

Abstract
DiGeorge critical region 8 (DGCR8) is essential for maturation of microRNAs (miRNAs) in animals. In the cleavage of primary transcripts of miRNAs (pri-miRNAs) by the Drosha nuclease, the DGCR8 protein directly binds and recognizes pri-miRNAs through a mechanism currently controversial. Our previous data suggest that DGCR8 trimerizes upon cooperative binding to pri-mir-30a. However, a separate study proposed a model in which a DGCR8 molecule contacts one or two pri-miRNA molecules using its two double-stranded RNA binding domains. Here, we extensively characterized the interaction between DGCR8 and pri-miRNAs using biochemical and structural methods. First, a strong correlation was observed between the association of DGCR8 with pri-mir-30a and the rate of pri-miRNA processing in vitro. Second, we show that the high binding cooperativity allows DGCR8 to distinguish pri-miRNAs from a nonspecific competitor with subtle differences in dissociation constants. The highly cooperative binding of DGCR8 to a pri-miRNA is mediated by the formation of higher-order structures, most likely a trimer of DGCR8 dimers, on the pri-miRNA. These properties are not limited to its interaction with pri-mir-30a. Furthermore, the amphipathic C-terminal helix of DGCR8 is important both for trimerization of DGCR8 on pri-miRNAs and for the cleavage of pri-miRNAs by Drosha. Finally, our three-dimensional model from electron tomography analysis of the negatively stained DGCR8–pri-mir-30a complex directly supports the trimerization model. Our study provides a molecular basis for recognition of pri-miRNAs by DGCR8. We further propose that the higher-order structures of the DGCR8–pri-miRNA complexes trigger the cleavage of pri-miRNAs by Drosha.
Keywords: microRNA processing, molecular recognition, RNA binding protein, DiGeorge syndrome, ribonuclease III
INTRODUCTION
MicroRNAs (miRNAs) are a class of non-protein-coding RNAs that function extensively in development and cell physiology (Bushati and Cohen 2007), and are involved in diseases such as cancer (Croce, 2009). They target specific gene expression through either translational repression or mRNA degradation (Filipowicz et al. 2008; Bartel 2009). Mature miRNAs are very short, only about 22 nucleotides (nt). In contrast, primary miRNAs (pri-miRNAs) are usually quite long and need to be processed through serial cleavage steps prior to their activation in gene regulation (Lee et al. 2002; Faller and Guo 2008; Kim et al. 2009). In the first step, a pri-miRNA is cleaved in the nucleus into an intermediate called the precursor miRNA (pre-miRNA) by the RNase III family enzyme Drosha (Lee et al. 2003). Drosha is assisted by the RNA binding protein DGCR8 (Denli et al. 2004; Gregory et al. 2004; Han et al. 2004; Landthaler et al. 2004). Drosha and DGCR8 copurify from cellular extracts and therefore are collectively called the Microprocessor complex (Denli et al. 2004; Gregory et al. 2004). The pre-miRNA is then exported by Exportin-5 to the cytoplasm, where it is further cleaved into a miRNA duplex by another RNase III enzyme, Dicer. The miRNA duplex is then incorporated into an effector complex called the miRNA-induced silencing complex (miRISC), or the mi-ribonucleoprotein (miRNP) complexes (Mourelatos et al. 2002), and is unwound into a single-stranded active form.
There are over 700 human miRNAs that have been confirmed experimentally and are registered in the miRBase database (Griffiths-Jones et al. 2008). The requirement for pri-miRNAs to undergo extensive processing raises an important question: How are their primary transcripts specifically recognized by the processing machinery? It is clear from bioinformatic analyses that no sequence is universally conserved to all human pri-miRNAs (Berezikov et al. 2005). Thus, the processing proteins have to recognize the structural features of pri-miRNAs. Most pri-miRNAs contain characteristic hairpin secondary structures about 30 base pairs (bp) in length (Lagos-Quintana et al. 2001; Lau et al. 2001; Lee and Ambros 2001; Lee et al. 2003). Previous characterizations showed that the hairpin loop should be longer than 9 nt (Zeng et al. 2005); the 6–11 nt immediate flanking a pri-miRNA hairpin (could be on either the 5′ or the 3′ side) should be unstructured (Zeng and Cullen 2005); and the cleavage site is determined mainly by the distance (∼11 bp) from the stem–ssRNA junction (Han et al. 2006). However, pri-miRNAs differ from each other with various types and locations of unpaired regions; and a lot of other RNA molecules also contain secondary structures that resemble those of pri-miRNAs (Lai et al. 2003; Lim et al. 2003). The question of how the DGCR8 and Drosha proteins specifically recognize pri-miRNAs remains largely unanswered.
DGCR8 is essential for processing of pri-miRNAs both in vitro and in vivo (Denli et al. 2004; Gregory et al. 2004; Han et al. 2004; Landthaler et al. 2004; Wang et al. 2007; Yi et al. 2009). Drosha does not cleave pri-miRNAs without it (Gregory et al. 2004). DGCR8 contains an N-terminal region required for nuclear localization, a central heme binding domain (HBD), two double-stranded RNA binding domains (dsRBDs), and a conserved C-terminal tail (here we named it CTT) that was shown to be required for co-immunoprecipitation (coIP) with Drosha (Fig. 1A; Yeom et al. 2006; Faller et al. 2007). C-terminal fragments of DGCR8, including the two dsRBDs and the CTT, are sufficient for directly binding pri-miRNAs and for triggering their cleavage by Drosha (Yeom et al. 2006; Faller et al. 2007). A crystal structure of the tandem dsRBDs of DGCR8 showed that the two dsRBDs interact with each other through hydrophobic interactions and hydrogen bonds and the RNA-binding surfaces of these domains point in opposite directions (Sohn et al. 2007). Based on these structural and biochemical data, Sohn et al. (2007) proposed an RNA recognition model in which each DGCR8 protomer binds to one or two pri-miRNA hairpins using both dsRBDs.
FIGURE 1.

The rate of pri-miRNA processing correlates with cooperative binding of DGCR8. (A) Schematic drawing of all the DGCR8 truncation constructs used in this study. The domain structure of the full-length DGCR8 is shown on the top as a reference. (B) Kinetic assays to measure the initial velocity of pri-mir-30a processing. The pri-mir-30a RNA uniformly labeled with [α-32P]UTP was annealed and incubated with recombinant Drosha (10–20 nM) and heme-bound NC1 (50 nM counting the number of protomers) for indicated time. (C) The accumulation of pre-mir-30a, as a processing product, was quantified, converted to the molar equivalent of pri-mir-30a using the ratios of U residues they contain, and was normalized to the amount of starting pri-mir-30a substrate. The initial rate of the reaction was the slope of the linear regression of the data. (D) The initial rates of pri-mir-30a processing were determined at a series of NC1 concentrations. The results are averages of two independent repeats, with bars representing the range of the rates. (E) The 45-min time points of the processing reactions in C were analyzed using a single denaturing polyacylamide gel. (F) The initial rates of pri-mir-30a processing were measured at a series of NC9 protein concentrations.
