

Abstract
The molecular mechanisms underlying cytoskeleton-dependent Golgi positioning are poorly understood. In mammalian cells, the Golgi apparatus is localized near the juxtanuclear centrosome via dynein-mediated motility along microtubules. Previous studies implicate Cdc42 in regulating dynein-dependent motility. Here we show that reduced expression of the Cdc42-specific GTPase-activating protein, ARHGAP21, inhibits the ability of dispersed Golgi membranes to reposition at the centrosome following nocodazole treatment and washout. Cdc42 regulation of Golgi positioning appears to involve ARF1 and a binding interaction with the vesicle coat protein coatomer. We tested whether Cdc42 directly affects motility, as opposed to the formation of a trafficking intermediate, using a Golgi capture and motility assay in permeabilized cells. Disrupting Cdc42 activation or the coatomer/Cdc42 binding interaction stimulated Golgi motility. The coatomer/Cdc42-sensitive motility was blocked by the addition of an inhibitory dynein antibody. Together, our results reveal that dynein and microtubule-dependent Golgi positioning is regulated by ARF1-, coatomer-, and ARHGAP21-dependent Cdc42 signaling.
Keywords: Golgi apparatus, microtubule, dynein, Cdc42, ARF1, coatomer, ARHGAP21
Introduction
Unlike many organelles of mammalian cells that are dispersed throughout the cytoplasm, the Golgi apparatus is often present as a compact ribbon-like structure that is localized near the juxtanuclear centrosome. The purpose for centrosomal localization of the Golgi apparatus is not fully understood, but it was found recently to contribute to directed secretion and cell polarity during wound healing (1). Centrosomal localization requires the minus-end-directed microtubule motor protein dynein. Inhibiting dynein or disrupting microtubules causes the Golgi apparatus to lose centrosomal localization and disperse throughout the cytoplasm (2–4). One way that dynein contributes to Golgi positioning is by directing Golgi-targeted trafficking intermediates toward microtubule minus ends. For example, dynein is necessary for the movement of vesiculotubular ER-to-Golgi transport intermediates (5–7), as well as for protein transport from early endosomes to the Golgi apparatus (8). Distinct dynein complexes contribute to each of these trafficking steps (9).
The capacity of Golgi membranes and Golgi-derived vesicles to undergo motor-dependent motility along microtubules has been demonstrated directly using permeabilized cells (3) and reconstitution with isolated microtubules (10, 11). These experiments indicated that Golgi membranes can undergo both dynein and kinesin mediated transport. Dynein provides a direct motile force on Golgi stacks that opposes kinesin-mediated dispersion (12). The dynein-binding proteins LIS1, NDE1 and NDEL1 were shown recently to be required for normal dynein recruitment and Golgi positioning (13). In addition to directing membrane transport toward the Golgi apparatus, Golgi-associated microtubules and dynein-dependent tubulation is necessary to generate ribbon-like Golgi morphology (14, 15).
Golgi positioning is not static; for example, the Golgi apparatus repositions during cell polarization and Golgi membranes undergo dispersion and reassembly during the cell cycle. The mechanisms for coordinating microtubule-motor function with cell processes such as mitosis and polarization remains only partially understood but may be dependent on golgins (1, 16) and GTP-binding proteins. GTP-binding proteins were implicated directly in membrane motility by the observation that GTPγS inhibited microtubule-dependent Golgi motility in vitro (10). In this regard, the ARF, Rab and Rho families of GTP-binding proteins have each been implicated in the regulation of cytoskeleton-mediated membrane motility (17, 18).
We have found previously that ARF1 and the Rho-family protein Cdc42 regulate dynein recruitment to Golgi membranes in vitro and that these GTP-binding proteins influence dynein-dependent trafficking events in intact cells (5, 19). Our previous studies suggest that Cdc42 regulates dynein-dependent transport at three distinct steps connected to the Golgi apparatus: ER-to-Golgi transport, retrograde Shiga-toxin trafficking from endosomes to the Golgi complex, and microtubule/dynein-dependent Golgi positioning (5, 8, 19). Our studies revealed that Cdc42 influences dynein function through a mechanism that involved changes in actin dynamics suggesting crosstalk between microtubule and actin-dependent intracellular motility.
Cdc42 is localized to the Golgi apparatus via a binding interaction with the ARF1-dependent vesicle-coat protein, coatomer (20–22). ARF1 stimulates the recruitment of coatomer to the membrane and thus affects the localization of coatomer/Cdc42 complexes. ARF1 also regulates Cdc42 function by binding the Cdc42-specific GAP ARHGAP21 (also referred to as ARHGAP10) (23, 24). ARHGAP21 regulates ARF1-dependent Cdc42 activity both at the Golgi apparatus and at the cell surface (23, 25). In this study, we used reconstitution of Golgi motility and intact-cell studies to show that ARHGAP21-sensitive Cdc42 activity regulates microtubule and dynein-dependent Golgi motility and positioning.
Results
Microtubule-dependent Golgi positioning involves ARHGAP21-regulated Cdc42 function
Golgi membranes undergo kinesin-dependent dispersal following microtubule disruption with nocodazole (12). When nocodazole is removed, the Golgi stacks return to the centrosome in a dynein-dependent manner. We showed previously that cells transfected with constitutively active Cdc42(Q61L) have defects in Golgi repositioning following nocodazole washout (5). Our interpretation is that the coatomer-associated Cdc42 on the Golgi membranes regulates the dynein-dependent translocation during reassembly.
As an additional test of this model, we have now examined the consequences of knocking down the expression of the Golgi-localized Cdc42 GAP, ARHGAP21 (23), which should mimic the effects of expressing the constitutively active Cdc42(Q61L) mutation (Figure 1). Consistent with previous reports (19, 23, 25), two siRNAs directed against human ARHGAP21 each caused an 80–90% reduction in transcript levels in HeLa cells when measured by quantitative PCR (not shown). Reduced ARHGAP21 protein levels were apparent by immunofluorescence in Hela cells stably expressing shRNA that targeted ARHGAP21 (Figure S1A). Golgi membranes were partially dispersed in cells when ARHGAP21 levels were reduced by RNA interference but not in control cells (Figures 1A and S1B). We expressed a GFP-tagged fragment of ARHGAP21 containing the ARF-binding and GAP domains (19, 23, 24) but missing the siRNA target site in the siRNA-transfected cells to confirm that the phenotype of the knockdown cells did not represent an off-target effect. The GFP-tagged ARHGAP21 fragment was localized to the Golgi apparatus as expected (Figure 1B). Cells expressing the truncated GFP-ARHGAP21 protein, but not GFP alone, displayed compact Golgi membranes demonstrating that the dispersed Golgi phenotype can be rescued.
Figure 1. Dynein-dependent Golgi positioning involves ARHGAP21.

(A) Hela cells were transfected with siRNA against ARHGAP21 or a control siRNA against luciferase as indicated. The cells were treated with nocodazole (20 μM) for 5 hours to disperse the Golgi apparatus, and the nocodazole was washed away for the indicated time. At each time point, cells were fixed and Golgi membranes were labeled with GM130 antibody. Shown are confocal micrographs. (B) Hela cells were transfected with siRNA as in (A). After 24 hours the cells were transferred to new plates and transfected with plasmids encoding GFP-ARFBD/GAP domain of ARHGAP21 or GFP alone as indicated. The cells were fixed after 24 additional hours and decorated with antibodies against the Golgi marker, GM130. The size bar equals 5 μm. (C) The fraction of cells containing a compact Golgi apparatus at 1 hour after nocodazole washout was determined for ARHGAP21 and luciferase siRNA expressing HelA cells. The GM130-labeled Golgi complex was defined as compact if it could fit inside a circle with a diameter of 10 μm. Shown is the average from 3 experiments; the bars indicate standard error. (D) The number of Golgi particles per cell was determined using a particle counter plugin for ImageJ. Shown is the average from 3 experiments of 10 cells each; the bars indicate standard error. There was no significant change in the average number of particles/cell before nocodazole washout (p=0.3245). After the one hour nocodazole washout, cells treated with ARHGAP21 siRNA had significantly more particles than cells treated with luciferase siRNAs (p<0.006).
