

Abstract
The haploid human genome contains approximately 29 million CpGs that exist in a methylated, hydroxymethylated or unmethylated state, collectively referred to as the DNA methylome. The methylation status of cytosines in CpGs and occasionally in non-CpG cytosines influences protein–DNA interactions, gene expression, and chromatin structure and stability. The degree of DNA methylation at particular loci may be heritable transgenerationally and may be altered by environmental exposures and diet, potentially contributing to the development of human diseases. For the vast majority of normal and disease methylomes however, less than 1% of the CpGs have been assessed, revealing the formative stage of methylation mapping techniques. Thus, there is significant discovery potential in new genome-scale platforms applied to methylome mapping, particularly oligonucleotide arrays and the transformative technology of next-generation sequencing. Here, we outline the currently used methylation detection reagents and their application to microarray and sequencing platforms. A comparison of the emerging methods is presented, highlighting their degrees of technical complexity, methylome coverage and precision in resolving methylation. Because there are hundreds of unique methylomes to map within one individual and interindividual variation is likely to be significant, international coordination is essential to standardize methylome platforms and to create a full repository of methylome maps from tissues and unique cell types.
Keywords: DNA methylation, MeDIP, methylation-sensitive restriction enzyme, microarray, MRE, next-generation sequencing, reduced representation bisulfite sequencing, RRBS
DNA methylation is essential for a properly functioning genome through its roles in the maintenance of chromatin structure, chromosome stability and transcription [1–4]. DNA methylation involves the transfer of a methyl group to cytosine in a CpG dinucleotide via DNA methyl-transferases that create or maintain methylation patterns. Methylation of cytosines outside of CpG is also found in mammalian genomes [5–7], and in Arabidopsis the non-CG cytosine methylation has significant biological function. Hydroxymethylation of cytosines has also been reported, though its biological significance and tissue specificity are unknown [8,9]. Unlike 5-methyl-cytosine, reagents to measure hydroxymethylation in specific genes are not yet available.
Considering the 29 million CpGs in the haploid genome, DNA methylation patterns in normal human cells are largely unexplored [10]. Several general methylation patterns are recognized. First, 7% of all CpGs are within CpG islands (CGIs), a majority of which are unmethylated [10]. Second, normally methylated sequences include promoter CGI on the inactive X chromosome, one allele of imprinted genes, tissue-specific genes and intragenic regions; although the function of intragenic methylation has been controversial [11,12]. Third, approximately 45% of CpGs are within repetitive elements and presumed constitutively methylated [11]. This large portion of the methylome has been inaccessible by microarray-based methods, but may well have a role in gene regulation. For example, a stunning observation in agouti mice demonstrated that the labile methylation status of a specific repeat element influences expression of a nearby gene and specific phenotypes in mice, including hair color and susceptibility to complex disease [13,14]. Thus, there is much more to be known about the epigenetic regulation of these abundant but enigmatic sequences, and how our genotype, environment and diet influence epigenetic regulation. There is even less known about how much and where DNA methylation patterns differ in different cell types, between genders or between genetically distinct populations. Advances in genome-wide methylation mapping therefore hold immense discovery potential.
Reagents
Reagents to detect or enrich for cytosine methylation include methylation-sensitive restriction enzymes (MREs), a 5-methylcytosine antibody, methylated DNA binding proteins or proteins that primarily bind unmethylated DNA, and chemicals such as bisulfite and hydrazine (Figure 1). There are approximately 50 unique MREs, though very few of these have a matching methylation-insensitive isoschizomer. By contrast to MREs, which are inhibited by methylation, the restriction enzyme McrBC instead recognizes and cleaves methylated DNA at two half-sites of (G/A)mC separated by as much as 3 kb, though the optimal separation is 55–103 base pairs. The majority of restriction enzymes used for methylation profiling are precise and inexpensive, though only those methylation events within the MRE recognition sites are assayed. However, when multiple nonre-dundant MREs are used in parallel, this limitation is effectively mitigated. MREs can resolve the methylation status in local regions or at individual CpGs, depending on the platform used following MRE digestion. Rare-cutting MREs with 6–8 bp recognition sites, such as AscI or NotI, are also useful for detection of DNA methylation when combined with digital karyotyping [15,16]
Figure 1. Reagents, platforms and analysis for genome-scale detection of DNA methylation.
