

Abstract
Chemically modified nucleic acids (CNAs) are widely explored as antisense oligonucleotide or small interfering RNA (siRNA) candidates for therapeutic applications. CNAs are also of interest in diagnostics, high-throughput genomics and target validation, nanotechnology and as model systems in investigations directed at a better understanding of the etiology of nucleic acid structure as well as the physical-chemical and pairing properties of DNA and RNA and for probing protein-nucleic acid interactions. In this article we review research conducted by our laboratory over the past two decades with a focus on crystal structure analyses of CNAs and artificial pairing systems. We highlight key insights into issues ranging from conformational distortions as a consequence of modification to the modulation of pairing strength and RNA affinity by stereoelectronic effects and hydration. Although crystal structures have only been determined for a subset of the large number of modifications that were synthesized and analyzed in the oligonucleotide context to date, they have yielded guiding principles for the design of new analogs with tailormade properties, including pairing specificity, nuclease resistance and cellular uptake. And, perhaps less obviously, crystallographic studies of CNAs and synthetic pairing systems have shed light on fundamental aspects of DNA and RNA structure and function that would not have been disclosed by investigations solely focused on the natural nucleic acids.
1. Introduction
The demonstration that synthetic oligonucleotides could be used to interfere with biological information transfer [1,2] spawned an intense interest in chemically modified nucleic acids (CNAs) in the early 1990s. Chemical modification was expected to benefit so-called antisense oligonucleotides (AONs) in multiple ways, i.e. protect them against degradation by cellular exo- and endonucleases, enhance their affinity for mRNA targets and improve uptake and possibly pharmacokinetics and pharmacodynamics [3–10]. Among the first generation modifications, phosphorothioate DNAs (PS-DNAs, Fig. 1) were widely pursued as AON candidates with anti-cancer, -viral and -inflammatory indications [11–14]. But PS-DNAs ultimately showed several limitations with regard to in vivo therapeutic applications [15,16], and only a single phosphorothioate-based drug received approval by the US Food and Drug Administration to date (Vitravene® in 1998 [17]). Around the time that synthetic efforts to chemically modify oligonucleotides gained momentum, one of us (M.E., 1989) joined the laboratory of Alexander Rich at MIT to pursue postdoctoral studies. By then the determination of crystal structures of DNA oligomers was well underway and dozens of structures of A-, B- and Z-form duplexes and DNA-drug complexes had been reported [18–28]. However, structures of oligonucleotides that featured a chemical modification were rare and included Br5C [21,22], base methylation (Me5C [23,24] or N6-MeA [25]), 2-amino-A [26] and a DNA hexamer with an alternating RP-phosphorothioate/phosphate backbone [27]. Moreover, only two crystal structures of RNA oligonucleotides had been reported by 1989 [28–30]. The only chemically modified DNA ultimately crystallized during those postdoctoral years was a hexamer with a central RP TpsA phosphorothioate step in complex with the anticancer drugs 11-deoxydaunomycin [31] and nogalamycin [32]. This oligonucleotide had been provided by the laboratory of Jacques H. van Boom at Leiden University [33]. However, one area of interest at the time that is clearly of relevance for the structure and function of synthetic analogs of nucleic acids concerned the conformational differences between DNA and RNA and the structure of chimeric DNA-RNA oligonucleotides. More systematic studies of the structures of CNAs were subsequently begun after the return of the first author to the ETH in Zürich as part of independent research carried out in the Laboratory for Organic Chemistry. The present contribution reviews insights into the structure and function of CNAs gained from crystallographic analyses conducted in the Egli laboratory and spanning almost two decades (Table).
Fig. 1.

Structures of DNA, RNA and phosphorothioate DNA (PS-DNA).
Table.
Overview of Crystal Structures of Chemically Modified Nucleic Acids Determined in the Egli Laboratorya)
| NDB codeb) | Modification | No. of mods. | Length | Context | Duplex form | Ref. |
|---|---|---|---|---|---|---|
| AD0007 | 2′-O-methyl-3′-methylenephosphonate-T (MP) | 1 | 10 | DNA | A | [50] |
| AD0011 | 2′-O-aminooxyethyl-T (AOE), Ba2+ form | 1 | 10 | DNA | A | [51] |
| AD0012 | 2′-O-fluoroethyl-T (FE), medium Na + form | 1 | 10 | DNA | A | [51] |
| AD0013 | 2′-O-methyl-3′-methylenephosphonate-T (MP), high Cs+ form | 1 | 10 | DNA | A | [51] |
| AD0014 | 2′-O-methyl-[tri(oxyethyl)]-T (TOE), medium Cs+ form | 1 | 10 | DNA | A | [51] |
| AD0015 | 2′-O-fluoroethyl-T (FE), high Rb+ form | 1 | 10 | DNA | A | [51] |
| AD0016 | 2′-O-methyl-[tri(oxyethyl)]-T (TOE), medium Rb+ form | 1 | 10 | DNA | A | [51] |
| AD0017 | 2′-O-methyl-3′-methylenephosphonate-T (MP), high K+ form | 1 | 10 | DNA | A | [51] |
| AD0018 | 2′-O-methyl-3′-methylenephosphonate-T (MP), medi. K+ form | 1 | 10 | DNA | A | [51] |
| AD0019 | 2′-O-methyl-3′-methylenephosphonate-T (MP), medi. Na+ form | 1 | 10 | DNA | A | [51] |
| AD0020 | 2′-O-4′-C-methylene-T (LNA) | 1 | 10 | DNA | A | [83] |
| AD0025 | guanyl G-clamp (C analog) | 1 | 10 | DNA | A | [133] |
| AD0027 | 2′-Se-methyl-U | 1 | 10 | DNA | A | [84,85] |
| AD0028 | 2′-O-[2-(methylthio)ethyl]-T (MTE) | 1 | 10 | DNA | A | [81] |
| AD0029 | 2′-O-[2-[2-(N,N-dimethylamino)ethoxy]ethyl]-T (DMAEOE) | 1 | 10 | DNA | A | [80] |
| AD0030 | (L)-α-threofuranosyl (3′-2′) T (TNA) | 1 | 10 | DNA | A | [122] |
| AD0031 | 2′-O-[2-(guanidinium)ethyl]-T (GE) | 1 | 10 | DNA | A | [79] |
| AD0034 | 2′-O-[2-[hydroxy(methyleneamino)oxy]ethyl]-T (AOE) | 1 | 10 | DNA | A | [77] |
| AD0035 | 2′-O-butyl-T (BTL), Mg2+ form | 1 | 10 | DNA | A | [77] |
| AD0036 | 2′-O-butyl-T (BTL), Sr2+ form | 1 | 10 | DNA | A | [77] |
| AD0037 | 2′-O-[2-(imidazolyl)ethyl]-T (IME) | 1 | 10 | DNA | A | [77] |
| AD0038 | 2′-O-[2-(fluoro)ethyl]-T (FE) | 1 | 10 | DNA | A | [77] |
| AD0039 | 2′-O-allyl-T (ALY) | 1 | 10 | DNA | A | [77] |
| AD0040 | 2′-O-propargyl-T (PRG) | 1 | 10 | DNA | A | [77] |
| AD0041 | 2′-O-[2-(trifluoro)ethyl]-T (TFE) | 1 | 10 | DNA | A | [77] |
| AD0042 | 2′-O-propyl-T (PRL) | 1 | 10 | DNA | A | [77] |
| AD0043 | 2′-O-[2-(N,N-dimethylaminooxy)ethyl]-T (DMAOE) | 1 | 10 | DNA | A | [77] |
| AD0044 | 2′-O-[2-(benzyloxy)ethyl]-T (BOE) | 1 | 10 | DNA | A | [77] |
| AD0057 | 2′-O-[2-(methoxy)ethyl]-2-thio-T (MOE) | 1 | 10 | DNA | A | [135] |
| AD0060 | 2′-O-methyl-A/2′-arabino-U | 2 | 10 | DNA | A | [91] |
| AD0061 | 2′-O-methyl-A/2′-deoxy-2′-fluoroarabino-T | 2 | 10 | DNA | A | [91] |
| ADLS105 | N3′→ P5′ phosporamidate DNA (NP-DNA) | 12 | 12 | — | A | [129] |
| AH0002 | 2′-O-(ethoxymethyl)-T (EOM) | 1 | 10 | DNA | A | [74] |
| AH0003 | 2′-O-[2-(methoxy)ethyl]-T (MOE) | 1 | 10 | DNA | A | [74] |
| AH0004 | 2′-O-methyl-[tri(oxyethyl)]-T (TOE) | 1 | 10 | DNA | A | [74] |
| AH0006 | 2′-O-(3-aminopropyl)-U (AP) | 1 | 10 | DNA | A | [78] |
| AH0021 | five-atom amide-based backbone modifications (TamT dimer) | 1 | 9 | DNA:RNA | A | [132] |
| AHJS55 | 2′-O-methyl-A | 1 | 10 | DNA | A | [44] |
| AR0015 | 2′-O-[2-(methoxy)ethyl)]-RNA (MOE, pH 6.2, 50 mM Mg2+, 120K) | 12 | 12 | — | A | [75] |
| AR0016 | 2′-O-[2-(methoxy)ethyl)]-RNA (MOE, pH 7.5, 50 mM Mg2+, 120K) | 12 | 12 | — | A | [75] |
| AR0017 | 2′-O-[2-(methoxy)ethyl)]-RNA (MOE, pH 7.5, 100 mM Mg2+, 120K) | 12 | 12 | — | A | [75] |
| AR0018 | 2′-O-[2-(methoxy)ethyl)]-RNA (MOE, pH 7.5, 100 mM Mg2+, rt) | 12 | 12 | — | A | [75] |
| AR0030 | phenyl-ribonucleotide | 1 | 8 | RNA | A | [137] |
| AR0063 | 4′-S-ribo-C | 1 | 8 | RNA | A | [92] |
| AR0068 | 2,4-difluorotoluyl-ribonucleotide (rDFT; rDFT:A pairs) | 1 | 12 | RNA | A | [140] |
| AR0080 | 2,4-difluorotoluyl-ribonucleotide (rDFT; rDFT:G pairs) | 1 | 12 | RNA | A | [141] |
| AR0097 | N2,N2-dimethyl-G (m22 G; m22G:A pairs) | 1 | 13 | RNA | A | [143] |
| BD0020 | 2′-deoxy-2′-fluoroarabino-T (2.2 Å resolution, Ca2+ form) | 1 | 12 | DNA | B | [49] |
| BD0030 | 2′-deoxy-2′-fluoroarabino-T (0.95 Å resolution, Mg2+ form) | 1 | 12 | DNA | B | [50] |
| BD0038 | [3.3.0]bicyclo-arabino-T | 1 | 12 | DNA | B | [89] |
| BD0060 | (L)-α-threofuranosyl (3′-2′) T (TNA) | 1 | 12 | DNA | B | [121] |
| BD0091 | 2′-arabino-U (T7 replacement; CGCGAAUTCGCG) | 1 | 12 | DNA | B | [91] |
| BD0092 | 2′-arabino-U (T8 replacement; CGCGAATUCGCG) | 1 | 12 | DNA | B | [91] |
| BD0104 | tricyclo-dA (tcDNA) | 1 | 12 | DNA | B | [106] |
| BDLB84 | 2′-deoxy-2′-fluoroarabino-T | 2 | 12 | DNA | B | [87] |
| BDLB85 | 2′-deoxy-2′-fluoroarabino-T (1.55 Å resolution, Mg2+ form) | 1 | 12 | DNA | B | [87] |
| BDLS79 | 2′-deoxy-6′-α-methyl-carbocyclic-T | 2 | 12 | DNA | B | [54] |
| BDLS80 | 2′-deoxy-6′-α-hydroxy-carbocyclic-T | 2 | 12 | DNA | B | [54] |
| DD0019 | trans-stilbenediether base-pair analog (3.2 Å resol., Sr2+ form) | 1 | 6 | DNA | B | [138] |
| DD0058 | trans-stilbenediether base-pair analog (1.5 Å resol., Mg2+ form) | 1 | 6 | DNA | B | [139] |
| PD0080 | E. coli DNA polI Klenow:2′-O-(3-aminopropyl)-U (AP) | 1 | 4 | DNA | single strand | [78] |
| PR0016 | E. coli DNA polI Klenow:2′-O-(3-aminopropyl)-A/U/C (AP) | 3 | 6 | DNA | single strand | [78] |
| PD1139c) | B. halodurans RNase H: 12mer DNA duplex | - | 12 | DNA | B | [90] |
| UD0056 | 2′-O-(N-methylcarbamate)-T (NMC) | 1 | 10 | DNA | A | [76] |
| UD0057 | 2′-O-[2-(methylamino)-2-oxoethyl]-T (NMA) | 1 | 10 | DNA | A | [76] |
| UD0070 | (6′→4′) oligo-(2′,3′-dideoxyglucopyranosyl) n.a. (homo-DNA) | 8 | 8 | — | — | [111] |
| UDBS38 | 2′-deoxycytidylyl-(3′-5′)-2′-deoxycytidine with intra-nucleo-sidyl C(3′)-C(5′) ethylene bridges (bicyclo DNA, bcDNA) | 2 | 2 | — | parallel-stranded | [101] |
| URBP33 | dimethylenesulfone-linked RNA analog [r(Gso2C)]2 | 2 | 2 | — | A-like | [123] |
| ZD0015 | phosphoroselenoate-DNA (PSe-DNA) | 1 | 6 | DNA | Z | [113] |
| 3EY2d) | 2′-S-methyl-U | 1 | 10 | DNA | A | [82] |
| 3EY3d) | 2′-S-methyl-U | 2 | 12 | DNA | intermediate A/B | [82] |
| 3EY1d) | B. halodurans RNase H: 12mer DNA with 2′-S-methyl-U | 2 | 12 | DNA | intermediate A/B | [82] |
| 2VA2d) | S. solfataricus Dpo4: DNA complex, 2,4-difluorotoluyl-2′-deoxy-ribonucleotide (DFT; DFT:A pair) | 1 | 18/13 | DNA/ddCTP | post-insertion | [142] |
| 2V9Wd) | S. solfataricus Dpo4: DNA complex, 2,4-difluorotoluyl-2′-deoxy-ribonucleotide (DFT; DFT:A pair) | 1 | 18/13 | DNA/dGTP | post-insertion | [142] |
| 2VA3d) | S. solfataricus Dpo4: DNA complex, 2,4-difluorotoluyl-2′-deoxy-ribonucleotide (DFT; DFT:G pair) | 1 | 18/13 | DNA/ddCTP | post-insertion | [142] |
Several other structures of CNAs have been determined in the context of an analysis of damage bypass by DNA trans-lesion polymerases; these are not listed here
Nucleic Acid Database [30]: http://ndbserver.rutgers.edu
Complex with native DNA duplex; complexes with chemically modified DNAs were determined, i.e. 2′-S-methyl-U, but some have not been deposited yet
PDB ID codes: http://www.rcsb.org
2. Nucleic Acid Structure – A Tale of Two Puckers
The 2′-deoxyribose in DNA is intrinsically flexible and the sugar is therefore not restricted to a particular conformational state (Fig. 2). Among the many conformations (puckers) that are possible, two, referred to as C2′-endo (South) and C3′-endo (North) are frequently observed and give rise to the A- and B-forms, respectively, of the DNA double helix [34–36] (Fig. 3). The presence of the ribose 2′-hydroxyl group affects the sugar conformational equilibrium through altered stereoelectronic and steric effects [37] and limits the ribose pucker to the Northern region (C3′-endo). Therefore, RNA duplexes adopt the A-form and a switch to the B-form is prevented by steric conflicts between the 2′-OH and both the 3′-phosphate group and the adjacent nucleobase.
