

Abstract
Biological processes such as circadian rhythms, cell division, metabolism, and development occur as ordered sequences of events. The synchronization of these coordinated events is essential for proper cell function, and hence the determination of critical time points in biological processes is an important component of all biological investigations. In particular, such critical time points establish logical ordering constraints on subprocesses, impose prerequisites on temporal regulation and spatial compartmentalization, and situate dynamic reorganization of functional elements in preparation for subsequent stages. Thus, building temporal phenomenological representations of biological processes from genome-wide datasets is relevant in formulating biological hypotheses on: how processes are mechanistically regulated; how the regulations vary on an evolutionary scale, and how their inadvertent disregulation leads to a diseased state or fatality. This paper presents a general framework (GOALIE) to reconstruct temporal models of cellular processes from time-course gene expression data. We mathematically formulate the problem as one of optimally segmenting datasets into a succession of “informative” windows such that time points within a window expose concerted clusters of gene action whereas time points straddling window boundaries constitute points of significant restructuring. We illustrate here how GOALIE successfully brings out the interplay between multiple yeast processes, inferred from combined experimental datasets for the cell cycle and the metabolic cycle.
Keywords: model building and model-checking, temporal data analysis, yeast cell cycle, yeast metabolic cycle, Kripke structures
Cells and organisms can be viewed as progressing through sequences of states, as a result of discrete mechanisms. Defining these states and identifying the underlying mechanisms are critical to how we understand biological processes and how we may treat metabolic and developmental disorders. Central to such analysis tools are algorithms for time series analysis using temporal logic formalisms that were originally developed with engineering and computer and systems sciences applications in mind (1, 2, 3).
The yeast species Saccharomyces cerevisiae, which has been researched extensively to understand the biology of eukaryotic microorganisms, is a good model organism to illustrate the ideas in this paper. To understand the systems biology of yeast, one may study temporal expression profiles of genes involved in a particular function—for instance, cellular (4) division or metabolism (5)—and create models of the state space dynamics in terms of labeled states and state transition relations. An illustration of this process in shown in Fig. 1. A yeast cell cycle (YCC) model can be created using data generated by Spellman et al. (6) and similarly, a yeast metabolic cycle (YMC) model can be created by combining data generated separately by two other research groups: Tu et al. (5), Klevecz et al. (7). These labeled state transition models are shown in the two insets in Fig. 1; formally, they can be viewed as Kripke structures (8), with atomic propositional labels corresponding to the Gene Ontology (GO) functional categories, thus enabling temporal logic model-checking to extract complex global properties of these modules. For instance, we learn from the Kripke structure of the cell cycle that for cytokinesis to lead to DNA replication, the cell size must have enlarged sufficiently for division.
Fig. 1.

Temporal process models reconstructed from segmentation algorithm. States are identified through the segmentation algorithm and edges are labeled by the experimental conditions under which the transitions are observed. (left) YCC. (right) YMC.
A key goal of this type of analysis is to be able to formulate models without preexisting hypotheses. For instance, how would the system behave when subjected to multiple perturbations? As an illustration, in Fig. 2, we computationally integrate data from the distinct YCC and YMC experiments along with data from other perturbations (e.g., by hydrogen peroxide (HP) or menadione (MD) treatments) into a more complex combined model. Such integration is possible even though the data sources for each experiment and perturbation were gathered independently. The combined model, created by this metaanalysis, reveals insightful and complex temporal properties of the combined system, not visible in the individual component models: for instance, the exit from cell cycle under HP perturbation is inferred as fundamentally different from that under MD treatment, in that under the latter the cells complete one full cycle before being arrested.
Fig. 2.

Combined temporal process model of the YCC, YMC, and exposure of yeast cells to HP and MD treatment.
To create Kripke structure models as shown in Fig. 2, we require algorithms to extract states and state transitions from the data and subsequently, to label the states. Our contribution here is of a methodological nature: we devise a mathematically rigorous method (GOALIE) to reconstruct temporal models of cellular processes from time-course gene expression data.
GOALIE: A Mathematical Method to Infer Temporal Models
GOALIE aims to assist an experimentalist to track which genes are under coordinated temporal regulation, how their expression persists and dynamically varies over time, hence revealing insight into the progression of events constituting a given process.
