

Abstract
Reverse turns are a major class of protein secondary structure; they represent sites of chain reversal and thus sites where the globular character of a protein is created. It has been speculated for many years that turns may nucleate the formation of structure in protein folding, as their propensity to occur will favor the approximation of their flanking regions and their general tendency to be hydrophilic will favor their disposition at the solvent-accessible surface. Reverse turns are local features, and it is therefore not surprising that their structural properties have been extensively studied using peptide models. In this article, we review research on peptide models of turns to test the hypothesis that the propensities of turns to form in short peptides will relate to the roles of corresponding sequences in protein folding. Turns with significant stability as isolated entities should actively promote the folding of a protein, and by contrast, turn sequences that merely allow the chain to adopt conformations required for chain reversal are predicted to be passive in the folding mechanism. We discuss results of protein engineering studies of the roles of turn residues in folding mechanisms. Factors that correlate with the importance of turns in folding indeed include their intrinsic stability, as well as their topological context and their participation in hydrophobic networks within the protein’s structure.
Keywords: reverse turns, protein folding, peptide models
INTRODUCTION
Reverse turns constitute one of the most common structural features in globular proteins.1 Despite their prevalence, they have always been more challenging to categorize than α-helices or β-sheets because of their nonperiodic nature and the heterogeneity of their structures. They are broadly defined as those regions of the polypeptide where a change of chain direction occurs.2 Because chain reversals allow a protein to fold onto itself, forming a compact globular state, they may play a key role in protein folding. The focus of this article is to explore this proposed role of reverse turns. The local nature of turns has led to many studies of their structural features and sequence preferences using peptide models. These have been complemented by protein engineering experiments in which turn residues were mutated to examine the impact on global thermodynamic stabilities and folding mechanisms. Our goal is to concisely review the available studies of turns based on peptide models and how they shed light on factors that govern intrinsic turn stabilities, and then to discuss the resulting findings in light of research in our own and other labs on the roles of turns in protein folding. Our motivation is to examine the extent to which the roles and behaviors of turns in protein folding reflect their stabilities and features in model peptides.
NOMENCLATURE AND DEFINITIONS
Reverse turns comprise the widely distributed β-turns, as well as the less prevalent γ-turns and α-turns, and may include well-defined loops, such as Ω-loops. A stringent definition of turn types was originally proposed based on a computational search for the different conformations of a polypeptide chain that can be sampled by short (three to four) amino acid segments and stabilized by an intra-turn backbone hydrogen bond.3 In current practice, the classification of a turn is based on the preferred φ,ψ angles of the chain backbone, the position of the stabilizing hydrogen bond, and the length of the chain where the turn occurs.4 The most common type of turn is the β-turn where the hydrogen bond occurs between the i and i + 3 residues. Venkatachalam’s analysis proposed two major classes of the four-residue β-turn, Types I and II, and this classification has been in use up to the present. The difference between Types I and II is mainly the relative orientation of the peptide bond between the i + 1 and i + 2 residues (so-called “corner residues”), which consequently affects the positions of their side chains. Another turn type is the G1 β-bulge turn, which is a Type I turn with an additional residue at position i + 3 that samples αL-space, causing the i and i + 4 positions to be hydrogen bonded.5,6 The term “open turn” refers to cases where no hydrogen bond is present and the φ,ψ angles are within 30° of the standard turn types.2 Different β-turn conformations observed in proteins have been systematically classified by Thornton and coworkers.7,8 A β-turn flanked by two β-strands makes up a β-hairpin, the basic building block of a β-sheet.6
The number of residues required to change the overall direction of the polypeptide chain varies for turns and loops: three for γ-turns, four for β-turns (or five, if a β-bulge is present), and more than five for loops. One or more reverse turns may be present in a loop to facilitate the change in chain direction, but this does not imply that turns are just smaller versions of loops.9 Unlike loops, turns have backbone groups that are in close proximity, consequently constraining the side-chains to protrude outwards and allowing hydrogen bonding of the backbone. In contrast, hydrogen bonding in a loop’s backbone is not regular, and the longer loops have more relaxed limitations on side-chain orientations, allowing the side-chains freedom to pack inside the loop’s core. An idealized loop backbone conformation is exemplified by an Ω-loop, where the distance between the termini is less than any α–α carbon separation in the loop, creating a structural feature that resembles the Greek letter omega (Ω) in which the termini “neck-in”.9 The classification of turns and loops is not straightforward since both lack the regular backbone dihedral angles and repetitive hydrogen bonding patterns so easily recognizable in α-helices and β-strands. However, careful observation reveals regular conformations as well as sequence and positional preferences.
FROM PEPTIDE MODELS TO PROTEINS
The short, self-contained nature of reverse turns has led to their extensive study using peptide models. In an approach pioneered by the Blout laboratory, cyclic peptides have served as constrained models for reverse turns.10 Numerous crystal structures from the Karle lab,11–14 and others,15 as well as NMR studies of cyclic peptides in solution from the Blout,16–19 Kopple,20–25 Tonelli,26,27 and Ivanov and Ovchinnikov28 groups, confirmed the original characterization of β-turn types and provided illustrations of γ-turns and α-turns. The similarity of these local structures in cyclic peptides to those observed in proteins gave early credence to the importance of local energetic contributions to turn conformations. Because of the conformationally constrained nature of cyclic peptides, they have also been useful in defining spectroscopic signatures of turns.15,16,29–31
These crystallographic and spectroscopic parameters in turn helped in the next logical step in characterizing turn formation: extension to small linear peptide models, which have considerably greater conformational latitude. Crystal structures (many from the labs of Aubry and Marraud32) revealed that β-turn conformations were among the most stable states for N-α and carboxy-protected linear dipeptides (which mimic tetrapeptides) of favorable sequence. Landmark work by Dyson, Wright, and Lerner showed that certain sequences within turns yielded strong conformational propensities as well in solution, such that turn conformations were highly populated in short peptides in water.33 This and follow-up work established an early set of “rules” for turn likelihood in otherwise flexible peptides and paved the way for a wealth of research on short linear peptides.34 Looking back, this work foreshadowed the current view that unfolded states of proteins are not randomly sampling all areas of φ,ψ space but instead are best described as populating an interconverting set of native-like local conformations, with occasional long-range native-like contacts or hydrophobic clusters.35 Also, this work and related studies in other labs supported an early proposal that β-turns may nucleate protein folding.36–39
Over the past two decades, considerable research has been focused on the next stage in a hierarchical build-up of structure from a β-turn: β-hairpins. These structural features are stabilized by virtue of turn propensities of amino acid residues as well cross-strand interactions between the sequences flanking the turns. Favorable energetic contributions to β-hairpin stability have been demonstrated to include cross-strand aromatic-aromatic, aromatic-polar, hydrogen-bonding, hydrophobic, and salt-bridge interactions,40–45 in combination with loop conformational propensity and entropy terms dependent on loop length.43,46
RESIDUE PREFERENCES IN TURNS AND PROTEIN STABILITY
