

Abstract
The survival of most patients with acute myelogenous leukemia (AML) remains poor, and novel therapeutic approaches are needed to improve outcomes. Given that the fraction of AML with mutated p53 is small (∼ 10%), it appears rational to study MDM2 inhibitors as therapy for AML. Here, we report results of a detailed characterization of sensitivity and resistance to treatment ex vivo with the MDM2 inhibitor MI219 in AML blasts from 109 patients. In line with previous observations, all AML cases with mutated p53 were resistant to MI219. Importantly, approximately 30% of AML cases with unmutated p53 also demonstrated primary resistance to MI219. Analysis of potential mechanisms associated with MI219 resistance in AML blasts with wild-type p53 uncovered distinct molecular defects, including low or absent p53 protein induction after MDM2 inhibitor treatment or external irradiation. Furthermore, a separate subset of resistant blasts displayed robust p53 protein induction after MI219 treatment, indicative of defective p53 protein function or defects in the apoptotic p53 network. Finally, analysis of very sensitive AML cases uncovered a strong and significant association with mutated Flt3 status (Flt3-ITD), which for the first time identified a clinically high-risk group of AML that may particularly benefit from MDM2 inhibitor treatment.
Introduction
The therapeutic outcome in adult acute myelogenous leukemia (AML) remains unsatisfactory, and novel treatment approaches are needed to improve the prognosis of affected patients. One promising approach involves chemical activation of p53 through use of drugs that interfere with the binding of p53 and MDM2 (MDM2 inhibitors): a nongenotoxic approach of inducing cancer cell apoptosis. Various compounds that directly interfere with the binding of p53 and MDM2, including the Nutlins and the MI-series of MDM2 inhibitors, have been developed.1–5 Currently available evidence indicates that induction of p53 through MDM2 inhibition by Nutlins or MI-series compounds results in the elevation of p53 protein levels, followed by p53-mediated apoptosis or p53/p21-mediated cell cycle arrest.3,6–9 For reasons that remain largely unknown, noncancerous cells are relatively resistant to MDM2 inhibitor-mediated apoptosis and usually undergo transient cell cycle arrest.10,11 Equally unclear is the nature and exact contribution of various p53 network/effector molecules to MDM2 inhibitor-induced apoptosis; thus, it remains unknown whether individual p53 effector genes or signaling pathways are absolutely necessary for MDM2 inhibitor-induced apoptosis to occur.6,12–15 Evidence for involvement of intrinsic and extrinsic apoptosis pathways in MDM2 inhibitor-induced apoptosis as well as direct effects of the p53 protein on mitochondrial apoptosis molecules has been provided, and it is thus possible that MDM2 inhibitor-mediated apoptosis uses functionally redundant apoptotic pathways.16–21
Studies into resistance mechanisms to MDM2 inhibitors in various cell systems have proven that intact p53 is necessary for MDM2 inhibitor-induced apoptosis to occur.10,22,23 What is less clear is how often and under what cellular circumstances wild-type p53 status is alone sufficient as a predictor for sensitivity, or what other sensitivity/resistance determinants may be operational.24–26 Two of the proposed regulators of p53-mediated apoptosis are MDM2 and MDMX, and elevated levels of these proteins have been shown to influence MDM2 inhibitor sensitivities in various experimental settings. Nonetheless, the available evidence in support of a critical role of these proteins is neither conclusive nor consistent across experimental systems; thus, it remains possible that these p53 regulatory molecules are not critical determinants of MDM2 inhibitor efficaciousness in all human tumors.27–29
As part of broad-based efforts to study the therapeutic potential of MDM2 inhibitors in hematologic malignancies (which are generally characterized by only a small fraction of cases with mutated p53), we have studied ex vivo apoptosis induction in more than 100 human AML samples. Through the studies detailed herein, we have identified a previously unsuspected large fraction of primary human AML samples that displayed primary resistance to MDM2 inhibitors despite wild-type p53 exons 5-9 gene status. Initial investigations into this phenomenon provide evidence for multiple distinct mechanisms of resistance: one centered on insufficient p53 protein induction and another centered on defective p53 protein or defective p53-regulated effector pathways. These novel findings substantially complicate transition of MDM2 inhibitors into clinical AML applications and motivate further study to achieve optimal efficaciousness of these drugs in the clinical setting. Finally, through correlative analysis, we have identified, for the first time, a significant and strong association between mutated Flt3 status (presence of Flt3-ITD) and heightened sensitivity to MDM2 inhibitors, thus providing a novel and practical rationale for MDM2 inhibitor trial design, patient subgroup selection and trial data interpretation in AML.
Methods
Patients
The 109 AML cases analyzed in this study were enrolled at the University of Michigan Comprehensive Cancer Center between March 2005 and October 2009. The study was approved by the University of Michigan Institutional Review Board (IRBMED 2004-1022), and written informed consent was obtained from all patients before enrollment in accordance with the Declaration of Helsinki. Samples from consecutively enrolled patients were analyzed as long as sufficient material was available for the various analyses described herein.
Cell isolation
Cell purification.
Mononuclear cells from blood or marrow from AML patients were isolated by Ficoll gradient centrifugation (GE Healthcare), aliquoted into fetal calf serum with 10% dimethyl sulfoxide (DMSO), and cryopreserved in liquid nitrogen. For purification of AML blasts using negative selection, cryopreserved cells were washed and recovered by centrifugation and then treated with anti–human CD3 (Miltenyi Biotec 130-050-101), anti–human CD14 (if blasts were negative for CD14 expression; Miltenyi Biotec 130-050-201), anti–human CD19 (if blasts were negative for CD19 expression; Miltenyi Biotec 130-050-301), and anti–human CD235a (Miltenyi Biotec 130-050-501) microbeads per the manufacturer's recommendations. Cell suspensions were run through Miltenyi MACS separation LS columns (130-042-401) to negatively enrich for AML blasts. All blast preps were analyzed by cytospins for purity. This schema always resulted in greater than 90% blast purity.
AML blast DNA used for SNP array, Version 6.0 profiling was extracted from samples that were further purified as follows: post-Miltenyi column samples were washed and stained with fluorescein isothiocyanate–conjugated anti-CD33, phycoerythrin-conjugated anti-CD13, and allophycocyanin-conjugated anti-CD45 (all antibodies: eBioscience). After final washing, propidium iodide (PI) was added to a concentration of 1 μg/mL to discriminate dead cells. Sorting of cells was done on a FACSAria high-speed flow cytometer (BD Biosciences). Live cells (PI-negative) were gated for blasts by identifying those cells with intermediate-intensity staining for CD45 and low- to moderate-intensity side scatter.30 CD33 and CD13 were then used to further discriminate blasts versus erythroid lineage and mature myeloid lineage cells.
Ex vivo AML blasts MDM2 inhibitor apoptosis assays
Blasts enriched to more than 90% purity using methods detailed in “Cell purification” were incubated in serum-supplemented RPMI medium at 2.5 × 105 cells in 100 μL final volume in the presence of various concentrations of the MDM2 inhibitors MI-219 and MI-63 (range, 0.625-20μM) for 40 hours. Apoptosis and necrosis were measured for each treated blast aliquot using annexin V/PI flow cytometry–based readouts, and values were subsequently normalized to spontaneous death rates in untreated parallel cultures according to the formula (% alive = % mean alive treated samples/% mean alive paired nontreated samples).