Our previous investigation suggested a different pri-miRNA recognition model (Faller et al. 2007). We found that a truncated DGCR8 construct containing the HBD, dsRBDs, and CTT (called NC1) (Fig. 1A), is expressed in Escherichia coli as both a heme-bound dimer and a heme-free monomer. The heme-bound NC1 dimer is more active than the heme-free monomer in reconstituted pri-miRNA processing assays. Interestingly, the heme-bound NC1 dimer seems to trimerize to form a hexamer upon binding to a pri-mir-30a RNA (150 nt), whereas the heme-free NC1 appears to form a cooperative dimer when associated with pri-mir-30a. In addition, a further truncated DGCR8 that contains the dsRBDs and CTT (residues 499–751) but not the HBD (called NC9) (Fig. 1A), is active in pri-miRNA processing and displays cooperativity and formation of higher-order structures upon binding to pri-miRNAs, similar to NC1. Therefore, trimerization of DGCR8 may be an important step in the processing of pri-miRNAs. However, there has been no structural study of DGCR8–pri-miRNA complexes so far, and the nature of this higher-order structural organization of DGCR8 in recognition of pri-miRNAs is unclear.
Here we systematically investigate the molecular mechanism of pri-miRNA recognition by DGCR8 using biochemical assays and electron tomography. First, we developed a procedure to quantify the pri-miRNA processing activity of DGCR8 in reconstituted assays. We show that binding of DGCR8 to pri-mir-30a correlates with processing in vitro. Second, the highly cooperative binding to pri-mir-30a allows DGCR8 to preferentially associate with this pri-miRNA over a nonspecific RNA even when the differences in their dissociation constants (KD) are very subtle. Third, the correlated pri-miRNA binding and processing properties of DGCR8 described above are not limited to pri-mir-30a. Furthermore, we reveal that a helix in the CTT of DGCR8 is required for highly cooperative binding of DGCR8 to pri-miRNAs and for their processing. Finally, the formation of this higher-order structure of DGCR8 in complex with pri-mir-30a is confirmed by electron tomography reconstructions of negative stained complexes. Our data suggest a model in which DGCR8 specifically recognizes pri-miRNAs through its cooperative trimerization upon association with the RNA, and the formation of proper higher-order structure of DGCR8–pri-miRNA complexes in turn triggers the cleavage of pri-miRNAs by Drosha.
RESULTS
Cooperative binding of DGCR8 to pri-miRNAs correlates with processing
To understand the molecular mechanism of pri-miRNA processing, we developed a procedure to measure the rate of pri-miRNA processing in reconstituted assays. Several groups previously reported in vitro processing of pri-miRNAs using Microprocessor complexes either reconstituted from recombinant Drosha and DGCR8 proteins or purified from human cells (Gregory et al. 2004; Han et al. 2004; Zeng and Cullen 2005). We overexpressed Drosha in insect cells and purified it to an extent that was nearly free of other nucleases. The recombinant Drosha protein, together with highly purified active DGCR8, displayed robust processing activity of pri-miRNAs that allowed us to perform semiquantitative kinetic measurement of these reactions. The pre-miRNAs, a product of pri-miRNA processing, accumulated nearly linearly within the initial 30 or 45 min, allowing the initial rate of the reaction to be measured (Fig. 1B,C).
To dissect the specific function of DGCR8, the initial rates of pri-miRNA processing were measured at different DGCR8 protein concentrations, with the Drosha concentration held constant (Fig. 1D). We found that the initial rate of processing increased from 0.003 min−1 to 0.013 min−1 when the NC1 (DGCR8276–751, the numbers in superscript represent the range of amino acid residues) concentration was increased from 10 nM to 50 nM. This jump of processing efficiency versus DGCR8 concentration could also be easily observed when the processing reactions were analyzed at a single 45-min time point (Fig. 1E). The increase of processing rate occurred in the same DGCR8 concentration range in which NC1 binds cooperatively to pri-mir-30a (K = 24 nM, where K is the protein concentration at which 50% of RNA is bound in a filter binding assay as reported previously) (Faller et al. 2007). A similar activation of processing as a function of DGCR8 concentration was also observed for another active DGCR8 construct NC9 (Fig. 1F). NC9 binds pri-mir-30a cooperatively with K = 22 nM. Cooperative binding of both active DGCR8 proteins to pri-miRNAs indicates that the DGCR8 proteins form higher-order structures upon association with a pri-miRNA (Faller et al. 2007). The fact that binding and processing of pri-mir-30a are activated in the same concentration range of DGCR8 suggests that the higher-order structure of a DGCR8–pri-miRNA complex could act as a trigger for the cleavage by the Drosha nuclease.
Specific recognition of pri-miRNAs through highly cooperative binding by DGCR8
To understand how DGCR8 distinguishes pri-miRNAs from nonspecific RNAs, we investigated its interaction with the P4-P6 domain of the Tetrahymena ribozyme, a self-splicing intron from the preribosomal RNA of Tetrahymena thermophila. The P4-P6 domain contains 160 nt, similar in size to the pri-mir-30a RNA. It contains extensive paired regions, tertiary interactions, as well as single-stranded regions (Fig. 2A). Our filter binding study showed that the heme-bound NC1 dimer binds the P4-P6 RNA with an affinity (K = 50 nM) only slightly lower that of the pri-mir-30a (Fig. 2B). However, the cooperativity of this binding reaction is reduced. The Hill plot indicated a straight line in the binding transition, with a Hill coefficient of 1.4 (Fig. 2C). Consistent with the Hill plot, the binding curve is best fit with a cooperative dimer model, in which two DGCR8 molecules bind each RNA cooperatively (Fig. 2B).
FIGURE 2.

Specific recognition of pri-miRNAs by DGCR8. (A) The secondary structure of the P4-P6ΔC209 RNA, which is used here to represent nonspecific RNAs. (B) Results of filter binding assays as a function of NC1 concentration. The data were fit using a cooperative dimer model, in which two NC1 molecules bind one RNA cooperatively. (C) Hill plot of the RNA binding data shown in B. Only the region in the binding transition is shown. (D) Competitive pri-mir-30a binding assays. Trace amount of 32P-labeled pri-mir-30a RNA was mixed with unlabeled competitor RNA, either the pri-mir-30a or the P4-P6 RNA, at various concentrations prior to the addition of the NC1 protein. (E) Schematic drawing of a variety of pri-miRNA fragments used in additional competitive pri-mir-30a binding assays. (F) Results of the competitive binding assays using unlabeled RNA shown in E.