Because they occur between regions of regular secondary structure, turns are frequently located at the surfaces of globular proteins. As a consequence, turns are primarily composed of hydrophilic residues.47 This characteristic, plus the higher than random proportion of glycine, proline, asparagine, and other small polar amino acids arising from the requirement to sample αL φ,ψ regions, has led to significant success in the prediction of turn propensity from amino acid sequence. Current β-turn prediction programs incorporate evolutionary information, secondary structure propensities, hydrophobicity scales, and statistical and neural network analysis in order to classify β-turn types.47–53 Gratifyingly, the turn propensities of amino acids at different positions of various protein β-turn types obtained through statistical analysis by directed evolution and phage-display correlate well with work on model peptides in showing glycine, proline, asparagine, and aspartic acid to be the most common β-turn-forming residues.8,54,55
Turns can influence the stability of a protein’s native state both by their intrinsic preference to sample favorable φ,ψ space and by their side-chain packing interactions and local environment.56 Since β-turns are mostly surface-exposed, they are well-suited to participate in ligand binding, molecular recognition, protein–protein or protein–nucleic acid interactions, thus modulating protein functions and intermolecular interactions; additionally, they are frequently sites of post-translational modifications such as phosphorylation and glycosylation, which are used to tune interactions.2 However, there is a fine balance between turn interactions that optimize activity and those that influence the stability of proteins. Indeed, there must be some evolutionary pressure to select for turn sequences that confer thermodynamic stability.57 For example, when a central turn in plastocyanin was mutated in a combinatorial fashion, creating a library of 98 mutants, most substitutions led to stably expressed products (i.e., compact and resistant to degradation) that had lost the ability to bind their metal cofactor,58 implying that this turn is important in defining both the function and the fold of the protein. In one recent study, the conformational stability of microbial ribonuclease Sa was increased by substituting certain nonproline and nonglycine turn residues with proline and glycine respectively, suggesting that protein stability can be modulated by introducing mutations based on positional turn propensities with the stipulation that other electrostatic, hydrophobic, and active site interactions are not compromised.59 When backbone flexibility, loop length, and amino acid turn potential within certain turns are altered, these are usually accompanied by changes in equilibrium free energy.60,61
There are cases when stabilizing turn interactions have deleterious effects on binding activity, such as the Pin1 WW domain, where ligand binding was greatly reduced when loop 1 was shortened and engineered to have higher turn propensity and increased thermodynamic stability.62 Similarly, mutations in α-lytic protease that lead to increased kinetic stability of the protein by improving the packing interactions of the β-hairpin cause a decrease in proteolytic activity.63
Turn propensities not only affect the stability of native states but are also implicated in the folding process itself. Many of the mutations that cause temperature-sensitive phenotypes in the bacteriophage P22 tailspike protein, a β-helical protein, are located at reverse turns.64 When grown at non-permissive conditions, the mutant proteins aggregate suggesting that the perturbation of these turns cause temperature sensitive folding (tsf) defects where the protein becomes vulnerable to aggregation. Further examination of peptides corresponding to a tsf turn showed that the aggregation-prone mutants have sequences with reduced turn propensities relative to the wild-type P22 tailspike protein, supporting the idea that the conformational stability imparted by turns is influential in populating productive folding intermediates.65
TURNS AND FOLDING
Turns have been proposed to be important in folding because they are capable of initiating productive structure formation without a large loss in chain entropy since the interactions involved in turn formation are largely local.36–39,66 This view is consistent with a hierarchical folding model in which certain turns containing residues with high turn propensities (such as glycine and aspartic acid) serve as active nucleation sites for structure formation, originating from the corner residues and propagating toward the flanking β-strands. The turn as the site for chain reversal becomes a nucleation point that facilitates cooperative formation of neighboring interactions (Figure 1). A contrasting view would emphasize the formation of sequence-encoded long-range hydrophobic contacts and a consequent global hydrophobic collapse. Turns in this case might be required to act passively in folding, serving only as flexible structural connectors. In this case, other folding events such as chain collapse or stable tertiary interactions promote structure formation, and turns form only as a consequence of structure consolidation from other regions of the protein.67
FIGURE 1.
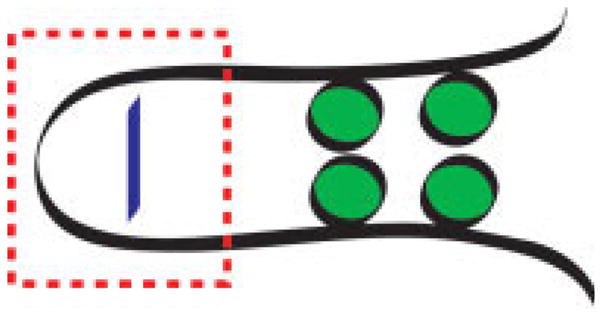
Schematic showing that β-hairpin formation can be initiated at the turn corner residues (red box). Turn formation can promote cooperative favorable interactions in the flanking regions of sequence (green circles). Alternatively, the presence of the favorable interactions on the flanking strands can drive turn formation provided the sequence in the turn allows the chain to adopt the required φ, ψ angles.
Thus, turns can play two different roles in the folding reaction of a protein: They can be either folding-active and serve as initiation sites, or folding-passive elements that form only after other regions develop. These differing roles are likely to arise from the relative importance of various interactions in forming the native states of different proteins. Proteins containing regions with high secondary structure propensities may have increased tendency to fold in a hierarchical manner consistent with the framework model of folding where the native structure propagates from a stable secondary structural element. In these cases, a structure-nucleating β-turn can play an active role in folding. In contrast, when secondary structures have low propensity to form, their formation relies on cooperative tertiary interactions. Certain features of the hydrophobic collapse model are consistent with this, since folding is viewed to begin with the very early collapse, accompanied by long-range interactions and followed by secondary structure formation. Here the β-turn likely acts as a passive folding element where turn formation is a consequence of the association of other nonlocal interactions in the flanking strands. Delineating the factors that control whether a turn is folding-active or passive will provide insight into the underlying principles that govern protein folding and may also be applied to protein design and engineering. Evidence for the two potential roles of turns in folding has been obtained in different proteins and protein domains. We discuss some specific examples in the following sections (and see Table I).
Table I.
Turn Properties and Roles in Protein Foldinga
| Protein (PDB ID) | Turnb | Conservationc | Predicted Turn Propensityd | Properties of Peptide Model | Ref. | |
|---|---|---|---|---|---|---|
| Protein G B1 Domain (1gb1) | (1) 9GKTL12 | 4.75 | 2.244 | Strong native propensity of the second turn fragment | 71,73 | |
| (2) 47DATK50 | 7.00 | 4.494 | ||||
| Protein L B1 Domain (1k50) | (1) 13ANGS16 | 5.25 | 3.385 | Weak native-like propensities of both turns | 66,71,74 | |
| (2) 51ADKG54 | 1.00 | 2.961 | ||||
| Ubiquitin (1ubq) | (1) 7TLTG10 | 7.00 | 2.838 | N-terminal hairpin stable in aqueous methanol and in water | 79,80,84,85 | |
| (2) 18EPSD21 | 2.00 | 4.423 | ||||
| (3) 38PDQQ41 | 4.50 | 1.834 | ||||
| (4) 45FAGK48 | 5.50 | 1.599 | ||||
| (5) 51EDGR54 | 3.75 | 2.532 | ||||
| (6) 62QKES65 | 3.50 | 2.533 | ||||
| Titin I band 27th Domain (1tit) | (1) 14FVGE17 | 4.50 | 2.811 | — | 87,88 | |
| (2) 36LKGQ39 | 6.50 | 5.037 | ||||
| (3) 44SPDC47 | 4.00 | 2.494 | ||||
| (4) 51EDGK54 | 4.75 | 3.800 | ||||
| (5) 65LGMT68 | 5.00 | e | ||||
| (6) 75AANA78 | 3.00 | 2.650 | ||||
| Pin1 WW Domain (1pin) | (1) 17RSSG20 | 5.50 | 7.322 | — | 89,92,93, 94 | |
| (2) 26NHIT29 | 5.50 | 6.226 | ||||
| SH3 Domain (1srm) | (1) 20TETD23 | 3.75 | 2.388 | Diverging turn (27–30) and distal hairpin (49–52) fragments have native-like conformations | 69,70,96, 98,101,102 | |
| (2) 27KKGE30 | 5.00 | 2.594 | ||||
| (3) 38TEGD41 | 4.75 | 3.187 | ||||
| (4) 49TTGQ52 | 2.75 | 2.417 | ||||
| CRABP I (1cbr) | (1) 46DGDQ49 | 4.50 | 2.490 | 123–125 | ||
| (2) 56TTVR59 | 6.25 | 2.786 | Strong native-like propensity of 3rd and 4th hairpin fragments | |||
| (3) 66KVGE69b | 6.25 | 1.197 | ||||
| (4) 75TVDG78 | 7.80 | 2.269 | ||||
| (5) 89NENK92 | 4.50 | 2.387 | ||||
| (6) 102GDGP105 | 3.25 | 3.454f | ||||
| (7) 114ANDE117 | 2.75 | 2.244 | ||||
| (8) 124ADDV127 | 2.75 | 2.426 | ||||
Turns shown by protein engineering to form before the transition state in bold italic font.