Ex vivo AML blasts epigenetic and MDM2 inhibitor apoptosis assays
Blasts enriched to more than 90% purity using methods detailed in “Cell purification” were incubated with either DMSO or 0.5μM 5-azacytidine (A2385; Sigma-Aldrich) for 48 hours (with 5-azacytidine replenished every 24 hours). During the last 12 hours of incubation, blasts were further aliquoted and treated with either 0.3μM trichostatin A (9950; Cell Signaling Technology) or DMSO. At the end of the 48-hour incubation, each of the 4 differentially treated subgroups of blasts was treated with MI-219 at final concentrations of 0, 2.5, 5, and 10μM for 40 hours, followed by annexin V/PI FACS-based analysis of apoptosis. Aliquots of blasts in parallel were cultured in a 48-well plate at 106 cells per well in 1 mL of medium and treated with 10μM MI-219 or solvent for 8 hours. Blasts were harvested, lysed, and protein prepared for immunoblotting as described.
Measurement of p53, MDM2, and MDMX mRNA expression using quantitative PCR
RNA was prepared from FACS-sorted blasts from AML cases using the Trizol reagent and resuspended in 50 μL diethyl pyrocarbonate-treated water. A total of 20 μL complementary DNA was made from approximately 50 ng of RNA using the Superscript III first strand synthesis kit (Invitrogen) and random priming. Primers and TaqMan-based probes were purchased from Applied Biosystems (Primers-on-demand). Primer/probe mixtures included: p53 (Hs_00153349_m1), MDM2 (Hs_01066930_m1), MDM4 (Hs_00159092_m1), and Hu PGK1.
Duplicate amplification reactions included primers/probes, TaqMan 2x Universal PCR Master Mix, No AmpErase UNG, and 1 μL of cDNA in a 20-μL reaction volume. Reactions were done on an ABI 7900HT machine. Normalization of relative copy number estimates for the mRNA of the gene of interest was done with the threshold cycle (Ct) values for PGK1 as reference (Ct mean gene of interest − Ct mean PGK1). Comparisons between AML subgroups were performed though subtractions of means of normalized Ct values.
AML blast treatment ex vivo and immunoblotting procedures
Primary AML cases with wild-type p53 exons 5 to 9 were ranked according to the 50% inhibitory concentration (IC50) values for MI-219 and 15 cases with high IC50 values (IC50 > 10μM) and 15 cases with low IC50 values (IC50 < 2μM) selected for further analysis. Blasts were purified as outlined in “Cell purification” and subsequently cultured for 8 hours with 10μM MI-219, 10μM Nutlin-3, solvent, or treated with 5 Gy of ionizing radiation. Cells were harvested after treatment, lysed in detergent lysis buffer (50mM Tris, pH 7.5, 100mM NaCl, 2mM ethylenediaminetetraacetic acid, 2mM ethyleneglycoltetraacetic acid, 1% Triton X-100, 20mM NaF, 1mM sodium orthovanadate [13721-39-6 Alfa Aesar], 1mM phenylmethylsulphonylfluoride [Pierce Chemical], phosphatase inhibitor cocktail I [P2850; Sigma-Aldrich], and protease inhibitor cocktail [P8340; Sigma-Aldrich]), protein fractionated, and prepared for immunoblotting with antibodies directed against p53 (Ab-6, clone DO-1; Calbiochem) and actin (AC-15; Sigma-Aldrich). Positive control lysates were generated from the AML cell line MOLM-13 treated with MI-219 at 10μM for 8 hours, and aliquots of these lysates were run side-by-side with lysates of the primary cases on every immunoblot. Thus, these MOLM-13 lysates served as internal standards for blot-to-blot band intensity comparisons. Leftover lysates from these experiments were subsequently prepared for immunoblotting using antibodies against human MDMX (A300-287A; Bethyl Laboratories), MDM2 (Ab-1, clone IF2; Calbiochem), p21 (clone SX118; BD Biosciences), and actin.
SNP 6.0 array analysis of AML blast DNA and paired buccal DNA
The SNP 6.0 assay was performed following the manufacturer's recommended protocols. Affymetrix CEL files for each blast and buccal sample were analyzed using Affymetrix Genotyping Console software for initial quality control, followed by use of the Affymetrix “Birdseed” algorithm to generate tab-delimited SNP call files in text format. Call rates for the entire group of samples included in this report were between 94.93% and 99.45%, with a mean call rate of 98.33%. Sample copy number heatmap displays were obtained from CEL files through use of the freely available software dChip (build date February 25, 2010)31 adapted to a 64-bit operating system environment. To generate functional and practical displays of loss of heterozygosity (LOH), a 2-step, internally developed, Java-based software analysis system was used. The Pre-LOH Unification Tool served to align all individual patient SNP calls to their respective dbSNP rs ID numbers and genomic physical positions before incorporation into the LOH tool, Version 2, an updated version of the LOH tool able to accommodate Affy SNP 6.0 array data.32 For LOH analysis between paired samples, a filter setting within the LOH tool, Version 2 was used, allowing visualization of individual paired SNP calls as LOH only if present within 3000 bp of another such call. This step filtered out many false, sporadically distributed single LOH calls because of platform noise.
Exon resequencing of NPM1, Flt3, and p53
Primers to amplify and sequence exon 12 of human NPM1, exons 13 to 15 and 20 of human Flt3, and exons 5 to 9 of human p53 and adjacent intronic sequences were designed using the primer 3 program (http://frodo.wi.mit.edu/primer3/). Polymerase chain reaction (PCR) products were generated using Repli-g (QIAGEN)–amplified DNA from highly pure blast cells as templates. Amplifications were done using Taq polymerase. PCR amplicons were prepared for direct sequencing with internal nested sequencing primers using the exonuclease/shrimp alkaline phosphatase method (USB). Mutation Surveyor (SoftGenetics LLC) software was used to compare experimental sequences against Refseq GenBank or genomic sequences as well as by visual inspection of sequence tracings. Mutations were confirmed using paired patient buccal DNA as templates.
Statistical methods
Associations between binary classifications (eg, between drug sensitivity and gene mutation status) were assessed using log odds ratios. Mean IC50 values were compared between groups of samples using 2-sample t tests. Results for all statistical tests are reported as Z-scores and 2-sided P values.
Results
Patient characteristics
Detailed clinical and genomic characteristics of the 109 AML patients analyzed in this study are summarized in Table 1 and supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Of the 109 AML cases analyzed, 90 (83%) were previously untreated and 19 (17%) were relapsed at study enrollment (postanalysis review of relapsed cases demonstrated that none of the cases displayed complex karyotypes, probably as a consequence of the small sample size). A total of 66%, 16%, and 18% were primary, secondary or treatment-related AML, respectively, and 12 cases had p53 exons 5 to 9 mutations.
Table 1.