Fig. 2.

The pseudo-rotation phase angle (P) cycle and conformations of the sugar moiety. The 4′-oxygen is highlighted as a red sphere.
Fig. 3.

Idealized, diamond lattice models of (a) RNA with the sugar adopting a C3′-endo pucker (A-DNA similar) and (b) DNA with the sugar adopting a C2′-endo pucker. Space filling models of (c) A- and (d) B-form DNA duplexes.
We were interested in the conformational properties of chimeric DNA-RNA oligonucleotides and whether the two nucleic acid species would coexist and both adopt their preferred conformation or whether RNA would dominate DNA in terms of conformation. In collaboration with Nassim Usman we prepared a series of mixed DNA-RNA sequences using the 2′-t-butyl dimethylsilyl protection group protocol for RNA phosphoramidites in conjunction with solid phase synthesis [38]. Crystal structures of chimeric decamer duplexes, in some cases featuring just one ribonucleotide per strand [i.e. rGd(CGTATACGC) or dGrCd(GTAATCGC)], all displayed a canonical A-form conformation [39]. Although a particular DNA sequence (i.e. those rich in C:G pairs) may have an intrinsic preference to adopt the A-conformation, we concluded that it was possible for a single RNA nucleotide to convert the entire DNA sequence to the A-form. Indeed, work carried out in Muttaiya Sundaralingam’s laboratory later confirmed this observation by demonstrating that DNA decamer duplexes crystallized in the B-form whereas oligomers of identical sequence but containing a single ribonucleotide adopted the A-form in the crystal [40,41]. Moreover, a so-called Okazaki fragment, a chimeric RNA-DNA paired to DNA also assumed an A-conformation in the crystal [42], providing further evidence that RNA appears capable of controlling the conformation of several neighboring 2′-deoxyribonucleotides [42]. Despite these results, there remains the possibility that the interplay of sequence, crystal packing and dehydration can influence the conformational equilibrium, for example, by favoring the A- over the B-type duplex conformation. Interestingly, all DNA:RNA hybrids crystallized in the absence of a protein (i.e. RNase H) adopt a more or less canonical A-form duplex. This supports the above notion that crystals appear to trap an RNA-like conformation and that we may thus miss more subtle conformational features of chimeric DNA-RNA oligonucleotides or DNA:RNA hybrids seen in solution structures (i.e. ref. [43]). We shall revisit this issue in subsequent sections on 2′-modified analogs and the recognition of substrates by RNase H.
3. Template Sequences for Crystallization
The DNA decamer d(GCGTATACGC) turned out to be useful as a crystallization template for studying conformation and hydration of modified ribonucleotides and many other CNAs. Residues at various sites in the decamer can be replaced by modified nucleotides whose conformation and hydration can then by analyzed in the A-form context [44–46]. We also relied on other template sequences, among them the so-called Dickerson-Drew Dodecamer (DDD [45,47]) with sequence d(CGCGAATTCGCG) to analyze numerous nucleic acid modifications, both 2′-deoxyribonucleotide and ribonucleotide analogs. Ideally, template sequences yield crystals of the native and modified forms that diffract to high resolution, allowing detailed insights into the water structure and the coordination sites and modes of mono- and divalent metal cations and the polyamine spermine [48–56] (Fig. 4, Molecular graphics images were produced using the UCSF Chimera package [57]). Gleaning the conformational perturbations potentially induced by chemical modification is the primary goal of a crystal structure determination, but changes that concern water structure or the ionic environment of a nucleic acid fragment also need to be taken into account in order to correlate structure and stability of a CNA in a meaningful way [58].
Fig. 4.

Space filling models of DDD duplexes with thymidines replaced by (a) 2′-deoxy-6′-α-hydroxy- and (b) 2′-deoxy-6′-α-methyl-carbocyclic Ts. The views are into the central portion of the minor groove and carbon, oxygen, nitrogen and phosphorus atoms are colored gray, red, blue and orange, respectively. The exocyclic O6′ hydroxyl oxygen and C7′ methyl carbon atoms are highlighted in magenta and green, respectively, and water molecules are depicted as cyan spheres. Note the differences in the minor groove width in the two duplexes; to accommodate the four methyl groups the minor groove is expanded in the duplex on the right and a water molecule is trapped between them.
4. RNA Hydration and Pairing Stability
RNA duplexes exhibit higher thermodynamic stability than the corresponding DNA duplexes of the same sequence. The key difference between DNA and RNA is the 2′-hydroxyl group in the latter that can act both as a hydrogen bond donor and acceptor. We used the crystal structure of the RNA octamer duplex [r(CCCCGGGG)]2 at 1.45 Å resolution to visualize the water structure and to assess the potentially stabilizing role of the ribose 2′-OH moieties [59,60]. The water structure enveloping the octamer duplex is remarkably regular and clear patterns emerge in the major and minor grooves and around individual 2′-hydroxyl groups (Fig. 5). Each hydroxyl group establishes four contacts to water molecules, resulting in clusters that are either wedged between the ribose and the nucleobase or the ribose and the phosphate, or located above the 2′-OH’s sugar ring or near the 3′-adjacent ribose (Fig. 5a). Two bands of pentagons wind down on both sides of the major groove, whereby water molecules that are either hydrogen bonded to N4(C), N7(G) or phosphate oxygens occupy four corners of individual pentagons (these are fused along one edge), with a phosphate oxygen positioned at the fifth (Fig 5b). In the central part of the groove, water molecules bridge O6 keto groups from adjacent Gs. In line with the relatively tight spacing of phosphates in an A-form duplex - the average intra-strand P···P distance amounts to ca. 6 Å - neighboring phosphates are linked by a single water molecule on the border of the major groove. In the minor groove, 2′-hydroxyl groups serve as bridgeheads for water tandems that cross the groove, whereby waters form a hydrogen bond to either O2(C) or N3(G) (Fig. 5c). The ordered water networks encountered in both grooves and involving ribose 2′-hydroxyls and phosphates would be expected to affect the thermodynamic parameters associated with pairing. Indeed, UV melting experiments demonstrated that the Tm of the [r(CCCCGGGG)]2 duplex is significantly higher than that of the corresponding DNA duplex [d(CCCCGGGG)]2. More importantly, the increased stability relative to DNA is largely due to the enthalpic contribution whereas the entropic contribution is unfavorable [45,60]. This simple example demonstrates convincingly the importance of water in the pairing stability of nucleic acids and that a seemingly minor difference (2′-OH vs. 2′-H) can result in a profound difference in the pairing affinities of RNA and DNA. Because AONs and siRNAs are both targeted against RNA and a high target affinity is considered beneficial, the potential effects a modification on the hydration need to be taken into account when designing a CNA or interpreting its stability and activity in vitro and/or in vivo.
Fig. 5.
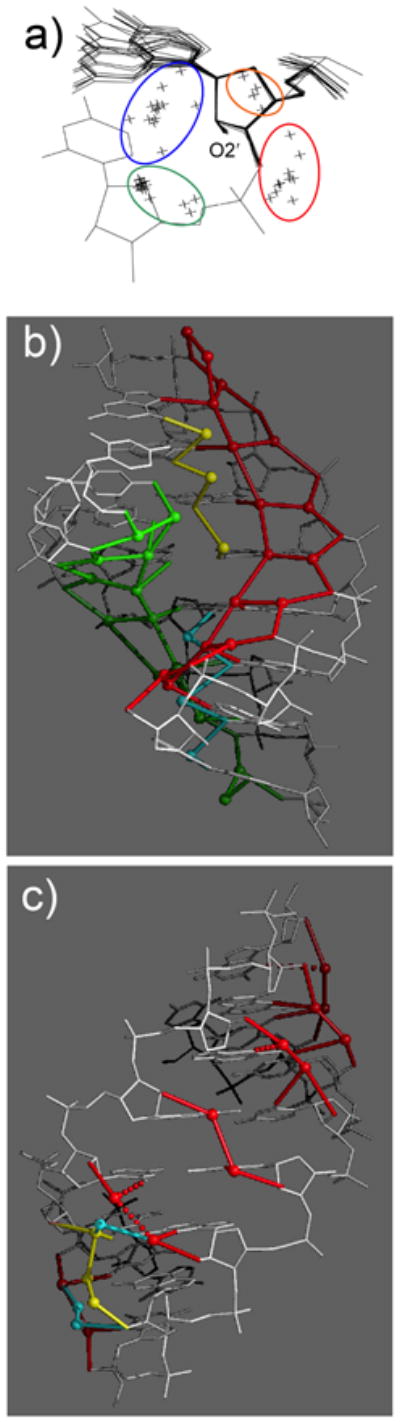
RNA hydration (adapted from [60]). (a) Superimposition of 16 nucleotides from the crystal structure of [r(CCCCGGGG)]2 reveals four groups of water molecules (crosses, highlighted with ellipsoids) around ribose 2′-hydroxyl groups. The blue group (water molecules linking O2′ to N3(G) or O2(C)), the red group (water molecules linking O2′ to phosphates) and the green group (water molecules located in the directed of the 3′-adjacent ribose) together with the O2′-C2′ bond form approximate tetrahedral geometries. (b) Major groove hydration; fused ribbons of pentagons involving four water molecules and a phosphate oxygen are highlighted in green and red and water molecules bridging O6 from adjacent guanines are highlighted in yellow. (c) Minor groove hydration; formation of tandem-water bridges across the groove whereby 2′-hydroxyl groups serve as bridgeheads. A more irregular pattern in the lower half of the duplex (spheres highlighted in cyan and yellow) arises as a consequence of inter-duplex lattice contacts.