Average of the conservation scores of the individual turn residues obtained using ConSurf.134
Propensities to form a turn calculated using COUDES.48
Not predicted as a turn in COUDES.
Predicted turn sequence is within the Ω-loop.
The contribution of turns in folding has been explored in two major ways: through conformational analysis of excised peptide fragments and through site-directed mutagenesis of different proteins and protein domains containing one or more β-hairpins. The former approach necessarily focuses on the extent to which a turn is independently stable with its stability governed by intra-turn contributions. The independent stability of a turn should provide insight into the likelihood that the turn plays an active role in folding and is potentially a feature of the unfolded state or early folding intermediates. Protein engineering analysis68 enables the exploration of the contribution of a turn sequence in the context of the entire protein.69–71 Briefly, the extent to which engineered mutations alter the stability of the protein or its energy landscape of folding reveals the role of the substituted residue in the native state, intermediates, or transition states. The contribution of a mutated residue to the transition state can be measured by comparing the folding kinetics and stabilities of the mutant and the wild-type protein. In a two-state process, the φ-value is conventionally used to represent the ratio of the change in the activation free energy for folding with the change in equilibrium free energy caused by a mutation.68 When the mutation causes a change in equilibrium stability but the transition states of the mutant and the wild-type protein are energetically equivalent, then interactions involving the mutated residue develop late in folding, i.e., only after the transition state. In cases where the transition state is perturbed as much as the native state, interactions involving the mutated residue must be present in the transition state ensemble. Most φ-value analysis experiments are based on fluorescence and CD spectroscopies, typically employing stopped-flow and temperature-jump kinetics experiments. More recently, a relaxation-dispersion NMR-based method has also been developed to look at φ-values involving intermediate states, which could be used not only to measure energetic contributions at the transition state but potentially provide structural information of the different states.72
Proteins L and G: Interchangeable Turns as Folding Nucleation Sites
Peptide fragment studies and protein engineering analysis on the B1 immunoglobulin domains of proteins L and G show that the intrinsic turn propensity as well as the protein topology are important in defining the role of a given turn in folding. The immunoglobulin B1 domain of protein G contains an α-helix and four β-strands connected by two β-turns (Figure 2a). The NMR and CD spectra of the isolated β-hairpins showed that the second hairpin fragment, but not the first, populates native-like conformations.73 The contribution of the turns to folding was revealed by introducing mutations throughout the protein.71 φ-value analysis indicated that the noncovalent interactions of the second β-turn were already intact at the transition state. Similar fragment and mutagenesis studies were performed on the protein G homolog, protein L, which is structurally similar to protein G (Figure 2b) but has only 15% sequence identity.66,71,74 In contrast to protein G, fragments of protein L do not significantly populate stable secondary structures.74 A more detailed energetic analysis using the protein engineering approach showed that the first turn of protein L is largely formed at the transition state along with the putative hydrophobic core of the protein.66 This is contrary to the case of protein G where it is the second turn that appears to promote formation of the transition state, and the first turn contributes only at the later stages of folding. Moreover, using combinatorial mutagenesis by phage-display, it was found that the second turn in protein L can tolerate more sequence variation than the first turn, suggesting that the first turn is likely to have a conserved, sequence-dependent role in the protein.75 This bias to mutations also extends to the folding rate where mutations in the first turn have larger effects on the folding rate of the intact protein L compared to those in the second turn. The dissimilarity in the folding mechanism in relation to the hairpins of these two proteins can be caused by: (1) different intrinsic propensities of the sequence to form hairpins; (2) different hydrogen bonds and side-chains pattern of assembly and; (3) different packing interactions with the helix.76
FIGURE 2.

Proteins in which the role of turns in folding has been investigated: a, protein G B1 domain; b, Protein L B1 domain; c, ubiquitin; d, 27th domain of titin I-band; e, WW domains (the Pin1 WW domain is shown as an example); f, SH3 domains (the src-SH3 domain is shown as an example); and g, cellular retinoic acid-binding acid protein I (CRABP I). The turns for each protein/protein domain are numbered and are classified based on their structure. The PDB IDs for the proteins shown are listed in Table I. The turns colored red are those that play an active role in folding based on peptide models and protein engineering. All other predicted turns are colored dark blue. The third turn in CRABP I (labeled 3, and referred to as “Turn III” in the text and in the literature) is colored green. This turn has a high propensity to take up a native-like conformation as a peptide but is proposed to play a passive role in folding (see text).
The mechanism of folding for this family of proteins depends on the formation of the β-hairpin with the lower free energy at the transition state, making it possible to inter-convert the folding pathway of proteins G and L by switching the intrinsic turn propensities of the two hairpins.77 A redesigned protein G mutant was constructed by stabilizing the first turn with an optimized backbone conformation and amino acid sequence; and by destabilizing the second turn with a mutation that removes a hydrogen bond. Folding kinetics of the intact, redesigned protein G showed that the first turn now forms at the transition state while the second turn remains unstructured, effectively making protein G fold like protein L. This study shows that the intrinsic stability of turns can modulate the mechanisms of protein folding; in particular, stable turns that bring together favorable contacts can act as initiation points for folding.
Ubiquitin: A Folding-Active Turn Involved in Both Native and NonNative Interactions
Turns also affect protein folding through the presence of specific structural features like the β-bulge, as observed in another protein ubiquitin. The structure of ubiquitin is in fact very similar to the B1 domain of protein G and L except that the second hairpins of the Ig domains are replaced by a longer loop in ubiquitin (Figure 2c). NMR and CD studies of synthetic peptide fragments corresponding to the different parts of ubiquitin showed that the protein’s N-terminal hairpin can populate native-like structures in 40–60% aqueous methanol.78 Further characterization of the excised N-terminal turn peptide showed that it also retains a glycine β-bulge to prevent stable non-native interactions.79 The importance of this β-bulge is also supported by the finding that glycine deletion decreases the observed residual structure of the peptide denatured state as well as the folding and unfolding rates of the intact protein.80 These results point to a model where turn formation occurs very early in folding, perhaps even contributing to the stability and structure of the denatured state. Changing the N-terminal Type I β-bulge into a Type I turn also destabilizes ubiquitin. Although this change introduces a non-native β-strand conformation in the isolated hairpin fragment, it was not sufficient to induce non-native interactions in the full-length protein.81 The β-bulge in this turn favors sampling of stable β-hairpin conformations and prevents formation of non-native hydrophobic interactions in ubiquitin. This implies that the local structure of β-turns affects the stability of the intact protein, and that the contribution of tertiary interactions is important in β-sheet formation as well. More recently, the terminal hairpin residues (away from the corner residues), specifically those that can form hydrophobic clusters, were shown to contribute to the stability of the N-terminal hairpin fragment.82 It was further suggested that this hydrophobic “cap” not only stabilizes the hairpin but also plays a crucial role in formation of tertiary interactions during folding, since these residues also are in contact with other regions in the intact protein that define the native topology. The information obtained using this fragment of ubiquitin is consistent with φ-value analysis on the full-length protein, where the N-terminal hairpin was found to be partially formed and accompanied by an intact helix in the transition-state ensemble.83 When an autonomously forming β-turn sequence was inserted into the N-terminal hairpin, the stability of ubiquitin increased.84 However, this stabilization was accompanied by the formation of an additional collapsed folding intermediate not present in the wild-type protein. It appears that over-stabilization of the local hairpin interactions promotes formation of either an off-pathway or an obligate misfolded intermediate, which then requires another rearrangement step prior to folding. This is similar to the case where a premature collapse is seen as a stabilizing hairpin is added to the more conformationally flexible C-terminal hairpin region.85 These studies show that turns can change the folding landscape based on their ability to promote stabilization of native or non-native interactions.