As studied here, time-course analysis (9) has become an important tool for the study of organism development, disease progression, and cyclical biological processes, e.g., the cell cycle (6), metabolic cycle (5), and even entire life cycles (10). The growing affordability of transcriptional profiling screens has fostered the generation of numerous time series datasets. When the number of time points is sufficiently large, researchers have utilized continuous representations to smooth out noise (11), applied hidden Markov models to guide clustering (12), and used static measurements to “fill in the gaps” in the time series data (13). When the number of time points is small, researchers have studied the role played by sampling rates (14) and proposed the use of model profiles (15) to guide clustering. Recently, researchers have begun to characterize periodicity in transcriptional profiles (16), quantify timing differences in gene expression (17, 18), to extract binary signals embedded in the data (19), and to reconstruct regulatory relationships (20).
One of the attractions of time series analysis is its promise to reveal temporal relationships underlying biological processes (21): which process occurs before what, what are the “checkpoints” that must be satisfied (and when), and whether there can be alternative pathways of time series progression. There are many temporal modeling formalisms available: linear time logic uses a single time line from start to end whereas branching time logics allow multiple possibilities. In this paper, we primarily use linear time logics for modeling individual experimental conditions (stresses) yielding Gantt chart representations. The interplay between the stresses and potential branching behavior is then inferred indirectly through concurrent notations such as Kripke diagrams. A direction of future work is to directly model branching time behavior using data mining techniques.
Given gene expression time-course data, we identify segments of the time course as follows: Within a segment, genes organize into groups depicting concerted behavior but across segments there is a significant “regrouping” of genes. Although such analysis can be conducted by tracking individual genes whose function is known, in GOALIE we desire to automatically mine, in an unsupervised manner, temporal relationships involving groups of genes, which are not a priori defined. Similarly motivated research can be found in, e.g., ref. 22, which models the activity level of genes as a mixture model of activity levels of processes but these works are not directly concerned with revealing the temporal coordination of processes—the focus of this paper. Biclustering algorithms such as presented in refs. 23 and 24 can reveal patterns that preserve order of the time course, but such biclusters do not yield a partition of the entire time course as is done here. We focus on deriving a complete segmentation from which we can construct a finite state model where the states (nodes) correspond to the segments and transitions (edges) denote observed state-state boundaries. Furthermore, propositions that hold true in each state (e.g., the biological processes enriched in the corresponding segment) can be used to label the corresponding node in the model. Models from multiple stresses are then combined by coalescing nodes and factoring transitions.
Information-Theoretic Framework
Given multiple vectors of measurements 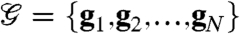 , where each gi is a time series over
, where each gi is a time series over 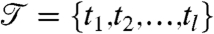 , the problem of segmentation is to find a sequence of segments,
, the problem of segmentation is to find a sequence of segments, 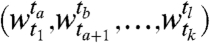 where each segment
where each segment 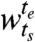 , ts ≤ te, is a set of consecutive time points beginning at time point ts and ending at time point te. The segment boundaries are identified such that, were we to cluster genes in neighboring windows separately, we would find highly dissimilar sets of clusters. Consider the case of segmentation into just two windows: i.e., we seek to find segments
, ts ≤ te, is a set of consecutive time points beginning at time point ts and ending at time point te. The segment boundaries are identified such that, were we to cluster genes in neighboring windows separately, we would find highly dissimilar sets of clusters. Consider the case of segmentation into just two windows: i.e., we seek to find segments 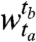 and
and 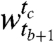 , with r clusters in the window
, with r clusters in the window 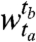 and c clusters in the window
and c clusters in the window 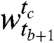 . Let α and β represent the cluster random variables for the windows
. Let α and β represent the cluster random variables for the windows 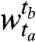 and
and 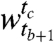 respectively. (The set of possible values for α is {1,…,r} and for β is {1,…,c}.)
respectively. (The set of possible values for α is {1,…,r} and for β is {1,…,c}.)
We begin by measuring the similarity of the clusters across the windows using a r × c contingency table. Entry nij in the (i,j)th cell of the table represents the overlap between the genes clustered together in cluster i of window 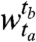 and in cluster j of window
and in cluster j of window 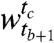 . The sizes of the clusters in
. The sizes of the clusters in 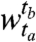 are given by the column-wise sums across each row:
are given by the column-wise sums across each row: 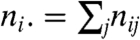 , while the sizes of clusters in
, while the sizes of clusters in 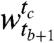 are given by row-wise sums down each column:
are given by row-wise sums down each column: 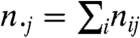 .