Baseline clinical and genomic characteristics of patients
| Characteristic | Treatment-naive at enrollment, no. (%) | Relapsed at enrollment, no. (%) |
|---|---|---|
| Sample size (N = 109) | 90 (83) | 19 (17) |
| Age, y | ||
| Median | 62 | 60 |
| Range | 20-85 | 24-79 |
| Sex | ||
| Male | 53 (59) | 11 (58) |
| Female | 37 (41) | 8 (42) |
| Pathogenesis | ||
| De novo | 60 (66) | 15 (80) |
| Prior myelodysplasia | 14 (16) | 2 (10) |
| Treatment-related | 16 (18) | 2 (10) |
| WHO classification | ||
| AML with recurrent genetic abnormalities | 10 (11) | 0 (0) |
| t(8;21)(q22;q22) | 2 (3) | 0 (0) |
| inv(16)(p13q22) or t(16;16)(p13;q22) | 4 (4) | 0 (0) |
| 11q23 abnormalities | 4 (4) | 0 (0) |
| AML with multilineage dysplasia | 16 (18) | 2 (11) |
| Antecedent MDS | 14 (15) | 2 (100) |
| Without antecedent MDS | 2 (3) | 0 (0) |
| AML, therapy related | 16 (18) | 2 (11) |
| AML, not otherwise specified* | 48 (53) | 15 (78) |
| AML, minimally differentiated | 7 (8) | 0 (0) |
| AML without maturation | 7 (8) | 4 (21) |
| AML with maturation | 7 (8) | 1 (5) |
| Acute myelomonocytic leukemia | 20 (21) | 6 (32) |
| Acute monoblastic leukemia | 3 (3) | 3 (16) |
| Acute erythroid leukemia | 0 (0) | 0 (0) |
| Acute megakaryoblastic leukemia | 0 (0) | 0 (0) |
| Acute basophilic leukemia | 0 (0) | 0 (0) |
| FAB classification† | ||
| M0 | 10 (11) | 0 (0) |
| M1 | 13 (14) | 4 (21) |
| M2 | 14 (16) | 2 (11) |
| M3 | 0 (0) | 0 (0) |
| M4 | 32 (36) | 6 (32) |
| M5 | 6 (7) | 3 (16) |
| M6 | 0 (0) | 0 (0) |
| M7 | 0 (0) | 0 (0) |
| Cytogenetic class‡ | ||
| Favorable | 6 (7) | 0 (0) |
| Intermediate | 48 (53) | 17 (90) |
| Unfavorable | 36 (40) | 2 (10) |
| No. of karyotypic abnormalities | ||
| 3 or more | 19 (21) | 0 (0) |
| Fewer than 3 | 71 (79) | 19 (100) |
| 5q− status | ||
| Present | 17 (19) | 0 (0) |
| Absent | 73 (81) | 19 (100) |
| 7q− status | ||
| Present | 6 (7) | 2 (10) |
| Absent | 84 (93) | 17 (90) |
| p53 exons 5 to 9 status | ||
| Mutated | 12 (13) | 0 (0) |
| Wild-type | 78 (87) | 19 (100) |
| Flt3 ITD status | ||
| ITD present | 12 (13) | 7 (37) |
| TK-835 mutation present | 2 (3) | 0 (0) |
| Wild-type | 76 (84) | 12 (63) |
| NPM1 status§ | ||
| Mutated | 12 (13) | 11 (58) |
| Wild-type | 78 (87) | 8 (42) |
| Induction type at diagnosis | ||
| Intensive therapy | 0 | 19 (100) |
| Anthracycline plus cytarabine only | 17 (90) | |
| Amonafide plus cytarabine | 1 (5) | |
| Other | 1 (5) |
MDS indicates myleodysplastic syndrome; and FAB, French-American-British.
The subtype of AML, not otherwise specified was unavailable in 5 cases.
The FAB classification was unspecified in 17 cases.
Based on the SWOG S0106 classification.
The NPM1 mutation frequency in normal karyotype AML was 40%
Primary resistance to MDM2 inhibitor treatment is common in adult AML
To evaluate the efficacy of MDM2 inhibitor-mediated apoptotic cell kill in AML blasts ex vivo, we purified blasts from 109 AML specimens (97 with wild-type p53 and 12 with mutant p53 by exon 5-9 exon sequence analysis) to more than 90% purity and incubated cell aliquots for 40 hours with escalating concentrations of the MDM2 inhibitors MI219 (N = 109) and MI63 (N = 60). The apoptotic cell fraction in treated samples was subsequently quantitated through annexin V-PI–based FACS analysis and normalized to measurements in paired untreated cells.
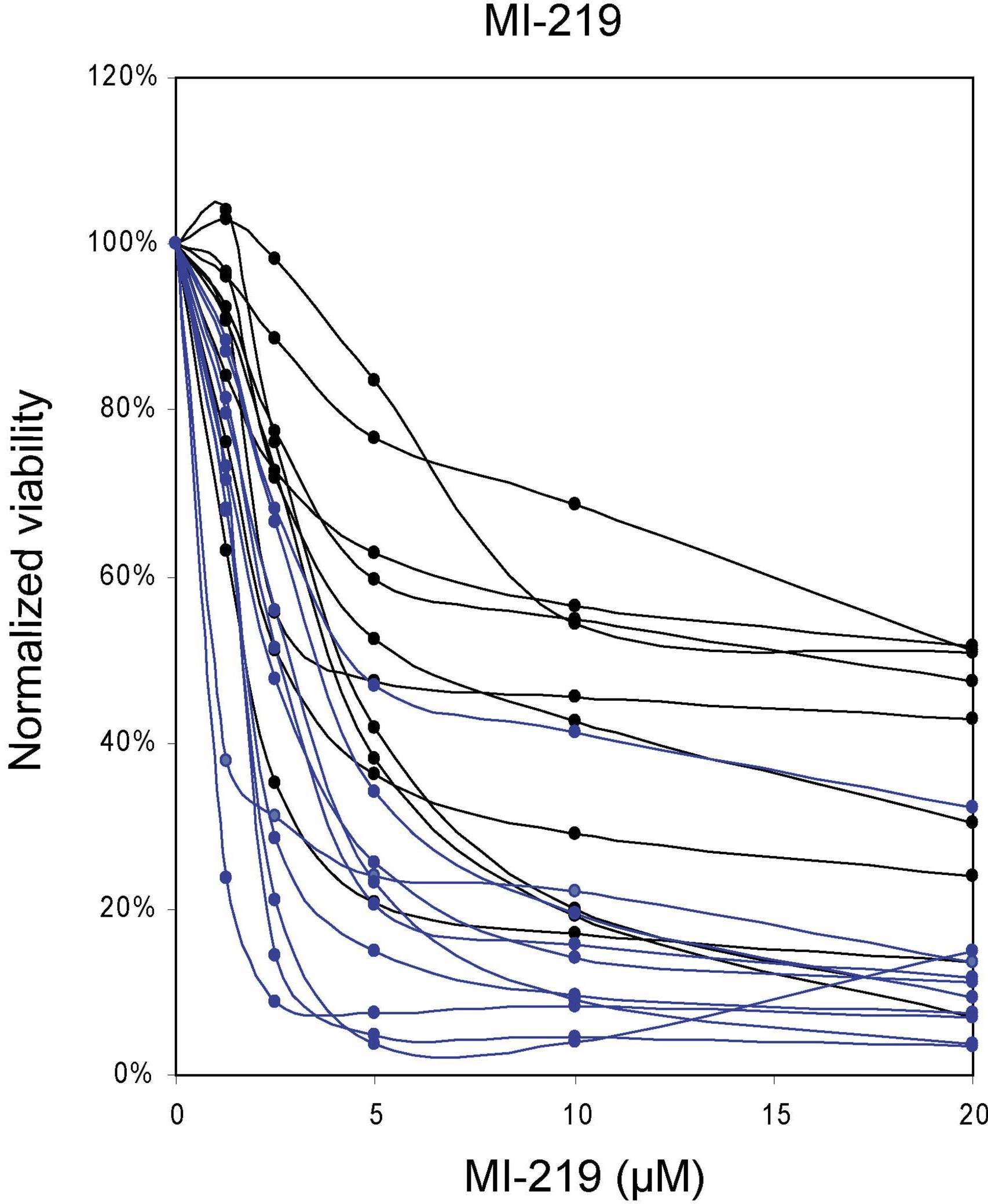
As can be seen in Figure 1, all AML cases with mutant p53 exons 5 to 9 (red) or absent p53 mRNA expression (green) displayed resistance to MDM2-inhibitor treatment, consistent with previous findings.20,22 Although many AML cases with wild-type p53 exons 5 to 9 (black) were very sensitive (IC50 < 2μM; 32 of 97 = 33%) or sensitive (IC50 ≥ 2μM to < 5μM; 33 of 97 = 34%) to MI-219 or MI-63, a substantial fraction of cases with wild-type p53 displayed various resistance levels (MI219 IC50 > 5μM for 32 of 97 = 33% and IC50 > 10μM for 21 of 97 = 22% of cases, respectively). Thus, unlike the situation in CLL, these data demonstrate that a substantial subset of AML cases with wild-type p53 exons 5 to 9 sequence displays primary resistance to MDM2 inhibitors ex vivo.