Titin I-band Domain 27: Topology-Driven Folding
Not all protein domains show turn-dependent folding: One example in which turns are present but not active in folding is the 27th domain of the titin I-band protein (TI I27), a structure composed of two anti-parallel β-sheets made of 4 β-strands (Figure 2d).86 From φ-value analysis, a cluster of residues that form both inter- and intra-strand interactions and not associated with any turns was shown to be present in the transition state ensemble.87 This nucleation region is stabilized by nonlocal interactions, which facilitate the formation of a transition state that resembles an expanded version of the native state. In this case, the turns in the TI I27 domain do not play a significant role in the transition state; they appear to act as folding-passive elements since the rate-limiting step appears to be the formation of long-range contact between strands. In this protein fold, the requirements of the native architecture seem to predominate in the folding mechanism over locally encoded structures. However, under nonphysiological conditions, such as high denaturant concentrations, the diffuse transition state can be altered into one in which most of the transition state interactions are clustered in one region.88 This alternative transition state is characterized by more localized interactions within one of the central strands of TI I27, suggesting a more polarized transition state ensemble.
WW Domains: Nucleation Through Turn Hydrogen Bonding
The importance of hydrogen bonding, dynamics and turn formation was studied using the all β-sheet WW domains. The WW domain, which is comprised of three anti-parallel β-strands (Figure 2e), binds proline-rich sequences. From extensive folding kinetics and thermodynamics studies done on the Pin1 WW domain, it appears that the first turn is significantly populated at the transition state of folding.89 The contribution of backbone hydrogen bonds to the transition state was also measured by amide to ester or amide to olefin mutations.90,91 When combined with the traditional protein engineering approach which probes the contributions of side-chains, these studies gave a detailed view of the earliest structure formed at the transition state and indicated that the hydrogen bond of the first turn forms before the consolidation of stabilizing interactions of the side-chains.91,92 Similar results were obtained for the FBP28 WW domain where a combination of MD and φ-value analysis showed that the region of the first β-turn is formed at the transition state of folding.93 A recent study looked at the dynamics of hydrogen bond formation in turns using NMR and showed that turn I of the FBP28 WW domain is both dynamic and solvent-exposed.94 The authors proposed that even though this turn was previously thought to be a nucleation site, it is dynamic and can possibly sample two types of conformations: one that allows productive folding, and the other that leads to a more aggregation-prone conformation. It was suggested that although this turn has been considered to be a nucleation site for folding, it may play a passive role where it simply allows structure formation, in contrast to an active role in which the turn initiates and promotes folding.
SH3 Domains: Polarized Transition State
The relationship of protein topology and secondary structure in the context of folding has been investigated in detail using the SH3 domain. Like the WW domain, the SH3 domain binds proline-rich regions and is part of a large and ubiquitous family of proteins that typically have five or six β-strands forming two orthogonal anti-parallel β-sheets connected by the RT loop, the n-src loop, the distal loop and a short 310 helix (Figure 2f).95 From conformational studies done on peptide fragments spanning the α-spectrin SH3 domain, peptides that contain the distal loop showed some weak native-like interactions.96 Inserting an autonomous turn folding sequence (BH19) at the distal hairpin stabilized the protein as well as increased the refolding rate.97 However, introducing BH19 in place of the distal hairpin of SH3 contributed some conformational frustration during folding, as evidenced by the presence of a roll-over in the chevron plot at low urea concentration that was presumed to arise from an additional collapsed folding intermediate.97
From φ-value analysis of both α-spectrin and src-SH3 (36% sequence similarity), the distal hairpin was found to be considerably formed at the folding transition state.70,98 It was proposed that this could be due to the conformational restriction or “stiffness” of the distal and n-src loops compared to the RT loop; the stiffer regions are likely to act as nucleating regions and limit the conformational heterogeneity of the transition state ensemble.99 Here, loop stiffness is related to the bending energy and the number of interstrand contacts of the different SH3 hairpins. The rigidity of this loop was also said to be important in limiting the conformational space sampled by the hydrophobic core of the protein thus promoting more efficient folding.100
In addition to the distal loop, a conserved 7-residue diverging β-turn was also shown to sample native-like conformations in isolation.101,102 It is interesting to note that this turn is stable as an isolated peptide, even though it does not form a tight hairpin in the full-length src-SH3 domain. Instead, it serves as the transition point from the front to the back sheet.101 This argues that perhaps the local transient interactions of the diverging β-turn enables it to establish the correct protein topology, which is critical in folding.69 Interestingly, this turn is at the base of the RT loop responsible for ligand binding. Perhaps this turn also allows the protein to present the correct binding site.
In terms of the effects of local and nonlocal interactions, when strong non-native helical mutations in the α-spectrin SH3 were introduced in the flexible region flanking the RT-loop, a region known to participate only at the later stages of folding, the protein still folded into the correct native structure but was compromised with respect to the stability and the folding rates.103 This suggests that in the case of α-spectrin SH3, local secondary contacts in the RT region can be compensated in part by tertiary interactions.
The SH3 domain shows a transition state biased towards formation of the diverging turn and distal hairpin. This polarized transition state is characterized by an uneven distribution of φ-values throughout the protein, showing that different clusters of interactions contribute to varying extents at different stages in folding. It is also possible that the polarized transition state has partial structure when most native interactions are not yet fully realized.
INTRACELLULAR LIPID-BINDING PROTEINS
Members of the intracellular lipid-binding protein (iLBP) family have been used as model systems to study folding of multi-phasic β-sheet proteins.104–115 The iLBP fold is a β-barrel formed by two anti-parallel, five-stranded β-sheets (Figure 2g). β-turns link all strands except for Strand 1 and 2, which are connected by a helix-turn-helix motif (containing a conserved Schellman motif116), and Strands 7 and 8, which are connected by a variable Ω-loop (6 in Figure 2g). Our laboratory has extensively investigated the details of the folding mechanism of one member of this family, cellular retinoic acid-binding protein I (CRABP I).117–122 To further understand how local interactions influence the conformational stability of a protein, peptide fragments corresponding to the seven β-turns of CRABP I were studied.123,124 Both NMR and CD analysis showed that peptide fragments comprising Turns III (65 FKVGEG70) and IV (74 ETVDGRK80) from CRABP I populate native-like conformations in aqueous solution, with the isolated Turn IV fragment adopting the most stable native-like turn. None of the other turn peptides from CRABP I folded into native-like structures to any significant extent. The combined 24-residue hairpin that contains both Turns III and IV also diplayed native-like conformational propensity, even in the presence of 7 M urea, suggesting that this region of the protein may be structured in the unfolded state and may therefore be important in narrowing the conformational space at the earliest stages in folding.125 This result is consistent with studies done on another member of the iLBP family, intestinal fatty acid-binding protein (IFABP), where residual structure was found in regions spatially close to Turn III (in Strands 3–7) and Turn IV (Schellman motif and Strands 9 and 10).105,126,127
To further explore the role that turns play in folding of an iLBP β-sheet protein, we have carried out an alanine-scanning mutagenesis study on Turns III and IV of CRABP I. (A. M. C. Marcelino, O. Bilsel and L. M. Gierasch, manuscript in preparation.) The rationale for the alanine substitutions is to eliminate side-chain interactions without substantially perturbing conformational flexibility of the polypeptide backbone. Results show that the two turns affect two different folding phases: Evidence suggests that Turn IV interactions influence early steps in folding and that the turn is formed in the rate-determining transition state ensemble. By contrast, Turn III forms late and is not present at the rate-determining transition state. The proposed folding pathway for this protein as established in our laboratory is illustrated in Figure 3, showing that refolding occurs via two intermediates. The refolding rates of Turn III mutants are similar to those of the wild-type protein, indicating that interactions involving Turn III are formed after the last energy barrier. Therefore, Turn III can be considered to play a passive role in folding, stabilizing the protein at the later stages. The late contribution of Turn III to folding was also suggested in IFABP: The side-chain of Leu 64 in IFABP (which corresponds to Val 67 in CRABP I) is specifically thought to stabilize the native state through its favorable packing interactions with the hydrophobic cluster in that region.128 This cluster of hydrophobic residues near Turn III is conserved throughout the iLBP family and is shown to include residues in the different strands both in the front and back sheets.129,130
FIGURE 3.