.
Interpreting each row and column as a probability distribution, ideally highly dissimilar clusters result in a total of (r + c) uniform distributions across the rows and columns of the contingency table. To capture the deviation of these distributions from the uniform distribution, we define r random variables Ri,i = 1,…,r occurring with probability 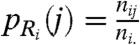 corresponding to each row. Similarly, we define c random variables Cj,j = 1,…,c occurring with probability
corresponding to each row. Similarly, we define c random variables Cj,j = 1,…,c occurring with probability 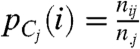 corresponding to each column. We capture the deviation of these distributions from the uniform distributions over the rows (
corresponding to each column. We capture the deviation of these distributions from the uniform distributions over the rows (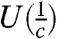 ) and columns (
) and columns (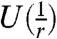 ) by
) by
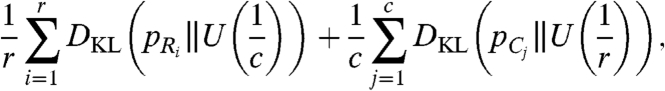 |
[1] |
where 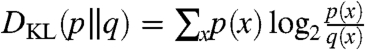 is the Kullback-Leibler (KL) divergence between two probability distributions p(x) and q(x). We can thus cluster the adjacent windows using this objective function, minimizing it in order to yield highly dissimilar clusters across the windows. Since the KL divergence of any distribution with respect to the uniform distribution differs from its negative entropy by a constant (when the sizes of the supports of the distributions are fixed), [1] can be equivalently expressed as
is the Kullback-Leibler (KL) divergence between two probability distributions p(x) and q(x). We can thus cluster the adjacent windows using this objective function, minimizing it in order to yield highly dissimilar clusters across the windows. Since the KL divergence of any distribution with respect to the uniform distribution differs from its negative entropy by a constant (when the sizes of the supports of the distributions are fixed), [1] can be equivalently expressed as
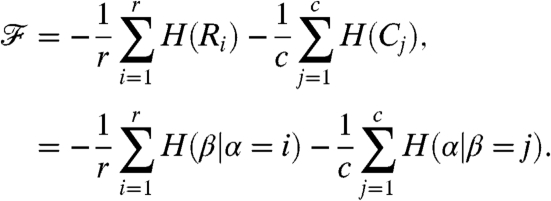 |
[2] |
Here H(X) (resp. H(X|Y)) denotes the entropy (resp. relative entropy) of a probability mass function p(x) (resp. p(x|y)) for X (resp. X relative to Y). Thus the function 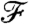 captures the mutual information between the clusterings in adjacent windows.
captures the mutual information between the clusterings in adjacent windows.
Our goal is to minimize 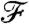 and obtain clusters that are local within each segment (similar to a k-means algorithm) but have high dissimilarity when compared with clusterings from the neighboring segment. We achieve this by parameterizing
and obtain clusters that are local within each segment (similar to a k-means algorithm) but have high dissimilarity when compared with clusterings from the neighboring segment. We achieve this by parameterizing 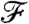 in terms of cluster prototypes, defining the cluster random variables to capture locality in their respective spaces, and optimizing
in terms of cluster prototypes, defining the cluster random variables to capture locality in their respective spaces, and optimizing 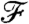 using an augmented Lagrangian algorithm (see SI Appendix for details).
using an augmented Lagrangian algorithm (see SI Appendix for details).
To identify the segments we employ a dynamic programming algorithm. Using minimum and maximum segment length constraints, we consider all possible “tilings” of the time course where every pair of neighboring tiles reduces to the problem above, i.e., where the evaluation consists of applying our clustering framework and determining the minimized value of 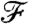 . These objective function values are then summed over an entire segmentation and used to evaluate one segmentation over another. Computationally, this reduces to a shortest path algorithm where each edge length is given by the minimized value of
. These objective function values are then summed over an entire segmentation and used to evaluate one segmentation over another. Computationally, this reduces to a shortest path algorithm where each edge length is given by the minimized value of 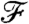 .
.
Results
Specific strains of Saccharomyces cerevisiae have been shown to have two robust biological cycles occurring simultaneously, e.g., the metabolic and cell cycles (25, 26). The GOALIE framework is validated through analysis of five yeast gene expression datasets, including two YMC time courses involving two different strains grown under two different conditions (YMC1: CEN.PK122 diploid strain, glucose-limited cultures (5) and YMC2: IFO 0233 diploid strain, not glucose limited (7)), a YCC dataset after release from α-factor synchronization (YCC: DBY8724 strain (6)), and observations of the cell cycle under treatment of (HP (27) and (MD (27).