ARHGAP21-regulated Cdc42 activity likely affects both actin- and microtubule-dependent cellular processes. In order to test more specifically whether ARHGAP21 was involved in the microtubule/dynein-dependent positioning of the Golgi apparatus, we assessed Golgi morphology during the recovery from nocodazole treatment. Both ARHGAP21- and luciferase-siRNA-treated cells displayed fully dispersed Golgi membranes when treated with nocodazole (Figure 1A). More than 35% of luciferase-siRNA control cells displayed compact Golgi morphology and juxtanuclear Golgi localization within 1 hour of nocodazole washout (Figure 1A,C). In the ARHGAP21 RNAi-treated cells, by contrast, the Golgi membranes remained widely dispersed at this time point. Both the control and ARHGAP21 RNAi-treated cells displayed Golgi morphology and distribution similar to that observed before nocodazole treatment by 4 hours after washout. Similar results were obtained with the shRNA-expressing stable cell line (Figure S1B).
As an alternative approach to quantify the effects of ARHGAP21 knockdown on Golgi morphology, we determined the number of distinct Golgi puncta before and after recovery from nocodazole treatment using automated particle counting (Figure 1D). The number of dispersed Golgi puncta was similar after nocodazole treatment, before the washout, irrespective of siRNA treatment. By contrast, the number of Golgi puncta was significantly higher in ARHGAP21 siRNA-treated cells when the cells were allowed to recover for one hour following nocodazole washout. The dispersal of Golgi membranes and the slow coalescence at the centrosome upon ARHGAP21 knock down (Figures 1 and S1) are similar to the effects observed upon expressing Cdc42(Q61L) (5). Together, the results are consistent with a role for ARHGAP21-sensitive Cdc42 activity in the regulation of dynein-based positioning.
The expression of mutant Cdc42 (5) or RNAi-mediated knockdown of ARHGAP21 (Figure 1) requires experiments that are carried out over a relatively long time course (i.e. greater than 24 hours). During this time period, indirect effects of Cdc42 disruption may become more pronounced or cells could undergo adaptations, such as changes in gene expression, that affect the consequences of Cdc42 disruption. With this in mind, we sought to determine if a pharmacological agent that inhibits Cdc42 acutely had an effect on microtubule-dependent Golgi repositioning following nocodazole treatment. For these experiments, NRK cells were incubated with the Rho-family inhibitor, C. difficile toxin B during the nocodazole washout (Figure 2A,B). In contrast to the restoration of normal Golgi morphology within 1 hour in the absence of toxin B, the Golgi membranes remained dispersed when toxin B was present (Figure 2A). We next quantified the rate of Golgi repositioning in living NRK cells by labeling the Golgi membranes with NBD-C6-ceramide and measuring the loss of peripheral fluorescence as a function of time (see Materials and Methods). Whereas peripheral fluorescence in control cells decreased in a linear fashion after the washout, it persisted in the toxin B-treated cells (Figure 2B). If the Golgi positioning is Cdc42-regulated, we expected that it should be sensitive to toxin B but resistant to the Rho-specific inhibitor C3 transferase. Indeed, we observed that the C3 transferase had no effect (Figure 2C). Our characterization of Golgi repositioning following nocodazole washout supports a role for Cdc42 and ARHGAP21 during microtubule-dependent positioning of Golgi membranes.
Figure 2. Microtubule-dependent Golgi positioning requires Rho GTPases.
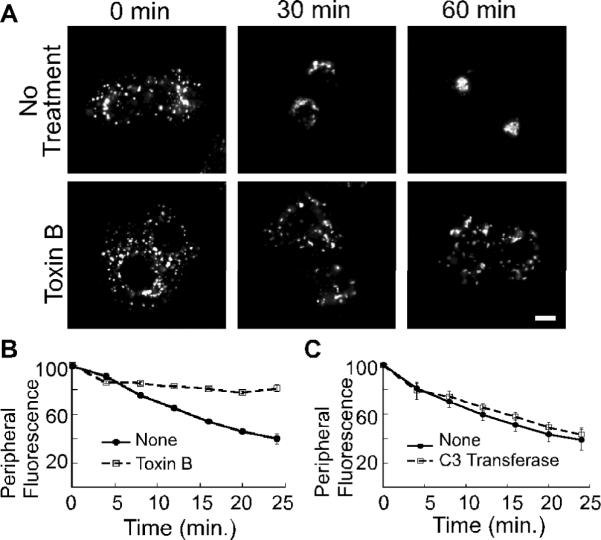
(A) NRK cells were treated with nocodazole for 2 hours. 100 ng/ml Toxin B was added as the nocodazole was washed out. Shown are confocal micrographs of cells that were fixed at the indicated time points and decorated with anti-GM130 antibody. The size bars represent 5 μm. (B–C) Golgi motility in living NRK cells was quantified by incubation with NBD-C6 ceramide prior to nocodazole addition. 100 ng/ml Toxin B (B) or 2 μg/ml C3 transferase (C) was added during the nocodazole wash out. A peripheral region of interest was defined for each cell and the change in peripheral fluorescence was recorded for 25 minutes at 37°C. The mean fluorescence intensity within the region of interest is plotted as a function of time; n=3 experiments for (B,C). The standard error in each case is indicated by bars.
Golgi positioning depends on ARF1 activity and coatomer
ARF proteins are essential regulators of vesicular transport out of the Golgi apparatus (26). ARF1 directs Cdc42 localization (20) and binds to the Cdc42-specific GAP, ARHGAP21 (24). Furthermore, we showed previously that ARF1 activation is required for dynein recruitment to Golgi membranes in vitro (5). Given the central role of ARF proteins in regulating Golgi dynamics and the relationships among ARF1, ARHGAP21, and Cdc42, we expected that the recovery of Golgi membranes from nocodazole washout should also be sensitive to ARF activity. We tested whether ARFs are involved in Golgi repositioning by inhibiting the GTP-exchange reaction with brefeldin A. Kinetic analysis shows that acute addition of brefeldin A, just prior to the nocodazole washout, caused the Golgi membranes to persist in the cell periphery (Figure 3 A,B). Thus, both Cdc42 and ARF1 activation appear to be necessary for Golgi repositioning to the centrosome.
Figure 3. Golgi positioning depends on ARF1 activity and coatomer.

(A) NRK cells were treated with nocodazole for 2 hours. 10 μM brefeldin A (BFA) was added as the nocodazole was washed out. Shown are confocal micrographs of cells that were fixed at the indicated time points and decorated with anti-GM130 antibody. The size bars represent 20 μm. (B–C) Golgi motility in living NRK cells was quantified by incubation with NBD-C6 ceramide prior to nocodazole addition. 10 μM brefeldin A (B), or 50 μM BAPTA-AM (C) was added during the nocodazole wash out. A peripheral region of interest was defined for each cell and the change in peripheral NBD fluorescence was recorded for 25 minutes at 37°C. The mean fluorescence intensity within the region of interest is plotted as a function of time. The number of experiments is n=3 for (B and C). The standard error in each case is indicated by bars. (D) Shown are confocal micrographs of NRK cells transfected with pEGFP-C1, pEGFP-p23 (199–212), and pEGFP-p23(199–212, KK-AA). Cells expressing the GFP constructs are marked with an asterisk. The cells were pretreated for 2 hours with 20 μM nocodazole. Nocodazole was washed off, and the cells were incubated for 20 minutes before fixation and decoration with a mouse polyclonal antibody against the Golgi marker, GM130. The bar represents 10 μm. (E) The percentage of cells with juxtanuclear Golgi membranes at the 20-minute time point was determined after cells were scored in a blind manner. The average of 4 experiments is plotted. The bars indicate standard error. The percentage of cells displaying juxtanuclear Golgi is significantly increased in the presence of GFP-p23 (p<0.05).