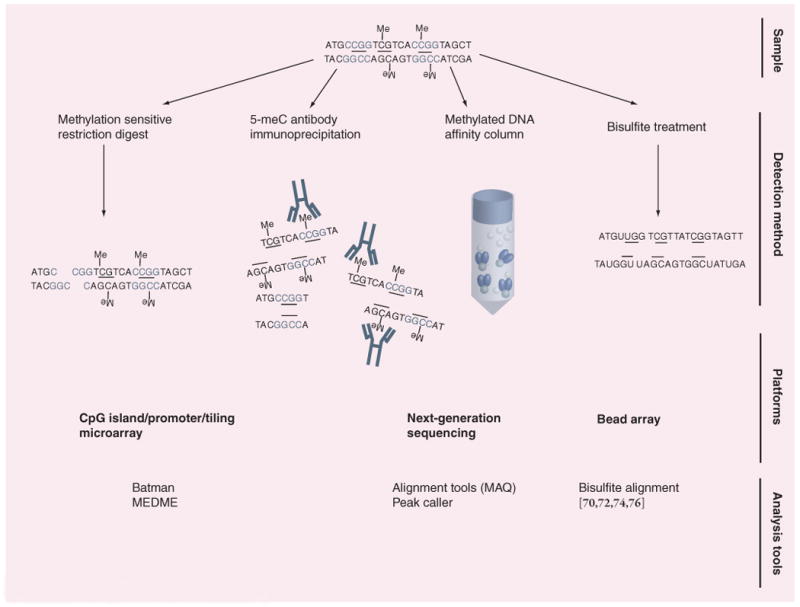
A DNA fragment with HpaII sites (denoted in gray) and DNA methylation (denoted with –Me) is used to illustrate how different reagents are used to detect DNA methylation. Reagents for detecting/enrichment of DNA methylation include methylation-sensitive restriction digest, immunoprecipitation with an antibody against 5-methyl-cytosine, binding of the DNA to a methyl DNA affinity column or treating the DNA with sodium bisulfite. After treatment of DNA with one of these methods, DNA microarrays, next-generation sequencing or bead arrays are used to detect the DNA methylation. Software analysis tools are then needed to determine the location and levels (either absolute or relative) of DNA methylation.
CG sites (before and after bisulfite treatment) are underlined.
Batman: Bayesian tool for methylation analysis; MAQ: Mapping and Assembly with Quality; MEDME: MeDIP enrichment.
By contrast to MREs, the monoclonal antibody against 5-methylcytosine and methylated DNA-binding proteins (domains of MBD2 alone, or in combination with MBD3L) allow enrichment for methylated DNA independent of DNA sequence (Figure 1) [17–20]. Enrichment is greater for methylated regions with higher CpG content relative to methylated regions with lower CpG content. These reagents are commercially available and simple to use. The lower limit of resolution of methylation status using the antibody or affinity columns is determined initially by the size range of randomly sheared DNA prior to enrichment, generally 100–300 bp, and subsequently by the platform used to assess the enrichment.
Chemicals including sodium bisulfite and hydrazine react differentially with unmethylated versus methylated cytosine and allow DNA methylation mapping at single CpG resolution (Figure 1) [21–23]. Of these, sodium bisulfite is the most commonly used chemical reagent as it results in a positive display of methylation, among other advantages. Sodium bisulfite allows for the conversion of cytosine to uracil, which is replaced by thymine during PCR amplification and conventional sequencing. By contrast, methylated cytosines are nonreactive, and remain as cytosine after bisulfite treatment. Sequencing of individual clones of the PCR product allows assessment of methylation status of contiguous CpGs on a single PCR allele derived from one genomic DNA fragment. Bisulfite has many advantages, including single CpG resolution, detection of strand specific methylation, and detection of cytosine methylation outside of CpGs. Unlike other methylation-detection reagents, bisulfite may provide quantification of absolute rather than relative DNA methylation levels, depending on the platform used. The reduced sequence complexity of the genome following bisulfite treatment complicates its application to oligonucleotide arrays [24].