Yeast Cell Cycle.
We computed the optimal segmentation for the YCC α-factor synchronization experiment of Spellman et al. (6) using GOALIE’s dynamic programming algorithm. This dataset comprises two cycles, one of which is explained in detail in Fig. 3 and both cycles are summarized in the complete segmentation (SI Appendix). To understand the temporal nature of the underlying dataset, in Fig. 3 we label each window with only functions from the cluster whose mean expression peaks during the window. We make several qualitative observations from the segmentation. First, from Fig. 3, observe how clusters within each window offer significant enrichments of biological processes (contingency tables in the first row) whereas there is significant regrouping of genes across neighboring windows (contingency tables in the second row). Second, GOALIE’s segmentation brings out the cyclic nature of the dataset—alternating M/G1, {G1,S}, {G2,M} phases—without explicit instruction. By studying the processes enriched in each segment of Fig. 3, the careful coordination of the cell cycle is easily seen. As stated in (6), the YCC time-course data spans approximately two points each for phases M/G1, G1, and S and spans only one time point for the G2 phase. Because our minimum window length is three (set so that we recover significant clusterings and regroupings), we cannot precisely resolve these short-lived phases with this dataset. A possible approach is to use continuous representations such as spline fits to gain greater resolution of data sampling (15). Nevertheless, the key events occurring in these segments are retrieved with high specificity (p < 10-7).
Fig. 3.

Preview of results from segmenting the YCC dataset. Only one cycle is shown here. The YCC involves the staged coordination of several phases (M/G1, time points [1–3]; G1,S, time points [4–6]; and G2,M, time points [7–9]). (A) Mean expression profiles for each group of genes depict the changing emphasis across the three phases. Contingency tables capture the concerted grouping of genes within segments (B, first row) as well as the regroupings between segments (B, second row). Observe that the contingency tables in the first row involve significant enrichments whereas the tables in the second row approximate a uniform distribution. Gantt chart views (C) depict the temporal coordination of biological processes underlying the dataset. Only some of the enriched functions are displayed, for lack of space.
Yeast Metabolic Cycle.
While the YCC has been well studied, the timing relationships in the YMC have only recently become elucidated. For instance, a main result of ref. 5 is the existence of three key clusters of expression patterns that oscillate coordinatively through the metabolic cycle phases, influenced by careful transcriptional control. GOALIE is able to recover the underlying temporal relationships in both the YMC datasets studied here. For YMC1, eight segments were inferred (Fig. 4A). These segments correspond to the successive reductive building (R/B), charging (R/C), and oxidative (Ox) phases of the metabolic cycle (5). The gene ontology (GO) categories enriched (p < 10-7) are clearly cyclic in nature. The same analysis applied to the YMC2 dataset yields nine segments (Fig. 4B), corresponding to three successive R/C, Ox, and R/B phases. The overlap in GO categories between YMC1 and YMC2 is fairly dramatic, especially with regards to processes associated with cell division. Clearly more GO categories were associated with the R/B segment of YMC2 growth relative to YMC1. Such differences may be related to differences in growth conditions as well as the strain employed.
Fig. 4.

Segmentation resulting from the GOALIE analysis of transcriptional profiling datasets evaluating the rhythmical growth of S. cerevisiae (YMC1: diploid CEN.PK122, nutrient-limited conditions; YMC2: diploid IFO0233, not nutrient limited). The time line of each experiment is shown with each hash mark indicating a sampling point. GOALIE accurately determined the G1, S, and G2/M phases of the cell cycle, respectively. Note that the genes associated with each segment were culture and strain-dependent.
Hydrogen Peroxide and Menadione Oxidative Stress.