Cdc42 localizes to the Golgi apparatus through a binding interaction with the ARF1-dependent coat complex, coatomer (21, 22). We found previously that the ability of Cdc42 to regulate dynein recruitment at the Golgi complex requires its binding interaction with coatomer (5). Based on these findings, we anticipated that Golgi-bound coatomer would be necessary for Cdc42 regulated Golgi positioning. We first tested this using the cell-permeant calcium chelator BAPTA-AM, which causes rapid dissociation of coatomer from the Golgi apparatus (27). Addition of BAPTA-AM to cells shortly before the nocodazole washout significantly inhibited Golgi repositioning from the periphery (Figure 3C). This indicates that coatomer could contribute to Golgi positioning.
We tested more specifically if coatomer-bound Cdc42 regulates Golgi positioning by expressing a fusion protein between GFP and the C-terminal cytosolic coatomer-binding motif (residues 199–212) of the p23 cargo receptor (Figure 3D,E). Cdc42 and p23 compete for the same dibasic-motif-binding site on the γ-COP subunit of coatomer, and the 13-amino-acid C-terminal cytosolic domain of p23 effectively dissociates the coatomer/Cdc42 complex (5, 21, 22, 28). NRK cells were transfected with GFP, GFP-p23(199–212), or GFP-p23(199–212, KK-AA), in which the lysines that comprise the p23 dibasic motif are mutated to alanines. A compact Golgi morphology was observed in NRK cells expressing GFP-p23(199–212) more frequently than in GFP-expressing control cells or cells expressing the lysine mutations (Figure S2A,B). Increased dynein function is one possible explanation for the compact Golgi phenotype.
To test this possibility further, we assayed Golgi repositioning using nocodazole treatment and washout. When observed at 20 minutes after the washout, the Golgi membranes in untransfected and GFP-expressing cells were still mostly dispersed (Figure 3D). In cells expressing GFP-p23(199–212), by contrast, the Golgi membranes were already relocalized to a more compact structure at one side of the nucleus. Quantification by scoring cells in a blind manner confirmed the effects of GFP-p23(199–212) on Golgi distribution (Figure 3E). We found that Golgi repositioning was not affected by expression of GFP-p23(199–212, KK-AA) (Figure 3D,E). The effects of brefeldin A, BAPTA-AM, GFP-p23(199–212) expression, taken together, implicate the coatomer/Cdc42 complex in Golgi morphology and positioning.
Surprisingly, the compounds expected to affect Cdc42 or the coatomer/Cdc24 complex acutely, (toxin b, brefeldin A, and BAPTA-AM) had inhibitory effects on Golgi repositioning whereas constitutive disruption of the coatomer/Cdc42 binding interaction with GFP-p23(199–212) expression had a stimulatory effect on Golgi repositioning. One possibility is that GFP-p23(199–212), containing the C-terminal dilysine motif of the Golgi cargo receptor, is more selective for Golgi apparatus-specific functions of Cdc42 than the pharmacological inhibitors. We examined this by testing the effects of GFP-p23(199–212) on the retrograde transport of Shiga toxin. We showed previously that intracellular trafficking of this bacterial exotoxin from endosomes to the Golgi complex involves dynein and is regulated by Cdc42 (8, 19). Whereas C. difficile toxin B and mutant Cdc42 expression inhibited retrograde Shiga toxin transport (19), we were unable to observe any overt effect of GFP-p23(199–212) expression on Shiga toxin internalization and trafficking (Figure S2C). An effect on Golgi positioning but not Shiga toxin trafficking suggests that GFP-p23(199–212) could be more selective for Cdc42 function at the Golgi apparatus. Importantly, the finding that Golgi repositioning is accelerated in the presence of GFP-p23(199–212), is consistent with our conclusions from previous studies (5, 19) that active Cdc42 plays an inhibitory role in dynein function.
Exogenous Golgi membranes exhibit microtubule-dependent capture and motility in permeabilized cells
The effects of Cdc42 disruption on Golgi positioning could be explained as an inability to form a trafficking intermediate, such as a vesicle or tubule that is necessary for repositioning. Alternatively, Cdc42 could directly regulate the dynein-dependent motility of membranes from the periphery to the centrosome. In order to differentiate between these possibilities, we examined the microtubule-dependent capture of exogenous Golgi membranes into permeabilized cells as demonstrated previously (3, 29). Golgi membranes were incubated with cytosol under conditions that can support actin polymerization and dynein recruitment (5, 21, 28, 30). The membranes were labeled by incubating with bodipy-C5-ceramide. The Golgi membranes were reisolated and added together with fresh cytosol to permeabilized NRK cells. We confirmed that the exogenous Golgi membranes bound to permeabilized cells but not intact cells (Figure 4A). It is unlikely that the exogenous Golgi membranes were captured through a binding interaction with the endogenous Golgi apparatus as they were not typically colocalized (Figure 4B).
Figure 4. Exogenous Golgi membranes bind permeabilized cells.
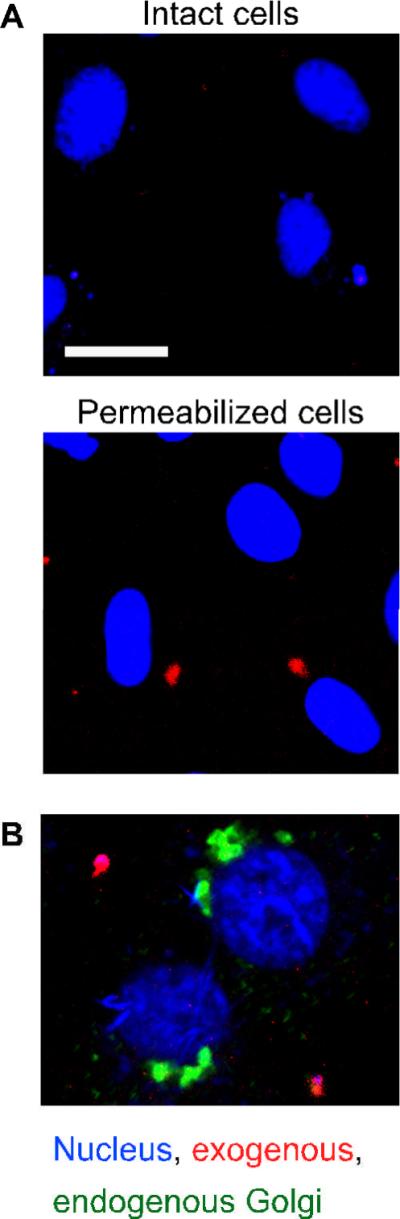
(A) NRK cells were mock treated or permeabilized by a freeze/thaw cycle as indicated. Rat-liver Golgi particles were incubated with cytosol, GTPγS, and an ATP-regenerating system then labeled with bodipy-C5-ceramide (red), and added to the permeabilized cells at 37°C. The nuclei were labeled using DRAQ5 (blue). In two independent experiments, no exogenous membranes were found bound to intact cells (n=20 cells) while an average of 0.6 membrane particles was observed per permeabilized cell (n=19 cells). The bar represents 10 μm. (B) Permeabilized Vero cells were incubated with rat-liver Golgi membranes (red). The cells were fixed and the endogenous Golgi membranes were decorated with a primate-specific antibody against giantin (green). The nuclei were labeled using DRAQ5 (blue).
We determined the suitability of this system for measuring microtubule-dependent Golgi membrane capture and motility by labeling cytoskeletal components during the assay. Although microtubule and microfilament levels were reduced following cell permeabilization (not shown), both microtubules (Figure 5A) and actin stress fibers (Figure 5B) were present after cells were incubated with the cytosol-containing reaction mixture used for the exogenous Golgi capture assay. Endogenous Golgi morphology was only slightly affected by permeabilization and incubation (Figure 5A). We tested whether microtubule reassembly was required for the ability of exogenous Golgi membranes to associate with cells by treating the NRK cells with nocodazole just prior to permeabilization. Consistent with the previous study (3), we observed that nocodazole treatment greatly reduced the number of cell-associated exogenous Golgi particles (Figure 5C,D) indicating that binding to the permeabilized cells is indeed microtubule dependent. Although bodipy-C5-ceramide-labeled rat-liver Golgi membranes were used for the majority of these experiments, we obtained similar results with GFP-labeled Golgi membranes isolated from CHO cells (Figure S3A).
Figure 5. Exogenous Golgi membranes undergo microtubule-dependent entry into permeabilized cells.