The proposed folding pathway for CRABP I proceeds through an early hydrophobically collapsed intermediate, I1 (τ= ~250 μs); followed by formation of an intermediate, I2, which has native topology and a functional ligand-binding cavity (τ= ~100 ms); and then by establishment of a hydrogen-bonding network and specific tertiary interactions to form the native state, N (τ= ~1 s). The proposed hydrophobic collapse involves interactions among a network of residues (schematically indicated by the gray oval). Productive folding is facilitated locally by turns when their formation brings together essential long-range interactions. Note that Turn IV is at the hub of several topology determining long-range interactions (represented by red circles), and its early formation may be key to folding (folding-active). Turn III is also involved in key long-range interactions (colored green), but these are part of the native hydrophobic packing network and by contrast may drive the formation of this turn instead of the reverse. The sequence of Turn III is thus evolutionarily conserved to allow the native structure to form (folding-passive).
In contrast to Turn III, mutations in Turn IV generally perturb the refolding properties of CRABP I (A. M. C. Marcelino, O. Bilsel and L. M. Gierasch, manuscript in preparation). A glycine to alanine mutation in Turn IV (position 78) slows the 100 ms kinetic phase in which native topology forms (I1 to I2), suggesting that this turn makes an energetic contribution during this early phase, either due to the conformational flexibility conferred by the glycine or due to its steric roles enabling sampling of turn-required φ,ψ space, both of which can affect favorable formation of turn conformations. In addition, the CRABP I variant harboring a Val to Ala mutation at position 76 in Turn IV showed an increase in the burst phase amplitude, which implies that the interactions involving Val 76 are present in the early folding phase. Moreover, Val 76 is also part of a small evolutionarily coupled network of hydrophobic interactions,130 which supports the idea that early turn formation coupled with productive organization of long range interactions is used to establish the correct protein topology (see Figure 3).
Since both turns in CRABP I are conserved, have propensities to sample native-like conformations as isolated fragments, and are associated with networks of hydrophobic interactions, what then is the basis for their disparate functions in protein folding? This difference may be due to the stability of the isolated fragments as a consequence of the type of conformation that these turns sample. Turn III is a diverging turn (irregular Type II), a conserved feature in the iLBP family which serves to connect two strands that are not hydrogen bonded except at the turn itself. On the contrary, Turn IV is a tighter turn (Type I with a glycine bulge) as evidenced by the number of sequential NOE signals among the turn residues. This greater number of contacts in the backbone of Turn IV may provide a more conducive environment in which the β-turn can propagate to form a stable β-hairpin and eventually promote productive longer-range interactions. Since these two turns share a common β-strand, it is possible that the region between Turn III and IV can act as a folding microdomain,125 wherein structure is nucleated at Turn IV, this initial structure promotes interactions between its flanking strands, and eventually the resulting partial hairpin acts as a staging area for the formation of the conserved hydrophobic core near the region of Turn III. Turn IV also connects the front and back sheets of CRABP I. The importance of establishing native-like interactions and topology early in this region may act as a negative-design feature131 that would prevent misfolding or possible aggregation of the protein.
CONCLUSIONS
Turns are implicated in protein folding in two quite contrasting ways: In the first, turns are active agents in folding due to the restrictions they impose on the conformational entropy of the polypeptide chain. Here, turns act as nucleation sites for folding. Hence, the formation of the turn promotes formation of adjacent secondary or super-secondary structure, such as a β-hairpin. Long-range or noncontiguous interactions are fundamental in β-sheet structure, and formation of β-turns between strands is crucial since they can actively facilitate cooperative formation of the β-strand. Although turns can play such active roles in structure formation, they can also have more passive roles in folding. This situation arises when extra-turn, generally longer range interactions are driving forces for folding, and the turn region is an “enabler,” allowing the polypeptide chain to reverse its path and take up the kinked conformation required for the longer range interactions to form. The relative importance of these distinct roles reveals inherent features of the folding mechanism of particular protein families, since the balance is dependent on topology and secondary structure propensity, as well as other factors like negative-design elements and functionality.
The likelihood that a turn would act as nucleation site for folding should be related to its intrinsic turn propensity and sequence conservation. Turns for the examples discussed in this review are listed in Table I and are assigned based on their 3D structure accompanied by secondary structure prediction programs like DSSP132 and Stride.133 The intrinsic turn propensity was calculated using the COUDES program,48 which gives weighted turn propensity scores for tetrapeptide sequences in coiled regions. Conservation scores for each position were obtained using ConSurf,134 and the overall conservation score of the turn secondary structure was calculated from the sum of the scores of each turn position divided by the number of turn residues. The turns that appear to be formed at the transition state determined from protein engineering analysis are in bold italics.
The folding-active turns in the B1 domain of proteins G and L, and the Pin1 WW domain are those that have a high predicted turn propensity and are highly conserved. In ubiquitin, the SH3 domain and CRABP I, the most conserved turns are also the folding-active turns suggesting considerable effects of evolutionary constraints. Moreover, in all of these cases, peptide fragments corresponding to these conserved, predicted turns indeed adopt native-like conformations. It is gratifying that the correlation of intrinsic properties and roles in folding is high in these examples. However, this does not apply to all cases, for example the 36 LKGQ39 turn in the TI 27th domain has high conservation and turn propensity scores but it does not appear to nucleate folding. Other networks of interactions like those in helices or in collapsed states may be the initiation points for folding.
There are most certainly other factors that distinguish active from passive turn roles, such as proximity to other conserved or functional regions. Turns spatially associated with regions that can define the native topology may have a greater likelihood to act as a nucleation site for folding, since they can both limit conformational space and establish the correct native contacts early on.
The combination of peptide fragments and protein engineering studies have allowed us to look at turn folding functions—their contributions to thermodynamic, kinetic, and conformational stability during folding. Our perceptions of turns have come a long way from simply viewing them as a stable set of conformations sampled by short segment of sequence. We have seen that turns are crucial for protein stability and folding, acting either as regions that promote structure formation or as passive elements that allow folding to proceed. The emerging concepts from these studies include the intimate relationship between local and long-range interactions during β-sheet folding. Proteins need to perform a balancing act with respect to these interactions; on the one hand over-stabilization of local contacts can cause frustration in a folding pathway, while substantial destabilization of locally encoded features can lead to misfolding. We also see evidence that turns can be used as initiation points for folding as well as mere structural connectors, reflecting their active and passive roles in folding. It is therefore important to consider the context of a turn with respect to folding mechanisms: the nature of its flanking interactions, its proximity to functional groups, and how it fits into the overall protein topology.
Most of the studies done on turns have focused on two-state folders or small proteins and protein domains that have simple topologies. More extensive study is needed of the roles in larger, multi-state β-sheet folders like the iLBPs. It appears that there are turns that form early and turns that form late. Is this general? Are there features that determine which turns will be folding-active or folding-passive? Are there stable turn conformations in unfolded states? What about the contributions of turns to the stabilities and topologies of intermediate states? In a large β-sheet protein where aggregation competes with folding, can a turn-facilitated early chain collapse prevent edge strand interactions that expose regions prone to self-association? These and other questions will be answered by increasingly detailed mechanistic study of a growing set of larger proteins with more complex topologies using ever more powerful methods.