Figure 1.

A substantial subset of primary AML displays primary resistance to MDM2 inhibitors. A total of 109 (MI219) and 60 (MI-63) AML samples were enriched to more than 90% blast purity through negative selection and incubated for 40 hours with various concentrations of either MI-219 or MI-63. Samples were prepared for annexin V and PI staining and analyzed by flow cytometry, and the residual live and nonapoptotic cell fraction was calculated for each concentration by comparison with the untreated control aliquots. (A) MI-219 assay results. Red represents p53 sequence mutants; green, cases with absent p53 mRNA; and black, wild-type p53 status. (B) MI-63 assay results. Red represents p53 sequence mutants; green, cases with absent p53 mRNA; and black, wild-type p53 status.
Further, the mean IC50 values for MI219 in primary, secondary, and treatment-related AML (exclusive of cases with p53 exon 5-9 mutations), were 6.1μM, 7.9μM, and 4.8μM, respectively. The mean IC50 values for MI219 in previously untreated versus relapsed AML cases (exclusive of cases with p53 exon 5-9 mutations) was 6.6μM versus 4.4μM, respectively.
Given that we had used highly purified blasts in the analysis outlined in this subsection, we became interested in measuring MDM2 inhibitor IC50 values for AML bone marrow-derived mononuclear cell mixtures (after Ficoll gradient purification). The analysis of 10 such cases disclosed invariably higher IC50 values than found in paired highly pure blasts, indicative of substantial resistance of nonblast cells (including nonmalignant bone marrow cells) to MDM2 inhibitor-induced apoptosis. These data are summarized in supplemental Figure 1 and supplemental Table 2.
Various degrees of sensitivity and resistance to MDM2 inhibitors in AML cell lines
Next, we assessed the ability of MI-219 to induce apoptosis in 19 AML-derived cell lines. Data are summarized in Figure 2 and Table 2. As can be seen in Figure 2, all AML cell lines with mutant p53 (red) were resistant to MI219, whereas the cell lines with wild-type p53 (black) displayed various degrees of sensitivity/resistance to MI-219, reminiscent of the findings in primary AML blasts presented under “Primary resistance to MDM2 inhibitor treatment is common in adult AML.”
Figure 2.

Sensitivities of 19 AML cell lines to MI219. Nineteen AML cell lines (Table 2) were incubated for 40 hours with various concentrations of MI-219. Samples were prepared for annexin V and PI staining and analyzed by flow cytometry, and the residual live and nonapoptotic cell fraction was calculated for each concentration by comparison with the untreated control incubation. Red represents p53 sequence mutants; and black, wild-type p53 status.
Table 2.
Summary of AML cell-line analysis and MDM2 inhibitor treatment IC50 values
| AML cell line | FAB type | p53 (exon 5-9) status | Immunoblot (MI-219 10μM 8 hours/16 hours) | IC50/MI-219, μM | IC50/MI-63, μM | IC50/Nutlin, μM |
|---|---|---|---|---|---|---|
| SKM-1 | sM5 | mut | Noninducible | > 20 | > 10 | > 10 |
| OCI-M2 | sM6 | mut | Noninducible | > 20 | > 10 | > 10 |
| MONO-MAC-6 | sM5 | mut | Noninducible | > 20 | > 10 | > 10 |
| GF-D8 | M1 | mut | Noninducible | > 20 | > 10 | > 10 |
| MOLM-16 | M0 | mut | Noninducible | > 20 | > 10 | > 10 |
| Kasumi-3 | M0 | mut | Noninducible | > 20 | > 10 | > 10 |
| Kasumi-1 | M2 | mut | Noninducible | > 20 | > 10 | > 10 |
| ME-1 | M4eo | mut | Null | > 20 | > 10 | > 10 |
| AML-193 | M5 | mut | Null | > 20 | > 10 | > 10 |
| KG-1 | M6 | mut | Null | > 20 | > 10 | > 10 |
| HL60 | M2 | deleted | Null | > 20 | > 10 | > 10 |
| OCI-AML-3 | M4 | wt | Inducible | 18.176 | > 10 | > 10 |
| OCI-AML-5 | M4 | wt | Inducible | 13.846 | > 10 | > 10 |
| ML-2 | sM4 | wt | Inducible | 7.038 | 7.468 | 10.447 |
| OCI-AML-2 | M4 | wt | Inducible | 5.248 | 4.735 | 7.216 |
| AP-1060 | M3 | wt | Inducible | 4.224 | 4.029 | 6.768 |
| SIG-M5 | M5a | wt | Inducible | 3.436 | 3.801 | 4.727 |
| MOLM-13 | sM5a | wt | Inducible | 2.657 | 2.855 | 4.044 |
| MUTZ-2 | M2 | wt | Inducible | 1.283 | 1.17 | 0.951 |
mut indicates mutated; and wt, wild-type.
Evidence for distinct mechanisms of primary resistance to MDM2 inhibitors in AML with wild-type p53 exons 5 to 9
Given the central importance of intact p53 to MDM2 inhibitor sensitivity, we proceeded with analysis of p53 protein expression levels in primary AML blasts. We ranked all primary AML cases with wild-type p53 exons 5 to 9 according to the IC50 values for MI-219 and selected 15 cases with high IC50 values (IC50 > 10μM) and 15 cases with low IC50 values (IC50 values < 2μM) for further analysis. Purified blasts were either left untreated or treated for 8 hours with MI219 (10μM), Nutlin 3 (10μM), or a one-time dose of 5 Gy of external irradiation. Cellular lysates made from these blasts were prepared for immunoblotting with anti-p53 and antiactin antibodies. Further, aliquots of lysates from the MI219-treated AML cell line MOLM13 were analyzed on each blot to permit blot-to-blot comparisons of band intensities.
As can be seen in Figure 3, all sensitive AML blasts demonstrated induction of p53 protein after MDM2 inhibitor treatment or external irradiation, albeit to different absolute levels. Importantly, analysis of p53 protein levels in resistant blasts disclosed 2 subsets: (1) blasts with absent or very low p53 expression after MDM2 inhibitor treatment or external irradiation and (2) blasts with baseline and induced p53 levels essentially equal to the levels measured in sensitive blasts. Thus, resistance to MDM2 inhibitors in AML with wild-type p53 exons 5 to 9 is associated with at least 2 distinct molecular defects: (1) low/absent p53 protein expression or (2) apoptotic p53 network defects (including the possibility of aberrant p53 proteins) in the setting of normal p53 protein levels.