Is DGCR8 capable of binding preferentially to the pri-miRNAs versus nonspecific RNAs, such as the P4-P6 domain? To address this question, we performed a competition binding experiment, in which a trace amount of 32P-labeled pri-mir-30a was mixed with unlabeled competitor RNAs, either pri-mir-30a or the P4-P6 RNA, prior to the addition of DGCR8 protein. In these assays, the DGCR8 protein was held at a constant concentration of 50 nM, above the K value (∼20 nM) for pri-mir-30a, so that the fraction of radiolabeled RNA bound to protein was around 90% in the absence of unlabeled RNA. As expected, the unlabeled pri-mir-30a competes efficiently with the fraction of radiolabeled RNA bound to protein, dropping to 50% at a pri-mir-30a concentration between 3 and 10 nM (Fig. 2D). In contrast, the P4-P6 RNA competed poorly with pri-mir-30a, with the fraction of 32P-labeled pri-mir-30a bound to protein remaining above 50% at 200 nM of P4-P6 RNA (Fig. 2D). This result demonstrated that DGCR8 has an extraordinary ability to distinguish RNAs with subtle differences in binding affinity. DGCR8 achieves this great molecular recognition through highly cooperative binding to pri-miRNAs.
What are the elements on pri-miRNAs that are essential for tight binding and specific recognition by DGCR8? This question was investigated by Kim and colleagues (Han et al. 2006) previously, using competitive binding assays based on UV cross-linking of Flag-tagged full-length DGCR8 expressed in mammalian cells and a radiolabeled pri-mir-16-1 RNA fragment. They found that the unlabeled miR-16 hairpin was unable to efficiently compete with pri-mir-16-1. Here we investigated this question using our quantitative competition assay and radiolabeled pri-mir-30a. The miR-30a hairpin competitor RNA contains the 81-nt stem–loop, but not the 25-nt and 44-nt stem–loops, in the 5′ and 3′ neighboring regions, respectively (Fig. 2E). The miR-30a hairpin was able to compete with pri-mir-30a to some extent but much less efficiently than the unlabeled pri-mir-30a. At least 75 nM of unlabeled miR-30a hairpin RNA was needed to reduce the fraction of 32P-labeled pri-mir-30a bound to protein to below 50% (Fig. 2F). The hairpin of miR-9-1 was even less efficient in the competition assays than the miR-30a hairpin (Fig. 2E, F). Our observations, along with these previous reports, suggest that the 5′ and 3′ regions beyond a miRNA hairpin are involved in the formation of higher-order complexes with DGCR8. In addition, the preferential binding to pri-miRNAs over miRNA hairpins demonstrated using both the full-length DGCR8 expressed in mammalian cells and the truncated heme-bound DGCR8 expressed in E. coli, indicating that the results described here using the latter protein preparation method are relevant to the physiological interactions.
It was shown that the junction of the hairpin and its immediately neighboring single-stranded regions are critical for recognition by the DGCR8 protein (Han et al. 2006). We were curious whether a simple junction like the miR-30a junction RNA shown in Figure 2E would be sufficient for tight binding and recognition by DGCR8. The pri-mir-30a junction RNA contains ∼20 bp of the bottom portion of the hairpin and ∼10 nt of the flanking single-stranded regions. The junction formed readily upon annealing of the two strands, as indicated by size exclusion chromatography (SEC) of either strand or their complex (Supplemental Fig. S1). Our result showed that the pri-mir-30a junction RNA was incapable of competing with pri-mir-30a even at 200 nM concentration (Fig. 2F). Thus, DGCR8 must make extensive contacts with a pri-miRNA, which likely cover regions further beyond those previously recognized as critical for processing.
DGCR8 cooperatively associates with pri-mir-21, likely through trimerization
Could the highly cooperative binding of DGCR8 to pri-mir-30a and their formation of higher-order structures be features to other pri-miRNAs? To answer this question, we first screened through a number of pri-miRNA fragments using in vitro processing assays and looked for those that are efficiently processed. Surprisingly, all the short pri-miRNA fragments, which typically include less than 30 nt on each side of the pre-miRNA region, were not processed as efficiently as pri-mir-30a in our assays. Together with the competitive DGCR8-binding assays described in the previous section, our results suggest that the regions on a pri-miRNA beyond those previously reported to be important for processing may enhance the efficiency of processing through strengthening the interaction with DGCR8. Additional cofactors might also be required for processing different subsets of miRNAs (Guil and Caceres 2007; Trabucchi et al. 2009).
In a test of longer pri-miRNA fragments, we identified a pri-mir-21 construct as being efficiently processed in our reconstituted assays (Fig. 3A). This pri-mir-21 contained the precursor (pre-mir-21, 59 nt), 44 nt in the 5′ neighboring region of pre-mir-21 and 37 nt on the 3′ side of pre-mir-21. Similar to pri-mir-30a, the processing of pri-mir-21 depended on the presence of both Drosha and DGCR8 proteins. A recent study showed that the mature miR-21, unlike most other miRNAs, is only partially depleted by the knockout of either DGCR8 or Dicer, suggesting that there might be an alternative maturation pathway for miR-21 (Yi et al. 2009). Our data indicate that pri-mir-21 can be processed by the Microprocessor complex, like most other pri-miRNAs.
FIGURE 3.

Processing of pri-mir-21 in a DGCR8-dependent fashion and trimerization of DGCR8 upon association with pri-mir-21. (A) Reconstituted pri-mir-21 processing assays. The reactions were incubated for 45 min at 37°C. Recombinant Drosha protein (10–20 nM) was present in all reactions, except the “no Drosha” sample, which only contained 100 nM of NC1. (B) Results of the filter binding assays. The upper panel indicates the fitting of the data using a cooperative trimer model, in which three NC1 molecules bind one RNA cooperatively. The lower panel shows the fitting using a cooperative dimer model. (C) Hill plot of the RNA binding data shown in B. Only the region in the binding transition is shown. (D) The molecular weight and protein–RNA ratio of the NC1–pri-mir-30a complex were estimated using SEC. The left y-axis indicates the absorption at 260, 280, and 450 nm. The right y-axis indicates the molar ratios of NC1 and the pri-mir-21 RNA estimated using either the A260/A280 ratios (shown in green) or based on the A450 values (red). (E) The SEC chromatogram of the pri-mir-21 RNA in the absence of proteins.
The interaction between DGCR8 and pri-mir-21 was characterized using filter binding assays and SEC. The filter binding assays showed that the heme-bound NC1 binds pri-mir-21 with high cooperativity (Fig. 3B), similarly to pri-mir-30a. The data were best fit using the cooperative trimer model (described above) with an R2 of 0.9985. The R2 value is a fraction between 0.0 and 1.0 and quantifies goodness of fit. In contrast, fitting using the cooperative dimer model gave a lower R2 (0.9915) and the fitted curve systematically deviated from the data: predicting more RNA bound to NC1 in the lower half of the binding transition and less RNA bound to protein in the higher half of the binding transition (Fig. 3B, lower panel). The Hill plot of the data indicated a straight line in the range of the binding transition (Fig. 3C), further demonstrating that the binding cooperativity is high. The slope of the fitted line (Hill coefficient, n) is 2.7 ± 0.2, consistent with DGCR8 forming trimer or higher oligomers upon association with pri-mir-21. The x-intercept of the Hill plot was used to calculate the K value as 13 ± 2 nM, slightly lower than that of pri-mir-30a. The high affinity of DGCR8 for pri-mir-21 provides an explanation of why pri-mir-21 is efficiently processed even at 10 nM NC1 concentration (Fig. 3A).
