot analysis showed that Wip1 expression could be seen as early as 4 hours after IR and increased at 8 and 16 hours after IR in the doxycycline treated cells, which correlated with a decrease in IR-induced γ-H2AX levels. Similar results were seen with immunofluorescence (Fig. 3C). At one hour after irradiation, no Wip1 expression could be detected in the doxycycline treated cells, and there was no difference between the levels of IR-induced γ-H2AX foci in the untreated and the doxycycline treated cells at this time point. However, at 4 and 8 hours after IR, lower levels of γ-H2AX foci were seen in doxycycline treated cells that showed Wip1 expression (white arrows, Fig. 3C). Theses data indicate that Wip1 has no effect on γ-H2AX before IR and, instead, removes γ-H2AX after irradiation.
Figure 3. Induction of Wip1 after IR exposure reduces IR-induced γ̃H2AX levels.
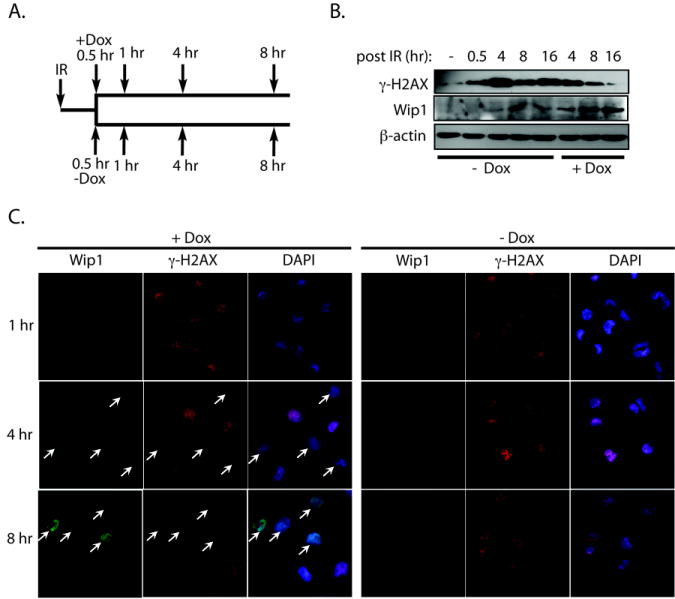
A) Schematic of the experimental design. H1299 cells were exposed to IR, and either untreated (“-Dox”) or treated with doxycycline to induce Wip1 expression (“+Dox”) 30 minutes after IR. Cells were harvested at the indicated time points after IR to measure levels of γ̃H2AX and Wip1. B) Immunoblot analysis and C) immunofluorescence of Wip1 and γ̃H2AX levels showed that cells expressing Wip1 had reduced γ̃H2AX levels after IR. β-actin is used as a loading control. White arrows indicate Wip1 expressing cells. (Wip1, green; γ̃H2AX, red; DAPI, blue)
Deletion of Wip1 enhances basal and IR-induced γ-H2AX in oncogenic stress conditions
We next sought to evaluate the effect of Wip1 on γ-H2AX levels in cells undergoing oncogenic stress since some strong oncogenes such as c-myc or H-Ras induce γ-H2AX foci, which are referred to as oncogene-induced DNA damaged foci (6, 7). γ-H2AX foci were evaluated by immunofluorescence in mouse embryonic fibroblasts (MEFs) transformed with the oncogenes E1A and H-Ras and harvested from either wild type or Wip1-/- mice (“wt E1A/Ras MEF” and “Wip1-/- E1A/Ras MEF,” respectively) (40, 42). As shown in Figure 4A, Wip1-/- E1A/Ras MEF have an increased frequency of basal γ-H2AX foci compared to wt E1A/Ras MEF. Immunoblot analysis showed similar results – γ-H2AX levels in the Wip1-/- E1A/Ras MEFs were high compared to the wt control (“wt E1A/Ras MEFs,” Fig. 4B). As shown in Fig. 4B, γ-H2AX levels remain high after IR in the Wip1-/- E1A/Ras MEFs compared to the wt E1A/Ras MEFs. Additionally, the wt E1A/Ras MEFs showed increased γ-H2AX levels after IR as expected (Fig. 4B and Supplementary Fig. 3a). However, the Wip1-/- E1A/Ras cells showed no change in γ-H2AX levels after IR, suggesting that the high basal γ-H2AX levels in these cells reduced the effect of γ-H2AX after IR exposure (Fig. 4B).