The effects of HP and MD on yeast strain DBY8724 were evaluated recently through temporal transcriptional profiling (27). In the case of the peroxide treatment, cells were synchronized with α-factor, exposed to HP for a set period of time, and subsequently released from the oxidative stress. GOALIE analysis of this dataset returned time segmentations that corresponded to the three main phases of the cell cycle (Fig. 5). Segments I, II, and IV are the time frames that cell cycle analyses indicated the G1, S, G2/M phases predominated (27). Also note that GOALIE accurately determined the length of peroxide treatment. Segment III appears to be an intermediate phase of growth resulting from the removal of oxidative stress, exit from the extended S-phase and continued passage through the replication process (G2/M). Cell cycle analysis of this segment indicated that the percent contribution of each phase of the cell cycle were approximately the same. MD treatment results in G1 arrest (27), and the segmentation obtained by GOALIE corresponded to the G1, S, G2/M, and G1 phases of the cell cycle (four segments), with accurate identification of entry into G1 arrest (Fig. 5). Observe that one of the inferred segments (iron-sulfur cluster assembly) is aligned to the timepoint when MD was added.
Fig. 5.

Segmentation resulting from the GOALIE analysis of a transcriptional profiling dataset evaluating the exposure of S. cerevisiae (BY8743) to HP (0.2 mM) and MD (2 mM). The time line of the experiment is shown; each hash mark indicates a sampling point, and the duration of the treatment is above the time line. GOALIE accurately assigned Segments I, II, and IV of the peroxide dataset to the times when the cells are predominantly in G1, S, and G2/M phases of the cell cycle, respectively. Segment III putatively represents the combined transition between phases of the cell cycle as well as the release from oxidative stress. Note the prevalence of genes associated with core metabolic processes including sulfur metabolism. GOALIE analysis of the MD treatment again resulted in the assignment of the cell cycle stages (I–III) as well as the G1 arrest.
Process Modeling with GOALIE.
A combined, dynamic, temporal process model inferred from all datasets is shown in Fig. 2. This model captures the interplay between the YMC and YCC, and the cyclic nature of their time courses. The exit of cells from the cell cycle due to HP treatment and subsequent cell cycle arrest is also captured. Note that these transitions involve the cysteine and glutathione metabolic processes that drive the transition to cell cycle arrest as indicated in (27). The transitions involving MD do not indicate a similar exit because the cells complete one full cycle before getting arrested.
Through our temporal models, we have shown that S. cerevisiae acts in a somewhat unified fashion, with cell cycles based on core metabolism and cell division. Connections between the YMC and the YCC have been under intense investigation, which has generated interesting hypotheses involving biochemical process compatibility versus coordinated metabolic “bursts” (25). The metaphor that emerges from this analysis is that the metabolic state of the cell is essentially a fuel gauge, assessing whether or not other key biological processes (e.g., reproduction, regulation, etc.) should continue or not. The underlying assumption that, choreographed by these two predominant cycles, the availability of energy controls whether a yeast cell divides or not, motivates many other important questions: What are the major intracellular and extracellular molecules that control an individual cell and its decision to divide? Can we use gene knockouts and/or growth condition modifications to separate the YMC and YCC so that they are independent of one another?
For example, our investigation of the transcriptional profiling associated with peroxide stress identified a time segment that corresponded to an “intermediate stage” (Fig. 5, Segment 3) where the yeast cells were recovering from peroxide stress. The GO categories enriched in this segment were related to core metabolic processes (ethanol, TCA, glycogen), sulfur metabolism, and inositol lipid-mediated signaling, as well as chromatin silencing and nuclear pore organization/biogenesis. While sulfur metabolism can be associated directly with the oxidative stress response, the linkage to inositol lipid-mediated signaling genes and chromatin silencing is a bit more remote. Nevertheless, our tools bring out the nature of temporal “hardwiring” manifest in biological processes. In particular they open up questions related to whether it would be possible to manipulate the system to adopt an aberrant cell state or make it proceed along a desired temporal order. For instance, the identification of well-defined transcriptional states such as found in Segment III of the peroxide treatment suggests that at this stage in the cell growth regime it may be possible to force the organism to adopt aberrant states. For example, exit from peroxide treatment results in entry into the G2/M state. What would be the effect of adding alpha factor to the growth medium directly after release from peroxide stress? Would the cells continue through the cell cycle once before entering into G1, or move directly to G1?
Discussion
This work builds upon our prior research (2, 3) to make two key contributions. First, it provides successful inferences from multiple yeast time-course datasets, demonstrating the wide applicability of our information-theoretic methods. Second, unlike prior research, it focuses on teasing out relationships across stresses and summarizing process-level relationships in an integrated temporal model. In particular, we have uncovered the stages of peroxide stress response and situated them in relation to the YCC and metabolic cycle response.