Figure 3.

Results of p53 immunoblotting in sensitive and resistant primary AML blasts after MI219 or Nutlin3 treatment or external irradiation. AML blasts were purified through negative selection and either left untreated or treated for 8 hours with MI219 (5μM), Nutlin 3 (5μM), or one-time external irradiation (5 Gy). After 8 hours, cells were lysed and protein fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Each gel was also loaded with an aliquot of a MOLM13 AML cell line lysate as an internal standard (loaded as 1.25, 2.5, and 5 μg of MI219-treated lysate or 5 μg of untreated lysate [UT], respectively). Protein was transferred to membrane and prepared for immunoblotting with an anti-p53 and anti-actin antibody. Films for both p53 and actin were developed together. IC50 values for MI219 are indicated in brackets.
To gain further insights into the mechanisms of low/absent p53 expression in resistant AML blasts, we measured normalized p53 mRNA levels in total RNA purified from FACS-sorted AML blast samples. This analysis disclosed that a few AML cases at baseline displayed absent p53 mRNA (see Figure 5E; AML cases 7, 80, and 120). Thus, resistance to MDM2 inhibitors in a small fraction of AML blasts is the result of absent p53 transcription and suggests an acquired p53 gene defect. However, the majority of resistant AML blasts with low/absent p53 protein did express p53 mRNA (AML cases 98, 138, 191, 36, 40, 101, and 100), thus implying posttranscriptional mechanisms for low p53 protein levels.
Figure 5.

p53 mutations or absent p53 mRNA expression in AML is associated with frequent copy-neutral LOH (acquired uniparental disomy) at 17p/p53 locus. Files generated through use of the Affymetrix program Genotyping Console for all patients were imported into the LOH tool, Version 2, using our software tool Pre-LOH Unification Tool, and all individual positions of LOH between buccal DNA and paired tumor DNA were graphed as a blue tick mark across the length of the chromosomes. Copy number estimates for all SNP positions for all patients were generated through dChipSNP, as described, and displayed across the length of the chromosomes. Copy losses are displayed with blue colors, copy gains with red colors. (A-B) Heatmap display of chromosomal copy number changes at 17p based on SNP 6.0 array profiling. Blue represents copy loss; and red, copy gain. (A) Buccal DNA. (B) AML blast DNA. (C) LOH analysis at 17p comparing paired blast and buccal DNA. Red numbering indicates copy-neutral LOH (acquired uniparental disomy); and black, LOH with copy loss. (D) p53 exon 5 to 9 mutation analysis results. (E) Normalized p53 mRNA expression in AML blasts grouped by MI219 IC50 values as indicated. Red diamonds represent AML blasts with mutated p53.
The treatment of resistant blasts with absent p53 expression using trichostatin A and azacytidine does not induce p53 expression
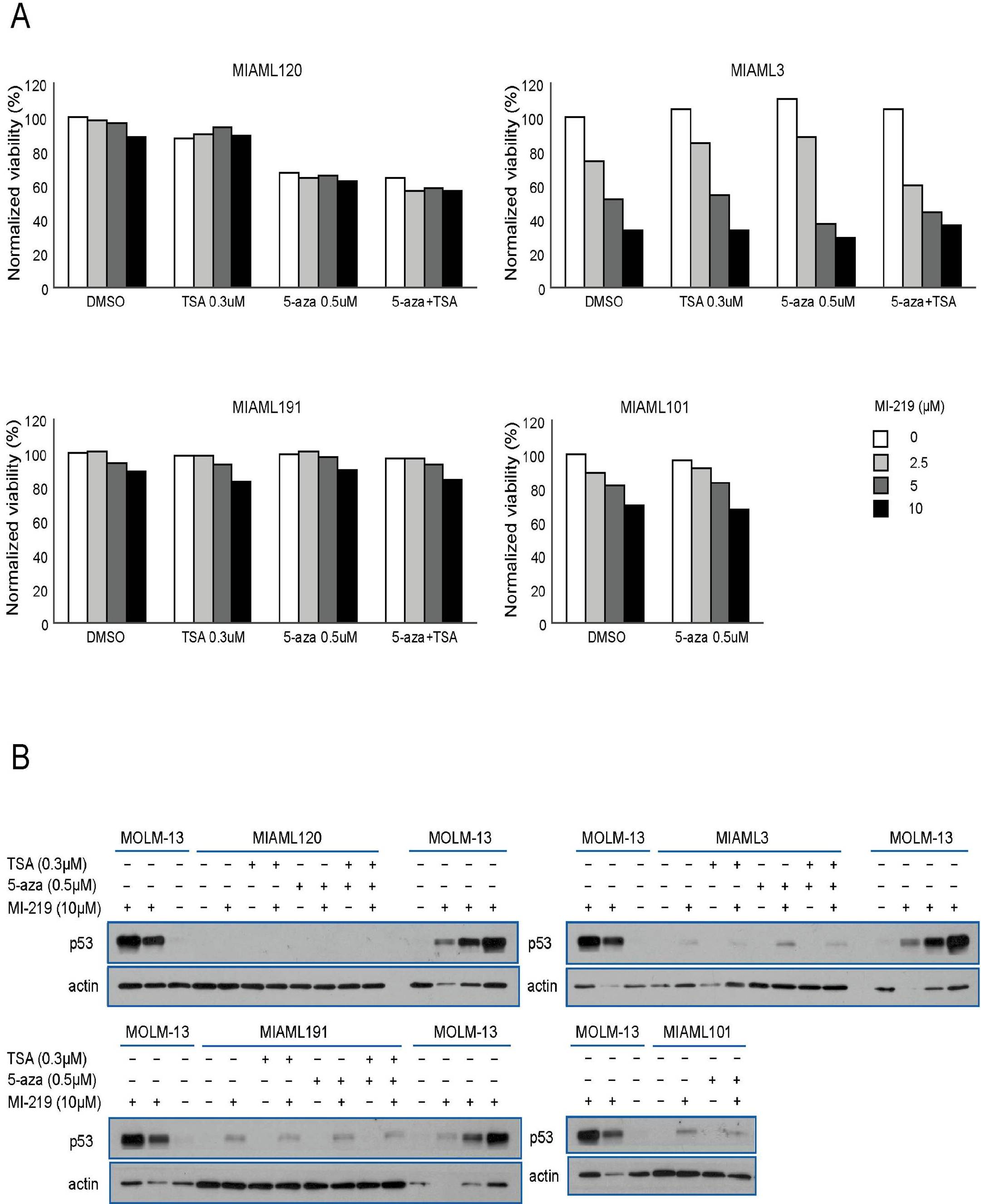
Attempting to obtain evidence for epigenetic p53 gene silencing in AML with absent p53 mRNA expression, we selected 4 AML cases based on the availability of cryopreserved cells with absent or very low p53 mRNA for further analysis and treated purified blasts with trichostatin A and azacytidine (alone or in combination) followed by treatment with MI219. The readout for these experiments was the fraction of blasts undergoing apoptosis and posttreatment p53 protein levels. As detailed in supplemental Figure 2, evidence for reversible epigenetic p53 gene silencing in these blasts was not found.