5. 2′-Carbohydrate Modifications
Modifications at the 2′-position of the sugar have been extensively investigated [16,61,62] and their synthetic preparation is relatively straightforward in most cases. A large subgroup of analogs comprises those with 2′-O-modifications (Fig. 6) and the structure and function of many have been analyzed in great detail (Table). These compounds are attractive for antisense purposes because 2′-substitution preorganizes the oligonucleotide for the RNA target thanks to stabilizing a C3′-endo sugar conformation. Particular substitutions can modulate RNA affinity, nuclease resistance and uptake via electrostatics (i.e. positively charged moieties), hydration, hydrophobic interactions and various stereoelectronic effects. On the other hand, 2′-O-substituted oligonucleotides paired with RNA constitute inhibitors of RNase H [63,64], an endonuclease that is considered a key player in antisense activity due to its ability to degrade the RNA portion of DNA:RNA or PS-DNA:RNA hybrids [11,65–70]. This limitation can be overcome to some extent by the use of gap-mer oligonucleotides with central PS-DNA windows and 2′-O-modifications in the flanks [71].
Fig. 6.

Structures of selected 2′-O-modifications.
In an initial study we analyzed the structure of a DNA decamer duplex containing 2′-O-methyl-adenosines at medium resolution [44]. We observed that the torsion angle C3′-C2′-O2′-CH3 adopts an antiperiplanar (ap) conformation, with the methyl group directed away from the ribose and into the minor groove. The 2′-oxygen is well hydrated although water is no longer able to bridge the 2′-oxygen to the minor groove edge of the base [60] as the methyl moiety is in the way. 2′-O-Methylated RNA can be considered a ‘super’ RNA as pairing between 2′-O-methyl RNA strands is thermodynamically favorable relative to self-pairing of RNA. However, our structural data didn’t provide an obvious explanation of the enhanced thermodynamic stability of 2′-O-methyl-RNA. A subsequent crystal structure of a fully 2′-O-methylated RNA hexamer duplex at high resolution [72] in conjunction with MD simulations led to the conclusion that the methyl groups stabilize a clathrate-like water structure that may in part explain the favorable thermodynamic behavior [73]. Together with Pierre Martin and Karl-Heinz Altmann at Ciba Ldt.’s Central Research Laboratories (Basel, Switzerland) we analyzed the conformations of the 2′-O-[2-(methoxy)ethyl]-, 2′-O-methyl-[tri(oxyethyl)]- and 2′-O-(ethoxymethyl)-RNA modifications (MOE-, TOE- and EOM-RNA, respectively) [74]. Three separate crystal structures of A-form DNA decamer duplexes, each containing a different modified nucleotide of the above type per strand, demonstrated convincingly that the 2′-O-MOE and 2′-O-TOE substituents are conformationally preorganized as a result of (multiple in the case of TOE) gauche effects that govern the torsion angles of ethylene glycol moieties. In addition, the 3′-, 2′- and the substituent’s outer oxygen atom provide a stable cavity for coordination of a water molecule (MOE and TOE) that is expected to provide a favorable contribution to the RNA affinities of 2′-O-MOE- and 2′-O-TOE-RNA relative to DNA or RNA. Even the longer 2′-O-TOE substituents (10 atoms including O2′) are well ordered in the crystal structure and snake along the sugar-phosphate backbone in a 5′ to 3′ direction, evidently tied down by interactions between C-H moieties from the sugar and lone pairs of oxygen atoms from the 2′-O-substituent [74]. Moreover, the structural data also provided insight into the unfavorable pairing affinity to RNA seen with 2′-O-EOM RNA relative to the two other analogs. The methylene spacer between O2′ and the ethoxy-oxygen of the substituent does not afford an effective conformational preorganization via the gauche effect and the more flexible EOM substituent induced a shift between base pairs at the site of the modification that is consistent with a loss of stacking. In collaboration with Muthiah Manoharan, then at Isis Phamaceuticals Inc. (Carlsbad, CA), we were later able to determine crystal structures of a fully 2′-O-MOE-modified RNA dodecamer duplex and confirm the conformational and hydration features of this analog [22 out of 24 2′-O-MOE substituents exhibited a synclinal (sc+ or sc−) conformation] [75] (Fig. 7a).
Fig. 7.

Structures of the (a) 2′-O-MOE-, (b) 2′-O-NMA-, (c) 2′-O-NMC and (d) 2′-O-DMAEOE-RNA modifications. Carbon, oxygen, nitrogen and phosphorus atoms are colored green, red, blue and orange, respectively. Carbon atoms of the 2′-O-substituents are colored in gray and water molecules are shown as cyan spheres.
A further structural analysis of a pair of carbohydrate modifications, 2′-O-[2-(methylamino)-2-oxoethyl]-RNA [2′-O-(N-methylacetamide)-RNA] and 2′-O-(N-methylcarbamate)- (NMA- Fig. 7b and NMC-RNA Fig. 7c, respectively] provided a clear understanding of the origins of the drastic difference between the RNA affinities of the two analogs despite the minor difference in terms of their chemistries (an additional CH2 moiety in the NMA substituent) ([76] and cited refs.). DNA oligonucleotides with residues carrying 2′-O-NMA modifications show an increased pairing stability with RNA that amounts to +2.5°C per modification. Compared to 2′-O-NMA-modified strands, the loss in the melting temperature of the corresponding 2′-O-NMC-modified oligonucleotides paired to RNA amounts to 5°C. The structural data reveal a preorganized conformation for the NMA substituent that is compatible with an A-form duplex (Fig. 7b). The NMA keto oxygen is rotated toward the 2′- and 3′-oxygen atoms from the substituted sugar, thus trapping a water molecule between sugar and phosphate that is reminiscent of the hydration pattern encountered with 2′-O-MOE-RNA. By contrast, the NMC keto oxygen is rotated away from the ribose, which results in a short contact to the O2 of pyrimidines in the minor groove (ca. 2.8 Å) (Fig. 7c). Although the keto oxygen was found to form hydrogen bonds to water molecules, the cradle-like water binding site between sugar and substituent seen in 2′-O-MOE- and 2′-O-NMA-RNA cannot be established with NMC due to the particular orientation assumed by the keto group relative to the ribose 2′- and 3′-oxygen atoms.
In a comprehensive analysis of ten different 2′-O-RNA modifications we compared their RNA affinities, resistances against degradation by exonucleases and crystal structures [77]. This work established clear correlations between activity/stability and structure and among the trends that were revealed is the superior nuclease resistance (≫PS-DNA) afforded by ribose substituents such as 2′-O-(3-aminopropyl) (AP, [78]), 2′-O-[2-(imidazolyl)ethyl]-T (IME) and 2′-O-[2-(N,N-dimethylaminooxy)ethyl]-T (DMAOE) (Table) that are positively charged. The RNA affinity of all modifications was increased relative to DNA, but shorter and in some cases electronegative substituents afforded smaller increases compared with longer substituents (i.e. 2′-O-MOE) or those carrying a cationic charge. As expected, shorter substituents also fared worse as far as nuclease resistance was concerned, although the bulky 2′-O-[2-(benzyloxy)ethyl]-T (BOE) modification constituted an exception both in terms of its relatively low nuclease resistance and the unexpectedly high RNA affinity. We concluded that conformational preorganization and electrostatic interactions with backbone atoms could account for the robust RNA affinity exhibited by 2′-O-BOE-modified oligoribonucleotides. The surprisingly low nuclease resistance despite considerable steric bulk can be rationalized by the lack of a close association between the BOE and phosphate moieties found in the crystal structure. Conversely, the 2′-O-IME substituent is located in close vicinity of the phosphate, thus potentially presenting an electrostatic and steric obstacle for an exonuclease.
To probe the origins of the nuclease resistance exhibited by an RNA modification with a positively charged 2′-O-substituent, we cocrystallized single-stranded DNA oligonucleotides carrying one or more 2′-O-AP residues at their 3′-end with the E. coli DNA polymerase I Klenow fragment [78]. The structures revealed that the ammonium moiety of the substituent was lodged at the exonuclease active site and thereby displaced one of the two metal ions required for catalysis. A further zwitteronic analog that was evaluated in terms of RNA affinity and nuclease resistance is 2′-O-[2-(guanidinium)ethyl]-RNA (2′-O-GE-RNA). This analog shows exceptional resistance against nuclease degradation and increases in Tm/modification for duplexes and triplexes of between 2.5 and 4.1°C [79]. However, these stabilizing effects are only achieved with modified oligonucleotides that feature dispersed 2′-O-GE-RNA residues. Consecutive placement of modified residues results in a slight destabilization, presumably because of repulsions between positively charged substituents from adjacent residues in the minor groove. This limitation was overcome with the creation of the 2′-O-[2-[2-(N,N-dimethylamino)ethoxy]ethyl]-RNA modification (2′-O-DMAEOE-RNA, Fig. 7d) that combines a conformationally preorganized tether with a positively charged dimethylamino group [80]. Even antisense oligonucleotides with consecutively placed 2′-O-DMAEOE-ribonucleotides exhibit higher affinity for RNA relative to unmodified DNA reference strand (ΔTm/mod. = 1.2°C).
6. Additional 2′-Ribo Modifications
The 2′-O-[2-(methylthio)-ethyl]-RNA (2′-O-MTE-RNA) modification is a variation of 2′-O-MOE-RNA and shows enhanced binding to proteins, i.e. human serum albumin, that improves the analog’s pharmacokinetic and biodistribution properties [81]. Like 2′-O-MOE-RNA, the MTE modification provides enhanced RNA affinity relative to DNA and PS-DNA, but is inferior in terms of its nuclease resistance compared with the MOE modification. In the crystal structure of an A-form DNA duplex with 2′-O-MTE-modified thymidines, the substituents display higher flexibility than MOE moieties and a diminished hydration. The reduced conformational preorganization and the somewhat greater hydrophobicity of the MTE relative to the MOE substituent - the latter property may limit MTE’s ability to interfere with a metal ion binding site as seen in the crystal structure of the complex with 2′-O-AP-RNA lodged at the exonuclease active site of the DNA Pol I Klenow fragment - are two potential reasons for the limited ability of 2′-O-MTE-RNA to dodge exonuclease degradation.
Another 2′-carbohydrate thio-modification studied structurally is the 2′-S-methyl-RNA analog. Replacing the electronegative 2′-oxygen by sulfur could potentially reduce the conformational constraints of the furanose and alter its preference for a C3′-endo pucker. We incorporated 2′-S-methylated uridines (U*) into 10mer A-DNA and 12-mer B-DNA (DDD; CGCGAAU*U*CGCG) templates and determined their crystal structures at atomic resolution [82]. Not unexpectedly, the pucker of the modified residues was C3′-endo in the A-form environment. But in the DDD duplex that adopts a B-form conformation in the native state, the 2′-SMe-Us also preferred the C3′-endo pucker, indicating that this modification retains the intrinsic conformational preference of the ribose. The structure of the modified DDD duplex was first determined in complex with B. halodurans RNase H and the refined model was then used to phase the diffraction data from the crystal of the duplex alone. Interestingly, the duplex reveals a structural transition as a result of residues opposite U* and in the outer portions of the duplex adopting C2′-endo or C1′-exo (Southern) sugar puckers, whereas U* sugars are all in the Northern conformation as pointed out above (Fig. 8). The sugars of cytidines located 3′-adjacent to the two U* residues in each strand are also flipped to the C3′-endo state. As a consequence of the opposing puckers of paired residues in the center of the modified DDD, the usually narrow A-tract (AATT) minor groove is widened by ca. 5 Å on average, and the bulky 2′-SMe substituents are thus easily accommodated.