Acknowledgments
Contract grant sponsor: NIH (GM027616 and OD000945)
Footnotes
This article is dedicated to the late Elkan R. Blout whose mentorship started one of us (LMG) on the road to her scientific career. His warmth, wisdom, excellent critical eye, and worldly view of science were inspirational.
References
- 1.Richardson JS. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 2.Rose GD, Gierasch LM, Smith JA. Adv Protein Chem. 1985;37:1–109. doi: 10.1016/s0065-3233(08)60063-7. [DOI] [PubMed] [Google Scholar]
- 3.Venkatachalam CM. Biopolymers. 1968;6:1425–1436. doi: 10.1002/bip.1968.360061006. [DOI] [PubMed] [Google Scholar]
- 4.Sibanda BL, Blundell TL, Thornton JM. J Mol Biol. 1989;206:759–777. doi: 10.1016/0022-2836(89)90583-4. [DOI] [PubMed] [Google Scholar]
- 5.Richardson JS, Getzoff ED, Richardson DC. Proc Natl Acad Sci USA. 1978;75:2574–2578. doi: 10.1073/pnas.75.6.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rotondi KS, Gierasch LM. Biopolymers. 2006;84:13–22. doi: 10.1002/bip.20390. [DOI] [PubMed] [Google Scholar]
- 7.Sibanda BL, Thornton JM. Nature. 1985;316:170–174. doi: 10.1038/316170a0. [DOI] [PubMed] [Google Scholar]
- 8.Hutchinson EG, Thornton JM. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leszczynski JF, Rose GD. Science. 1986;234:849–855. doi: 10.1126/science.3775366. [DOI] [PubMed] [Google Scholar]
- 10.Rizo J, Gierasch LM. Annu Rev Biochem. 1992;61:387–418. doi: 10.1146/annurev.bi.61.070192.002131. [DOI] [PubMed] [Google Scholar]
- 11.Karle IL. Int J Pept Protein Res. 1986;28:420–427. doi: 10.1111/j.1399-3011.1986.tb03274.x. [DOI] [PubMed] [Google Scholar]
- 12.Karle IL, Gibson JW, Karle J. J Am Chem Soc. 1970;92:3755–3760. doi: 10.1021/ja00715a037. [DOI] [PubMed] [Google Scholar]
- 13.Karle IL, Karle J. Acta Crystallogr. 1963;16:969–975. [Google Scholar]
- 14.Karle IL, Urry DW. Biopolymers. 2005;77:198–204. doi: 10.1002/bip.20214. [DOI] [PubMed] [Google Scholar]
- 15.Ramakrishnan C, Paul PKC, Ramnarayan K. Proc Int Symp Biomol Struct Interact Suppl J Biosci. 1985;8:239–251. [Google Scholar]
- 16.Gierasch LM, Deber CM, Madison V, Niu CH, Blout ER. Biochemistry. 1981;20:4730–4738. doi: 10.1021/bi00519a032. [DOI] [PubMed] [Google Scholar]
- 17.Pease LG, Deber CM, Blout ER. J Am Chem Soc. 1973;95:258–260. doi: 10.1021/ja00782a056. [DOI] [PubMed] [Google Scholar]
- 18.Torchia DA, Di Corato A, Wong SC, Deber CM, Blout ER. J Am Chem Soc. 1972;94:609–615. doi: 10.1021/ja00757a048. [DOI] [PubMed] [Google Scholar]
- 19.Torchia DA, Wong SC, Deber CM, Blout ER. J Am Chem Soc. 1972;94:616–620. doi: 10.1021/ja00757a049. [DOI] [PubMed] [Google Scholar]
- 20.Kopple KD, Ohnishi M, Go A. Biochemistry. 1969;8:4087–4095. doi: 10.1021/bi00838a028. [DOI] [PubMed] [Google Scholar]
- 21.Kopple KD, Go A, Logan RH, Jr, Savrda J. J Am Chem Soc. 1972;94:973–981. doi: 10.1021/ja00758a042. [DOI] [PubMed] [Google Scholar]
- 22.Kopple KD, Go A, Schamper TJ, Wilcox CS. J Am Chem Soc. 1973;95:6090–6096. doi: 10.1021/ja00799a042. [DOI] [PubMed] [Google Scholar]
- 23.Kopple KD, Schamper TJ, Go A. J Am Chem Soc. 1974;96:2597–2605. doi: 10.1021/ja00815a046. [DOI] [PubMed] [Google Scholar]
- 24.Kopple KD, Go A. J Am Chem Soc. 1977;99:7698–7704. doi: 10.1021/ja00465a047. [DOI] [PubMed] [Google Scholar]
- 25.Kopple KD, Parameswaran KN. Int J Pept Protein Res. 1983;21:269–280. doi: 10.1111/j.1399-3011.1983.tb03104.x. [DOI] [PubMed] [Google Scholar]
- 26.Tonelli AE, Brewster AI. J Am Chem Soc. 1972;94:2851–2854. doi: 10.1021/ja00763a052. [DOI] [PubMed] [Google Scholar]
- 27.Tonelli AE, Brewster AI. Biopolymers. 1973;12:193–200. doi: 10.1002/bip.1973.360120118. [DOI] [PubMed] [Google Scholar]
- 28.Ovchinnikov YA, Ivanov VT. Tetrahedron. 1975;31:2177–2209. [Google Scholar]
- 29.Woody RW. Methods Enzymol. 1995;246:34–71. doi: 10.1016/0076-6879(95)46006-3. [DOI] [PubMed] [Google Scholar]
- 30.Madison V. Biopolymers. 1973;12:1837–1852. doi: 10.1002/bip.1973.360120811. [DOI] [PubMed] [Google Scholar]
- 31.Bush CA, Sarkar SK, Kopple KD. Biochemistry. 1978;17:4951–4954. doi: 10.1021/bi00616a015. [DOI] [PubMed] [Google Scholar]
- 32.Marraud M, Aubry A. Biopolymers. 1996;40:45–83. doi: 10.1002/(sici)1097-0282(1996)40:1<45::aid-bip3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Dyson HJ, Cross KJ, Houghten RA, Wilson IA, Wright PE, Lerner RA. Nature. 1985;318:480–483. doi: 10.1038/318480a0. [DOI] [PubMed] [Google Scholar]
- 34.Dyson HJ, Rance M, Houghten RA, Lerner RA, Wright PE. J Mol Biol. 1988;201:161–200. doi: 10.1016/0022-2836(88)90446-9. [DOI] [PubMed] [Google Scholar]
- 35.Dill KA, Shortle D. Annu Rev Biochem. 1991;60:795–825. doi: 10.1146/annurev.bi.60.070191.004051. [DOI] [PubMed] [Google Scholar]
- 36.Dyson HJ, Wright PE. Curr Opin Struct Biol. 1993;3:60–65. [Google Scholar]
- 37.Wright PE, Dyson HJ, Lerner RA. Biochemistry. 1988;27:7167–7175. doi: 10.1021/bi00419a001. [DOI] [PubMed] [Google Scholar]
- 38.Lewis PN, Momany FA, Scheraga HA. Proc Natl Acad Sci USA. 1971;68:2293–2297. doi: 10.1073/pnas.68.9.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmerman SS, Scheraga HA. Proc Natl Acad Sci USA. 1977;74:4126–4129. doi: 10.1073/pnas.74.10.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiehna SE, Waters ML. Protein Sci. 2003;12:2657–2667. doi: 10.1110/ps.03215403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hughes RM, Waters ML. Curr Opin Struct Biol. 2006;16:514–524. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 42.Ciani B, Jourdan M, Searle MS. J Am Chem Soc. 2003;125:9038–9047. doi: 10.1021/ja030074l. [DOI] [PubMed] [Google Scholar]
- 43.Dyer RB, Maness SJ, Peterson ES, Franzen S, Fesinmeyer RM, Andersen NH. Biochemistry. 2004;43:11560–11566. doi: 10.1021/bi049177m. [DOI] [PubMed] [Google Scholar]
- 44.Blanco F, Ramirez-Alvarado M, Serrano L. Curr Opin Struct Biol. 1998;8:107–111. doi: 10.1016/s0959-440x(98)80017-1. [DOI] [PubMed] [Google Scholar]