Figure 4. Deletion of Wip1 enhanced basal and IR-induced γ̃H2AX levels in cells undergoing oncogenic stress.
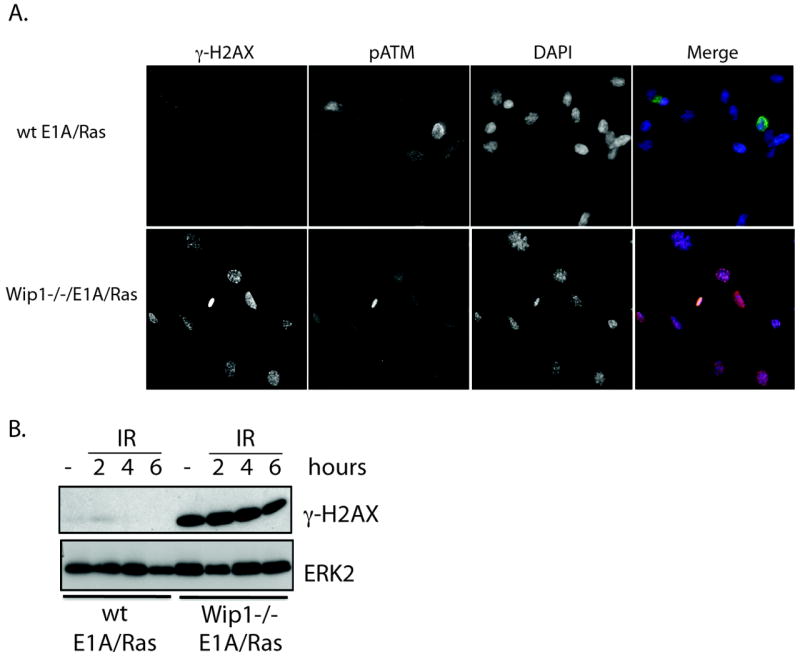
A) Immunocytochemistry of Wip1-/- E1A/Ras MEFs showed enhanced basal γ̃H2AX levels compared to wt E1A/Ras MEFs in the absence of p-ATM staining. B) Immunoblot analysis of γ-H2AX showed that Wip1-/- E1A/Ras MEFs had elevated γ-H2AX levels basally and after IR at the indicated time points compared to the wild type control (“wt E1A/Ras MEFs”). ERK2 is used as a loading control.
Additionally, the presence of γ-H2AX is independent of ATM activity. Phosphorylated ATM at serine 1981, the active form of ATM, can be detected by immunofluorescence with a specific antibody (Supplementary Fig. 2a) (4, 43). There was no phospho-ATM signal detected in Wip1-/- E1A/Ras MEFs that showed γ-H2AX foci (Fig. 4A), indicating that Wip1 does not act through ATM. Collectively, these data demonstrate that Wip1 reduces γ-H2AX levels in cells undergoing oncogenic stress, genotoxic stress, or both.