Platforms
A variety of platforms have been developed to increase the number of CpGs for which methylation can be assessed (Figure 1 & Table 1). The first breakthrough platform was the MRE-based 2D gel termed restriction landmark genome scanning (RLGS) [25]. RLGS allows assessment of 2000 or more unique CpG sites per enzyme combination, and has been used to investigate DNA methylation in a wide-variety of cellular and tissue studies, and in multiple organisms [26]. Additional PCR and gel-based methods [27–29], as well as pioneering work in methylation detection by microarrays [30] were developed in the late 1990s. Many other valuable methods have been developed, but are not discussed here. Below, the discussion is focused on the most current DNA methylation platforms in the mid- to large-scale range.
Table 1. Parameters of DNA methylation platforms.
| Method | Coverage of CpGs | Resolution* (bp) | Starting amount | Treatment of DNA | Ability to assess repeats |
|---|---|---|---|---|---|
| Sequenom Epityper™ | 103 | 1 | 10 ng | Bisulfite | No |
| Digital karyotyping | 104 | 1 | 1-5 μg | Restriction digest | Yes |
| SNP arrays | 104 | 1 | 500 ng | Restriction digest | No |
| Infinium™/Goldengate® | 104 | 1 | 500 ng | Bisulfite | No |
| MeDIP-Chip (promoter) | 105 | 1000 | 1–4 μg | Sonication/immunoprecipitation | No |
| MIRA-Chip (promoter) | 105 | 1000 | 500 ng–1 μg | Sonication/methyl-binding column | No |
| MeDIP-Chip (whole-genome tiling) | 106 | 1000 | 5–35 μg | Sonication/immunoprecipitation | No |
| MIRA-Chip (whole-genome tiling) | 106 | 1000 | 2 μg | Sonication/methyl-binding column | No |
| McrBC-Chip/CHARM | 106 | 1000 | 5 μg | Restriction digest | No |
| Methyl-Seq/HELP-Seq/MSCC | 105–106 | 1 | 10 ng–1 μg | Restriction digest | Yes |
| RRBS | 106 | 1 | 1 μg | Restriction digest/bisulfite | Yes |
| Shotgun BS | 107 | 1 | 5 μg | Sonication/bisulfite | Yes |
| MeDIP-Seq | 107 | 200 | 1 μg | Sonication/immunoprecipitation | Yes |
Approximations.
BS: Bisulfate sequencing; CHARM: Comprehensive high-throughput arrays for relative methylation; HELP: HpaII tiny fragment enrichment by ligation-mediated PCR; MeDIP: Methylated DNA Immunoprecipitation; MIRA: Methylated CpG Island Recovery Assay; MSCC: Methyl-sensitive cut counting; RRBS: Reduced representation bisulfite sequencing.
Bead arrays
Illumina (CA, USA) methylation assays are mid-range platforms that quantitatively interrogate 1505 individual CpG sites in 807 genes (GoldenGate®) or 27,578 individual CpG sites in approximately 14,000 genes (Infinium™, Illumina) [31]. These methods use bisulfite conversion and bead arrays to detect relative methylation levels at individual CpGs selected based on potential relevance (e.g., known cancer genes, promoter CpGs). Bead-bound oligonucleotides corresponding to the methylated and unmethylated states of a single CpG site are hybridized to bisulfite-converted DNA, along with a third locus-specific bead-bound oligonucleotide, which is designed to anneal near to, but not overlapping the CpG site. Each of the three oligonucleotides contains a different universal PCR primer site, and the locus-specific oligonucleotide also contains a unique ‘address’ sequence for a particular bead type. Allele-specific extension is performed and the extended oligonucleotide is ligated to the adjacent locus-specific oligonucleotide. The extension products are amplified by PCR with Cy3- and Cy5-labeled primers and competitively hybridized to the bead array. The full bead array contains an average of 30 beads specific for each locus. Each bead is coated with probes complementary to a particular locus-specific oligonucleotide, and both Cy3- and Cy5- labeled products can hybridize to each bead. The methylation level is determined by the ratio of Cy3 and Cy5 fluorescence.
The GoldenGate assay allows 12, 16 or 96 sample formats and single-site CpG resolution for assaying relative methylation levels, though relatively few CpGs in total [31]. Ladd-Acosta et al. measured methylation using the Illumina GoldenGate Methylation Cancer Panel I platform [32], and discovered DNA methylation signatures that distinguish human brain regions. Custom CpG panels on the GoldenGate platform are particularly useful for follow-up studies of genome-wide screens, or for assaying select types and sets of genomic loci.