Fig. 8.
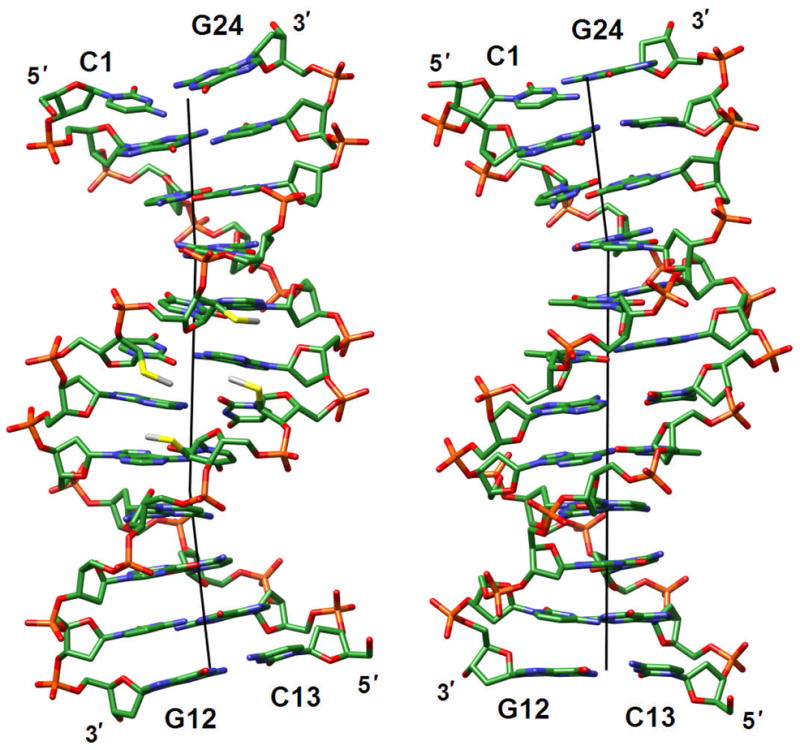
(a) The [d(CGCGAA)-U*U*-d(CGCG)]2 duplex with 2′-S-methylated uridines (U*) viewed into the central minor groove. (b) The native DDD duplex [d(CGCGAATTCGCG)]2 viewed roughly along the same direction. The color code is the same as in Fig. 7 and carbon and sulfur atoms of modified residues are colored in gray and sulfur, respectively. Thin solid lines represent the helical axis.
Two further structural studies concerned a 2′-O-4′-C-methylene-RNA (locked nucleic acid, LNA) that was found to adopt the expected C3′-endo sugar pucker [83] and the 2′-Se-methyl RNA analog that was introduced to facilitate the phasing of RNA crystal structures [84–86]. The selenated analog is chemically stable and replacement of the ribose 2′-oxygen by selenium may not fundamentally alter the secondary or tertiary structures of most RNAs. Thus, the modification should be easily tolerated in a double helical context [85]. However, it is noteworthy that our attempts to determine the crystal structure of the above 2′-S-methyl-U modified DDD by replacing first one, then the other, or both of the U* residues with 2′-Se-methyl-U all failed because crystals of the Se-modified 12mer could not be grown anymore. This example may serve as a ‘cautionary tale’ of the potentially adverse consequences of a seemingly benign modification (S → Se). Although selenium and sulfur differ slightly in terms of atomic radius and electronegativity, it came as a surprise that incorporation of a single 2′-SeMe-U in place of 2′-SMe-U prevented crystal formation, particularly since the change was subtler than replacement of oxygen by selenium (the 2′-Se-methyl moiety was designed to replace the RNA 2′-hydroxyl group).
7. 2′-Arabinonucleic Acid Modifications
Prior to the structure determination of the DDD with incorporated 2′-deoxy-2′-fluoroarabino-thymidines (FANA-T), it was assumed that the analog would display a preference for the C2′-endo pucker (South, Fig. 2). We crystallized the modified dodecamer DNA in collaboration with Victor Marquez at NIH-NCI and found that all four FANA residues in the duplex adopted an Eastern O4′-endo pucker [87]. Besides the preference for this somewhat unusual conformation of the five-membered sugar ring, FANA has another remarkable property: it belongs to only a handful of modifications whose hybrids with RNA are recognized as substrates by RNase H [88]. In the arabino configuration the 2′-substituent is pointing into the major groove and therefore poses no problem for RNase H that contacts the hybrid duplex from the minor groove side. However, hybrids between another arabinonucleic acid (ANA) modification, [3.3.0]bicyclo-ANA [89], and RNA do not elicit RNase H action. Although this bicyclic sugar also exhibits an O4′-endo pucker the bulkier modification most likely interferes with RNase H binding. An O4′-endo pucker was also proposed as the likely pucker adopted by sugars in a DNA strand paired to RNA [43]. But it is clear now that DNA:RNA hybrids exhibit a range of conformations depending on environment and sequence (i.e. ref. [70]) and 2′-deoxyriboses opposite RNA at the active site of RNase H were found to prefer a South C2′-endo pucker [69]. Moreover, the enzyme binds DNA and RNA duplexes but is unable to cleave either [66] (a crystal structure of an RNase H:DNA complex has provided insight on the role of double-stranded DNA as an inhibitor [90]). FANA:RNA duplexes are of higher stability than both the corresponding DNA:RNA or PS-DNA:RNA duplexes ([91] and cited refs.) and a duplex between an all-C3′-endo RNA strand and an all-O4′-endo FANA strand can be readily built [89]. To address the conformational boundaries of ANA and FANA we determined A- and B-form DNA duplexes with incorporated ANA and FANA residues and determined their crystal structures at high resolution (Fig. 9) ([87], B-form FANA; [91]). The structural data reveal clear differences in the conformational preferences of ANA and FANA. Accordingly, ANA residues populate the Southeastern region in the pseudorotation cycle whereas FANA residues are limited to the Northeast quadrant. RNAs opposite FANA are more efficiently cleaved by RNase H than those opposite ANA strands and the activity differences may to some degree reflect the conformational preferences of these arabinonucleic acids. For now it appears likely that the enzyme tolerates a range of conformations by the sugars in the strand that is paired with RNA, but a more definitive answer has to await the determination of a crystal structure of a complex between RNase H and a FANA:RNA duplex.
Fig. 9.

Conformations of (a) FANA- and (b) ANA-modified duplexes exhibiting different degrees of bending into the major groove. The color code is the same as in Fig. 7 and 2′-fluorine and 2′-oxygen atoms are highlighted as green and red spheres, respectively.
8. 4′-Thio-RNA
Together with collaborators at SIRNA Pharmaceuticals, we prepared all four 4′-thio phosphoramidite building blocks for solid phase synthesis of modified RNAs [92]. Alternative approaches to the generation of 4′-thio-RNA were published by Imbach and coworkers [93,94] and Matsuda and coworkers [95,96]. 4′-Thio-RNA displays enhanced nuclease resistance and also shows favorable pairing stability compared with RNA. Incorporation of one or more 4′-thio-cytidines into an RNA duplex increased its melting temperature by about 1°C per modified residue on average relative to native RNA [92]. The crystal structure of an RNA octamer duplex with a single 4′-thio-C per strand revealed only minor differences as a consequence of the larger sulfur in the sugar ring and the local helical parameters such as rise and twist changed only minimally. The most obvious change concerns the longer C-S bonds (ca. 1.84 Å compared to 1.44 Å for C-O [92]). 4′-Thio-sugars adopted C2′-exo and C3′-endo puckers (North) as expected for a ribose analog. Interestingly, investigations with 4′-thio-DNA indicated an RNA-like behavior of this analog [97], although an earlier X-ray crystal structure of the DDD with two 4′-thio-Ts demonstrated that the sugar pucker of modified residues was in the Southern region [98]. However, Walker reported many years ago that the 4′-thio-DNA modification renders the RNA in modified RNA:DNA hybrids more resistant to degradation by RNase H [99]. This could be taken as evidence that the 4′-thio-DNA strand assumes a more A-like conformation compared with native DNA, a feature that is known to hamper the activity of the endoribonuclease.
9. Bicyclo- and Tricyclo-DNA
DNA analogs with bicyclic and tricyclic sugar moieties were studied by the Leumann group (University of Bern) to analyze the effects of restricted conformational flexibility on nucleic acid pairing stability [100]. Provided that the analog’s conformation is preorganized for the target strand, the stability of the duplex formed should be affected favorably (i.e. by reducing the loss of entropy upon duplex formation). In bicyclo-DNA (bcDNA) the C3′ and C5′ centers are bridged by an ethylene moiety. In tricyclo-DNA (tcDNA) the C3′ and C5′ centers are connected by an ethylene that is fused to a cyclopropane ring. Thus, these modifications will not simply affect the sugar conformation but they might alter backbone torsion angles as well (Fig. 10). The crystal structure of a bicyclo-DNA CC dimer [bcd(CC)]2 was studied at atomic resolution and revealed a parallel-stranded dimer with hemiprotonated C:C+ base pairs under formation of three hydrogen bonds [101,102]. The helical rise amounts to 3.25 Å and the twist is 34°. The conformation of this parallel-stranded duplex differs significantly from the so-called C-rich i-motif that consists of two parallel-stranded self-intercalated duplexes [103]. Although the sugar puckers are essentially those expected for B-form DNA (C1′-exo and C2′-endo), the β and γ backbone torsion angles differ distinctly from those encountered in B-DNA. In bcDNA, both β and γ fall into the anticlinal (ac) angle range; by comparison, in B-DNA the β angle is always ap and γ is sc.
Fig. 10.

Crystal structures of (a) bicyclo-DNA and (b) tricyclo-DNA.
Whereas the bicyclic modification is therefore not fully compatible with the conformational preferences of B-form DNA, tcDNA exhibits enthalpically and entropically favorable self-pairing relative to DNA and all-tcDNA oligonucleotides show an increased affinity to both complementary DNA and RNA strands [104]. Perhaps not unexpectedly, the tcDNA modification also confers superb protection against nuclease degradation [105]. We determined the crystal structure of the DDD with incorporated tcdAs at high resolution and found an unusual compensatory effect of the cyclopropane ring on torsion angles β and γ [106]. Accordingly, the β angles of tcdA residues fall into the sc range and γ angles into the ap range. As mentioned above, β is ap and γ is sc in canonical B-form duplexes. Moreover, the conformations of the tcDNA sugars (C2′-exo) and the glycosydic torsion angles (ca. −160°) are consistent with an A-type conformation. This observation provides a rationalization for the increased RNA affinity displayed by tcDNA relative to DNA. Modeling studies suggest that the cyclopropane ring in tcDNA may cause an unfavorable steric interaction at exo- and endonuclease active sites [106], thus explaining the higher protection against degradation afforded by the tricyclic modification.
10. Homo-DNA and TNA
(6′→4′) Oligo-(β-D-2′,3′-dideoxyglucopyranosyl)-nucleotides (homo-DNA, Fig. 11) were studied as an early model system in research directed at an etiology of nucleic acid structure [107,108]. In homo-DNA the sugar is 2′,3′-dideoxyglucopyranose that differs from the standard 2′-deoxyribose by a single methylene group in the ring. However, this seemingly minor change has far-reaching consequences as far as the pairing behavior and structure of homo-DNA are concerned. Homo-DNA constitutes an autonomous pairing system and thus far no other pairing system, natural or synthetic, has been found to hybridize with homo-DNA [109,110]. Self-pairing of homo-DNA oligomers is entropically stabilized compared with DNA and RNA. The pairing priorities in the natural nucleic acids (G:C ≫ A:T) are altered in homo-DNA, whereby adenine and guanine exhibit strong self-pairing of the reverse-Hoogsteen type (G:C > A:A ≈ G:G > A:T > AC).
Fig. 11.

Comparison between the overall geometries of (a) the homo-DNA [(β-D-2′,3′-dideoxyglucopyranosyl)] nucleic acid duplex [dd(CGAATTCG)]2 (viewed into the major groove) and a canonical B-form DNA duplex of the same sequence (viewed across the major and minor grooves). (b) Structure of homo-DNA. (c) Structure of (L)-α-threofuranosyl (3′→2′) nucleic acid (TNA).