Wip1 directly binds to and dephosphorylates H2AX
Since Wip1 reversed γ-H2AX in an ATM-independent manner and in conditions where H2AX is not phosphorylated by a known Wip1 target (i.e. after UVR), Wip1 most likely dephosphorylates γ-H2AX. If this is the case, then inhibition of γ-H2AX by Wip1 should diminish when Wip1 phosphatase activity is disrupted. H1299 cells were stably transfected with either a pBabe retroviral vector that is empty, encodes wild type Wip1, or encodes a phosphatase dead Wip1 which has an alanine substituted for the aspartic acid at position 314 as described previously (44). The cells were harvested 20 hours after UVR, and γ-H2AX levels were then analyzed by immunoblot analysis. There is less of an increase in γ-H2AX levels in cells over-expressing Wip1 (“Wip1”, Fig. 5A), and over-expression of the phosphatase dead Wip1 mutant (“Wip1DA,” Fig. 5A) not only relieves this inhibition but also enhances the γ-H2AX level compared to the empty vector control (“-,” Fig. 5A). These results indicate that Wip1 phosphatase activity is required to reduce γ-H2AX levels.
Figure 5. Wip1 directly dephosphorylates γ̃H2AX.
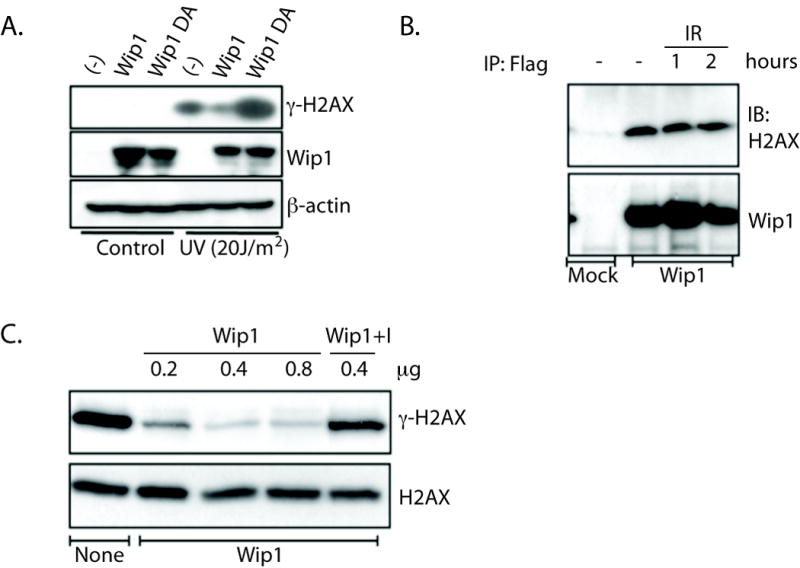
A) Immunoblot analysis demonstrated reduced or enhanced γ̃H2AX levels in UVR exposed H1299 cells stably overexpressing Wip1 (“Wip1”) or a phosphatase dead mutant of Wip1 (“Wip1DA”), respectively. β-actin is used as a loading control. B) γ̃H2AX was detected by immunoblot (“IB”) in Flag-immunoprecipitates (“IP”) from sham (“-”) and IR exposed (time points indicated) HCT116 cells transfected with a Flag-Wip1 construct (“Wip1”), but not in cells transfected with an empty vector (“mock”). C) In vitro phosphatase reactions analyzed by immunoblot showed an increasing loss of γ̃H2AX levels with the indicated increasing concentrations of recombinant Wip1 protein. γ̃H2AX levels were restored with a Wip1 inhibitor (“I”, Compound M from {Belova et al., 2005, Cancer Biol Ther, 4, 1154-8}, 0.5 μM).
Direct dephosphorylation of γ-H2AX by Wip1 also requires that Wip1 interacts with H2AX in vivo, which is shown in Figure 5B. HCT116 cells were transiently transfected with either an empty flag vector (“Mock”) or a flag-tagged Wip1 vector (“Wip1”), and the cells were harvested for flag-immunoprecipitation after sham irradiation (“-”) or IR exposure. Immunoblot analysis of the precipitated proteins showed that H2AX complexes with Wip1, since H2AX immunoprecipitates from lysates of cells transfected with flag-tagged Wip1 and not the mock transfected cells (Fig. 5B). Additionally, H2AX immunoprecipitates with Wip1 after IR at levels similar to basal levels (Fig. 5B). Therefore, Wip1 seems to reside in a complex with H2AX basally and remains associated with H2AX up to 2 hours post-IR.