The filter binding assay does not directly detect the higher-order structures (an RNA molecule is measured as either bound to protein or free). In principle, the Hill coefficient provides the lower limit of the oligomerization state of the protein. SEC complements the filter binding assays and provides information regarding the stoichiometry of DGCR8 and pri-miRNA in four independent ways. First, the NC1–pri-mir-21 complex eluted from the column as a single peak (Fig. 3D), suggesting that it is present as a single species. Second, the complex eluted at a larger volume (9.66 mL) than the 480-kDa apoferritin (8.34 mL), indicating that the molecular weight of the complex is lower than 480 kDa. This result rules out the possibility that each NC1–pri-mir-21 complex contains more than one RNA molecule and the corresponding number of NC1 protomers (six times the number of RNA molecules). Based on a standard curve, the elution volume of the complex provides a rough estimate of its molecular weight, 332 kDa, consistent with that prediction based on the cooperative trimer model (370 kDa). While estimation of molecular weights based on elution volumes can be complicated by the nonglobular shapes of the macromolecules, the following two methods take advantage of the UV-vis absorption of the complex at different wavelengths and they are independent of the shapes of protein–RNA complexes. Third, the A260/A280 ratio was used to estimate the molar ratio of the protein and RNA in their complexes in SEC peaks (details described in Materials and Methods). The molar ratio of NC1 and pri-mir-21 calculated based on A260/A280 ratios remains around 5.5 across the peak of the NC1–pri-mir-21 complex (Fig. 3D, green curve) close to the value of 6.0 expected for the cooperative trimer model. Fourth, the absorption at 450 nm in the chromatogram of the NC1–pri-mir-21 complex resulted from the heme cofactor bound to NC1 and provided an independent estimate of the NC1 protein concentration. After the A260 contributed by NC1 was subtracted, the pri-mir-21 RNA concentration was calculated. The molar ratio of NC1 and pri-mir-21 was calculated to be around 5.0 across the peak (Fig. 3D, red curve), again consistent with the cooperative trimer model within experimental errors. Due to the lower reading of A450, this method is less accurate than that based on A260/A280 ratios in the conditions used in this study.
In summary, our RNA-binding assays indicate that the NC1 dimer likely trimerizes upon association with pri-mir-21, and this binding correlates with efficient processing of pri-mir-21 by Drosha, similar to the data for pri-mir-30a. Thus, the formation of DGCR8 trimers (of dimers) upon association with pri-miRNAs is not confined to a single pri-miRNA but could be a general feature for miRNA biogenesis in animals. Our trimerization model depends on the assumption that the NC1 dimer remains stable in the process of binding to pri-miRNAs. Our data do not rule out the possibilities that DGCR8 and pri-miRNA are present in their complexes at molar ratios subtly different from 6:1, e.g., 5:1.
The C-terminal helix of DGCR8 is important for its trimerization upon binding to pri-miRNAs
To further understand how DGCR8 binds to pri-miRNAs to form cooperative trimers (of dimers), we examined two DGCR8 truncation constructs. One construct (dsRBD1 or DGCR8499–589) contained the sequence for the first dsRBD, and the other (dsRBD2-CTT, or DGCR8590–751) contained the sequences of the second dsRBD and the CTT of DGCR8 (Fig. 1A). Using the filter binding assays, we found that dsRBD1 did not strongly bind to pri-mir-30a within the protein concentration range tested (up to 15 μM), whereas dsRBD2-CTT associated with pri-mir-30a with a moderate affinity (K = 262 nM) (Fig. 4A). Importantly, dsRBD2-CTT retained the high cooperativity of binding to pri-mir-30a, with a Hill constant of 4.1 (Fig. 4B). Although the Hill constant of dsRBD2-CTT was higher than that (3.0) of the NC1 dimer, these results encouraged us to further examine the region containing residues 590–751 to look for structures important for cooperative binding to pri-miRNAs. In reconstituted pri-miRNA processing assays, dsRBD1 was inactive to support the cleavage (data not shown), whereas dsRBD2-CTT was able to trigger, albeit very weakly, the cleavage of pri-mir-30a by Drosha at a protein concentration (500 nM) higher than its K value for cooperative binding to this RNA (Fig. 4C). This result provides additional support to the model that cooperative binding of DGCR8 to pri-miRNA is essential for triggering the cleavage reaction by Drosha.
FIGURE 4.

The dsRBD2 and CTT of DGCR8 retain the ability to bind cooperatively to pri-mir-30a. (A) Filter binding assays of DGCR8590–751. The data were fit using the cooperative trimer model. (B) Hill plot of the data shown in A. Only the region in the binding transition is indicated. (C) Weak but detectable pri-mir-30a processing activity of DGCR8590–751 at a protein concentration above the binding transition.
We next estimated the contribution of the conserved CTT of DGCR8 to its pri-miRNA binding affinity and cooperativity by generating a series of C-terminal truncations in the context of NC1 (with no affinity tag): DGCR8276–700, DGCR8276–726, and DGCR8276–738 (Fig. 1A). These mutants were expressed in E. coli as heme-bound forms and analyzed using pri-miRNA binding and processing assays (Fig. 5). Consistent with the previous report that residues 739–750 are required for pri-miRNA processing and for interaction with Drosha (Yeom et al. 2006), all three truncated DGCR8 constructs failed to reconstitute the Microprocessor activity (data not shown). Our filter binding assays showed that these mutants bound to the pri-mir-30a RNA with affinities similar to, or higher than, that of wild-type NC1 dimer (Fig. 5A), suggesting that residues 701–751 of DGCR8 are not required for high-affinity binding to pri-miRNAs. However, we notice that the truncated DGCR8 proteins bind the pri-mir-30a with reduced cooperativity (Fig. 5A,B): The Hill constants were reduced to ∼2, and their binding data were best fit with a cooperative dimer model, in which two NC1 dimers bind to one pri-mir-30a cooperatively. This evidence raised an intriguing possibility that these C-terminal truncations of DGCR8 may have failed to form the proper higher-order structure upon association with pri-miRNAs and consequently failed to trigger their cleavage by Drosha.
FIGURE 5.

The C-terminal helix of DGCR8 is required for trimerization of DGCR8 upon association with pri-mir-30a. (A) The values of K, Hill coefficient, and pri-mir-30a processing activity of the C-terminal truncation constructs. (B) Filter binding results of DGCR8276–700 are shown as an example. The panel on the left is the binding data fit with a cooperative dimer model. Hill plot of the data in the binding transition is indicated on the right. (C) The residues in the α729–749 helix are plotted on an helical wheel. The conserved residues are drawn in bold. Sequence alignment of DGCR8 homologs in this helical region is shown on the right. (D) Reconstituted pri-mir-30a processing assays. The NC1 mutants are present at 50 nM concentration. The reactions were incubated for 45 min at 37°C. (E) Summary of the pri-mir-30a binding results.