To gain insight into the unique properties of homo-DNA we decided to determine its crystal structure. Crystals of a self-complementary octamer dd(CGAATTCG) had been grown as early as 1992 by Christian Leumann who was then in Albert Eschenmoser’s laboratory at the ETH-Zürich. Subsequent attempts to crystallize other homo-DNA oligonucleotides, in particular sequences giving rise to purine-purine pairs all failed. Remarkably, it took another 15 years to crack the structure of the above octamer duplex [111]; the thorny path leading to the final solution of the puzzle has been described in an article in the Chem. Soc. Rev. [112]. To phase the homo-DNA structure, we developed a new derivatization approach that is based on replacement of one of the non-bridging phosphate oxygens by selenium [113,114]. The crystal structure [111] and crystal morphology and packing [115] have been analyzed and insights with regard to pairing gained from the structure have been extensively reviewed [116].
One striking feature of the homo-DNA crystal structure was its conformational heterogeneity. Each of the eight nucleotides per strand displayed a rather different conformation (Fig. 11). This observation alone may help answer the question whether homo-DNA could have served as an alternative molecular framework for storing the genetic blueprint. Other hallmarks of the structure are the strongly inclined backbone-base axes and the virtual absence of intra-strand stacking which we are familiar with from B-form DNA. At one location a nucleobase is pulled out from the stack and replaced with a base from an adjacent duplex that inserts itself opposite the remaining base in a reverse-Hoogsteen mode. In the crystal two duplexes cross each other at an angle of around 60°, whereby the crossing is so tight that bases need to be extruded in order to avoid a steric clash. As expected sugars adopt the chair conformation; at a single location the electron density is consistent with an equilibrium between a chair and a boat. Adoption of the relatively rigid chair conformation is of course consistent with homo-DNA pairing being entropically favored relative to DNA or RNA pairing.
The homo-DNA duplex is right-handed and the average twist is less than half of that in a canonical B-form duplex. The average helical rise of 3.8 Å is somewhat higher than in native DNA but considerably smaller than was expected based on models. Unlike in a tightly wound RNA duplex the limited helical twist in homo-DNA does not obscure the strong inclination of the backbones relative to the base-pair planes. In fact this backbone-base inclination significantly exceeds that in double-stranded RNA and, more importantly, it is of the opposite sign. Backbone-base inclination angles can be easily calculated independent of the structure a nucleic acid and relative angle values provide an indication of whether two systems can pair with each other or not [117]. For example, RNA features a negative backbone-base inclination of ca. −30° and homo-DNA’s inclination amounts to about +45°. Therefore it is clear that the two cannot possibly pair without one of them undergoing a drastic conformational change. However, neither RNA nor homo-DNA possess that conformational flexibility. Conversely, DNA in a B-form conformation lacks a significant inclination between backbone and bases, but it can adapt to the constraints provided by RNA and switch to an A-type conformation to hybridize efficiently with the latter. One recent example of the usefulness of the backbone-base inclination concept concerns glycol nucleic acid (GNA [118]). The crystal structure of (S)-GNA revealed a right-handed duplex with a considerably reduced helical twist compared with DNA or RNA and backbones that are negatively inclined relative to the base-pair axes [119]. (S)-GNA’s negative backbone-base inclination explains the cross-pairing between (S)-GNA and RNA and the inability of (R)-GNA to do so. Although (R)-GNA has a positively inclined backbone it cannot pair with homo-DNA because their twists [left-handed for (R)-GNA and right-handed for homo-DNA] would not match. And (R)-GNA cannot pair with left-handed Z-DNA because the backbone of the latter does not exhibit an appreciable inclination relative to the base-pair planes. In conclusion it is satisfying that two simple geometric parameters (relative twist and relative backbone-base inclination) allow one to explain the existence or absence of hybridization between natural and/or artificial nucleic acid pairing systems.
The (L)-α-threofuranosyl (3′→2′) nucleic acid (Fig. 11c) analog (TNA) is another system studied within the context of nucleic acid etiology. Remarkably, TNA despite its tetrose sugar and a backbone that is shortened by one atom relative to DNA and RNA cross-pairs with both under formation of stable duplexes [120]. TNA duplexes in some cases exhibit higher thermodynamic stability than DNA or RNA duplexes of the same sequence. We studied the conformational properties of isolated TNA residues in the contexts of both B-form [121] and A-form DNA [122]. The crystallographic data revealed a unique C4′-exo pucker of the threose sugar that was present independent of the overall conformation of the duplex. The combined structural data indicated a relatively rigid conformational behavior of TNA and variations in the five backbone torsion angles for residues embedded in the B- and A-form DNA sequences were limited to subtle fluctuations in ε and ζ. The distance between phosphates attached to the 2′ and 3′ centers amounted to about 5.8 Å independent of whether TNA was incorporated into an A- or B-form duplex. This distance matches that between adjacent intra-strand phosphates in duplex RNA and may explain why TNA pairs more strongly with RNA than with DNA.
11. DNA- and RNA-Based Backbone Modifications
One of the earliest structures analyzed in our laboratory concerned a backbone modification that involved the loss of the negative charge on the phosphate: dimethylene sulfone-linked RNA [123]. We determined the crystal structure of the dimer r(Gso2C) that formed a mini-duplex with standard Watson-Crick base pairs. At first sight the geometry of the dimer duplex differs only little from that of the native RNA duplex [r(GpC)]2 [124,125]. The base pairs are inclined relative to the helical axis and exhibit the negative slide characteristic for an A-type duplex and both riboses (the duplex sits on a dyad) adopt the C3′-endo pucker. However, a closer look also reveals that the slide between adjacent base pairs is more pronounced in the dimethylene sulfone-linked RNA (−3.2 Å vs. −1.3 Å in RNA), most likely as a result of the S-C bonds that are ca. 0.2 Å longer than P-O bonds and steric conflicts due to the presence of methylene hydrogen atoms. Nevertheless, the duplex diameters vary only minimally (S···S = 18.0 Å vs. P···P = 17.7 Å). The most striking difference concerns the helical twist which is drastically reduced in the [r(Gso2C)]2 duplex compared to the native RNA (20.8° vs. 34.7°). When an extended duplex is built based on helical parameters extracted from the dimer, it becomes apparent that dimethylene sulfone RNA features a wide-open major groove, and has a more ribbon-like appearance and ca. 17 residues per helical turn instead of the 11 residues in canonical A-RNA [45]. Thus, it is clear that the loss of the negative charge in RNA goes along with some profound changes at the structural level.
Another RNA mimic, N3′→ P5′ phosporamidate DNA (3′-NP-DNA) was the focus of a structural study in collaboration with Sergei Gryaznov, then at Lynx Therapeutics, CA. This analog shows strong self-pairing and high RNA affinity with gains in the melting temperature that amount to 2.3 to 2.6°C relative to the corresponding phosphodiester compounds [126–128]. Surprisingly, when the 5′-oxygen is replaced by an amino group, self-pairing and cross-pairing with RNA are abolished. An explanation as to why the consequences of the replacement of O5′ by an amino group should be so different from those of the replacement of O3′ was elusive [128]. The crystal structure of a fully modified 3′-NP-DNA dodecamer duplex provided answers to all questions regarding the analog’s pairing properties and the distinct behaviors of 3′-NP-DNA and 5′-NP-DNA [129] (Fig. 12). Although the phosphoramidate DNA lacks 2′-hydroxyl groups, its sugars adopt a C3′-endo conformation as a result of a weaker gauche effect between O4′ and N3′ relative to that between O4′ and O3′ in DNA.
Fig. 12.

(a) Anomeric effect between the N3′ lone electron pair (green) and the antibonding σ* orbital of the P-O5′ bond in N3′→P5′ phosphoramidate DNA. (b) Putative conjugation between the N5′ lone pair (green) and the antibonding σ* orbital of the P-O3′ bond with P→N5′ phosphoramidate DNA. Provided the furanose adopts an A-type pucker, this arrangement would result in a steric repulsion between the 5′-amino hydrogen and a 2′-hydrogen of the 2′-deoxyribose (red arrow).
The nitrogen represents a chiral center and although the resolution of the crystal structure did not allow us to distinguish between the lone pair and the amino hydrogen, packing features permitted assignment of the absolute stereochemistry. Thus, lone pairs were directed toward ammonium ions located in channels between three neighboring duplexes. And in turn 3′-NH groups were coordinated to chloride anions. This arrangement results in an antiperiplanar orientation of the amino lone pair and the antibonding P-O5′σ* orbital, clearly demonstrating the existence of an anomeric effect that constitutes the basis for the sc−/sc− (ζ/α) backbone conformation in the phosphoramidate and, by analogy, the phosphodiester moieties (Fig. 12a). Assuming that the anomeric effect is also operative in 5′-NP-DNA, the amino hydrogen can be expected to point inwards and therefore clash with H2′Si from the sugar in a Northern conformation (Fig. 12b). If one assumes an overall B-type conformation by the 5′-NP-DNA analog, steric conflicts between amino hydrogen and both O4′ and H6 of pyrimidines would arise. Either scenario is consistent with the observed absence of stable duplex formation with 5′-NP-DNA.
Besides these stereochemical constraints, the presence of the amino group in phosphoramidate DNA introduces a hydrogen bond donor into the backbone (the DNA sugar-phosphate backbone is devoid of H-bond donors). Indeed, the crystal structure revealed a superb hydration of the 3′-NP-DNA backbone and shallow groove, thus underlining the similarities between this analog and RNA. In the latter, the 2′-hydroxyl groups serve as bridgeheads for tandem water bridges across the minor groove (Fig. 5c) whereas amino groups take on a similar role in 3′-NP-DNA (the 2′-OH functionality can of course act both as a donor and an acceptor of hydrogen bonds) [129]. It is remarkable that RNA motifs made entirely of 3′-NP-DNA can recruit RNA-binding proteins with similar affinities as the parent RNA compounds do [130]. The structural data support the notion that 3′-NP-DNA mimics RNA not just in terms of its overall geometry but that it shares the latter’s conformational rigidity and more extensive water structure relative to DNA.
The example of TNA showed that a shorter backbone does not necessarily hamper the ability of an analog to stably hybridize to DNA and RNA. In collaboration with Karl-Heinz Altmann and Peter von Matt at Ciba’s Central Research Labs, we investigated the potential consequences for pairing with a series of analogs featuring longer backbones based on 5-atom amide linkages [131,132]. The different amides included structures with homochiral (*) linkers of the type X3′-C*H(CH3)-CO-NH-CH2 (X=O, CH2) as well as the corresponding analogs carrying methoxy groups at the 2′-position of the 3′-nucleosides. Interestingly, the longer backbone not only maintained pairing with both DNA and RNA, but it actually resulted in modest gains in terms of duplex stability in some cases. A crystal structure of a DNA:RNA hybrid with a single amide-linked dimer in the oligo-2′-deoxynucleotide strand manifested very minor changes in the local helical parameters as a consequence of the additional atoms in the backbone [132]. In particular, the helical rise appeared unaffected and there was no obvious kinking at the site of the asymmetrically linked dimer step. This work demonstrated that stable cross-pairing between two different types of nucleic acids does not require the numbers of atoms linking their individual residues to match.
12. Nucleobase Modifications
We determined crystal structures of DNA and RNA molecules with incorporated chemically modified nucleobases that were analyzed in very different contexts, including antisense (guanyl G-clamp [133,134], 2′-O-[2-methoxy)ethyl]-2-thiothymidine [135]) and ribozyme (phenyl ribonucleotide [136,137]) activity, electron transfer (conjugates with bis(2-hydroxyethyl)stilbene-4,4′-diether linkers [138,139]), the effects on the fidelity of DNA replication by a hydrophobic T isostere (2,4-difluoro-toluene; DFT [140–142]) and the control of the G:A mismatch pairing type by methylation (N2,N2-dimethylguanosine; m22G [143]). The guanyl G-clamp is a cytosine analog whose design was inspired by an Arg···G interaction, a frequently observed motif in protein-DNA complexes. The 9-(2-guanidino-ethoxy)-phenoxazine analog places a guandinium moiety opposite the major groove of G such that two hydrogen bonds can be formed to the Hoogsteen edge (O6 and N7) in addition to the three standard Watson-Crick hydrogen bonds (Fig. 13). A crystal structure at 1 Å resolution indeed revealed formation of five hydrogen bonds between guanine and the tricyclic G-clamp incorporated into a decameric A-form DNA duplex [133]. Besides the favorable electrostatic and stacking interactions, the extraordinary increase in Tm of ca. 16°C per incorporated G-clamp was consistent with extensive water networks between the modification and the sugar-phosphate backbone [134]. Phenoxazines alone were found to increase the stability of modified duplexes by between 2 and 7°C [144].