To confirm that Wip1 can directly dephosphorylate γ-H2AX, we performed an in vitro phosphatase assay using commercially available γ-H2AX as a substrate. Incubation of γ-H2AX with recombinant Wip1 protein, which has been used previously (39), significantly reduced the phosphorylation level of H2AX in a dose dependent manner (Figure 5C). A specific Wip1 inhibitor efficiently blocked Wip1 dephosphorylation of γ-H2AX (Figure 5C, last lane) (39). Thus, γ-H2AX is a Wip1 target since Wip1 binds to H2AX and dephosphorylates γ-H2AX in vitro.
Wip1 expression interferes with accumulation of DNA repair factors at damaged sites
A number of studies have shown that γ-H2AX is important for recruiting various DNA damage repair factors to damage sites, such as NBS1, 53BP1, Brca1, Rad50 and Mdc1 (21, 45-47). Therefore, premature removal of γ-H2AX due to Wip1 expression may affect the recruitment process of these DNA repair factors. To test this possibility, foci generated by various DNA repair proteins at damage sites were examined in tet-on Wip1 inducible H1299 cells (Fig. 6A, B). All cells expressing Wip1 after doxycycline treatment (white arrows) show a significant reduction of γ-H2AX foci compared to the non-Wip1 expressing cells (white arrow heads) as expected. In Wip1 expressing cells, there was a marked reduction of NBS1 nuclear foci compared to the non-Wip1 expressing cells (Figure 6A, second row). Wip1 expression also reduced the recruitment of Rad50, Mdc1, and 53BP1 (white arrows, Fig. 6A, last row).
Figure 6. Wip1 reduced foci formation of several DNA damage response proteins.
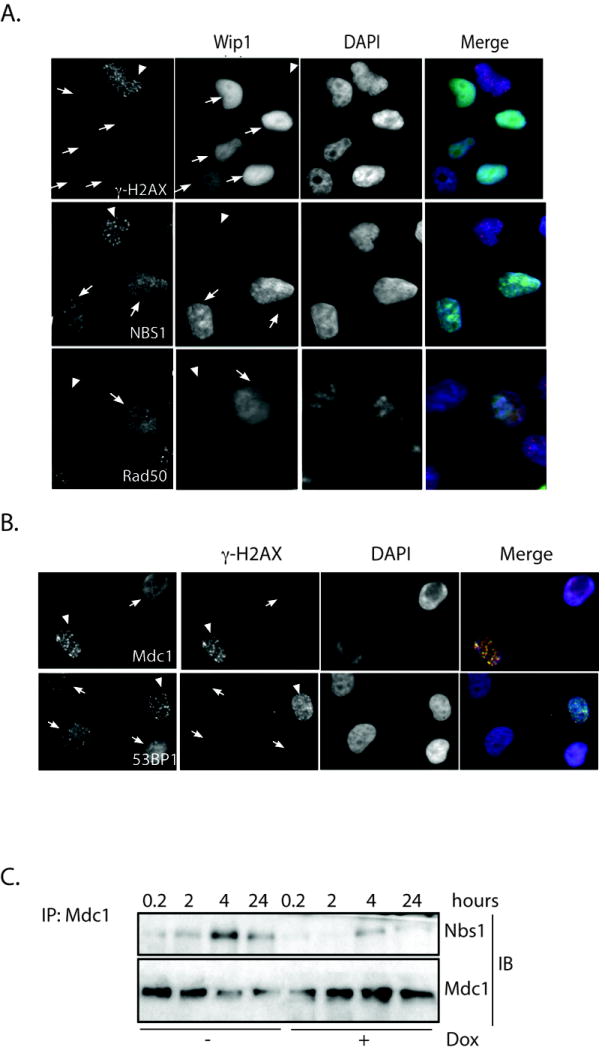
Immunocytochemistry showed decrease foci of A) γ̃H2AX, NBS1 and Rad50 (red) or B) Mdc1 and 53BP1 (red) in irradiated H1299 cells expressing Wip1 (white arrows) compared to the controls (white arrowheads). In B), cells that showed reduced γ̃H2AX foci are indicated as Wip1 expressing cells. (A, Wip1-green, DAPI-blue; B, γ̃H2AX-green, DAPI-blue). C) Decreased levels of NBS1 were detected by immunoblot (“IB”) in Mdc1 immunoprecipitates from H1299 cells expressing Wip1 (“+Dox”) compared to the controls (“-Dox”) at the indicated time points after IR exposure.