(A) Shown are confocal micrographs of NRK cells that were fixed at the indicated time points relative to permeabilization and incubation. The cells were decorated with antibodies against the Golgi marker GM130 (green) and microtubules (red). The nuclei were labeled using DRAQ5 (blue). (B) Shown are confocal micrographs indicating actin distribution at the indicated time points relative to the permeabilization and incubation. (C) NRK cells were either mock treated (control) or incubated with 20 μM nocodazole before and after permeabilization. Rat-liver Golgi membranes (red) and cytosol were added to the cells, and cell-associated membranes were visualized by confocal microscopy. The bar represents 20 μm. (D) NRK cells were treated as in (C) and the average number of Golgi particles per cell was determined from four independent experiments by blind counting with (n=85 cells) or without (n=111 cells) nocodazole treatment. The standard error is indicated by bars.
Time-lapse microscopy (Figure 6A, Supporting Information Movies S1 and S2) revealed that the exogenous Golgi membranes were not “captured” statically to microtubules, but instead underwent occasional movement. The motility we observed required factors present on the exogenous Golgi membranes, as fluorescent beads were never observed to undergo motility (Figure S3B, Movie S3). 0, 2.5, and 5-second time points were used to define a path and determine the distances traveled (Figure S4A) and velocities (Figure 6B) for each Golgi particle. The Golgi membranes were defined as motile if they moved at least 1 micrometer in 5 seconds. We found that in the presence of cytosol alone, approximately 30% of the Golgi particles were motile (Figures S4B). These controls indicated that the previously described capture assay would also be useful for characterizing the microtubule-dependent motility of isolated Golgi membranes.
Figure 6. p23 peptide, recombinant PBD, and cytochalasin D stimulate dynein-dependent Golgi motility.

(A) Shown are merged images of confocal micrographs taken at an initial time point (0 seconds; red), 2.5 seconds (yellow), and 5 seconds (green) after incubating rat-liver Golgi membranes with permeabilized NRK cells. Prior to their addition to the NRK cells, the membranes were incubated with p23 peptide or cytochalasin D as indicated. Nuclei were labeled with 10 μM DRAQ5. The bar represents 10 μm. (B) Golgi membranes were treated with or without the p23 peptide and incubated with permeabilized cells. The velocities of Golgi particles was calculated and plotted as a histogram representing the average from 3 independent experiments. A total of 109 particles associated with 53 cells (minus peptide) and 65 particles associated with 54 cells (plus peptide) were analyzed. The standard error is indicated by bars. (C) The fraction of motile Golgi particles during a five second interval was determined for recombinant PBD (5 μg/ml) or mock-treated membranes. Over three independent experiments, 56 Golgi particles associated with 45 cells (minus PBD) and 54 particles associated with 48 cells (plus PBD) were analyzed. PBD treatment significantly increased the fraction of motile membrane particles (p<0.04). (D) The fraction of motile Golgi membranes was determined following mock or cytochalasin D (CytoD) treatment. Over three independent experiments, 226 Golgi particles associated with 77 cells (minus CytoD), and 273 particles associated with 59 cells were analyzed. The standard error is indicated by bars. Cytochalasin D significantly increases the fraction of motile particles (p<0.05). (E) Rat-liver Golgi membranes were incubated using reaction conditions identical to those used for the motility assay shown in panels A-D. Following the incubation, the Golgi membranes were reisolated by centrifugation and processed for SDS-PAGE. Western blot analysis was used to determine the levels of β-COP, dynein, and Cdc42 as indicated. (F) The fraction of motile Golgi membrane particles was determined after mock treatment (176 particles associated with 100 cells), treatment with the inhibitory dynein antibody 70.1 (199 particles; 114 cells), treatment with the p23 peptide (235 particles; 123 cells), or treatment with both (206 particles; 130 cells). Plotted are the averages from three independent experiments. The standard error is indicated by bars. The anti-dynein antibody significantly inhibits p23-stimulated motility (p<0.05).
Disrupting Cdc42-dependent actin dynamics stimulates dynein-mediated Golgi-membrane motility
The consequences of disrupting Cdc42 signaling in intact cells and the results from membrane-binding experiments (5, 22) suggest that the coatomer/Cdc42 complex regulates microtubule-motor-mediated motility of Golgi membranes. We tested this model directly by dissociating Cdc42 from coatomer using the γ-COP-binding C-terminal p23 peptide containing residues 199–212 (5, 21, 22) and assaying the motility of the exogenous Golgi membranes in the permeabilized cells. We observed that the fraction of motile Golgi-membrane particles, the distances traveled, and the particle velocities were each increased when the membranes were incubated with the p23 peptide (Figures 6A,B, S4A,B, Movies S1 and S2); in the absence of the peptide approximately 30% of the particles moved during a 5-second observation, whereas in its presence about 60% were motile. A control peptide had no effect on membrane motility (Figure S4B).
Although effects of the p23 peptide have been previously characterized to result from dissociation of the coatomer/Cdc42 complex (5, 22), it is still possible that the peptide stimulates Golgi motility through a Cdc42 independent mechanism. Hence, we have also used the recombinant Cdc42-binding (PBD) fragment from PAK1 as an alternative method to inhibit Cdc42 activity (31). Addition, of recombinant PBD increased the fraction of motile Golgi particles (Figures 6C and S4C), Golgi particle velocity (Figure S4D), and travel distances (Figure S4E). The similar results obtained with the p23 peptide and the recombinant PBD are fully consistent with a role for Cdc42 in regulating the Golgi motility.
Our previous study demonstrated that ARF1 not only influences Cdc42 signaling at the Golgi apparatus, but that ARF1 activation was necessary for dynein recruitment to Golgi membranes (5). Hence, we examined the effects of ARF inhibition on Golgi motility. We found that brefeldin A completely blocked the ability of p23 to stimulate motility indicating that ARF activation is required (Figure S5). Low levels of the actin toxin cytochalasin D was found previously to stimulate dynein recruitment (5). This is consistent with the model that ARF1- and Cdc42-regulated actin dynamics influence microtubule-mediated motility. We find that cytochalasin D has a similar stimulatory effect on Golgi membrane motility (Figure 6A,D).
The ability to stimulate Golgi motility with the coatomer-binding p23 peptide, PBD, and cytochalasin D support the model that coatomer-bound Cdc42 regulates dynein-dependent motility of Golgi membranes. Furthermore, we calculated the average velocity of Golgi particle movement in the presence of the p23 peptide to be 664±35 nm/sec (n=30). This velocity is consistent with previous measurements for dynein-based motility in vitro (32). We used Western blot analysis of reisolated Golgi membranes to test directly whether p23 peptide addition affects the recruitment of dynein to Golgi membranes under the reaction conditions used for the motility assay. Consistent with our previous report (5), the peptide inhibited Cdc42 binding while stimulating dynein recruitment (Figure 6E). Finally, we used an inhibitory dynein antibody to determine if p23-peptide-stimulated motility was dynein-dependent. Importantly, the peptide no longer stimulated motility in the presence of the inhibitory dynein antibody (Figure 6F). A control antibody against kinesin did not inhibit the motility (not shown). The effects of p23 peptide on Golgi motility in vitro is consistent with the effects of GFP-p23(199–212) expression in intact cells and together support a model wherein dissociation or inactivation of coatomer-bound Cdc42 acts in a permissive manner to stimulate dynein recruitment and dynein-based motility of Golgi membranes.
Discussion
We found previously that activation of Cdc42, a Rho-family GTP-binding protein, inhibits the recruitment of dynein motors to membranes and that Cdc42 influences intracellular dynein-dependent trafficking (5, 19). We have now further characterized the contribution of Cdc42 signaling to dynein-dependent Golgi positioning at the centrosome. We find that both constitutive and acute disruption of the Cdc42 GTPase cycle affects Golgi repositioning after nocodazole treatment and washout. Notably, we find that expression of GFP-p23(199–212), which contains a dibasic motif expected to dissociate Cdc42 from the Golgi-coat protein coatomer, is able to increase the rate of Golgi repositioning at the centrosome. By contrast, activating Cdc42 using RNAi-based knockdown of ARHGAP21 expression decreased the ability of Golgi membranes to cluster at the centrosome. Taken together our results lead to the hypothesis that Cdc42 acts in an inhibitory manner to regulate dynein-based motility of intracellular membranes.