Fig. 13.

Crystal structure of the guanyl G-clamp (a cytosine analogue), confirming formation of five hydrogen bonds indicated by thin solid lines. Carbon atoms of the modification are colored in gray.
Incorporation of hydrophobic phenylribonucleotides into RNA leads to a drastic loss of stability [137], but the analog’s effects were mainly assessed in terms of ribozyme activity. In some cases substitutions of pyrimidines by the phenyl residue were of surprisingly little consequence or actually enhanced the cleavage rate [145,146]. In the crystal structure of an RNA octamer duplex with a single phenylribonucleotide per strand, slippage of the two strands places the two phenyls opposite each other in the center [137]. The two hydrophobic moieties are in van der Waals contact and only result in very minor deviations from the standard A-form geometry of the RNA duplex. Obvious changes in the vicinity of this Ph:Ph ‘pair’ include the absence of water molecules in the grooves and somewhat reduced stacking interactions with adjacent base pairs due to the lack of exocyclic functions with the phenyl moiety.
The situation regarding thermodynamic stability changes couldn’t be more different from the phenyl residue for another hydrophobic base or base-pair analog, the bis(2-hydroxyethyl)stilbene-4,4′-diether (Sd, Fig. 14a) linker that results in dramatically increased melting temperatures when it is used to cap DNA or RNA hairpins. For example, a hairpin formed by two G:C base pairs linked by Sd melts at >80°C in a buffered solution containing 0.1 M NaCl [138]. The Sd linker is strongly fluorescent in the absence of nucleobases but its fluorescence is quenched in hairpins with neighboring A:T or G:C pairs. This quenching is attributed to a photo-induced electron transfer process whereby singlet Sd serves as the electron donor and either T or A as the electron acceptor [147]. The high thermodynamic stability of Sd-linked DNA hairpins is consistent with stable structures in solution. In one crystal structure determined for a DNA hexamer hairpin capped by Sd with four independent molecules per asymmetric unit, all trans-stilbenes were stacked on the adjacent base pair (Fig. 14b) (face-to-face interaction) [138]. However, a subsequent structure of the same hairpin determined at higher resolution revealed significant conformational flexibility of the Sd moiety [139]. Thus the two phenyl rings were twisted relative to each other in one of the hairpins and the stilbene was detached from the adjacent base pair, effectively exhibiting an edge-to-face orientation (Fig. 14c). Such behavior by aromatic molecules is well known and electronic structure calculations at a high level of theory for the benzene dimer have indicated that offset face-to-face and edge-to-face geometries possess similar energies [148]. In addition to fluorescence, the trans-cis isomerization of the Sd linker that is observed in the absence of nucleobases is also strongly quenched in DNA hairpins. In view of the edge-to-face orientation of Sd in the crystal structure and the high level of twisting around the C=C bond it appears unlikely that restricted motion is responsible for the absence of photoisomerization. Instead one is tempted to attribute the prevention of isomerization to a fast electron-transfer quenching [139].
Fig. 14.

Comparison of DNA duplexes capped by a stilbenediether (Sd) linker. (a) Structure of Sd. (b) Face-to-face and (c) edge-to-face orientations of Sd relative to the adjacent G:C base pair. Carbon atoms of Sd are colored in gray.
The hydrophobic thymine isostere 2,4-difluorotoluene (DFT) was created to assess the relative importance of hydrogen bonding and shape in accurate replication [149] and has led some to the conclusion that shape may trump hydrogen bonding as the basis for insertion of the correct nucleotide triphosphate opposite a template base by so-called A-class replicative DNA polymerases [150]. Although a large body of work regarding DFT and other base analogs with bulkier substituents at the 2 and 4 positions (i.e. Cl, Br, I, etc.) has been gathered over the last decade, detailed structural information on DFT opposite the natural purines either in the context of DNA alone or at the active sites of DNA polymerases was unavailable. In collaboration with Muthiah Manoharan at Alnylam Pharmaceuticals Inc., we initially investigated the effects of the ribonucleotide analog of DFT, rDFT, inside the guide and passenger strands of siRNA duplexes [140,141]. We found that single rDFT:A pairs were tolerated by the Ago2 slicer enzyme of the RISC complex, even when they were placed adjacent to the cleavage site. However, incorporation of three consecutive rDFT:A pairs greatly attenuated downregulation as did the presence of a single rDFT:G or a U:G mismatch. The individual RNAi activities of oligoribonucleotides containing the hydrophobic isostere at various locations did not appear to be correlated with changes in the thermodynamic stability. Instead, structural data obtained for RNA duplexes with incorporated rDFT:A, rDFT:G (Fig. 15) or various mismatch pairs provided evidence that the extent of local conformational deviations from a standard Watson-Crick geometry seemed more important in this respect. We found that an rDFT:A pair displays a distinctly different geometry from a U:A pair [140], but, surprisingly, that rDFT:G and U:G closely resemble each other, including the distributions of water molecules around the major and minor groove base edges [141]. Unexpectedly, the rDFT:G mismatch is slightly more stable than the dDFT:A pair in RNA and osmotic stressing studies and computational simulations supported the notion that, although DFT constitutes a T analog, G appears to be a better match for DFT in RNA than A. This differs markedly from the results of thermodynamic studies in DNA where a DFT:G mismatch was found to be more destabilizing than a DFT:A pair [151]. Although the origin of these different behaviors in DNA and RNA remains somewhat unclear, the structural data at high resolution in combination with semiempirical calculations indicate that fluorine can act as a weak hydrogen bond acceptor in rDFT:G pairs (Fig. 15b) [141]. Kinetic and structural studies of the DFT analog with the Y-class trans-lesion DNA polymerase Dpo4 from S. solfataricus confirmed that DFT may well be an isostere of T but that the geometries of DFT:A or DFT:G pairs at the polymerase active site bear limited resemblance to those of T:A or T:G pairs [142]. An important although perhaps obvious lesson from our structural results is that shape and hydrogen bonding are intimately related and that the lack of hydrogen bonds in base pairs involving the DFT analog alters the shape of such pairs considerably. A further issue that may have been ignored to some extent in the analysis of the in vitro nucleotide insertion and extension data involving the DFT isostere concerns key differences between the active sites of high-fidelity and bypass DNA polymerases, in that the former probe the edges of base pairs at the replicative and post-replicative positions from the minor groove side with hydrogen bonding interactions. Replacing T with DFT will not just remove hydrogen bonds between the incoming nucleotide and the template base but also abolish H-bond formation between the hydrophobic analog and active site residues in some DNA polymerases.
Fig. 15.

Structures of (a) an RNA difluorotoluene (rDFT):A base pair (b) an RNA difluorotoluene (rDFT):G base pair.
13. Conclusions and Outlook
The results regarding structure, activity and stability of CNAs summarized in this contribution demonstrate the value of high resolution structural information for gaining insights into the consequences of chemical modification in regard to RNA affinity, nuclease resistance, interactions with key enzymes such as RNase H (antisense) or Ago2 (RNAi) and a host of other biochemical and biophysical issues. Structural information can provide useful principles for the design of new generations of CNAs with improved properties for putative therapeutic applications or as agents in diagnostics, materials science or high-throughout screening. Although structures sometimes merely confirm a conclusion that was reached using chemical, biochemical, thermodynamic or kinetic tools among others, there are many cases, where a structure provides truly novel insights that could not have been obtained with any other means. Examples that come to mind from our own work are the precise origin of the exceptional resistance to nuclease degradation exhibited by the 2′-O-(3-aminopropyl)-RNA modification or the structures of N3′→ P5′ phosporamidate DNA and homo-DNA that gave comprehensive answers to all puzzles regarding these analogs. We are continuing to explore the three-dimensional structures of CNAs alone and in complex with enzymes such as RNase H and DNA polymerases. Some current projects concern the unique RNAi activities of siRNAs with alternating 2′-OH/2′-F or 2′-F/2′-OMe sugar-phosphate backbones, third-generation antisense modifications with novel chemistries to preorganize the oligonucleotide for the mRNA target, so-called unlocked nucleic acid that modulates the RNAi activity, the pairing properties of glycol nucleic acid (GNA) and the geometries of RNA:CNA duplexes at the active site of RNase H.
Acknowledgments
The Principal Investigator is grateful to his current and former coworkers, Drs. F. Li, P. Lubini, G. Minasov, S. Portmann, R. Pattanayek, S. Sarkhel, M. Teplova, V. Tereshko, and C. J. Wilds, and longtime collaborators C. J. Leumann (University of Bern), M. Manoharan (Alnylam Pharmaceuticals Inc.), V. E. Marquez (NCI, NIH), T. P. Prakash (Isis Pharmaceuticals Inc.), and J. Wengel (University of Southern Denmark). We would like to thank the US National Institutes of Health, General Medical Sciences, for continuous financial support of research directed at the structure and function of nucleic acids (grant R01 GM55237).