The results described above clearly indicate that Wip1 expression impairs the recruitment of DNA repair factors to damaged DNA. We then hypothesized that Wip1-mediated premature loss of γ-H2AX could also alter protein-protein interactions between the DNA repair proteins formed at the damage site. We chose to monitor the effect Wip1 expression has on the interaction between NBS1 and Mdc1, since previous work has shown that Mdc1 interacts with NBS1 after IR (48), and direct interaction of Mdc1 and NBS1 with γ-H2AX is essential for the DNA damage response (46, 49). H1299 cells were exposed to IR and harvested at various time points (Fig. 6C) either with (“+dox”) or without (“-dox”) Wip1 expression. Mdc1 was immunoprecipitated, and then Mdc1 and NBS1 levels were analyzed by immunoblot analysis. As shown in Figure 6C, Mdc1 interacts with NBS1 in a time dependent manner after IR exposure. Furthermore, Nbs1 interaction with Mdc1 is reduced at time points after irradiation in Wip1-expressing cells compared to non-Wip1-expressing cells. Taken together, these results indicate that premature loss of γ-H2AX by Wip1 expression impairs DNA repair machinery by interfering with the recruitment and maintenance of DNA repair factors at the damaged site.
Premature dephosphoryation of γ-H2AX by Wip1 delays DSB repair
Since premature dephosphorylation of γ-H2AX by Wip1 inhibits the recruitment of repair factors to damage sites, Wip1 should also affect DNA repair. Therefore, we measured the level of DSBs at multiple time points after IR exposure in tet-on Wip1 inducible H1299 cells with the comet assay. As shown in Figure 7A, all of the cells show significant comet tails, which confirms the presence of DSBs, at 10 minutes after IR exposure regardless of Wip1 expression. This is consistent with our previous data, since γ-H2AX levels at this early time point are not affected by Wip1 expression (Fig. 2A and 2B). On the other hand, non-Wip1 expressing cells show a significant reduction in DSBs indicated by the absence of comet tails at 5 hours post-IR compared to the Wip1 expressing cells (Fig. 7A and 7B). These results indicate that Wip1 impedes DSB repair.
Figure 7. Wip1 delayed DNA repair after IR.
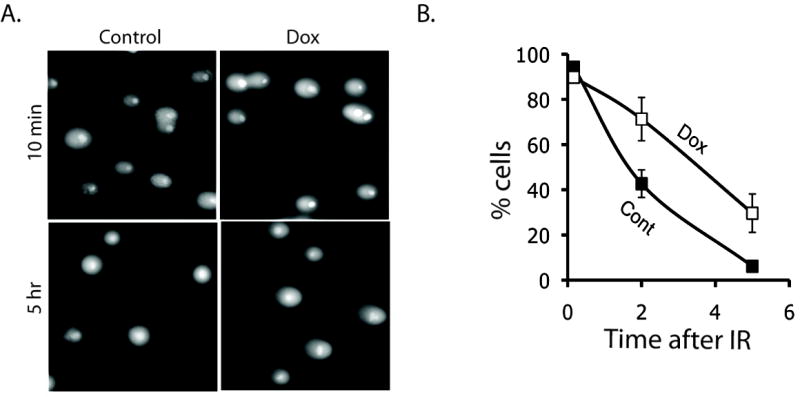
H1299 cells showed comet tails ten minutes after IR regardless of Wip1 expression. Comet tails of Wip1 expressing cells (“Dox”) persisted 5 hours after irradiation, unlike the control cells (“Control”). B) Quantification of comet-positive cells in Wip1 expressing (“Dox”) and control H1299 cells (“Control”) at ten minutes, two hours, and five hours after IR. (Percentage of comet-positive cells verses time after irradiation).