- 45.Gellman SH. Curr Opin Chem Biol. 1998;2:717–725. doi: 10.1016/s1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- 46.Du D, Zhu Y, Huang CY, Gai F. Proc Natl Acad Sci USA. 2004;101:15915–15920. doi: 10.1073/pnas.0405904101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rose GD. Nature. 1978;272:586–590. doi: 10.1038/272586a0. [DOI] [PubMed] [Google Scholar]
- 48.Fuchs PF, Alix AJ. Proteins. 2005;59:828–839. doi: 10.1002/prot.20461. [DOI] [PubMed] [Google Scholar]
- 49.de la Cruz X, Hutchinson EG, Shepherd A, Thornton JM. Proc Natl Acad Sci USA. 2002;99:11157–11162. doi: 10.1073/pnas.162376199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou PY, Fasman GD. Biochemistry. 1974;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 51.Shepherd AJ, Gorse D, Thornton JM. Protein Sci. 1999;8:1045–1055. doi: 10.1110/ps.8.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuhn M, Meiler J, Baker D. Proteins. 2004;54:282–288. doi: 10.1002/prot.10589. [DOI] [PubMed] [Google Scholar]
- 53.Kaur H, Raghava GP. Bioinformatics. 2004;20:2751–2758. doi: 10.1093/bioinformatics/bth322. [DOI] [PubMed] [Google Scholar]
- 54.Guruprasad K, Rajkumar S. J Biosci. 2000;25:143–156. [PubMed] [Google Scholar]
- 55.Hsu HJ, Chang HJ, Peng HP, Huang SS, Lin MY, Yang AS. Structure. 2006;14:1499–1510. doi: 10.1016/j.str.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 56.Predki PF, Agrawal V, Brunger AT, Regan L. Nat Struct Biol. 1996;3:54–58. doi: 10.1038/nsb0196-54. [DOI] [PubMed] [Google Scholar]
- 57.Zhou HX, Hoess RH, DeGrado WF. Nat Struct Biol. 1996;3:446–451. doi: 10.1038/nsb0596-446. [DOI] [PubMed] [Google Scholar]
- 58.Ybe JA, Hecht MH. Protein Sci. 1996;5:814–824. doi: 10.1002/pro.5560050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trevino SR, Schaefer S, Scholtz JM, Pace CN. J Mol Biol. 2007;373:211–218. doi: 10.1016/j.jmb.2007.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson ER, Meldrum JK, Bofill R, Crespo MD, Holmes E, Searle MS. Angew Chem Int Ed Engl. 2005;44:4939–4944. doi: 10.1002/anie.200500577. [DOI] [PubMed] [Google Scholar]
- 61.Simpson ER, Meldrum JK, Searle MS. Biochemistry. 2006;45:4220–4230. doi: 10.1021/bi052495g. [DOI] [PubMed] [Google Scholar]
- 62.Jager M, Zhang Y, Bieschke J, Nguyen H, Dendle M, Bowman ME, Noel JP, Gruebele M, Kelly JW. Proc Natl Acad Sci USA. 2006;103:10648–10653. doi: 10.1073/pnas.0600511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Truhlar SM, Agard DA. Proteins. 2005;61:105–114. doi: 10.1002/prot.20525. [DOI] [PubMed] [Google Scholar]
- 64.Villafane R, King J. J Mol Biol. 1988;204:607–619. doi: 10.1016/0022-2836(88)90359-2. [DOI] [PubMed] [Google Scholar]
- 65.Stroup AN, Gierasch LM. Biochemistry. 1990;29:9765–9771. doi: 10.1021/bi00494a002. [DOI] [PubMed] [Google Scholar]
- 66.Kim DE, Fisher C, Baker D. J Mol Biol. 2000;298:971–984. doi: 10.1006/jmbi.2000.3701. [DOI] [PubMed] [Google Scholar]
- 67.Dinner AR, Lazaridis T, Karplus M. Proc Natl Acad Sci USA. 1999;96:9068–9073. doi: 10.1073/pnas.96.16.9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Matouschek A, Kellis JT, Jr, Serrano L, Fersht AR. Nature. 1989;340:122–126. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 69.Grantcharova VP, Riddle DS, Santiago JV, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 70.Martinez JC, Serrano L. Nat Struct Biol. 1999;6:1010–1016. doi: 10.1038/14896. [DOI] [PubMed] [Google Scholar]
- 71.McCallister EL, Alm E, Baker D. Nat Struct Biol. 2000;7:669–673. doi: 10.1038/77971. [DOI] [PubMed] [Google Scholar]
- 72.Neudecker P, Zarrine-Afsar A, Davidson AR, Kay LE. Proc Natl Acad Sci USA. 2007;104:15717–15722. doi: 10.1073/pnas.0705097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blanco FJ, Rivas G, Serrano L. Nat Struct Biol. 1994;1:584–590. doi: 10.1038/nsb0994-584. [DOI] [PubMed] [Google Scholar]
- 74.Ramirez-Alvarado M, Serrano L, Blanco FJ. Protein Sci. 1997;6:162–174. doi: 10.1002/pro.5560060119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu H, Kim D, Baker D. J Mol Biol. 1997;274:588–596. doi: 10.1006/jmbi.1997.1374. [DOI] [PubMed] [Google Scholar]
- 76.Karanicolas J, Brooks CL., III Protein Sci. 2002;11:2351–2361. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nauli S, Kuhlman B, Baker D. Nat Struct Biol. 2001;8:602–605. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
- 78.Cox JP, Evans PA, Packman LC, Williams DH, Woolfson DN. J Mol Biol. 1993;234:483–492. doi: 10.1006/jmbi.1993.1600. [DOI] [PubMed] [Google Scholar]
- 79.Searle MS, Williams DH, Packman LC. Nat Struct Biol. 1995;2:999–1006. doi: 10.1038/nsb1195-999. [DOI] [PubMed] [Google Scholar]
- 80.Chen PY, Gopalacushina BG, Yang CC, Chan SI, Evans PA. Protein Sci. 2001;10:2063–2074. doi: 10.1110/ps.07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Platt GW, Simpson SA, Layfield R, Searle MS. Biochemistry. 2003;42:13762–13771. doi: 10.1021/bi030147d. [DOI] [PubMed] [Google Scholar]
- 82.Riemen AJ, Waters ML. Biopolymers. 2007 doi: 10.1002/bip.20840. in press. [DOI] [PubMed] [Google Scholar]
- 83.Went HM, Jackson SE. Protein Eng Des Sel. 2005;18:229–237. doi: 10.1093/protein/gzi025. [DOI] [PubMed] [Google Scholar]
- 84.Bofill R, Simpson ER, Platt GW, Crespo MD, Searle MS. J Mol Biol. 2005;349:205–221. doi: 10.1016/j.jmb.2005.03.048. [DOI] [PubMed] [Google Scholar]
- 85.Bofill R, Searle MS. J Mol Biol. 2005;353:373–384. doi: 10.1016/j.jmb.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 86.Improta S, Politou AS, Pastore A. Structure. 1996;4:323–337. doi: 10.1016/s0969-2126(96)00036-6. [DOI] [PubMed] [Google Scholar]
- 87.Fowler SB, Clarke J. Structure. 2001;9:355–366. doi: 10.1016/s0969-2126(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 88.Geierhaas CD, Best RB, Paci E, Vendruscolo M, Clarke J. Biophys J. 2006;91:263–275. doi: 10.1529/biophysj.105.077057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jager M, Nguyen H, Crane JC, Kelly JW, Gruebele M. J Mol Biol. 2001;311:373–393. doi: 10.1006/jmbi.2001.4873. [DOI] [PubMed] [Google Scholar]