References
- 1.Zamecnik PC, Stephenson ML. Proc Natl Acad Sci USA. 1978;75:280. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephenson ML, Zamecnik PC. Proc Natl Acad Sci USA. 1978;75:285. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uhlmann E, Peyman A. Chem Rev. 1990;90:543. [Google Scholar]
- 4.Milligan JF, Matteucci MD, Martin JC. J Med Chem. 1993;36:1923. doi: 10.1021/jm00066a001. [DOI] [PubMed] [Google Scholar]
- 5.De Mesmaeker A, Häner R, Martin P, Moser HE. Acc Chem Res. 1995;28:366. [Google Scholar]
- 6.Herdewijn P. Liebigs Ann. 1996:1337. [Google Scholar]
- 7.Szymkowski DE. Drug Discovery Today. 1996;1:415. doi: 10.1016/S1359-6446(04)03296-9. [DOI] [PubMed] [Google Scholar]
- 8.Bennett CF, Dean N, Ecker DJ, Monia BP. In: Methods in molecular medcine: antisense therapeutics. Agrawal S, editor. Humana Press; Totowa, NJ: 1996. pp. 13–46. [DOI] [PubMed] [Google Scholar]
- 9.Crooke ST, editor. Handbook of Experimental Pharmacology, Antisense research and application. 1. Vol. 131. Springer-Verlag; Berlin and Heidelberg: 2002. [Google Scholar]
- 10.Opalinska JB, Gewirtz AM. Nature Rev Drug Discov. 2002;1:503. doi: 10.1038/nrd837. [DOI] [PubMed] [Google Scholar]
- 11.Crooke ST. In: Therapeutic Applications of Oligonucleotides. Crooke ST, editor. Landes; Austin, TX: 1995. pp. 63–79. [Google Scholar]
- 12.Monia BP, Johnston JF, Geiger T, Müller M, Fabbro T. Nature Med. 1995;2:668. doi: 10.1038/nm0696-668. [DOI] [PubMed] [Google Scholar]
- 13.Tang J, Roskey A, Li Y, Agrawal S. Nucleosides & Nucleotides. 1995;14:985. [Google Scholar]
- 14.Koziolkiewicz M, Wojcik M, Kobylanska A, Karwowski B, Rebowska P, Guga B, Stec WJ. Antisense & Nucleic Acid Drug Dev. 1997;7:43. doi: 10.1089/oli.1.1997.7.43. [DOI] [PubMed] [Google Scholar]
- 15.Levin AA. Biochim Biophys Acta. 1999;1489:69. doi: 10.1016/s0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- 16.Manoharan M. Biochim Biophys Acta. 1999;1489:117. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 17.Stix G. Sci Am. 1998;279:46. doi: 10.1038/scientificamerican1198-46b. [DOI] [PubMed] [Google Scholar]
- 18.Kennard O, Hunter WN. Angew Chem Int Ed Engl. 1991;30:1254. [Google Scholar]
- 19.Dickerson RE. Methods Enzym. 1992;211:67. doi: 10.1016/0076-6879(92)11007-6. [DOI] [PubMed] [Google Scholar]
- 20.Egli M. In: Structure Correlation. Bürgi H-B, Dunitz JD, editors. Vol. 2. VCH Publishers Inc; Weinheim, Germany: 1994. pp. 705–749. [Google Scholar]
- 21.Fratini AV, Kopka ML, Drew HR, Dickerson RE. J Biol Chem. 1982;257:14686. [PubMed] [Google Scholar]
- 22.Chevrier B, Dock AC, Hartmann B, Leng M, Moras D, Thuong MT, Westhof E. J Mol Biol. 1986;188:707. doi: 10.1016/s0022-2836(86)80016-x. [DOI] [PubMed] [Google Scholar]
- 23.Wang AHJ, Hakoshima T, van der Marel GA, van Boom JH, Rich A. Cell. 1984;37:321. doi: 10.1016/0092-8674(84)90328-3. [DOI] [PubMed] [Google Scholar]
- 24.Frederick CA, Saal D, van der Marel GA, van Boom JH, Wang AHJ, Rich A. Biopol. 1987;26:S145. doi: 10.1002/bip.360260014. [DOI] [PubMed] [Google Scholar]
- 25.Frederick CA, Quigley GJ, van der Marel GA, van Boom JH, Wang AHJ, Rich A. J Biol Chem. 1988;263:17872. doi: 10.2210/pdb4dnb/pdb. [DOI] [PubMed] [Google Scholar]
- 26.Coll M, Wang AHJ, van der Marel GA, van Boom JH, Rich A. J Biomol Struct Dyn. 1986;4:157. doi: 10.1080/07391102.1986.10506337. [DOI] [PubMed] [Google Scholar]
- 27.Cruse WBT, Salisbury SA, Brown T, Cosstick R, Eckstein F, Kennard O. J Mol Biol. 1986;192:891. doi: 10.1016/0022-2836(86)90035-5. [DOI] [PubMed] [Google Scholar]
- 28.Dock-Bregeon AC, Chevrier B, Podjarny A, Moras D, deBear JS, Gough GR, Gilham PT, Johnson JE. Nature. 1988;335:375. doi: 10.1038/335375a0. [DOI] [PubMed] [Google Scholar]
- 29.Wang AHJ, Fujii S, van Boom JH, van der Marel GA, van Boeckel SAA, Rich A. Nature. 1982;299:601. doi: 10.1038/299601a0. [DOI] [PubMed] [Google Scholar]
- 30.Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. Biophys J. 1992;63:751. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Williams LD, Egli M, Ughetto G, van der Marel GA, van Boom JH, Quigley GJ, Wang AHJ, Rich A, Frederick CA. J Mol Biol. 1990;215:313. doi: 10.1016/S0022-2836(05)80349-3. [DOI] [PubMed] [Google Scholar]
- 32.Egli M, Williams LD, Frederick CA, Rich A. Biochem. 1991;30:1364. doi: 10.1021/bi00219a029. [DOI] [PubMed] [Google Scholar]
- 33.van der Marel GA, Ploegh H. Nature. 2004;431:755. doi: 10.1038/431755a. [DOI] [PubMed] [Google Scholar]
- 34.Altona C, Sundaralingam M. J Am Chem Soc. 1972;94:8205–8212. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- 35.Egli M. In: Nucleic Acids in Chemistry and Biology. 3. Blackburn GM, Gait MJ, Loakes D, Williams DM, editors. Royal Society of Chemistry; Cambridge, UK: 2006. pp. 13–75. [Google Scholar]
- 36.Neidle S. Principles of Nucleic Structure. Academic Press; London, UK: 2008. [Google Scholar]
- 37.Thibaudeau C, Acharya P, Chattopadhyaya J. Stereoelectronic Effects in Nucleosides and Nucleotides and Their Structural Implications. Uppsala University Press; Uppsala, SE: 1999. [Google Scholar]
- 38.Usman N, Egli M, Rich A. Nucleic Acids Res. 1992;20:6695. doi: 10.1093/nar/20.24.6695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egli M, Usman N, Rich A. Biochem. 1993;32:3221. [PubMed] [Google Scholar]
- 40.Ban C, Ramakrishnan B, Sundaralingam M. J Mol Biol. 1994;236:275. doi: 10.1006/jmbi.1994.1134. [DOI] [PubMed] [Google Scholar]
- 41.Wahl MC, Sundaralingam M. Nucleic Acids Res. 2000;28:4356. doi: 10.1093/nar/28.21.4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egli M, Usman N, Zhang S, Rich A. Proc Natl Acad Sci USA. 1992;89:534. doi: 10.1073/pnas.89.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fedoroff OY, Salazar M, Reid BR. J Mol Biol. 1993;233:509. doi: 10.1006/jmbi.1993.1528. [DOI] [PubMed] [Google Scholar]
- 44.Lubini P, Zürcher W, Egli M. Chem & Biol. 1994;1:39. doi: 10.1016/1074-5521(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 45.Egli M. Angew Chem Int Ed Engl. 1996;35:1894. [Google Scholar]
- 46.Egli M. In: Advances in Enzyme Regulation. Weber G, editor. Vol. 38. Elsevier; Oxford, UK: 1998. pp. 181–203. [DOI] [PubMed] [Google Scholar]
- 47.Tereshko V, Minasov G, Egli M. J Am Chem Soc. 1999;121:470. [Google Scholar]
- 48.Tereshko V, Minasov G, Egli M. J Am Chem Soc. 1999;121:3590. [Google Scholar]
- 49.Minasov G, Tereshko V, Egli M. J Mol Biol. 1999;291:83. doi: 10.1006/jmbi.1999.2934. [DOI] [PubMed] [Google Scholar]
- 50.Egli M, Tereshko V, Teplova M, Minasov G, Joachimiak A, Sanishvili R, Weeks CM, Miller R, Maier MA, An H, Cook PD, Manoharan M. Biopol (Nucleic Acid Sciences) 2000;48:234. doi: 10.1002/(SICI)1097-0282(1998)48:4<234::AID-BIP4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 51.Tereshko V, Wilds CJ, Minasov G, Prakash TP, Maier MA, Howard A, Wawrzak Z, Manoharan M, Egli M. Nucleic Acids Res. 2001;29:1208. doi: 10.1093/nar/29.5.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Egli M. Chem Biol. 2002;9:277. doi: 10.1016/s1074-5521(02)00116-3. [DOI] [PubMed] [Google Scholar]
- 53.Egli M, Tereshko V. Curvature and Deformation of Nucleic Acids: Recent Advances, New Paradigms. In: Stellwagen N, Mohanty U, editors. ACS Symp Ser. Vol. 884. 2004. p. 97. [Google Scholar]
- 54.Portmann S, Altmann KH, Reynes N, Egli M. J Am Chem Soc. 1997;119:2396. [Google Scholar]
- 55.Maltseva TV, Altmann KH, Egli M, Chattopadhyaya J. J Biomol Struct Dyn. 1999;16:569. doi: 10.1080/07391102.1998.10508270. [DOI] [PubMed] [Google Scholar]
- 56.Maier T, Przylas I, Strater N, Herdewijn P, Saenger W. J Am Chem Soc. 2005;127:2937. doi: 10.1021/ja045843v. [DOI] [PubMed] [Google Scholar]
- 57.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 58.Egli M. Antisense Nucleic Acid Drug Dev. 1998;8:123. doi: 10.1089/oli.1.1998.8.123. [DOI] [PubMed] [Google Scholar]
- 59.Portmann S, Usman N, Egli M. Biochem. 1995;34:7569. doi: 10.1021/bi00023a002. [DOI] [PubMed] [Google Scholar]
- 60.Egli M, Portmann S, Usman N. Biochem. 1996;35:8489. doi: 10.1021/bi9607214. [DOI] [PubMed] [Google Scholar]
- 61.Freier SM, Altmann KH. Nucleic Acids Res. 1997;25:4429. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cook PD. Annu Rep Med Chem. 1998;33:313. [Google Scholar]
- 63.Stein H, Hausen P. Science. 1969;166:393. doi: 10.1126/science.166.3903.393. [DOI] [PubMed] [Google Scholar]
- 64.Hostomsky Z, Hostomska Z, Matthews DA. In: Nucleases. 2. Linn SM, Lloyd SR, Roberts RJ, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1993. pp. 341–376. [Google Scholar]
- 65.Walder RT, Walder JA. Proc Natl Acad Sci USA. 1988;85:5011. doi: 10.1073/pnas.85.14.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lima WF, Crooke ST. Biochem. 1997;36:390. doi: 10.1021/bi962230p. [DOI] [PubMed] [Google Scholar]
- 67.Lima WF, Mohan V, Crooke ST. J Biol Chem. 1997;272:18191. doi: 10.1074/jbc.272.29.18191. [DOI] [PubMed] [Google Scholar]
- 68.Lima WF, Nichols JG, Wu H, Prakash TP, Migawa MT, Wyrzykiewicz TK, Bhat B, Crooke ST. J Biol Chem. 2004;279:36317. doi: 10.1074/jbc.M405035200. [DOI] [PubMed] [Google Scholar]
- 69.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Cell. 2005;121:1005. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 70.Loukachevitch LV, Egli M. Acta Cryst Sect F. 2007;63:84. doi: 10.1107/S1744309106055461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Geary RS, Watanabe TA, Truong L, Freier S, Lesnik EA, Sioufi NB, Sasmor H, Manoharan M, Levin AA. J Pharm Exp Therap. 2001;296:890. [PubMed] [Google Scholar]
- 72.Adamiak DA, Milecki J, Popenda M, Adamiak RW, Dauter Z, Rypniewski WR. Nucleic Acids Res. 1997;25:4599. doi: 10.1093/nar/25.22.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Auffinger P, Westhof E. Angew Chem Int Ed. 2001;40:4648. doi: 10.1002/1521-3773(20011217)40:24<4648::aid-anie4648>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 74.Tereshko V, Portmann S, Tay E, Martin P, Natt F, Altmann KH, Egli M. Biochem. 1998;37:10626. doi: 10.1021/bi980392a. [DOI] [PubMed] [Google Scholar]
- 75.Teplova M, Minasov G, Tereshko V, Inamati G, Cook PD, Manoharan M, Egli M. Nature Struct Biol. 1999;6:535. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 76.Pattanayek R, Sethaphong L, Pan C, Prhavc M, Prakash TP, Manoharan M, Egli M. J Am Chem Soc. 2004;126:15006. doi: 10.1021/ja044637k. [DOI] [PubMed] [Google Scholar]