Discussion
This study identifies Wip1 as a direct modulator of γ-H2AX levels. Not only do we demonstrate that Wip1 removes γ-H2AX after genotoxic stress (IR and UVR) and impairs the DDR by premature dephosphorylation of H2AX, but we also show that loss of Wip1 in cells undergoing oncogenic stress results in heightened γ-H2AX levels. These results show a novel mechanism of γ-H2AX removal and a new role of Wip1 in facilitating the return of a cell to homeostatic conditions after stress and potentially promoting tumorigenesis when aberrantly expressed1.
Our results indicate that Wip1 reverses γ-H2AX by direct dephosphorylation, and not indirectly by inhibiting upstream kinases such as ATM. We provide evidence for this in several ways. A time course analysis shows normal IR-induced γ-H2AX formation in Wip1 expressing cells, which is not consistent with inhibition of a γ-H2AX kinase (Fig. 2A and 2B). Induction of Wip1 expression after IR exposure resulted in a reduction of IR-induced γ-H2AX levels (Fig. 3). Wip1 expression dramatically reduces UVR-induced γ-H2AX (Fig. 2C and 2D), and phosphorylation of H2AX after UVR is dependent on other factors such as ATR (41), NER factors (3), and JNK (11), which are not Wip1 targets. Additionally, we show that Wip1 does not regulate γ-H2AX formation or γ-H2AX levels by inhibiting ATM in oncogenic stress conditions (Fig. 4 and Supplementary Fig. 5a). Additionally, Shreeram et al show that recombinant Wip1 protein dephosphorylates a γ-H2AX phosphopeptide with a low Km value similar to other known Wip1 substrates (37). Finally, Wip1 effectively dephosphorylates γ-H2AX in vitro (Fig. 5). Taken together, the evidence presented in this study indicates that Wip1 is a γ-H2AX phosphatase.
Previous studies indicate that a major physiological function of Wip1 is to reverse stress signaling when cellular damage is repaired, which is important for cell survival (34). Therefore, it is not surprising that Wip1 is ubiquitously expressed in tissues of the mouse (52). In addition, the phenotype of the Wip1-/- mouse is mild; there is only a dramatic phenotype when tissues from this mouse are challenged with an exogenous stress (37, 42, 53), which highlights the important physiological role of Wip1 to dampen stress signaling. Wip1 has been shown to inhibit several stress signaling proteins, such as p53 for example, after genotoxic stress exposure, which is necessary for the cell to escape cell cycle arrest and apoptosis initiation (34). Since γ-H2AX, like p53, is a major regulator of the DDR, γ-H2AX must be reverted back to unphosphorylated H2AX once repair is complete. In this light, Wip1 dephosphorylation of γ-H2AX during the DDR is an additional mechanism by which Wip1 returns the cell to homeostatic conditions.
Wip1 removal of γ-H2AX also has important implications for tumorigenesis by highlighting (i) an additional oncogenic function of Wip1 and (ii) an additional mechanism by which H2AX’s tumor suppressor-like feature is dysregulated. Wip1 has been shown to promote tumorigenesis by inhibiting various tumor suppressors (34, 37, 40, 54), and H2AX displays tumor suppressor properties. Evidence for this is the genomic instability phenotype of the H2AX-/- mice (21) and the function of H2AX in the DDR (55). Furthermore, γ-H2AX foci formed during oncogenic stress (such as those in the transformed E1A/H-Ras MEFs shown in this study) may play a role in maintaining genomic stability and protecting the cell from tumorigenesis (21, 25). We show in this study that deletion of Wip1 in cells undergoing oncogenic stress have elevated γ-H2AX levels. These results highlight another role whereby Wip1 can affect the cell’s tumor surveillance network. In conclusion, direct dephosphorylation of γ-H2AX by Wip1 describes a novel method of γ-H2AX removal, which is required to “turn-off” the DDR and is a function of Wip1 in tumorigenesis.