Various expression levels of MDM2 and MDMX do not account for the resistance to MDM2 inhibitors in AML
The expression levels of MDM2 and MDMX, 2 critical regulators of p53 protein, could account for the observed differences in IC50 values to MDM2 inhibitor treatment in AML cases with wild-type p53 exons 5-9 and for the observed differences in p53 protein levels. To test such hypotheses, we initially measured normalized MDM2 and MDMX mRNA levels across the entire AML cohort. Subsequently, we correlated these mRNA levels with IC50 values to MDM2 inhibitor-mediated apoptosis in all AML cases with wild-type p53 exons 5-9 (Figure 4); as can be seen in Figure 4, neither MDMX nor MDM2 levels correlated with MI-219 IC50 values.
Figure 4.

The mRNA levels of MDM2 and MDMX do not correlate with MI219 IC50 values. Normalized expression levels of MDM2 and MDMX mRNA were measured in cDNA made from RNA from FACS-sorted AML blasts. Displayed are δ Ct values (Ct mean MDM2 or MDMx − Ct mean PGK1) grouped by MI219 IC50 values as indicated. Red diamonds represent AML blasts with mutated p53.
Next, we measured MDMX, MDM2, and p21 protein levels in lysates from blasts derived from the experiment outlined in Figure 3. As can be seen in supplemental Figures 3 and 4, neither MDMX, nor MDM2, nor p21 protein levels demonstrated a clear association with MI-219 IC50 values.
Acquired uniparental disomy (copy-neutral LOH) is common in AML and is often associated with p53 mutations or absent p53 expression
To obtain additional information regarding p53 gene status in AML, we analyzed DNA samples from ultra-pure AML blast populations from 109 AML cases and paired buccal DNA for acquired chromosomal copy number alterations and LOH using ultra-high-density Affymetrix SNP 6.0 arrays.
In Figure 5, we display subchromosomal copy number status at 17p (Figure 5A, buccal DNA; Figure 5B, AML blast-derived DNA; p53 is located at ∼ 7.5 Mb physical position on 17p), LOH at 17p (Figure 5C), p53 exons 5 to 9 sequence data (Figure 5D), and normalized p53 mRNA data (Figure 5E). As can be seen, 17 of 109 (15%) AML cases displayed LOH involving parts or all of 17p that spans the p53 locus. Importantly, paired analysis for copy loss uncovered 2N status for nearly half (8) of these cases (red numbering): examples of copy-neutral LOH or acquired uniparental disomy at 17p. Of note, copy-neutral LOH is undetectable using conventional karyotyping or comparative genomic hybridization and is thus missed in routine clinical practice. Given that copy-neutral LOH is often associated with gene mutations, we compared LOH data with p53 sequence data and p53 mRNA data and found that 6 of 8 of these 17p-associated acquired uniparental disomy cases (red) displayed homozygous p53 mutations (AML 12, 41, 88, 117, 153, and 157, Figure 5D) and 1 of 8 cases (120) had very little p53 mRNA expression. Thus, acquired copy-neutral LOH is common at the p53 locus, is associated with p53 null states in the majority of cases, and is associated with resistance to MDM2 inhibitor treatment. High resolution copy number analysis of the p53 gene and the p53 promoter did not identify homozygous deletions in AML.
The presence of Flt3-ITD is associated with enhanced sensitivity to MDM2 inhibitor treatment in AML
Our analysis of ex vivo sensitivities to MDM2 inhibitors outlined under “Primary resistance to MDM2 inhibitor treatment is common in adult AML” disclosed many cases that were very sensitive to MDM2 inhibitor-mediated apoptosis. We therefore became interested in identification of markers that would correlate with increased MDM2 inhibitor sensitivity. Focusing on Flt3 and NPM1 (the 2 most commonly mutated genes in AML), we correlated the presence or absence of Flt3-ITD or NPM1 exon 12 mutations with MI-219 IC50 values initially in all AMLs with wild-type p53 exons 5 to 9. We repeatedly dichotomized the AML cohort at either the 25th or 50th percentile (corresponding to threshold IC50 values of 1.78μM and 3.2μM, respectively) and determined Z-scores for the presence of mutated Flt3 (Flt3-ITD) or NPM1, respectively. From this analysis, Flt3-ITD emerged as significantly enriched in sensitive AML cases, with Z-scores of 1.91 (P = .06) and 2.26 (P = .02) for the 25th and 50 percentile analysis, respectively. Eleven of 19 (58%) and 13 of 19 (68%) Flt3-ITD–mutated AML cases had IC50 values to MDM2 inhibitors of less than 2μM and less than 2.25μM, respectively.
We also performed a similar analysis for the comparison of Flt3-ITD positive cases versus all other cases (N = 90; including p53 exon 5-9 mutated cases). From this analysis, Flt3-ITD again emerged as significantly enriched in sensitive AML cases, with Z-scores of 2.42 (P = .02) and 2.64 (P = .01) for the 25th and 50th percentile analysis, respectively.
Analysis of AML cases with both Flt3-ITDs and NPM1 mutations (10 of 19 Flt3-ITD positive cases in this cohort were also NPM1 mutated) as well as cases with NPM1 mutations in the absence of Flt3-ITDs (supplemental Table 1) did not suggest a significant role of NPM1 mutations in conferring MDM2 inhibitor sensitivity (the NPM1 mutation frequency in this cohort was 40% in the subset of AML with normal karyotype).
Next, we graphically displayed IC50 values for all 109 AML cases in 3 mutually exclusive categories: (1) presence of p53 exons 5-9 mutations, (2) presence of Flt3-ITD, and (3) all others (Figure 6). As can be seen in Figure 6, most Flt3-ITD-positive AML cases displayed very low IC50 values and significantly lower mean IC50 values than the Flt3 wt and p53 wt group (P = .02). Thus, the majority of AML blasts with Flt3-ITD mutations are highly sensitive to MDM2 inhibitor treatment. Therefore, this analysis identifies, for the first time, a genomic biomarker for MDM2 inhibitor sensitivity with potential for clinical applications.
Figure 6.

AML cases with mutated Flt3 (Flt3-ITD) are very sensitive to MI219. MI219 IC50 values categorized by (1) p53 mutation status, (2) presence of Flt3-ITD, and (3) all others. Differences in the mean IC50 value between Flt3-ITD+ and all other cases are significant (P = .02).
Discussion
This report summarizes detailed studies of the molecular determinants in primary AML blasts (N = 109) and their influence on sensitivity or resistance to MDM2 inhibitor treatment ex vivo. One novel finding of this study is the description and quantitative analysis of primary resistance to MDM2 inhibitors in AML. Within this context, it was demonstrated that: (1) p53 mutations confer resistance to MI219, as expected; (2) low or absent p53 expression in the absence of p53 exon 5-9 mutations exists in a subset of AML blasts and is associated with MDM2 inhibitor resistance; and (3) MDM2 inhibitor resistance exists in subsets of AML despite wild-type p53 and robust p53 protein induction after MDM2 inhibitor treatment or irradiation, thus implying defects in the apoptotic p53 network or in the p53 protein. Together, these various AML blast-intrinsic defects result in primary resistance to MDM2 inhibitors in approximately one-third of all AML cases, a fraction much larger than previously appreciated.