- 77.Egli M, Minasov G, Tereshko V, Pallan PS, Teplova M, Inamati GB, Lesnik EA, Owens SR, Ross BS, Prakash TP, Manoharan M. Biochem. 2005;44:9045. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- 78.Teplova M, Wallace ST, Minasov G, Tereshko V, Symons A, Cook PD, Manoharan M, Egli M. Proc Natl Acad Sci USA. 1999;96:14240. doi: 10.1073/pnas.96.25.14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prakash TP, Püschl A, Lesnik E, Tereshko V, Egli M, Manoharan M. Org Lett. 2004;6:1971. doi: 10.1021/ol049470e. [DOI] [PubMed] [Google Scholar]
- 80.Prhavc M, Prakash TP, Minasov G, Egli M, Manoharan M. Org Lett. 2003;5:2017. doi: 10.1021/ol0340991. [DOI] [PubMed] [Google Scholar]
- 81.Prakash TP, Manoharan M, Fraser AS, Kawasaki AM, Lesnik E, Sioufi N, Leeds JM, Teplova M, Egli M. Biochem. 2002;41:11642. doi: 10.1021/bi020264t. [DOI] [PubMed] [Google Scholar]
- 82.Pallan PS, Prakash TP, Manoharan M, Egli M. Chem Comm. 2009:2017. doi: 10.1039/b822781k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Egli M, Minasov G, Teplova M, Kumar R, Wengel J. Chem Comm. 2001:651. [Google Scholar]
- 84.Du Q, Carrasco N, Teplova M, Wilds CJ, Kong X, Egli M, Huang Z. J Am Chem Soc. 2002;124:24. doi: 10.1021/ja0171097. [DOI] [PubMed] [Google Scholar]
- 85.Teplova M, Wilds CJ, Wawrzak Z, Tereshko V, Du Q, Carrasco N, Huang Z, Egli M. Biochimie. 2002;84:849. doi: 10.1016/s0300-9084(02)01440-2. [DOI] [PubMed] [Google Scholar]
- 86.Pallan PS, Egli M. Nature Protocols. 2007;2:647. doi: 10.1038/nprot.2007.75. [DOI] [PubMed] [Google Scholar]
- 87.Berger I, Tereshko V, Ikeda H, Marquez VE, Egli M. Nucleic Acids Res. 1998;26:2473. doi: 10.1093/nar/26.10.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Damha MJ, Wilds CJ, Noronha A, Brukner I, Borkow G, Arion D, Parniak MA. J Am Chem Soc. 1998;120:12976. [Google Scholar]
- 89.Minasov G, Teplova M, Nielsen P, Wengel J, Egli M. Biochem. 2000;39:3525. doi: 10.1021/bi992792j. [DOI] [PubMed] [Google Scholar]
- 90.Pallan PS, Egli M. Cell Cycle. 2008;7:2562. doi: 10.4161/cc.7.16.6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li F, Sarkhel S, Wilds CJ, Wawrzak Z, Prakash TP, Manoharan M, Egli M. Biochem. 2006;45:4141. doi: 10.1021/bi052322r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haeberli P, Berger I, Pallan PS, Egli M. Nucleic Acids Res. 2005;33:3965. doi: 10.1093/nar/gki704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Leydier C, Bellon L, Barascut JL, Morvan F, Rayner B, Imbach JL. Antisense Res Dev. 1995;5:167. doi: 10.1089/ard.1995.5.167. [DOI] [PubMed] [Google Scholar]
- 94.Leydier C, Bellon L, Barascut JL, Imbach JL. Nucleosides Nucleotide. 1995;14:1027. [Google Scholar]
- 95.Naka T, Minakawa N, Abe H, Kaga D, Matsuda A. J Am Chem Soc. 2000;122:7233. [Google Scholar]
- 96.Hoshika S, Minakawa N, Matsuda A. Nucleic Acids Res. 2004;32:3815. doi: 10.1093/nar/gkh705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inoue N, Minakawa N, Matsuda A. Nucleic Acids Res. 2006;34:3476. doi: 10.1093/nar/gkl491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boggon TJ, Hancox EL, McAuley-Hecht KE, Connolly BA, Hunter WN, Brown T, Walker RT, Leonard GA. Nucleic Acids Res. 1996;24:951. doi: 10.1093/nar/24.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Walker RT. R Soc Chem Spec Publ (Anti-Infectives) 1997;198:203. [Google Scholar]
- 100.Leumann CJ. Chimia. 2001;55:1041. [Google Scholar]
- 101.Egli M, Lubini P, Bolli M, Dobler M, Leumann C. J Am Chem Soc. 1993;115:5855. [Google Scholar]
- 102.Lubini P, Egli M. In: Structural Biology: State of the Art 1993: Proceedings of the Eighth Conversations. Sarma RH, Sarma MH, editors. Vol. 2. Adenine Press; Albany, NY: 1994. pp. 155–176. [Google Scholar]
- 103.Berger I, Egli M, Rich A. Proc Natl Acad Sci USA. 1996;93:12116. doi: 10.1073/pnas.93.22.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Renneberg D, Leumann CJ. J Am Chem Soc. 2002;124:5993. doi: 10.1021/ja025569+. [DOI] [PubMed] [Google Scholar]
- 105.Renneberg D, Bouliong E, Reber U, Schümperli D, Leumann CJ. Nucleic Acids Res. 2002;30:2751. doi: 10.1093/nar/gkf412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pallan PS, Ittig D, Heroux A, Wawrzak Z, Leumann CJ, Egli M. Chem Commun. 2008:883. doi: 10.1039/b716390h. [DOI] [PubMed] [Google Scholar]
- 107.Eschenmoser A. Chem Commun. 2004:1247. [Google Scholar]
- 108.Eschenmoser A, Dobler M. Helv Chim Acta. 1992;75:218. [Google Scholar]
- 109.Hunziker J, Roth HJ, Böhringer M, Giger A, Diederichsen U, Göbel M, Krishnan R, Jaun B, Leumann C, Eschenmoser A. Helv Chim Acta. 1993;76:259. [Google Scholar]
- 110.Eschenmoser A, Loewenthal E. Chem Soc Rev. 1993;21:1. [Google Scholar]
- 111.Egli M, Pallan PS, Pattanayek R, Wilds CJ, Lubini P, Minasov G, Dobler M, Leumann C, Eschenmoser A. J Am Chem Soc. 2006;128:10847. doi: 10.1021/ja062548x. [DOI] [PubMed] [Google Scholar]
- 112.Egli M, Lubini P, Pallan PS. Chem Soc Rev. 2007;36:31. doi: 10.1039/b606807c. [DOI] [PubMed] [Google Scholar]
- 113.Wilds CJ, Pattanayek R, Pan C, Wawrzak Z, Egli M. J Am Chem Soc. 2002;124:14910. doi: 10.1021/ja021058b. [DOI] [PubMed] [Google Scholar]
- 114.Pallan PS, Egli M. Nature Protocols. 2007;2:640. doi: 10.1038/nprot.2007.74. [DOI] [PubMed] [Google Scholar]
- 115.Pallan PS, Lubini P, Egli M. Chem Commun. 2007:1447. doi: 10.1039/b614983a. [DOI] [PubMed] [Google Scholar]
- 116.Egli M, Pallan PS. Annu Rev Biophys Biomol Struct. 2007;36:281. doi: 10.1146/annurev.biophys.36.040306.132556. [DOI] [PubMed] [Google Scholar]
- 117.Pallan PS, Lubini P, Bolli M, Egli M. Nucleic Acids Res. 2007;35:6611. doi: 10.1093/nar/gkm612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang L, Peritz A, Meggers E. J Am Chem Soc. 2005;127:4174. doi: 10.1021/ja042564z. [DOI] [PubMed] [Google Scholar]
- 119.Schlegel MK, Essen LO, Meggers E. J Am Chem Soc. 2008;130:8158. doi: 10.1021/ja802788g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schöning KU, Scholz P, Guntha S, Wu X, Krishnamurthy R, Eschenmoser A. Science. 2000;290:1347. doi: 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]
- 121.Wilds CJ, Wawrzak Z, Krishnamurthy R, Eschenmoser A, Egli M. J Am Chem Soc. 2002;124:13716. doi: 10.1021/ja0207807. [DOI] [PubMed] [Google Scholar]
- 122.Pallan PS, Wilds CJ, Wawrzak Z, Krishnamurthy R, Eschenmoser A, Egli M. Angew Chem Int Ed. 2003;115:5893. doi: 10.1002/anie.200352553. [DOI] [PubMed] [Google Scholar]
- 123.Roughton AL, Portmann S, Benner SA, Egli M. J Am Chem Soc. 1995;117:7249. [Google Scholar]
- 124.Day RO, Seeman NC, Rosenberg JM, Rich A. Proc Natl Acad Sci USA. 1973;70:849. doi: 10.1073/pnas.70.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rosenberg JM, Seeman NC, Day RO, Rich A. J Mol Biol. 1976;104:145. doi: 10.1016/0022-2836(76)90006-1. [DOI] [PubMed] [Google Scholar]
- 126.Gryaznov SM, Chen JK. J Am Chem Soc. 1994;116:3143. [Google Scholar]
- 127.Chen JK, Schultz RG, Lloyd DH, Gryaznov SM. Nucleic Acids Res. 1995;23:2661. doi: 10.1093/nar/23.14.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gryaznov SM, Lloyd DH, Chen JH, Schultz RG, DeDionisio LA, Ratmeyer L, Wilson WD. Proc Natl Acad Sci USA. 1995;92:5798. doi: 10.1073/pnas.92.13.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tereshko V, Gryaznov S, Egli M. J Am Chem Soc. 1998;120:269. [Google Scholar]
- 130.Rigl CT, Lloyd DH, Tsou DS, Gryaznov S, Wilson WD. Biochem. 1997;36:650. doi: 10.1021/bi961980w. [DOI] [PubMed] [Google Scholar]
- 131.Wilds CJ, Minasov G, von Matt P, Altmann KH, Egli M. Nucleosides, Nucleotides & Nucleic Acids. 2001;20:991. doi: 10.1081/NCN-100002475. [DOI] [PubMed] [Google Scholar]
- 132.Pallan PS, von Matt P, Wilds CJ, Altmann K-H, Egli M. Biochem. 2006;45:8048. doi: 10.1021/bi060354o. [DOI] [PubMed] [Google Scholar]
- 133.Wilds CJ, Maier MA, Tereshko V, Manoharan M, Egli M. Angew Chem. 2002;114:123. doi: 10.1002/1521-3773(20020104)41:1<115::aid-anie115>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 134.Wilds CJ, Maier MA, Manoharan M, Egli M. Helv Chim Acta. 2003;86:966. [Google Scholar]
- 135.Diop-Frimpong B, Prakash TP, Rajeev KG, Manoharan M, Egli M. Nucleic Acids Res. 2005;33:3965. doi: 10.1093/nar/gki823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Matulic-Adamic J, Beigelman L, Portmann S, Egli M, Usman N. J Org Chem. 1996;61:3909. doi: 10.1021/jo960091b. [DOI] [PubMed] [Google Scholar]
- 137.Minasov G, Matulic-Adamic J, Wilds CJ, Haeberli P, Usman N, Beigelman L, Egli M. RNA. 2000;6:1516. doi: 10.1017/s1355838200001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lewis FD, Liu X, Wu Y, Miller S, Wasielewski MR, Letsinger RL, Sanishvili R, Joachimiak A, Tereshko V, Egli M. J Am Chem Soc. 1999;121:9905. [Google Scholar]
- 139.Egli M, Tereshko V, Murshudov GN, Sanishvili R, Liu X, Lewis FD. J Am Chem Soc. 2003;125:10842. doi: 10.1021/ja0355527. [DOI] [PubMed] [Google Scholar]
- 140.Xia J, Noronha A, Li F, Rajeev KG, Akinc A, Braich R, Egli M, Manoharan M. Am Chem Soc Chem Biol. 2006;1:176. doi: 10.1021/cb600063p. [DOI] [PubMed] [Google Scholar]
- 141.Li F, Pallan PS, Maier MA, Rajeev KG, Mathieu SL, Kreutz C, Fan Y, Sanghvi J, Micura R, Rozners E, Manoharan M, Egli M. Nucleic Acids Res. 2007;35:6424. doi: 10.1093/nar/gkm664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Irimia A, Eoff RL, Pallan PS, Guengerich FP, Egli M. J Biol Chem. 2007;282:36421. doi: 10.1074/jbc.M707267200. [DOI] [PubMed] [Google Scholar]
- 143.Pallan PS, Kreutz C, Bosio S, Micura R, Egli M. RNA. 2008;14:2125. doi: 10.1261/rna.1078508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lin KY, Jones RJ, Matteucci MD. J Am Chem Soc. 1995;117:3873. [Google Scholar]
- 145.Burgin AB, Jr, Gonzalez C, Matulic-Adamic J, Karpeisky AM, Usman N, McSwiggen JA, Beigelman L. Biochem. 1996;35:14090. doi: 10.1021/bi961264u. [DOI] [PubMed] [Google Scholar]
- 146.Baidya N, Ammons GE, Matulic-Adamic J, Karpeisky AM, Beigelman L, Uhlenbeck OC. RNA. 1997;3:1135. [PMC free article] [PubMed] [Google Scholar]
- 147.Lewis FD, Letsinger RL, Wasielewski MR. Acc Chem Res. 2001;34:159. doi: 10.1021/ar0000197. [DOI] [PubMed] [Google Scholar]
- 148.Sinnokrot MO, Valeev EF, Sherrill CD. J Am Chem Soc. 2002;124:10887. doi: 10.1021/ja025896h. [DOI] [PubMed] [Google Scholar]
- 149.Morales JC, Kool ET. Nat Struct Biol. 1998;5:950. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 150.Kool ET, Sintim HO. Chem Commun. 2006:3665. doi: 10.1039/b605414e. [DOI] [PubMed] [Google Scholar]
- 151.Guckian KM, Schweitzer BA, Ren RX, Sheils C, Paris PL, Tahmassebi DC, Kool ET. J Am Chem Soc. 1996;118:8182. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]