Our analysis of the DGCR8 primary sequence predicted two α-helices in the conserved C-terminal region: α715–724 and α729–749 (Rost et al. 2004). The truncation analyses described above showed that α729–749 is required for trimerization of DGCR8 upon association with pri-miRNAs. Thus, we further examined the contribution of residues within α729–749 to the highly cooperative binding of DGCR8 to pri-miRNAs. Threading of the amino acid sequence of DGCR8 onto a helical wheel showed that this helix is amphipathic, with the hydrophobic residues being most conserved among DGCR8 homologs (Fig. 5C). This observation suggests that the hydrophobic patch on α729–749 may be important for the addition of the third NC1 dimer to pri-miRNAs. To test this conjecture, we constructed four single point mutations by replacing the conserved hydrophobic residues Leu732, Leu735, Met739, and Leu742 with alanine; again, in the context of NC1. Our initial pri-mir-30a processing assays showed that the L732A, L735A, and M739A mutants were nearly completely inactive, whereas the activity of the L742A mutant was comparable to that of the wild-type NC1 dimer (Fig. 5D). Our subsequent pri-miRNA binding assays showed no pronounced mutational effects on the affinity between DGCR8 and pri-mir-30a but did show a correlation between cooperative binding of the DGCR8 mutants to pri-mir-30a and the processing of this RNA (Fig. 5D,E). The Hill coefficients of binding of the inactive L732A, L735A, and M739A mutants are 2.0, 2.2, and 1.9, respectively, whereas that of the active L742A is 2.7. Consistent with the Hill coefficients, the binding curves of the L742A mutant fit better with the cooperative trimer model, whereas the curves of other mutants generally fit better with the cooperative dimer model (data not shown). Thus, we identified a hydrophobic patch of α729–749, containing L732, L735, and M739, to be important for trimerization of DGCR8 upon binding to pri-miRNAs. Since the C-terminal truncation and point mutants of DGCR8 dimerize readily upon binding to pri-mir-30a, the α729–749, the helix is likely to be necessary for addition of the third DGCR8 subunit to pri-miRNAs.
Electron tomography of the DGCR8–pri-mir-30a complex
We next examined the three-dimensional (3D) structure of DGCR8 in complex with the 150-nt pri-mir-30a RNA, using electron tomography. Heme-bound NC1 was incubated with pri-mir-30a, and the complex was further purified using SEC prior to their staining and fixation on EM grids using 2% uranyl acetate. The NC1–pri-mir-30a complex particles were largely homogenous (Fig. 6A). After the tomogram reconstructions of the negatively stained DGCR8–RNA complexes were obtained (Fig. 6B,C), individual particles were segmented out for comparison. We observed that the major species had a size of ∼20 nm in diameter (Fig. 6D), consistent with the expected 372-kDa complex. Particle-to-particle fitting showed a reasonable correlation between the particles, indicating a common structural arrangement (Fig. 6C). We further carried out subtomographic averaging to improve the signal-to-noise ratio and to minimize structural distortions caused by the missing wedge problem inherent in electron tomography (Fig. 6D).
FIGURE 6.

Three-dimensional (3D) structural analysis of the DGCR8–pri-mir-30a complex by electron tomography. (A) Representative tilt images from an electron tomography single tilt series of 141 images, showing the negatively stained DGCR8–pri-mir-30a complex particles imaged from −70° to +70°. Each tilt series was acquired with an angular interval of one degree of tilt between images. (B) A slice through the 3D tomogram reconstructed from the tilt series shown in A after alignment. (C) Illustration of subtomographic volume averaging of individual DGCR8–pri-mir-30a complex particles. Two particles selected from the 3D tomogram as indicated in B were segmented out, low pass filtered, and rotated to create particles suitable for averaging. Shown here are single pixel (7.4 Å) slices at three different z-heights of two particles segmented from the reconstruction in B and aligned in 3D. Particles aligned this way were averaged together to generate an averaged 3D density map of the complex to improve signal/noise ratio and to minimize distortions due to the missing wedge problem intrinsic to electron tomography. (D) Surface representation of the averaged density map of DGCR8–pri-mir-30a complex. As a comparison of sizes and shape, we also show a model of the double-stranded region of miR-30a hairpin and the crystal structure of the dsRBDs of DGCR8 (Sohn et al. 2007).
The averaged 3D density of the DGCR8–pri-mir-30a complex is ∼200 Å in height, 160 Å in width, and 110 Å in depth (Fig. 7A). There is sufficient space to accommodate the expected DGCR8 and RNA molecules, including a 30-bp pri-mir-30a hairpin and six copies of the dsRBD1-dsRBD2 domains (the crystal structure was previously determined) (Sohn et al. 2007), as well as the remaining residues in DGCR8 and pri-mir-30a (Figs. 6D, 7B). The DGCR8–RNA complex has an overall shape of a butterfly (Fig. 7A). A spine runs down the center of the particle with large, globular densities (the wings) on either side. Each wing is divided into two lobes that are connected to each other. The upper lobe (the forewing) is smaller than the lower lobe (the hind wing). The back of the complex has a concave surface with a central cleft formed by the spine and the globular “wing” lobes. Finally, on each side of the central cleft of the complex, there is a region absent of density located between the spine and the small and large lobes (Fig. 7A).
FIGURE 7.

Structural organization of the DGCR8–pri-mir-30a complex. (A) Surface representation of the averaged density map viewed from three orthogonal directions. (B) Plausible arrangement of protein molecules and RNA in the complex based on the dimension and volume of individual components. We suggest that the RNA helix is located in the central region of the complex. Six copies of the crystal structure of DGCR8 dsRBDs are placed in the density, solely for the purpose of demonstrating that there is sufficient room in the electron density for three DGCR8 dimer subunits and the RNA strand.
The 3D electron tomography reconstruction strongly supports the cooperative trimerization model in which six DGCR8 subunits, contributed by three dimeric NC1 molecules, bind one pri-miRNA and form a complex with specific higher-order structure, which in turn triggers the cleavage by the Drosha nuclease. Each DGCR8 subunit contains two RNA binding domains. A previous mutagenesis study showed that both dsRBDs of DGCR8 likely contact pri-miRNAs directly (Sohn et al. 2007). Thus, DGCR8 must make extensive direct interactions with the pri-mir-30a RNA in the complex. We propose that the pri-mir-30a hairpin is located in the spine of the complex (Fig. 7B). The location of the DGCR8 subunits cannot be determined. The Drosha nuclease could bind to the DGCR8–pri-miRNA complex either in the cleft or on the opposite surface of the complex.