- 90.Fu Y, Gao J, Bieschke J, Dendle MA, Kelly JW. J Am Chem Soc. 2006;128:15948–15949. doi: 10.1021/ja065303t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deechongkit S, Nguyen H, Powers ET, Dawson PE, Gruebele M, Kelly JW. Nature. 2004;430:101–105. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 92.Deechongkit S, Nguyen H, Jager M, Powers ET, Gruebele M, Kelly JW. Curr Opin Struct Biol. 2006;16:94–101. doi: 10.1016/j.sbi.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 93.Petrovich M, Jonsson AL, Ferguson N, Daggett V, Fersht AR. J Mol Biol. 2006;360:865–881. doi: 10.1016/j.jmb.2006.05.050. [DOI] [PubMed] [Google Scholar]
- 94.Sharpe T, Jonsson AL, Rutherford TJ, Daggett V, Fersht AR. Protein Sci. 2007;16:2233–2239. doi: 10.1110/ps.073004907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kuriyan J, Cowburn D. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- 96.Viguera AR, Jimenez MA, Rico M, Serrano L. J Mol Biol. 1996;255:507–521. doi: 10.1006/jmbi.1996.0042. [DOI] [PubMed] [Google Scholar]
- 97.Viguera AR, Serrano L. J Mol Biol. 2001;311:357–371. doi: 10.1006/jmbi.2001.4738. [DOI] [PubMed] [Google Scholar]
- 98.Riddle DS, Grantcharova VP, Santiago JV, Alm E, Ruczinski I, Baker D. Nat Struct Biol. 1999;6:1016–1024. doi: 10.1038/14901. [DOI] [PubMed] [Google Scholar]
- 99.Klimov DK, Thirumalai D. Proc Natl Acad Sci USA. 2000;97:2544–2549. doi: 10.1073/pnas.97.6.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Spagnolo L, Ventura S, Serrano L. Protein Sci. 2003;12:1473–1482. doi: 10.1110/ps.0302603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yi Q, Bystroff C, Rajagopal P, Klevit RE, Baker D. J Mol Biol. 1998;283:293–300. doi: 10.1006/jmbi.1998.2072. [DOI] [PubMed] [Google Scholar]
- 102.Larson SM, Davidson AR. Protein Sci. 2000;9:2170–2180. doi: 10.1110/ps.9.11.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prieto J, Wilmans M, Jimenez MA, Rico M, Serrano L. J Mol Biol. 1997;268:760–778. doi: 10.1006/jmbi.1997.0984. [DOI] [PubMed] [Google Scholar]
- 104.Ropson IJ, Gordon JI, Frieden C. Biochemistry. 1990;29:9591–9599. doi: 10.1021/bi00493a013. [DOI] [PubMed] [Google Scholar]
- 105.Ropson IJ, Frieden C. Proc Natl Acad Sci USA. 1992;89:7222–7226. doi: 10.1073/pnas.89.15.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ropson IJ, Dalessio PM. Biochemistry. 1997;36:8594–8601. doi: 10.1021/bi962983b. [DOI] [PubMed] [Google Scholar]
- 107.Burns LL, Dalessio PM, Ropson IJ. Proteins. 1998;33:107–118. doi: 10.1002/(sici)1097-0134(19981001)33:1<107::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 108.Dalessio PM, Ropson IJ. Arch Biochem Biophys. 1998;359:199–208. doi: 10.1006/abbi.1998.0908. [DOI] [PubMed] [Google Scholar]
- 109.Dalessio PM, Ropson IJ. Biochemistry. 2000;39:860–871. doi: 10.1021/bi991937j. [DOI] [PubMed] [Google Scholar]
- 110.Yeh SR, Ropson IJ, Rousseau DL. Biochemistry. 2001;40:4205–4210. doi: 10.1021/bi0155044. [DOI] [PubMed] [Google Scholar]
- 111.Burns LL, Ropson IJ. Proteins. 2001;43:292–302. doi: 10.1002/prot.1040. [DOI] [PubMed] [Google Scholar]
- 112.Kim K, Frieden C. Protein Sci. 1998;7:1821–1828. doi: 10.1002/pro.5560070818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hoeltzli SD, Frieden C. Biochemistry. 1998;37:387–398. doi: 10.1021/bi971962u. [DOI] [PubMed] [Google Scholar]
- 114.Chattopadhyay K, Zhong S, Yeh SR, Rousseau DL, Frieden C. Biochemistry. 2002;41:4040–4047. doi: 10.1021/bi012042l. [DOI] [PubMed] [Google Scholar]
- 115.Chattopadhyay K, Saffarian S, Elson EL, Frieden C. Biophys J. 2005;88:1413–1422. doi: 10.1529/biophysj.104.053199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Aurora R, Rose GD. Protein Sci. 1998;7:21–38. doi: 10.1002/pro.5560070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Clark PL, Liu ZP, Rizo J, Gierasch LM. Nat Struct Biol. 1997;4:883–886. doi: 10.1038/nsb1197-883. [DOI] [PubMed] [Google Scholar]
- 118.Liu ZP, Rizo J, Gierasch LM. Biochemistry. 1994;33:134–142. doi: 10.1021/bi00167a017. [DOI] [PubMed] [Google Scholar]
- 119.Sukumar M, Gierasch LM. Fold Des. 1997;2:211–222. doi: 10.1016/S1359-0278(97)00030-8. [DOI] [PubMed] [Google Scholar]
- 120.Clark PL, Weston BF, Gierasch LM. Fold Des. 1998;3:401–412. doi: 10.1016/s1359-0278(98)00053-4. [DOI] [PubMed] [Google Scholar]
- 121.Clark PL, Liu ZP, Zhang J, Gierasch LM. Protein Sci. 1996;5:1108–1117. doi: 10.1002/pro.5560050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eyles SJ, Gierasch LM. J Mol Biol. 2000;301:737–747. doi: 10.1006/jmbi.2000.4002. [DOI] [PubMed] [Google Scholar]
- 123.Rotondi KS, Gierasch LM. Biochemistry. 2003;42:7976–7985. doi: 10.1021/bi034304k. [DOI] [PubMed] [Google Scholar]
- 124.Rotondi KS, Gierasch LM. Biopolymers. 2003;71:638–651. doi: 10.1002/bip.10592. [DOI] [PubMed] [Google Scholar]
- 125.Rotondi KS, Rotondi LF, Gierasch LM. Biophys Chem. 2003;100:421–436. doi: 10.1016/s0301-4622(02)00296-x. [DOI] [PubMed] [Google Scholar]
- 126.Hodsdon ME, Frieden C. Biochemistry. 2001;40:732–742. doi: 10.1021/bi001518i. [DOI] [PubMed] [Google Scholar]
- 127.Rajabzadeh M, Kao J, Frieden C. Biochemistry. 2003;42:12192–12199. doi: 10.1021/bi0301688. [DOI] [PubMed] [Google Scholar]
- 128.Kim K, Ramanathan R, Frieden C. Protein Sci. 1997;6:364–372. doi: 10.1002/pro.5560060212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gunasekaran K, Hagler AT, Gierasch LM. Proteins. 2004;54:179–194. doi: 10.1002/prot.10520. [DOI] [PubMed] [Google Scholar]
- 130.Marcelino AM, Smock RG, Gierasch LM. Proteins. 2006;63:373–384. doi: 10.1002/prot.20860. [DOI] [PubMed] [Google Scholar]
- 131.Richardson JS, Richardson DC. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 133.Frishman D, Argos P. Proteins. 1995;23:566–579. doi: 10.1002/prot.340230412. [DOI] [PubMed] [Google Scholar]
- 134.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, Ben-Tal N. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]