Simply by extracting and analyzing the connections between the YMC and the YCC processes, which had remained latent in published data, it seems possible to refine hypotheses involving biochemical process compatibility versus coordinated metabolic “bursts,” which are currently under intense investigation (25). Temporal analysis of existing data points could lead to a systematic way to generate and experimentally refute (hitherto) nonobvious hypotheses.
Methods
Datasets and Data Preprocessing.
Our datasets came from a variety of sources (see SI Appendix). For each dataset we retained only genes that have an annotation in the GO biological process taxonomy (revision 4.205 of GO released on March 14, 2007), log transformed (base 10) their expression values and normalized them such that the mean expression of each gene across all time points is zero.
Dynamic Programming Algorithm for Optimal Segmentation.
As described elsewhere (3), we apply a dynamic programming algorithm for segmenting the various time series. We used different settings for the numbers of clusters and different thresholds for minimum and maximum possible window lengths to search in the space of possible segmentations. Besides the number of clusters in each segment, and minimum/maximum constraints on window lengths, we parameterized the segmentation algorithm with a parameter λ that controls the sizes of the clusters in the resulting segmentations and can be adjusted to yield approximately equal cluster sizes (see SI Appendix). After the segmentation reveals windows and clusters of genes in each window, we perform functional enrichment over the selected sets of genes. A hypergeometric p-value is calculated for each GO biological process term, and an appropriate cutoff is chosen using false discovery rate q-level of 0.01 (28). The time bounded enrichments are summarized as Gantt charts as presented earlier. We employ various statistical tests to assess the sensitivity of the segmentation to variations in the number of clusters (see SI Appendix).
Inferring Temporal Coordination of Processes.
We derive temporal process models from Gantt charts as follows: Given two neighboring segments, we assume that each of the processes enriched in the first segment precedes (i.e., has a state transition to) a process enriched in the second segment. We then find maximal sets of processes that are common across two or more datasets that obey the same precedence relationships.
Supplementary Material
Acknowledgments.
This work was supported in part by National Science Foundation Grants (ITR-0428344, CCF-0836649, CCF-0937133), and the Institute for Critical Technology and Applied Science (ICTAS) at Virginia Tech.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006283107/-/DCSupplemental.
References
- 1.Kleinberg S, Casey K, Mishra B. Systems biology via redescription and ontologies (I): Finding phase changes with applications to malaria temporal data. Systems and Synthetic Biology Journal. 2008;1:197–205. doi: 10.1007/s11693-008-9014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramakrishnan N, Antoniotti M, Mishra B. Reconstructing formal temporal logic models of cellular events using the GO process ontology; Proceedings of the Eighth Annual Bio-Ontologies Meeting; Detroit MI: ISMB (Intelligent Systems for Molecular Biology); 2005. p. 2. [Google Scholar]
- 3.Tadepalli S, Ramakrishnan N, Watson L, Mishra B, Helm R. Simultaneously segmenting multiple gene expression time courses by analyzing cluster dynamics. Journal of Bioinformatics and Computational Biology. 2009;7:339–356. doi: 10.1142/s0219720009004114. [DOI] [PubMed] [Google Scholar]
- 4.Chen K, et al. Integrative analysis of cell cycle control in budding yeast. Mol Biol Cell. 2004;15:3841–3862. doi: 10.1091/mbc.E03-11-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tu B, Kudlicki A, Rowicka M, McKnight S. Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science. 2005;310:1152–1158. doi: 10.1126/science.1120499. [DOI] [PubMed] [Google Scholar]
- 6.Spellman P, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klevecz R, Bolen J, Forrest G, Murray DB. A genomewide oscillation in transcription gates DNA replication and cell cycle. Proc Nat’l Acad Sci USA. 2004;101:1200–1205. doi: 10.1073/pnas.0306490101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke E, Grumberg O, Peled D. Model Checking. Boston, MA: MIT Press; 1999. [Google Scholar]