DISCUSSION
In this study, we focus on the role of the higher-order structure that DGCR8 forms upon association with pri-miRNAs in the Drosha cleavage step of miRNA biogenesis. Using quantitative binding and processing assays, we show that cooperative binding of DGCR8 to pri-mir-30a and processing coincide at the same DGCR8 concentration range, suggesting that the formation of a proper higher-order structure may serve as a trigger for cleavage of pri-miRNAs. The highly cooperative binding of DGCR8 to pri-miRNAs allows it to distinguish a pri-miRNA from other dsRNA-containing nonspecific RNAs with subtle differences in binding affinity. The trimerization of DGCR8 upon association with pri-miRNAs appears to be a feature common to at least a subset of miRNAs in animals. The C-terminal amphipathic helix of DGCR8 is required for the addition of the third DGCR8 molecule to pri-miRNAs. Electron tomogram reconstruction of the NC1–pri-mir-30a complex demonstrated a butterfly-shaped complex with dimensions consistent with the cooperative trimer model. It is likely that the pri-miRNA hairpin is located at the central spine of the complex with extensive contacts made by the DGCR8 protein.
The cleavage of pri-miRNAs by Drosha represents their entry of the maturation pathway (Faller and Guo 2008; Kim et al. 2009). At this step of miRNA processing, it is critical to make sure that only authentic pri-miRNAs enter the pathway. Otherwise, other RNAs could be cleaved by Drosha, resulting in undesired effects on their expression and biological functions. DGCR8 is the RNA binding partner of Drosha, whose major function is to specifically recognize the pri-miRNAs and trigger the cleavage by Drosha. Interestingly, our biochemical measurements show that pri-miRNAs have affinities only slightly higher than that of a nonspecific RNA that also contains double-stranded structures and junctions between double- and single-stranded regions (Fig. 2). Here we propose a simple model (called the cooperative recognition model) that provides an explanation of how DGCR8 recognizes pri-miRNAs through cooperative binding and formation of higher-order structures from three perspectives (Fig. 8). First, the highly cooperative binding of DGCR8 to a pri-miRNA results in a very sharp binding transition over the protein concentration (Figs. 3, 8A, upper panel). The pri-miRNAs, with K values lower than the active DGCR8 concentration in cells, are recognized and processed by the Microprocessor complex. On the other hand, nonspecific RNAs are unable to compete with pri-miRNAs in binding to DGCR8 even if their affinity for DGCR8 is only slightly lower than those of pri-miRNAs. Second, the highly cooperative binding of DGCR8 to pri-miRNAs is mediated by the formation of higher-order structures (Fig. 8B). The higher-order structure results in a more extensive protein–RNA interface, allowing more features on a pri-miRNA to be recognized by DGCR8. Third, higher-order complexes formed by DGCR8 and pri-miRNAs likely serve as the structural platform to activate the cleavage of pri-miRNAs by Drosha. The requirement for a pri-miRNA to form a proper structure in complex with DGCR8 during processing further ensures that only pri-miRNAs will enter the miRNA processing pathway. Therefore, the cooperative binding of DGCR8 upon association with pri-miRNAs and the formation of a higher-order DGCR8–pri-miRNA complex structure are the major molecular bases for recognition of pri-miRNAs. Note that this simplified model does not take into account the contribution of pri-miRNA recognition by Drosha and other RNA binding proteins that regulate the processing of specific miRNA families such as snRNP A1 (Guil and Caceres 2007), Lin28 (Heo et al. 2008; Newman et al. 2008; Viswanathan et al. 2008), and KSRP (Trabucchi et al. 2009). The recognition of a particular pri-miRNA could be more complex.
FIGURE 8.

Models of how DGCR8 recognizes pri-miRNAs. (A) The cooperative recognition of pri-miRNAs by DGCR8. (B) Formation of proper higher-order structure of DGCR8–pri-miRNA complexes. For details, see the main text.
Although our cooperative recognition model differs from that proposed by Sohn et al. (2007), our studies revealed some common features of the interaction between DGCR8 and pri-miRNAs. In their electrophoresis mobility shift assays (EMSAs), the binding of DGCR8M (residues 493–738) to pri-miR-16 is cooperative, with a Hill coefficient of 2. The Hill coefficient of their binding reaction is very similar to what we observed for DGCR8276–738 and pri-miR-30a (Fig. 5A). Interestingly, their DGCR8M and our DGCR8276–738 have identical C termini and are inactive in pri-miRNA processing due to the C-terminal truncation, but they differ in the presence of the HBD in DGCR8276–738. In addition, both studies show that inclusion of residues 739–750 (DGCR8L in Sohn et al. 2007) or 739–751 (in our study) does not cause large changes in affinity for pri-miRNAs (Fig. 5A). We report here that the presence of residues 739–751 facilitates the addition of the third DGCR8 molecule to a pri-miRNA. Unfortunately, the Hill coefficient of DGCR8L binding to pri-mir-16 was not provided (Sohn et al. 2007). The fact that both studies—using different DGCR8 constructs, different pri-miRNAs, and different methods at different conditions (such as salt concentrations)—demonstrated cooperative binding of DGCR8 to pri-miRNAs strongly argues that models of higher-order DGCR8–RNA complexes have to be considered.
Our cooperative recognition model predicts that the recognition of pri-miRNAs may work the best if the active DGCR8 concentration in cells is controlled at a level between its KD values for pri-miRNAs and nonspecific RNAs. This is consistent with the previous gene expression profiling showing that the DGCR8 mRNA is present at relatively low levels across different tissues and cell types (Shiohama et al. 2003; Thomson et al. 2006). Reduced expression of DGCR8 through RNA interference enhances cellular transformation and tumorogenesis (Kumar et al. 2007). Heterozygous deletion of the Dgcr8 gene results in abnormal miRNA biogenesis in mouse brains (Stark et al. 2008). Recent reports showed that the Microprocessor complex could cleave the miRNA-like hairpins in the mRNA of DGCR8, thus regulating its stability and the expression level of the DGCR8 protein (Han et al. 2009; Triboulet et al. 2009). In addition, the requirement for a steady DGCR8 activity may be assured through activation of its pri-miRNA processing activity by heme (Faller et al. 2007; Senturia et al. 2010). We showed previously that the heme-bound DGCR8 protein is more active in pri-miRNA processing in vitro than the heme-free form (Faller et al. 2007). More recently, we revealed that three heme-binding deficient mutants of DGCR8 are all defective in miRNA processing in cultured HeLa cells (M Gong, R Senturia, and F Guo, in prep.). Although heme contents vary greatly among animal tissues, it is well established that heme biosynthesis and degradation are tightly controlled through feedback mechanisms (Ponka, 1999). Free heme molecules activate oxygen molecules to generate reactive oxygen species and cause oxidative stress to cells. It is possible that DGCR8 uses heme availability as a reference to keep its own activity consistently low and to ensure proper pri-miRNA recognition through its cooperative binding.