We have tested this hypothesis in the second half of this study by measuring Golgi membrane capture and motility in permeabilized cells. Although we have previously shown effects of Cdc42 on dynein recruitment and intracellular trafficking (5, 19), heretofore, we have not shown that Cdc42 can directly affect membrane motility. We now show that treatments expected to dissociate Cdc42 and stimulate dynein recruitment to Golgi membranes greatly increase the fraction of Golgi membranes undergoing dynein-mediated transport in permeabilized cells.
Cdc42 functions downstream of ARF1 and ARHGAP21 to regulate Golgi motility
Cdc42 function at the Golgi apparatus is connected to ARF1 in at least two ways. First, Cdc42 localizes to the Golgi complex through a cargo-protein-sensitive binding interaction with the ARF1-regulated vesicle coat protein, coatomer (5, 21, 22). Second ARF1 binds to the Golgi-localized Cdc42-specific GAP ARHGAP21 (23, 24). We propose that ARF1 helps direct the localization of both ARHGAP21 and Cdc42 in order to regulate the activation state of Cdc42 on Golgi membranes.
Our previous work suggests that the coatomer/Cdc42 complex regulates dynein recruitment through a manner that involves changes in actin dynamics (5, 19). We have now shown that the motility of isolated Golgi membranes in vitro can be stimulated with either the actin toxin cytochalasin D or the p23 C-terminal peptide. Both of these treatments may disrupt the formation of a Cdc42-dependent actin-based structure that regulates dynein binding. In this respect, we showed previously that actin assembly occurring downstream of coatomer/Cdc42 leads to the recruitment of a distinct set of actin-binding proteins (21, 28, 30). Together our results are consistent with a model wherein ARF1- and ARHGAP21-regulated activation of coatomer-bound Cdc42 stimulates actin polymerization and the recruitment of distinct actin-binding proteins that act in a regulatory manner during dynein recruitment.
A link between Golgi membrane motility and vesicle assembly
Results from both biochemical fractionation and immuno-EM indicate that the microtubule motors are recruited at or near vesicles on the Golgi membrane (5, 33). The microtubule motors could be recruited for the subsequent motility of transport vesicles rather than for organelle motility. In this regard, it was unexpected that the positioning of the Golgi complex at the centrosome and the motility of isolated Golgi stacks in vitro are also dependent on ARF1, p23, and the coatomer/Cdc42 complex. However, coatomer coats have also been implicated recently in the mobility and tubulation of the ER-Golgi intermediate compartment (34). We propose that Golgi positioning and motility are mediated by vesicle-associated motors and that these motors are supplied either by nascent vesicle buds that have not completed the scission reaction or by completely assembled vesicles that are tethered to the Golgi stacks. Our findings imply that Golgi positioning in the cell is intimately connected to the regulation of vesicle formation.
Silencing the expression of cargo receptors in the early secretory pathway affects coatomer distribution, fragments the Golgi apparatus, and causes abnormal ERGIC morphology (35). Similarly, microinjecting the p23 and p24 C-terminal peptides affects the ability to form microtubule-dependent tubular ER-to-Golgi trafficking intermediates (36). However, the p23 peptides do not affect coatomer distribution in cells (36), nor the recruitment of coatomer to Golgi membranes in vitro (21). Together, these findings are consistent with the model that occupation of the cargo binding sites are important not only for coat recruitment and cargo selection, but also for signaling to microtubule motors to instruct Golgi positioning and trafficking to and from the Golgi complex.
Cdc42 may connect cell polarity with Golgi positioning
Golgi positioning and morphology in mammalian cells responds both to changes in cell polarity and to the cell cycle. Interestingly, phosphorylation of the Golgi protein grasp65 affects both cell-polarity-dependent Golgi positioning and the Golgi dispersion that occurs during mitosis (16, 37). ZW10 may also connect Golgi positioning to the cell cycle in that it is required both for microtubule chromosome segregation and dynein recruitment to the Golgi (38–40). Cdc42 was first isolated as a cell-division cycle (cdc) mutant in yeast with defects in cell polarity and morphology (41). Golgi-localized Cdc42 could be important for sensing the status of the Golgi during cell cycle signaling and for helping to direct Golgi positioning and morphology changes in response to cell cycle and cell polarity signaling. We conclude that dynein recruitment to Golgi membranes is directed by the coatomer/Cdc42 complex as part of these regulatory processes that connect Golgi trafficking, positioning, and morphology to the cell cycle and cell polarity.
Materials and Methods
Materials
The following antibodies were used in this study: mouse anti-dynein IC 70.1, rabbit anti-tubulin (Abcam, Cambridge, MA); rabbit anti-Cdc42 (Cell Signaling, Beverly, MA); rabbit anti-actin, mouse anti-β-COP (Sigma-Aldrich, St. Louis, MO); mouse anti-GM130 (BD Biosciences, San Jose, CA); mouse anti-dynein intermediate chain, 74.1 (Covance, Richmond, CA). NBD-C6-ceramide, bodipy-C5-ceramide, and BAPTA-AM were obtained from Invitrogen. Nocodazole, GTPγS, brefeldin A, and cytochalasin D, were obtained from Sigma-Aldrich (St. Louis, MO). Clostridium difficile toxin B was obtained from (List Biological Laboratories, Campbell, CA) and C3 transferase from (Cytoskeleton Inc). The peptide sequences are: p23, YLRRFFKAKKLIE; control peptide, CAPDGSEDEPPKDSDGEDSE (Sigma-Genosys, The Woodlands, TX).
Cell culture and immunofluorescence
Normal rat-kidney (NRK) were cultured in alpha-minimal essential medium (MEM). Vero and HeLa cells were cultured in DMEM. The media were supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin. Immuno-fluorescence was carried out using appropriate dilutions of the indicated antibodies as described previously (8). The cells were analyzed by confocal microscopy (Carl Zeiss MicroImaging, Thornwood, NY).
siRNA and shRNA-based knockdown of ARHGAP21 expression
siRNA duplexes targeting human ARHGAP21, 5'-GGAUCUGUGUCGCAGUUUAdTdT, (23), 5 '-GTCATTGTGCCTTCTGAGAdTdT (25), and firefly luciferase, 5'- CUUACGCUGAGUACUUCGAdTdT, were purchased from (Sigma-Proligo). HeLa cells at 20–30% confluence were transfected with the indicated siRNA (500nM) using Lipofectamine (Invitrogen, Carlsbad, CA). To measure ARHGAP21 depletion after 64 hours of siRNA treatment, a short-length (< 1kb) cDNA library was generated from total RNA by using the Aurum total RNA mini kit (Bio-RAD, Hercules, CA) and iScript cDNA Synthesis kit (Bio-RAD, Hercules, CA). ARHGAP21 primers, position 2390–2413 and position 2539-2516, were used for quantitative real-time PCR. Primers targeting the ARHGAP21 intron, actin, and GAPDH were used as controls. Primer sequences are available upon request. SYBR green supermix, MyiQ single-color PCR detection system, and optical system software (Bio-Rad) were used for real-time PCR (42). For the rescue experiment, cells were transfected after 24 hours with a pEGFP plasmid encoding amino acids 855 to 1346 of ARHGAP21 that is missing the siRNA target site.