- 9.Bar-Joseph Z. Analyzing time series gene expression data. Bioinformatics. 2004;20:2493–2503. doi: 10.1093/bioinformatics/bth283. [DOI] [PubMed] [Google Scholar]
- 10.Lund J, et al. Transcriptional profile of aging in C. elegans. Curr Biol. 2002;12:1566–1573. doi: 10.1016/s0960-9822(02)01146-6. [DOI] [PubMed] [Google Scholar]
- 11.Bar-Joseph Z, Gerber G, Gifford DK, Jaakkola T, Simon I. Continuous representations of time-series gene expression data. J Comput Biol. 2003;10:341–356. doi: 10.1089/10665270360688057. [DOI] [PubMed] [Google Scholar]
- 12.Schliep A, Schonhuth A, Steinhoff C. Using hidden Markov models to analyze gene expression time course data. Bioinformatics. 2003;19:i255–i263. doi: 10.1093/bioinformatics/btg1036. [DOI] [PubMed] [Google Scholar]
- 13.Simon I, Siegfried Z, Ernst J, Bar-Joseph Z. Combined static and dynamic analysis for determining the quality of time-series expression profiles. Nat Biotechnol. 2005;23:1503–1508. doi: 10.1038/nbt1164. [DOI] [PubMed] [Google Scholar]
- 14.Singh R, Palmer N, Gifford D, Berger B, Bar-Joseph Z. Active learning for sampling in time-series experiments with application to gene expression analysis; Proceedings of the Twenty-Second International Conference on Machine Learning (ICML’05); New York: Association for Computing Machinery (ACM); 2005. pp. 832–839. [Google Scholar]
- 15.Ernst J, Nau GJ, Bar-Joseph Z. Clustering short time series gene expression data. Bioinformatics. 2005;21:i159–i168. doi: 10.1093/bioinformatics/bti1022. [DOI] [PubMed] [Google Scholar]
- 16.Kudlicki A, Rowicka M, Otwinowski Z. SCEPTRANS: An online tool for analyzing periodic transcription in yeast. Bioinformatics. 2007;23:1559–1561. doi: 10.1093/bioinformatics/btm126. [DOI] [PubMed] [Google Scholar]
- 17.Rowicka M, Kudlicki A, Tu B, Otwinowski Z. High-resolution timing of cell cycle-regulated gene expression. Proc Nat’l Acad Sci USA. 2007;104:16892–16897. doi: 10.1073/pnas.0706022104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoneya T, Mamitsuka H. A hidden Markov model-based approach for identifying timing differences in gene expression under different experimental factors. Bioinformatics. 2007;23:842–849. doi: 10.1093/bioinformatics/btl667. [DOI] [PubMed] [Google Scholar]
- 19.Sahoo D, Dill D, Tibshirani R, Plevritis S. Extracting binary signals from microarray time-course data. Nucleic Acids Res. 2007;35:3705–3712. doi: 10.1093/nar/gkm284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi Y, Mitchell T, Bar-Joseph Z. Inferring pairwise regulatory relationships from multiple time series datasets. Bioinformatics. 2007;23:755–763. doi: 10.1093/bioinformatics/btl676. [DOI] [PubMed] [Google Scholar]
- 21.de Lichtenberg U, Jensen L, Brunak S, Bork P. Dynamic complex formation during the yeast cell cycle. Science. 2005;307:724–727. doi: 10.1126/science.1105103. [DOI] [PubMed] [Google Scholar]
- 22.Segal E, Battle A, Koller D. Decomposing gene expression into cellular processes; Proceedings of the Pacific Symposium on Biocomputing (PSB’03); Singapore: World Scientific Press; 2003. pp. 89–100. [DOI] [PubMed] [Google Scholar]
- 23.Madeira SC, Oliveira AL. A polynomial time biclustering algorithm for finding approximate expression patterns in gene expression time series. Algorithms Mol Biol. 2009;4:8. doi: 10.1186/1748-7188-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Supper J, Strauch M, Wanke D, Harter K, Zell A. 2007 EDISA: Extracting biclusters from multiple time-series of gene expression profiles. BMC Bioinformatics. 8:334. doi: 10.1186/1471-2105-8-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Futcher B. Metabolic cycle, cell cycle, and the finishing kick to start. Genome Biol. 2006;7:107. doi: 10.1186/gb-2006-7-4-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palkova Z, Vachova L. Life within a community: benefit to yeast long-term survival. FEMS Microbiol Rev. 2006;30:806–824. doi: 10.1111/j.1574-6976.2006.00034.x. [DOI] [PubMed] [Google Scholar]
- 27.Shapira M, Segal E, Botstein D. Disruption of yeast forkhead-associated cell cycle transcription by oxidative stress. Mol Biol Cell. 2004;15:5659–5669. doi: 10.1091/mbc.E04-04-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storey J, Tibshirani R. Statistical significance for genomewide studies. Proc Nat’l Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.