Supplementary Material
Acknowledgments
Financial Support: NIAID 1 U19 AI067773, KOSEF M20806000049-08B0600-04910
Footnotes
References
- 1.Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–94. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 5.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 6.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–8. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sankar N, Kadeppagari RK, Thimmapaya B. c-Myc-induced aberrant DNA synthesis and activation of DNA damage response in p300 knockdown cells. J Biol Chem. 2009;284:15193–205. doi: 10.1074/jbc.M900776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–17. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B: relationship to cell cycle phase and induction of apoptosis. Cell Cycle. 2005;4:339–45. [PubMed] [Google Scholar]
- 10.Limoli CL, Giedzinski E, Bonner WM, Cleaver JE. UV-induced replication arrest in the xeroderma pigmentosum variant leads to DNA double-strand breaks, gamma -H2AX formation, and Mre11 relocalization. Proc Natl Acad Sci U S A. 2002;99:233–8. doi: 10.1073/pnas.231611798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu C, Zhu F, Cho YY, et al. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–32. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–9. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Keogh MC, Kim JA, Downey M, et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- 14.Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–26. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heo K, Kim H, Choi SH, et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 16.Svetlova M, Solovjeva L, Nishi K, Nazarov I, Siino J, Tomilin N. Elimination of radiation-induced gamma-H2AX foci in mammalian nucleus can occur by histone exchange. Biochem Biophys Res Commun. 2007;358:650–4. doi: 10.1016/j.bbrc.2007.04.188. [DOI] [PubMed] [Google Scholar]
- 17.Downs JA, Allard S, Jobin-Robitaille O, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell. 2004;16:979–90. doi: 10.1016/j.molcel.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Kusch T, Florens L, Macdonald WH, et al. Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science. 2004;306:2084–7. doi: 10.1126/science.1103455. [DOI] [PubMed] [Google Scholar]
- 19.van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 2004;119:777–88. doi: 10.1016/j.cell.2004.11.033. [DOI] [PubMed] [Google Scholar]
- 20.Bassing CH, Suh H, Ferguson DO, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–70. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 21.Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bassing CH, Chua KF, Sekiguchi J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco S, Gostissa M, Zha S, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–14. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Celeste A, Difilippantonio S, Difilippantonio MJ, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–83. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin B, Bassing CH. The sticky business of histone H2AX in V(D)J recombination, maintenance of genomic stability, and suppression of lymphoma. Immunol Res. 2008;42:29–40. doi: 10.1007/s12026-008-8030-4. [DOI] [PubMed] [Google Scholar]
- 27.Castellino RC, De Bortoli M, Lu X, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245–56. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirasawa A, Saito-Ohara F, Inoue J, et al. Association of 17q21-q24 gain in ovarian clear cell adenocarcinomas with poor prognosis and identification of PPM1D and APPBP2 as likely amplification targets. Clin Cancer Res. 2003;9:1995–2004. [PubMed] [Google Scholar]
- 29.Li J, Yang Y, Peng Y, et al. Oncogenic properties of PPM1D located within a breast cancer amplification epicenter at 17q23. Nat Genet. 2002;31:133–4. doi: 10.1038/ng888. [DOI] [PubMed] [Google Scholar]
- 30.Rauta J, Alarmo EL, Kauraniemi P, Karhu R, Kuukasjarvi T, Kallioniemi A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res Treat. 2006;95:257–63. doi: 10.1007/s10549-005-9017-7. [DOI] [PubMed] [Google Scholar]