In this study, we show that a hydrophobic patch in the C-terminal amphipathic helix α729–750 is required for trimerization of DGCR8 upon binding to pri-miRNAs. Yeom et al. (2006) showed that the C-terminal half of the same helix, including residues 739–750, was critical for coIP of DGCR8 with Drosha. These intriguing observations further support a direct link between the assembly of a higher-order DGCR8 structure on pri-miRNAs and the cleavage of pri-miRNAs by Drosha. The Drosha protein concentration was estimated to be ∼10 nM in these assays (data not shown). Thus, a large excess of DGCR8 protein (50–300 nM) over Drosha surprisingly does not inhibit the pri-miRNA processing (Figs. 1D,F, 3A), as would be predicted by a model in which the Drosha/DGCR8 complex (the Microprocessor) is the only active species. It is possible that the higher-order complexes of DGCR8 and pri-miRNAs recruit Drosha or the Drosha/DGCR8 complex for cleavage under these experimental conditions.
Finally, the reconstituted pri-miRNA processing seems to be a rather inefficient reaction. Using semiquantitative reconstituted pri-miRNA processing assays, we measured the rate of cleavage of pri-mir-30a at various DGCR8 protein concentrations and found it to plateau at about 0.01 min−1 (Fig. 1D,F). This observation parallels the slow cleavage activity (0.5–1.0 mol of product per mole of enzyme in 30 min) of Dicer, another ribonuclease III family member that cleaves pre-miRNAs or dsRNAs into 21-nt duplexes (Zhang et al. 2002). The cleavage activity of Dicer is enhanced by the RNA protein TRBP (J Doudna, pers. comm.). Possibly, other RNA-binding proteins or helicases increase the pri-miRNA cleavage activity of Drosha in similar fashions.
MATERIALS AND METHODS
Plasmids, RNAs, and proteins
All the DGCR8 expression constructs were cloned into the pET-24a vector (Novagen) between NdeI and EcoRI sites. Site-directed mutagenesis was carried out using the standard 4-primer PCR method (Ausubel et al. 2007). All the pri-miRNA plasmid transcription templates were cloned, along with a T7 promoter, into the pUC19 vector using PCR methods, in which the human genomic DNA was used as a template. The coding sequences of all plasmids were verified by sequencing. The 32P-labeled pri-miRNAs and the unlabeled RNAs were transcribed in vitro using the T7 RNA polymerase. Synthetic DNA templates were used to transcribe the pri-mir-30a junction RNA. The concentrations of the unlabeled RNAs were determined using their absorbance at 260 nm.
The recombinant Drosha and NC1 proteins (wild-type, truncation, and point mutants) were expressed and purified as described (Faller et al. 2007). The DGCR8499–589 and DGCR8590–751 proteins were overexpressed and purified using the same procedures as that of NC1. These two proteins were concentrated in 20 mM Tris (pH 8.0), 400 mM NaCl, and 1 mM dithiothreitol (DTT).
pri-miRNA binding and processing assays
The filter binding assays, SEC characterization of the DGCR8–pri-miRNA complexes, and reconstituted pri-miRNA processing assays, were performed essentially the same as described previously (Faller et al. 2007). In the kinetic processing assays, recombinant Drosha and DGCR8 were preincubated with Mg2+, and the reactions were started by the addition of pri-miRNAs, which were uniformly labeled through addition of α-32P-UTP to the transcription reactions. At the indicated time points, the reactions were quenched using a 2× RNA loading buffer containing 20 mM of EDTA and 10 M of urea. The pri-miRNA substrates and their cleavage products were resolved using 15% denaturing PAGE, visualized using autoradiography, and quantified using either the program Quantity One (version 4.4.1, Bio-Rad), or ImageQuant (version 5.2, GE Healthcare). Nonspecific nuclease activity contributed significantly to the degradation of pri-miRNAs and the reaction products at later time points, and compromised their use in a complete reaction curve fitting (data not shown). The signals from the pre-miRNAs were used to calculate the initial rate of processing through conversion to pri-miRNAs, followed by normalization to the amount of starting substrate and linear regression. Since the pri-miRNAs were uniformly label using α-32P-UTP, the signal intensity of a pre-miRNA can be converted to that of its corresponding pri-miRNA by multiplying the ratio of U residues in the pri-miRNA and the pre-miRNA (2.625 for pri-mir-30a processing). The data were plotted and fit using the program PRIsM (version 4.0, GraphPad).
The R2 value given in the nonlinear regression for the filter binding data is defined as R2 = 1 − (SSreg/SStot), where SSreg is the sum-of-squares (SS) of the vertical distances of the data points from the fitted curve, and SStot is the SS of the distances of data points from a horizontal line through the mean of all Y values.
In the SEC characterization of the NC1–pri-mir-21 complex, the A260/A280 ratio (R) was used to estimate the molar ratio of NC1 and pri-mir-21. The R value of NC1 was measured to be 0.683, and that of pri-mir-21 was 2.72 (Fig. 3E; data not shown). The A260/A280 ratio of their complex is ∼1.98. The molar ratio of the protein and RNA in their complexes ([protein]/[RNA], where [protein] and [RNA] represent the concentrations of protein and RNA, respectively) was calculated using the following equation as described previously:
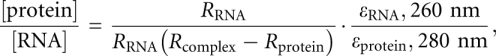 |
where εRNA, 260 nm is the extinction coefficient of the RNA, which is 1.22 × 106 M−1 cm−1 for the pri-miR-21 RNA, and εprotein, 280 nm is the extinction coefficient of the protein, which is 4.72 × 104 M−1 cm−1 for a NC1 protomer (half a dimer).
Tomography data acquisition
Electron tomography tilt series were collected on an FEI Tecnai TF20 instrument with an accelerating voltage of 200 kV as previous described (Peng et al. 2010). A Gatan cryoholder with a tilt range of −70 to +70 degrees was used and a 16-megapixel CCD camera was used to acquire the images. FEI Batch Tomography software was used to set up and acquire each tomography tilt series. The magnification used was 40,600×, with 2× binning, giving a CCD sampling pixel size of 7.4 Å/pixel. The underfocus value of the zero-tilt image was set to 3 μm.
Image processing and 3D reconstruction
Each saved tilt series was processed with a variety of programs to generate a 3D reconstruction. We started with Inspect3D to quickly obtain an initial rough alignment (translation, rotation, and tilt axis) of the tilt series. Inspect3D has a good graphical interface to allow visual inspection of the progress. The stacks were aligned using cross-correlation of the filtered tilt images. The filter was set up with the correct high- and low-pass limits to get a good cross-correlation peak in order to give good results. After alignment, the tilt axis was adjusted so that it was vertical and centered. This process was repeated a few times until there were no further significant changes in the tilt parameters. The aligned tilt series was then used to make an initial 3D reconstruction using the SIRT reconstruction algorithm in Inspect3D using GPU processing. Further refinements were performed by the ProTomo package (Winkler and Taylor 2006) in Linux computer clusters. The tilt series were unstacked and cleaned to remove any outlier hot pixels. The cleaned images were then precisely aligned iteratively by using ProTomo's refine and super-refine scripts until there was no detectable improvement in the alignment parameters. This process took about 1 d of a single core's computation time for each tilt series. The refined alignments were then used to reconstruct 3D models from the images by using a weighted back projection algorithm. The 3D reconstructions were saved as a stack of x-y plane images that are single pixel slices along the z-plane.