The pSUPER RNAi System (Oligoengine, Seattle, WA) was used to generate a viral vector pSR-shARHGAP21 to produce an shRNA to ARHGAP21. The oligonucleotides used for the shRNA were 5'-GATCCCCGTCATTGTGCCTTCTGAGATTCAAGAG ATCTCAGAAGGCACAATGACTTTTTGGAAA-3' and 5'AGCTTTTCCAAAAAGTCATTGTGCCTTCTGAGAT CTCTTGAATCTCAGAAGGCACAATGACGGG-3'. The oligonucleotide duplex was cloned into pSRgfp/neo by BglII/HindIII sites. A stable cell line expressing the shRNA to ARHGAP21 was generated using a retroviral spin infection. Retroviral supernatant was generated from 293T cells transfected with the pSR-shARHGAP21 with the pCL-Eco packaging construct and pCL-VSVG. Hela cells were grown to subconfluence in a 12-well dish. Cells and viral supernatant were centrifuged at 2500 r.p.m. for 2 h at 32°C with 8 μg/ml polybrene. A clonal population of GFP- expressing cells was obtained and analyzed for ARHGAP21 expression using a sheep polyclonal antibody that was generated against a GST-tagged fragment of ARHGAP21, amino acids 2–34 (Elmira Biologicals Inc., Iowa City, IA). Anti-ARHGAP21 antibodies from sheep serum were then used at a 1:100 dilution for immunostaining in Hela cells and Hela cells with the ARHGAP21 shRNA. The antibody specifically recognizes ARHGAP21 because no signal was detected by immunofluorescence after incubation with the ARHGAP21 fragment (amino acids 2–34) in Hela cells (data not shown).
Expressing the p23 cytosolic domain as a GFP fusion protein
Oligonucleotides encoding the C-terminal 13 amino-acid residues (199–212) of p23 or the same motif in which the lysine residues at the minus 4 and 5 positions are mutated to alanines were ligated in frame into the pEGFP-C1 plasmid. NRK cells were transfected with the plasmids using lipofectamine. Golgi morphology was determined by fixing cells 48 hours after the transfection and labeling with antibodies against GM-130. The effect of GFP-p23(199–212) expression on Shiga toxin trafficking in Vero cells was carried out using Cy3.5-labeled Shiga toxin B subunit as described previously (8, 19).
Golgi repositioning after nocodazole washout
Golgi membranes were labeled by incubating the cells with 5 μM NBD-C6-ceramide for 30 minutes followed by a 30 minute wash or by immunostaining. Nocodazole treatment (20 μM) and washout were carried out as described previously (8). At the time of the nocodazole washout, the cells were incubated with Toxin B, brefeldin A, BAPTA-AM, or C3 transferase as indicated. For cells treated with an ARHGAP21 siRNA or a luciferase siRNA the extent of Golgi repositioning after one hour was quantified using the particle counter plugin from ImageJ. The average number of particles per cell was determined for at least 3 experiments with 10 cells each. In addition, live-cell imaging was performed using an inverted confocal microscope and heated stage (Carl Zeiss MicroImaging). Kinetic analysis of labeled Golgi membranes was accomplished by measuring fluorescence changes in a defined region of interest at the cell periphery as described (8). For the experiments using GFP-p23(199–212) NRK cells were transfected 48 hours prior to nocodazole treatment.
Reconstitution of Golgi motility in permeabilized cells
Rat-liver (43) or GFP-GalT-labeled CHO-cell (44) Golgi membranes were isolated as described. The membranes (0.2 mg/ml) were incubated with bovine-brain cytosol (1.0 mg/ml), reaction buffer (25 mM HEPES pH 7.2, 2.5 mM magnesium acetate, 15 mM potassium chloride, 0.2 M sucrose, 50 μM ATP, 1 mM creatine phosphate, 8 units/ml creatine kinase, 250 μM UTP), and GTPγS (20 μM) for 20 minutes at 37°C; conditions known to promote vesicle assembly. The p23 peptide (250 μM), control peptide (250 μM), cytochalasin D (20 μg/ml), brefeldin A (400 μM), or Glutathione S-transferase (GST)-p21-binding domain (PBD) encoding amino acids 70–118 from Pak1 (5 μg/ml) were included in the incubation where indicated. When rat-liver Golgi membranes were used, the membranes were fluorescently labeled by including 2.5 μM bodipy-C5-ceramide in the reaction. Following the first incubation, the membranes were isolated by sedimentation and washed in (25 mM HEPES pH 7.2, 2.5 mM magnesium acetate, 50 mM potassium chloride, 0.2 M sucrose). The isolated membranes were then either resuspended in reaction buffer containing fresh cytosol for the motility experiments or processed for Western blotting. Anti-dynein 70.1 (0.6 μg/ml), was added to the resuspended membranes where indicated.
NRK cells were treated with 10 μM DRAQ5 for 5 minutes prior to the experiment to label the nuclei. The membrane/cytosol suspension was then added to NRK cells that had been permeabilized on a cover slip using a freeze/thaw cycle (45). The cells were incubated with the membrane suspension for approximately 1 minute at 37°C. The membrane suspension was then removed from the cells and replaced with fresh cytosol in reaction buffer. The NRK cells were viewed at 37°C using a confocal microscope equipped with a heated stage. Images were collected every 2.5 seconds. The data were averaged from 3 or 4 independent experiments. Standard error was calculated from the experimental averages and the significance determined using Student's t test.
Supplementary Material
(A) A Hela cell-line that stably expresses GFP and an shRNA targeting ARHGAP21 (asterisks) was mixed with untreated Hela cells. The mixed cell lines were fixed and labeled with a polyclonal anti-ARHGAP21 antibody (top panel). Note that ARHGAP21 levels appear lower in the GFP-labeled (ARHGAP21 shRNA-expressing) cells than in the adjacent control cells. (B) The ARHGAP21 shRNA-expressing cell line was treated with nocodazole (20 μM) for 5 hours to disperse the Golgi apparatus. The nocodazole was then washed away for two hours. At each time point, the cells were fixed and Golgi membranes were labeled with a GM130 antibody. The bar represents 10 μm.
(A) Shown are confocal micrographs of NRK cells transfected with pEGFP-C1, pEGFP-p23(199–212), and pEGFP-p23(199–212, KK-AA) as indicated. 48 hours after transfection, the cells were fixed, permeabilized, and labeled with antibodies against GM130. (B) The fraction of cells containing compact Golgi was determined for pEGFP-C1 (GFP) and pEGFP-p23(199–212) expressing NRK cells. The GM130-labeled Golgi complex was defined as compact if it could fit inside a circle with a diameter of 5 μm. Shown is the average from 3 experiments; the bars indicate standard error. (C) Vero cells were transfected with pEGFP-C1 or pEGFP-p23(199–212). Cy3.5 labeled Shiga toxin B subunit was bound to the cells by incubating for 1 min. at 0°C. The cells were washed with media and the Shiga toxin was then allowed to internalize by incubating for 10 min at 37°C.
(A) Golgi membranes were isolated from GFP-galactosyl transferase-expressing CHO cells. The membranes (green) were incubated with permeabilized NRK cells using reaction conditions identical to those used to assay membrane motility in Figure 6. The cells were fixed and decorated with an antibody against tubulin (red). Shown are two representative confocal micrographs. (B) Shown are merged confocal micrographs of permeabilized NRK cells incubated either with latex beads (3 μM) or with exogenous p23-peptide-treated Golgi membranes. An initial time point (time=0 seconds, red) and 5 seconds later (green) are shown. Nuclei were labeled with 10 μM DRAQ5. The cell surface was outlined (white). The bar represents 10 μm.
(A) Golgi membranes were treated with or without the p23 peptide and incubated with permeabilized cells. The Golgi particle travel distance during a five second incubation was calculated and plotted as a histogram representing the average from 3 independent experiments. The standard error is indicated by bars. (B) The fraction of motile rat-liver Golgi particles was determined using permeabilized NRK cells following mock treatment (143 particles associated with 100 cells), incubation with the C-terminal p23 peptide (141 particles; 102 cells) or a control peptide (171 particles; 98 cells). The bars indicate standard error. The p23 peptide significantly stimulates motility compared to the control peptide (p<0.05). (C) Shown are merged confocal micrographs of permeabilized NRK cell-associated Golgi membranes at an initial time point (red) and 5 seconds (green). The membranes were incubated with or without PBD (5 μg/ml), as indicated, prior to their addition to the NRK cells. The bar represents 10 μm. (D and E) Golgi membranes were treated with or without the recombinant PBD and incubated with permeabilized cells. The velocities (D) and travel distances (E) of Golgi particles were calculated and plotted as a histogram representing the average from 3 independent experiments. A total of 59 particles (plus PBD) and 49 particles (without PBD) were counted. The standard error is indicated by bars.