Regarding the p53 gene status of resistant AML blasts, multiple findings emerged: (1) the p53 exon 5-9 mutation frequency was 10%, which is in line with previous estimates,33,34 and is insufficient to explain MDM2 inhibitor resistance in the majority of AML cases; (2) p53 mutations frequently (∼ 50% of all p53 mutations) occurred in the setting of acquired uniparental disomy at 17p in AML; (3) some AML cases with 17p deletions that spanned p53 carried wild-type p53 and were sensitive to MI219; and (4) some AML cases with 17p deletions that spanned p53 lacked p53 mRNA expression and thus were resistant to MI219. Therefore, substantial combinatorial molecular diversity exists in the p53 gene status in AML with direct effects on the ability of MDM2 inhibitors to effect apoptotic AML blast death.20,35
Focusing on the resistant AML cases with wild-type p53 exons 5-9 and presence of p53 mRNA, 2 subsets emerged that displayed either: (1) low or absent p53 proteins or (2) preserved p53 protein levels. Regarding the molecular basis for low p53 protein in subsets of AML blasts, our initial analysis did not identify supportive evidence for reversible epigenetic changes. We are thus left with the possibility of a defective p53 gene (including alterations in the promoter or epigenetic changes that defied our pharmacologic attempts at reversal), impaired p53 mRNA translation, or reduced p53 protein stability that is independent of MDM2 or MDMX levels.36–42 Given the scarcity of the primary source material, this was not investigated further but raises questions regarding correlations between p53 status and p53 protein levels that should be evaluated in future studies. Regarding the AML blasts with robust induction of p53 protein after MDM2 inhibitor treatment or external irradiation, the 2 principal molecular defects are: (1) a defective p53 protein, possibly because of aberrant posttranslational modifications of p53, resulting in an altered ability of p53 to activate apoptotic signaling pathways43–45; or (2) defects in the p53-regulated apoptotic network. Regarding the latter possibility of defects in the p53 apoptotic network, it is important to note that a quantitative analysis of the relative importance of various p53 inducible genes in relation to the apoptotic response after p53 induction is not available. In the setting of the treatment of cells with the low-molecular-weight compound RITA (but not Nutlin treatment), it has recently been demonstrated that p21 down-regulation provides a switch between p53-induced apoptosis and cell cycle arrest.46 Our analysis of p21 protein levels before and after Nutlin or MI219 treatment also does not provide clear evidence for a unique role of p21 in AML blast fate decision after MDM2 inhibitor treatment. The initial analysis of gene expression changes in sensitive versus resistant blasts after treatment with MI219 (not shown) identified differential induction of a subset of classic p53-responsive genes, but interpretation of these data is hampered by the fact that the critical genes with importance to p53-mediated apoptosis in leukemia are not known. It is thus possible that genes other than classic p53-responsive genes are involved in conferring resistance to MDM2 inhibitors, and future analysis of such genes is of importance for a comprehensive understanding of MDM2 inhibitor effects on myeloid leukemia blasts.
One of the novel results from this analysis was the identification of AML blasts that were very sensitive to MI219 ex vivo. Approximately one-third of AML cases displayed IC50 values to MI219 of less than 2μM. Attempts at identification of determinants of such heightened sensitivity uncovered frequent and significant association with mutated Flt3 (presence of Flt3-ITDs). For instance, 58% and 84% of all AML samples with Flt3-ITD (N = 19) displayed MI219 IC50 values of less than 2μM and less than 5μM, respectively. Therefore, activated Flt3 appears to sensitize AML blasts to MDM2 inhibitor-mediated apoptosis, a finding that could be exploited for clinical applications of MDM2 inhibitors in AML.
Regarding the direct clinical implications of these findings for clinical trials with MDM2 inhibitors in AML, we offer the following conclusions: (1) p53 mutations confer absolute resistance to MDM2 inhibitors in AML; therefore, patients with p53 mutations should be excluded from such trials; (2) defective p53-mediated apoptosis is substantially more common in AML than p53 mutations; (3) 17p deletions (spanning p53) as detected through karyotyping in AML (or AML-fluorescence in situ hybridization as inferred from the 17p anatomy described here) capture only a small subset of p53 mutated cases; (4) complex karyotype is a good predictor of underlying p53 mutations but identifies only a subset of the total number of AML cases with primary MDM2 inhibitor resistance; (5) a subset of normal karyotype AML cases are absolutely resistant to MDM2 inhibitors; (6) the presence of FLT3-ITDs identifies a subset of AML for which the majority are very sensitive to MDM2 inhibitors (but many additional sensitive AML cases lack such a predictive molecular marker); and (7) no currently used clinical test is able to identify primary MDM2 inhibitor resistance in AML with high sensitivity or specificity. Based on these observations, one may therefore propose that ex vivo testing of purified AML blasts from patients may offer clinical benefits in the setting of MDM2 inhibitor treatment, and such testing may be incorporated into clinical trial designs.
In conclusion, this unique dataset, based on the analysis of more than 100 primary AML cases, quantitatively describes primary resistance to MDM2 inhibitors in AML and provides evidence for multiple distinct molecular mechanisms. These unexpected findings thus substantially complicate transition of MDM2 inhibitors into AML therapy and provide the rationale for further in-depth preclinical studies of resistance mechanisms in AML. Our studies also provide an additional, albeit incomplete, rationale for combination-targeted therapy in AML with primary resistance to MDM2 inhibitors in an attempt to overcome resistance.47 Conversely, our novel finding of an association of mutated Flt3 (Flt-ITD) and heightened sensitivity to MDM2 inhibitors was not expected based on published findings48 and may be important to AML therapy as: (1) Flt3-ITD AML cases tend to have short remission durations, (2) therapeutic blockage of Flt3 using Flt3 inhibitor monotherapy has not yet resulted in substantial clinical benefits to patients, and (3) application of MDM2 inhibitors to AML with mutated Flt3 and intact p53 may offer clinical benefits.49 This novel description of a genomic biomarker for MDM2 inhibitor sensitivity thus introduces the concept of “MDM2 inhibitor sensitizer gene mutations” and justifies ongoing searches for additional genes with similar effects. Finally, such MDM2 inhibitor sensitizer mutations may ultimately also offer explanations for the heightened sensitivity of neoplastic cells to MDM2 inhibitor treatment.
Supplementary Material
Acknowledgments
The authors thank the microarray core of the University of Michigan Comprehensive Cancer Center for services provided.
This work was supported by the National Institutes of Health (1R01 CA136537-01) (S.N.M.) and the Translational Research Program of the Leukemia and Lymphoma Society of America (S.N.M.), in part by the National Institutes of Health through the University of Michigan's Cancer Center (support grant 5 P30 CA46592), and in part through the Michigan Institute for Clinical and Health Research (translational grant UL1RR024986).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.L., P.O., and S.N.M. performed the laboratory research; B.P., H.E., D.B., and S.N.M. enrolled patients and contributed and analyzed clinical data; K.S. assisted with statistical analysis; S.N.M. and S.W. conceived the study and supervised the work; and J.L., P.O., S.W., and S.N.M. wrote the paper.
Conflict-of-interest disclosure: S.W. is a shareholder and founder of and a consultant to Ascenta Therapeutics Inc, and the principal investigator of a research contract between Ascenta Therapeutics Inc and the University of Michigan. The remaining authors declare no competing financial interests.
Correspondence: Sami N. Malek, Department of Internal Medicine, Division of Hematology and Oncology, University of Michigan, 1500 E Medical Center Dr, Ann Arbor, MI 48109-0936; e-mail: smalek@med.umich.edu; and Shaomeng Wang, Department of Internal Medicine, Division of Hematology and Oncology, University of Michigan, 1500 E Medical Center Dr, Ann Arbor, MI 48109-0936; e-mail: shaomeng@med.umich.edu.