3D visualization
The 3D reconstruction stack was then manipulated and processed using 3D imaging and processing software. We utilized the UCSF Chimera (Pettersen et al. 2004) and Amira (VSG3d.com) for 3D imaging and used the EMAN package (Ludtke et al. 1999) for processing and averaging. Chimera was used to view the 3D density map of the whole reconstruction. It was then utilized to crop sections from the map, and the sections were segmented using Amira. Particles that were segmented out were then compared and aligned in Chimera. The particles were oriented and aligned by fitting them inside one another (Fit-Map-to-Map). The rotation and translation parameters were then recorded and applied using Proc3D in the EMAN software package to make each particle align on top of one another in the correct orientation. Once the particles were aligned and oriented, they were averaged using avg3d (ZH Zhou, unpubl.), which averages the density of all of the particles. The outputs of averages of different sets of particles were compared visually using the UCSF Chimera. Several different sets of particles were averaged, each giving similar results.
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
This project is supported in part by grants from the National Institutes of Health (GM080563 to F.G.; GM071940 and AI069015 to Z.H.Z.) and Abraxis BioScience, Inc. We acknowledge the use of the cryoEM facility at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057 to Z.H.Z.).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2111310.
REFERENCES
- Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, Struhl K 2007. Current protocols in molecular biology Wiley-Interscience, New York [Google Scholar]
- Bartel DP 2009. MicroRNAs: Target recognition and regulatory functions. Cell 136: 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E 2005. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120: 21–24 [DOI] [PubMed] [Google Scholar]
- Bushati N, Cohen SM 2007. MicroRNA functions. Annu Rev Cell Dev Biol 23: 175–205 [DOI] [PubMed] [Google Scholar]
- Croce CM 2009. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10: 704–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ 2004. Processing of primary microRNAs by the Microprocessor complex. Nature 432: 231–235 [DOI] [PubMed] [Google Scholar]
- Faller M, Guo F 2008. MicroRNA biogenesis: There's more than one way to skin a cat. Biochim Biophys Acta 1779: 663–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller M, Matsunaga M, Yin S, Loo JA, Guo F 2007. Heme is involved in microRNA processing. Nat Struct Mol Biol 14: 23–29 [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N 2008. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat Rev Genet 9: 102–114 [DOI] [PubMed] [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R 2004. The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235–240 [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ 2008. miRBase: Tools for microRNA genomics. Nucleic Acids Res 36: D154–D158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guil S, Caceres JF 2007. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol 14: 591–596 [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN 2004. The Drosha–DGCR8 complex in primary microRNA processing. Genes Dev 18: 3016–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN 2006. Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 125: 887–901 [DOI] [PubMed] [Google Scholar]
- Han J, Pedersen JS, Kwon SC, Belair CD, Kim YK, Yeom KH, Yang WY, Haussler D, Blelloch R, Kim VN 2009. Post-transcriptional cross-regulation between Drosha and DGCR8. Cell 136: 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Cho J, Ha M, Han J, Kim VN 2008. Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell 32: 276–284 [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC 2009. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10: 126–139 [DOI] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T 2007. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39: 673–677 [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T 2001. Identification of novel genes coding for small expressed RNAs. Science 294: 853–858 [DOI] [PubMed] [Google Scholar]
- Lai EC, Tomancak P, Williams RW, Rubin GM 2003. Computational identification of Drosophila microRNA genes. Genome Biol 4: R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landthaler M, Yalcin A, Tuschl T 2004. The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 14: 2162–2167 [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294: 858–862 [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V 2001. An extensive class of small RNAs in Caenorhabditis elegans. Science 294: 862–864 [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN 2002. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J 21: 4663–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419 [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP 2003. The microRNAs of Caenorhabditis elegans. Genes Dev 17: 991–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W 1999. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82–97 [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, Rappsilber J, Mann M, Dreyfuss G 2002. miRNPs: A novel class of ribonucleoproteins containing numerous microRNAs. Genes Dev 16: 720–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Thomson JM, Hammond SM 2008. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 14: 1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L, Ryazantsev S, Sun R, Zhou ZH 2010. Three-dimensional visualization of gammaherpesvirus life cycle in host cells by electron tomography. Structure 18: 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Ponka P 1999. Cell biology of heme. Am J Med Sci 318: 241–256 [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J 2004. The PredictProtein server. Nucleic Acids Res 32: W321–W326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senturia R, Faller M, Yin S, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F 2010. Structure of the dimerization domain of DiGeorge Critical Region 8. Protein Sci (in press). doi: 10.1002/pro.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiohama A, Sasaki T, Noda S, Minoshima S, Shimizu N 2003. Molecular cloning and expression analysis of a novel gene DGCR8 located in the DiGeorge syndrome chromosomal region. Biochem Biophys Res Commun 304: 184–190 [DOI] [PubMed] [Google Scholar]
- Sohn SY, Bae WJ, Kim JJ, Yeom KH, Kim VN, Cho Y 2007. Crystal structure of human DGCR8 core. Nat Struct Mol Biol 14: 847–853 [DOI] [PubMed] [Google Scholar]
- Stark KL, Xu B, Bagchi A, Lai WS, Liu H, Hsu R, Wan X, Pavlidis P, Mills AA, Karayiorgou M, et al. 2008. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet 40: 751–760 [DOI] [PubMed] [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM 2006. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20: 2202–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucchi M, Briata P, Garcia-Mayoral M, Haase AD, Filipowicz W, Ramos A, Gherzi R, Rosenfeld MG 2009. The RNA-binding protein KSRP promotes the biogenesis of a subset of microRNAs. Nature 459: 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triboulet R, Chang HM, Lapierre RJ, Gregory RI 2009. Post-transcriptional control of DGCR8 expression by the Microprocessor. RNA 15: 1005–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI 2008. Selective blockade of microRNA processing by Lin28. Science 320: 97–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R 2007. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet 39: 380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler H, Taylor KA 2006. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy 106: 240–254 [DOI] [PubMed] [Google Scholar]
- Yeom KH, Lee Y, Han J, Suh MR, Kim VN 2006. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res 34: 4622–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, Sander C, O'Carroll D, Stoffel M, Tuschl T, et al. 2009. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci 106: 498–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR 2005. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J Biol Chem 280: 27595–27603 [DOI] [PubMed] [Google Scholar]
- Zeng Y, Yi R, Cullen BR 2005. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J 24: 138–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W 2002. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. EMBO J 21: 5875–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]