(A) Shown are merged confocal micrographs of permeabilized cell-associated rat-liver Golgi membranes taken at an initial time point (red) and 5 seconds later (green). The membranes were incubated with BFA and or the p23 peptide prior to being added to the NRK cells. Nuclei were labeled with 10 μM DRAQ5. The bar represents 10 μm. (B) The fraction of motile membranes was determined following mock treatment (131 particles associated with 98 cells), incubation with the C-terminal p23 peptide (203 particles; 114 cells), brefeldin A (BFA) (154 particles; 116 cells), or both (120 particles; 106 cells). Brefeldin A significantly inhibits p23-stimulated motility (p<0.05).
Images of exogenous rat-liver Golgi membranes bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.
Images of p23-peptide-treated rat-liver Golgi membranes bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.
Images of a latex bead bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.
Acknowledgements
We are grateful to Katrina Longhini, Susan Stamnes and Andrew Davis for technical assistance. We thank the University of Iowa Central Microscopy Research Facility and Dr. M. Bridget Zimmerman for assistance with the statistical analysis. We are grateful to Dr. Charles Yeaman for reading the manuscript. This work was supported by NIH grant RO1 GM068674 (M.S.), American Heart Association Grant-in-aid 0950167G (M.S.), an American Heart Association predoctoral fellowship (H.H.), and an NIH Interdisciplinary Research Fellowship 5 T32 HL 07638-23 (H.H.).
References
- 1.Yadav S, Puri S, Linstedt AD. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol Biol Cell. 2009;20(6):1728–1736. doi: 10.1091/mbc.E08-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139(2):469–484. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corthesy-Theulaz I, Pauloin A, Pfeffer SR. Cytoplasmic dynein participates in the centrosomal localization of the Golgi complex. J Cell Biol. 1992;118(6):1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ho WC, Allan VJ, van Meer G, Berger EG, Kreis TE. Reclustering of scattered Golgi elements occurs along microtubules. Eur J Cell Biol. 1989;48(2):250–263. [PubMed] [Google Scholar]
- 5.Chen JL, Fucini RV, Lacomis L, Erdjument-Bromage H, Tempst P, Stamnes M. Coatomer-bound Cdc42 regulates dynein recruitment to COPI vesicles. J Cell Biol. 2005;169(3):383–389. doi: 10.1083/jcb.200501157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389(6646):81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 7.Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat Cell Biol. 2005;7(1):48–55. doi: 10.1038/ncb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hehnly H, Sheff D, Stamnes M. Shiga toxin facilitates its retrograde transport by modifying microtubule dynamics. Mol Biol Cell. 2006;17(10):4379–4389. doi: 10.1091/mbc.E06-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer KJ, Hughes H, Stephens DJ. Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol Biol Cell. 2009;20(12):2885–2899. doi: 10.1091/mbc.E08-12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fullerton AT, Bau MY, Conrad PA, Bloom GS. In vitro reconstitution of microtubule plus end-directed, GTPgammaS-sensitive motility of Golgi membranes. Mol Biol Cell. 1998;9(10):2699–2714. doi: 10.1091/mbc.9.10.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wozniak MJ, Allan VJ. Cargo selection by specific kinesin light chain 1 isoforms. Embo J. 2006;25(23):5457–5468. doi: 10.1038/sj.emboj.7601427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minin AA. Dispersal of Golgi apparatus in nocodazole-treated fibroblasts is a kinesin-driven process. J Cell Sci. 1997;110(Pt 19):2495–2505. doi: 10.1242/jcs.110.19.2495. [DOI] [PubMed] [Google Scholar]
- 13.Lam C, Vergnolle MA, Thorpe L, Woodman PG, Allan VJ. Functional interplay between LIS1, NDE1 and NDEL1 in dynein-dependent organelle positioning. J Cell Sci. 2010;123(Pt 2):202–212. doi: 10.1242/jcs.059337. [DOI] [PubMed] [Google Scholar]
- 14.Judson BL, Brown WJ. Assembly of an intact Golgi complex requires phospholipase A(2) (PLA(2)) activity, membrane tubules, and dynein-mediated microtubule transport. Biochem Biophys Res Commun. 2009 doi: 10.1016/j.bbrc.2009.08.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller PM, Folkmann AW, Maia AR, Efimova N, Efimov A, Kaverina I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat Cell Biol. 2009;11(9):1069–1080. doi: 10.1038/ncb1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisel B, Wang Y, Wei JH, Xiang Y, Tang D, Miron-Mendoza M, Yoshimura S, Nakamura N, Seemann J. ERK regulates Golgi and centrosome orientation towards the leading edge through GRASP65. J Cell Biol. 2008;182(5):837–843. doi: 10.1083/jcb.200805045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hammer JA, 3rd, Wu XS. Rabs grab motors: defining the connections between Rab GTPases and motor proteins. Curr Opin Cell Biol. 2002;14(1):69–75. doi: 10.1016/s0955-0674(01)00296-4. [DOI] [PubMed] [Google Scholar]
- 18.Hehnly H, Stamnes M. Regulating cytoskeleton-based vesicle motility. FEBS Lett. 2007;581(11):2112–2118. doi: 10.1016/j.febslet.2007.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hehnly HA, Longhini KM, Chen JL, Stamnes M. Retrograde Shiga Toxin Trafficking Is Regulated by ARHGAP21 and Cdc42. Mol Biol Cell. 2009;20:4303–4312. doi: 10.1091/mbc.E09-02-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Erickson JW, Zhang C, Kahn RA, Evans T, Cerione RA. Mammalian Cdc42 is a brefeldin A-sensitive component of the Golgi apparatus. J Biol Chem. 1996;271(43):26850–26854. doi: 10.1074/jbc.271.43.26850. [DOI] [PubMed] [Google Scholar]
- 21.Fucini RV, Chen JL, Sharma C, Kessels MM, Stamnes M. Golgi vesicle proteins are linked to the assembly of an actin complex defined by mAbp1. Mol Biol Cell. 2002;13(2):621–631. doi: 10.1091/mbc.01-11-0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu WJ, Erickson JW, Lin R, Cerione RA. The gamma-subunit of the coatomer complex binds Cdc42 to mediate transformation. Nature. 2000;405(6788):800–804. doi: 10.1038/35015585. [DOI] [PubMed] [Google Scholar]
- 23.Dubois T, Paleotti O, Mironov AA, Fraisier V, Stradal TE, De Matteis MA, Franco M, Chavrier P. Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat Cell Biol. 2005;7(4):353–364. doi: 10.1038/ncb1244. [DOI] [PubMed] [Google Scholar]
- 24.Menetrey J, Perderiset M, Cicolari J, Dubois T, Elkhatib N, El Khadali F, Franco M, Chavrier P, Houdusse A. Structural basis for ARF1-mediated recruitment of ARHGAP21 to Golgi membranes. Embo J. 2007;26(7):1953–1962. doi: 10.1038/sj.emboj.7601634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumari S, Mayor S. ARF1 is directly involved in dynamin-independent endocytosis. Nat Cell Biol. 2008;10(1):30–41. doi: 10.1038/ncb1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.D'Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol. 2006;7(5):347–358. doi: 10.1038/nrm1910. [DOI] [PubMed] [Google Scholar]
- 27.Ahluwalia JP, Topp JD, Weirather K, Zimmerman M, Stamnes M. A role for calcium in stabilizing transport vesicle coats. J Biol Chem. 2001;276(36):34148–34155. doi: 10.1074/jbc.M105398200. [DOI] [PubMed] [Google Scholar]
- 28.Xu W, Stamnes M. The actin-depolymerizing factor homology and charged/helical domains of drebrin and mAbp1 direct membrane binding and localization via distinct interactions with actin. J Biol Chem. 2006;281(17):11826–11833. doi: 10.1074/jbc.M510141200. [DOI] [PubMed] [Google Scholar]
- 29.Corthesy-Theulaz I, Pfeffer SR. Microtubule-mediated Golgi capture by semiintact Chinese hamster ovary cells. Methods Enzymol. 1992;219:159–165. doi: 10.1016/0076-6879(92)19018-2. [DOI] [PubMed] [Google Scholar]
- 30.Fucini RV, Navarrete A, Vadakkan C, Lacomis L, Erdjument-Bromage H, Tempst P, Stamnes M. Activated ADP-ribosylation factor assembles distinct pools of actin on golgi membranes. J Biol Chem. 2000;275(25):18824–18829. doi: 10.1074/jbc.M000024200. [DOI] [PubMed] [Google Scholar]