References
- 1.Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105(10):3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13(1):23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 4.Ding K, Lu Y, Nikolovska-Coleska Z, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49(12):3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- 5.Shangary S, Wang S. Targeting the MDM2-p53 interaction for cancer therapy. Clin Cancer Res. 2008;14(17):5318–5324. doi: 10.1158/1078-0432.CCR-07-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137(4):609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387(6630):296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 8.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387(6630):299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 9.Kussie PH, Gorina S, Marechal V, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274(5289):948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 10.Secchiero P, Barbarotto E, Tiribelli M, et al. Functional integrity of the p53-mediated apoptotic pathway induced by the non-genotoxic agent nutlin-3a in B-cell chronic lymphocytic leukemia (B-CLL). Blood. 2006;107(10):4122–4129. doi: 10.1182/blood-2005-11-4465. [DOI] [PubMed] [Google Scholar]
- 11.Stuhmer T, Chatterjee M, Hildebrandt M, et al. Nongenotoxic activation of the p53 pathway as a therapeutic strategy for multiple myeloma. Blood. 2005;106(10):3609–3617. doi: 10.1182/blood-2005-04-1489. [DOI] [PubMed] [Google Scholar]
- 12.Tovar C, Rosinski J, Filipovic Z, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103(6):1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9(10):749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302(5647):1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 15.Shibue T, Takeda K, Oda E, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17(18):2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaseva AV, Marchenko ND, Moll UM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle. 2009;8(11):1711–1719. doi: 10.4161/cc.8.11.8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morselli E, Galluzzi L, Kepp O, Kroemer G. Nutlin kills cancer cells via mitochondrial p53. Cell Cycle. 2009;8(11):1647–1648. [PubMed] [Google Scholar]
- 18.Du W, Wu J, Walsh EM, Zhang Y, Chen CY, Xiao ZX. Nutlin-3 affects expression and function of retinoblastoma protein: role of retinoblastoma protein in cellular response to nutlin-3. J Biol Chem. 2009;284(39):26315–26321. doi: 10.1074/jbc.M109.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kojima K, Konopleva M, McQueen T, O'Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108(3):993–1000. doi: 10.1182/blood-2005-12-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima K, Konopleva M, Samudio IJ, et al. MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood. 2005;106(9):3150–3159. doi: 10.1182/blood-2005-02-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137(3):413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 22.Saddler C, Ouillette P, Kujawski L, et al. Comprehensive biomarker and genomic analysis identifies p53 status as the major determinant of response to MDM2 inhibitors in chronic lymphocytic leukemia. Blood. 2008;111(3):1584–1593. doi: 10.1182/blood-2007-09-112698. [DOI] [PubMed] [Google Scholar]
- 23.Coll-Mulet L, Iglesias-Serret D, Santidrian AF, et al. MDM2 antagonists activate p53 and synergize with genotoxic drugs in B-cell chronic lymphocytic leukemia cells. Blood. 2006;107(10):4109–4114. doi: 10.1182/blood-2005-08-3273. [DOI] [PubMed] [Google Scholar]
- 24.Secchiero P, Melloni E, di Iasio MG, et al. Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood. 2009;113(18):4300–4308. doi: 10.1182/blood-2008-11-187708. [DOI] [PubMed] [Google Scholar]
- 25.Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F-1 transcriptional activity is a critical determinant of Mdm2 antagonist-induced apoptosis in human tumor cell lines. Oncogene. 2008;27(40):5303–5314. doi: 10.1038/onc.2008.164. [DOI] [PubMed] [Google Scholar]
- 26.Kitagawa M, Lee SH, McCormick F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol Cell. 2008;29(2):217–231. doi: 10.1016/j.molcel.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 27.Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444(7115):61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- 28.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281(44):33030–33035. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 29.Francoz S, Froment P, Bogaerts S, et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci U S A. 2006;103(9):3232–3237. doi: 10.1073/pnas.0508476103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borowitz MJ, Guenther KL, Shults KE, Stelzer GT. Immunophenotyping of acute leukemia by flow cytometric analysis: use of CD45 and right-angle light scatter to gate on leukemic blasts in three-color analysis. Am J Clin Pathol. 1993;100(5):534–540. doi: 10.1093/ajcp/100.5.534. [DOI] [PubMed] [Google Scholar]
- 31.Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20(8):1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 32.Ross CW, Ouillette PD, Saddler CM, Shedden KA, Malek SN. Comprehensive analysis of copy number and allele status identifies multiple chromosome defects underlying follicular lymphoma pathogenesis. Clin Cancer Res. 2007;13(16):4777–4785. doi: 10.1158/1078-0432.CCR-07-0456. [DOI] [PubMed] [Google Scholar]
- 33.Fenaux P, Jonveaux P, Quiquandon I, et al. p53 gene mutations in acute myeloid leukemia with 17p monosomy. Blood. 1991;78(7):1652–1657. [PubMed] [Google Scholar]
- 34.Stirewalt DL, Kopecky KJ, Meshinchi S, et al. FLT3, RAS, and Tp53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97(11):3589–3595. doi: 10.1182/blood.v97.11.3589. [DOI] [PubMed] [Google Scholar]
- 35.Seifert H, Mohr B, Thiede C, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009;23(4):656–663. doi: 10.1038/leu.2008.375. [DOI] [PubMed] [Google Scholar]
- 36.Naski N, Gajjar M, Bourougaa K, Malbert-Colas L, Fahraeus R, Candeias MM. The p53 mRNA-Mdm2 interaction. Cell Cycle. 2009;8(1):31–34. doi: 10.4161/cc.8.1.7326. [DOI] [PubMed] [Google Scholar]
- 37.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32(2):180–189. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees JA. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc Natl Acad Sci U S A. 2008;105(30):10408–10413. doi: 10.1073/pnas.0805036105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123(1):49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 40.Asher G, Lotem J, Kama R, Sachs L, Shaul Y. NQO1 stabilizes p53 through a distinct pathway. Proc Natl Acad Sci U S A. 2002;99(5):3099–3104. doi: 10.1073/pnas.052706799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112(6):779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 42.Dornan D, Wertz I, Shimizu H, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429(6987):86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 43.Knights CD, Catania J, Di Giovanni S, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol. 2006;173(4):533–544. doi: 10.1083/jcb.200512059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Giovanni S, Knights CD, Rao M, et al. The tumor suppressor protein p53 is required for neurite outgrowth and axon regeneration. EMBO J. 2006;25(17):4084–4096. doi: 10.1038/sj.emboj.7601292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9(9):702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 46.Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell. 2009;15(3):171–183. doi: 10.1016/j.ccr.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 47.Kojima K, Shimanuki M, Shikami M, et al. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia. 2008;22(9):1728–1736. doi: 10.1038/leu.2008.158. [DOI] [PubMed] [Google Scholar]
- 48.Kojima K, Konopleva M, Tsao T, et al. Selective FLT3 inhibitor FI-700 neutralizes Mcl-1 and enhances p53-mediated apoptosis in AML cells with activating mutations of FLT3 through Mcl-1/Noxa axis. Leukemia. 2010;24(1):33–43. doi: 10.1038/leu.2009.212. [DOI] [PubMed] [Google Scholar]
- 49.Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters. An analysis of 3082 patients. Blood. 2008;111(5):2527–2537. doi: 10.1182/blood-2007-05-091215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}