- 31.Saito-Ohara F, Imoto I, Inoue J, et al. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003;63:1876–83. [PubMed] [Google Scholar]
- 32.Sinclair CS, Rowley M, Naderi A, Couch FJ. The 17q23 amplicon and breast cancer. Breast Cancer Res Treat. 2003;78:313–22. doi: 10.1023/a:1023081624133. [DOI] [PubMed] [Google Scholar]
- 33.Yu E, Ahn YS, Jang SJ, et al. Overexpression of the wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res Treat. 2007;101:269–78. doi: 10.1007/s10549-006-9304-y. [DOI] [PubMed] [Google Scholar]
- 34.Lu X, Nguyen TA, Moon SH, Darlington Y, Sommer M, Donehower LA. The type 2C phosphatase Wip1: an oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008;27:123–35. doi: 10.1007/s10555-008-9127-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan DS, Lambros MB, Rayter S, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res. 2009;15:2269–80. doi: 10.1158/1078-0432.CCR-08-2403. [DOI] [PubMed] [Google Scholar]
- 36.Fiscella M, Zhang H, Fan S, et al. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc Natl Acad Sci U S A. 1997;94:6048–53. doi: 10.1073/pnas.94.12.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shreeram S, Demidov ON, Hee WK, et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol Cell. 2006;23:757–64. doi: 10.1016/j.molcel.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Lee JS, Lee MO, Moon BH, Shim SH, Fornace AJJ, Cha HJ. Senescent growth arrest in mesenchymal stem cells is bypassed by Wip1-mediated downregulation of intrinsic stress signaling pathways. Stem Cells. 2009;27:1963–75. doi: 10.1002/stem.121. [DOI] [PubMed] [Google Scholar]
- 39.Belova GI, Demidov ON, Fornace AJJ, Bulavin DV. Chemical inhibition of Wip1 phosphatase contributes to suppression of tumorigenesis. Cancer Biol Ther. 2005;4:1154–8. doi: 10.4161/cbt.4.10.2204. [DOI] [PubMed] [Google Scholar]
- 40.Bulavin DV, Phillips C, Nannenga B, et al. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat Genet. 2004;36:343–50. doi: 10.1038/ng1317. [DOI] [PubMed] [Google Scholar]
- 41.Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28:2298–304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 42.Choi J, Nannenga B, Demidov ON, et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol Cell Biol. 2002;22:1094–105. doi: 10.1128/MCB.22.4.1094-1105.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 44.Fujimoto H, Onishi N, Kato N, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006;13:1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 45.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol. 2000;10:886–95. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 46.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 47.Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene. 2002;21:8967–80. doi: 10.1038/sj.onc.1206136. [DOI] [PubMed] [Google Scholar]
- 48.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–6. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 49.Kobayashi J, Tauchi H, Sakamoto S, et al. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol. 2002;12:1846–51. doi: 10.1016/s0960-9822(02)01259-9. [DOI] [PubMed] [Google Scholar]
- 50.Macurek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010 doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- 51.Moon SH, Lin L, Zhang X, et al. Wildtype p53-induced phosphatase 1 dephosphorylates histone variant {gamma}-H2AX and suppresses DNA double strand break repair. J Biol Chem. 2010 doi: 10.1074/jbc.M109.071696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi J, Appella E, Donehower LA. The structure and expression of the murine wildtype p53-induced phosphatase 1 (Wip1) gene. Genomics. 2000;64:298–306. doi: 10.1006/geno.2000.6134. [DOI] [PubMed] [Google Scholar]
- 53.Nannenga B, Lu X, Dumble M, et al. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol Carcinog. 2006;45:594–604. doi: 10.1002/mc.20195. [DOI] [PubMed] [Google Scholar]
- 54.Bulavin DV, Demidov ON, Saito S, et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat Genet. 2002;31:210–5. doi: 10.1038/ng894. [DOI] [PubMed] [Google Scholar]
- 55.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–9. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.