- 31.Zigmond SH, Joyce M, Borleis J, Bokoch GM, Devreotes PN. Regulation of actin polymerization in cell-free systems by GTPgammaS and Cdc42. J Cell Biol. 1997;138(2):363–374. doi: 10.1083/jcb.138.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319(5866):1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fath KR, Trimbur GM, Burgess DR. Molecular motors and a spectrin matrix associate with Golgi membranes in vitro. J Cell Biol. 1997;139(5):1169–1181. doi: 10.1083/jcb.139.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomas M, Martinez-Alonso E, Ballesta J, Martinez-Menarguez JA. Regulation of ER-Golgi intermediate compartment tubulation and mobility by COPI coats, motor proteins and microtubules. Traffic. 2010;11(5):616–625. doi: 10.1111/j.1600-0854.2010.01047.x. [DOI] [PubMed] [Google Scholar]
- 35.Mitrovic S, Ben-Tekaya H, Koegler E, Gruenberg J, Hauri HP. The cargo receptors Surf4, endoplasmic reticulum-golgi intermediate compartment (ERGIC)-53, and p25 are required to maintain the architecture of ERGIC and Golgi. Mol Biol Cell. 2008;19(5):1976–1990. doi: 10.1091/mbc.E07-10-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simpson JC, Nilsson T, Pepperkok R. Biogenesis of tubular ER-to-Golgi transport intermediates. Mol Biol Cell. 2006;17(2):723–737. doi: 10.1091/mbc.E05-06-0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutterlin C, Polishchuk R, Pecot M, Malhotra V. The Golgi-associated protein GRASP65 regulates spindle dynamics and is essential for cell division. Mol Biol Cell. 2005;16(7):3211–3222. doi: 10.1091/mbc.E04-12-1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirose H, Arasaki K, Dohmae N, Takio K, Hatsuzawa K, Nagahama M, Tani K, Yamamoto A, Tohyama M, Tagaya M. Implication of ZW10 in membrane trafficking between the endoplasmic reticulum and Golgi. Embo J. 2004;23(6):1267–1278. doi: 10.1038/sj.emboj.7600135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inoue M, Arasaki K, Ueda A, Aoki T, Tagaya M. N-terminal region of ZW10 serves not only as a determinant for localization but also as a link with dynein function. Genes Cells. 2008;13(8):905–914. doi: 10.1111/j.1365-2443.2008.01215.x. [DOI] [PubMed] [Google Scholar]
- 40.Vallee RB, Varma D, Dujardin DL. ZW10 function in mitotic checkpoint control, dynein targeting and membrane trafficking: is dynein the unifying theme? Cell Cycle. 2006;5(21):2447–2451. doi: 10.4161/cc.5.21.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson DI, Pringle JR. Molecular characterization of CDC42, a Saccharomyces cerevisiae gene involved in the development of cell polarity. J Cell Biol. 1990;111(1):143–152. doi: 10.1083/jcb.111.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Malhotra V, Serafini T, Orci L, Shepherd JC, Rothman JE. Purification of a novel class of coated vesicles mediating biosynthetic protein transport through the Golgi stack. Cell. 1989;58(2):329–336. doi: 10.1016/0092-8674(89)90847-7. [DOI] [PubMed] [Google Scholar]
- 44.Balch WE, Dunphy WG, Braell WA, Rothman JE. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39(2 Pt 1):405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- 45.Mardones G, Gonzalez A. Selective plasma membrane permeabilization by freeze-thawing and immunofluorescence epitope access to determine the topology of intracellular membrane proteins. J Immunol Methods. 2003;275(1–2):169–177. doi: 10.1016/s0022-1759(03)00015-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) A Hela cell-line that stably expresses GFP and an shRNA targeting ARHGAP21 (asterisks) was mixed with untreated Hela cells. The mixed cell lines were fixed and labeled with a polyclonal anti-ARHGAP21 antibody (top panel). Note that ARHGAP21 levels appear lower in the GFP-labeled (ARHGAP21 shRNA-expressing) cells than in the adjacent control cells. (B) The ARHGAP21 shRNA-expressing cell line was treated with nocodazole (20 μM) for 5 hours to disperse the Golgi apparatus. The nocodazole was then washed away for two hours. At each time point, the cells were fixed and Golgi membranes were labeled with a GM130 antibody. The bar represents 10 μm.
(A) Shown are confocal micrographs of NRK cells transfected with pEGFP-C1, pEGFP-p23(199–212), and pEGFP-p23(199–212, KK-AA) as indicated. 48 hours after transfection, the cells were fixed, permeabilized, and labeled with antibodies against GM130. (B) The fraction of cells containing compact Golgi was determined for pEGFP-C1 (GFP) and pEGFP-p23(199–212) expressing NRK cells. The GM130-labeled Golgi complex was defined as compact if it could fit inside a circle with a diameter of 5 μm. Shown is the average from 3 experiments; the bars indicate standard error. (C) Vero cells were transfected with pEGFP-C1 or pEGFP-p23(199–212). Cy3.5 labeled Shiga toxin B subunit was bound to the cells by incubating for 1 min. at 0°C. The cells were washed with media and the Shiga toxin was then allowed to internalize by incubating for 10 min at 37°C.
(A) Golgi membranes were isolated from GFP-galactosyl transferase-expressing CHO cells. The membranes (green) were incubated with permeabilized NRK cells using reaction conditions identical to those used to assay membrane motility in Figure 6. The cells were fixed and decorated with an antibody against tubulin (red). Shown are two representative confocal micrographs. (B) Shown are merged confocal micrographs of permeabilized NRK cells incubated either with latex beads (3 μM) or with exogenous p23-peptide-treated Golgi membranes. An initial time point (time=0 seconds, red) and 5 seconds later (green) are shown. Nuclei were labeled with 10 μM DRAQ5. The cell surface was outlined (white). The bar represents 10 μm.
(A) Golgi membranes were treated with or without the p23 peptide and incubated with permeabilized cells. The Golgi particle travel distance during a five second incubation was calculated and plotted as a histogram representing the average from 3 independent experiments. The standard error is indicated by bars. (B) The fraction of motile rat-liver Golgi particles was determined using permeabilized NRK cells following mock treatment (143 particles associated with 100 cells), incubation with the C-terminal p23 peptide (141 particles; 102 cells) or a control peptide (171 particles; 98 cells). The bars indicate standard error. The p23 peptide significantly stimulates motility compared to the control peptide (p<0.05). (C) Shown are merged confocal micrographs of permeabilized NRK cell-associated Golgi membranes at an initial time point (red) and 5 seconds (green). The membranes were incubated with or without PBD (5 μg/ml), as indicated, prior to their addition to the NRK cells. The bar represents 10 μm. (D and E) Golgi membranes were treated with or without the recombinant PBD and incubated with permeabilized cells. The velocities (D) and travel distances (E) of Golgi particles were calculated and plotted as a histogram representing the average from 3 independent experiments. A total of 59 particles (plus PBD) and 49 particles (without PBD) were counted. The standard error is indicated by bars.
(A) Shown are merged confocal micrographs of permeabilized cell-associated rat-liver Golgi membranes taken at an initial time point (red) and 5 seconds later (green). The membranes were incubated with BFA and or the p23 peptide prior to being added to the NRK cells. Nuclei were labeled with 10 μM DRAQ5. The bar represents 10 μm. (B) The fraction of motile membranes was determined following mock treatment (131 particles associated with 98 cells), incubation with the C-terminal p23 peptide (203 particles; 114 cells), brefeldin A (BFA) (154 particles; 116 cells), or both (120 particles; 106 cells). Brefeldin A significantly inhibits p23-stimulated motility (p<0.05).
Images of exogenous rat-liver Golgi membranes bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.
Images of p23-peptide-treated rat-liver Golgi membranes bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.
Images of a latex bead bound to permeabilized NRK cells were captured every 2 seconds. Shown are 14 frames.