The larger-scale methylation platform from Illumina is the Infinium methylation assay. The Infinium/HumanMethylation27 DNA Analysis BeadChip™ (Illumina) interrogates 12 samples at a time and includes probes from 1000 cancer-related genes and from putative promoters of 110 microRNAs, among others. While there are on average two CpG sites assayed per gene for the majority of genes, 150 genes known to exhibit aberrant tumor-specific methylation are assayed at 5–10 CpGs each. Infinium is applicable even when DNA amounts are limiting, as it requires only 500 ng DNA prior to bisulfite conversion. Overall, Infinium offers a balance of a greater number of CpG assessed but lower sample-throughput relative to GoldenGate. These methods do not assess multiple closely apposed CpGs individually, and such regions are generally avoided in the assay development. This bias is likely to impact biological insights drawn from extrapolations of this data. It is also worth noting that only approximately 0.1% of the 29 million CpGs in the human genome are assayed by Infinium.
Another bisulfite-based method, the Sequenom (CA, USA) EpiTyper™ assay, utilizes matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry to analyze RNA cleavage fragments derived from post-bisulfite PCR products that contain a promoter to drive transcription [33,34]. This unique assay allows high-throughput quantitative methylation analysis at hundreds of loci, usually at single CpG resolution, and is quite useful for candidate loci in many samples, or as a follow-up to genome-wide profiling.
Microarrays used in DNA methylation profiling
The use of microarrays for interrogating CpG methylation began with arrays of thousands of PCR products corresponding to CGIs [35] and has been extended to bacterial artificial chromosome arrays and large-scale whole-genome tiling arrays with hundreds of thousands of oligonucleotide probes [36–38]. While most methylation detection methods can be applied to microarrays, bisulfite modification is not amenable to commercial arrays due to the bisulfite-induced cytosine conversion. The detection method and the particular array platform used will determine the resolution at which DNA methylation is determined.
CpG island microarrays
CpG island microarrays were the first microarray platform used to identify genomic loci that exhibit differential methylation. CGI microarrays were originally fabricated using clones from libraries enriched for unmethylated CpG rich DNA [17,30,35,39], or through size selection of SmaI fragments [40]. However, the CGI PCR product arrays have low resolution, and variable hybridization kinetics that complicate data analysis. Limitations in consistency, resolution and methylome coverage of early CGI arrays were overcome to varying degrees by commercial oligonucleotide arrays, often with many probes spanning each CGI [41]. Several of the oligonucleotide arrays extend genome coverage beyond gene promoters to include intragenic and intergenic loci.
Promoter microarrays
Another microarray platform used to determine DNA methylation patterns is a gene-promoter microarray. These microarrays include CGI and non-CGI promoters, and contain oligonucleotide probes spanning 1–10.5 kb across transcription start sites. Probe size and spacing vary depending on the manufacturer, but generally are 50–75 nucleotides long and spaced 100–200 bp apart. Probe spacing does not translate directly to resolution level, since computation of methylation values can require merging values of up to ten consecutive probes to reduce noise. Promoter microarrays have been used to map DNA methylation patterns, histone modifications and the binding of transcription factors [42,43]. One important distinction is that, unlike CGI arrays, most promoter microarrays do not assess methylation in gene bodies and intergenic regions, common sites of tissue-specific DNA methylation. Conversely, promoter arrays assess nearly all annotated gene promoters, while CGI arrays include approximately half of all promoters. Recent array designs from commercial vendors combine promoter and CGI probes on a single platform to allow more comprehensive coverage of the methylome.
Tiling microarrays
Tiling arrays contain probes that cover the majority of the nonrepetitive portion of the genome (approximately 1 × 109 bp), and thus have greater methylome coverage than promoter and CGI microarrays. Repeat-masked probes are spaced 35–50 bp apart, with variable probe lengths. For smaller genomes such as Arabidopsis Thaliana, the tiled genome is contained on a single array chip/slide. For the larger genomes of the mouse and human, 7–10 array slides are necessary for complete genomic coverage. The first ‘complete’ methylome profile generated using a tiling array platform was that of A. Thaliana [37,38]. However, due to the number of arrays needed for complete coverage of the human genome, a larger amount of genomic DNA (5–35 μg depending on the arrays used) is required relative to promoter and CGI arrays [20,37,44]. Thus, whole-genome amplification [37], ligation-mediated PCR [20] or T7 based amplification [45] may be necessary after methylation detection but prior to hybridization in order to use tiling arrays with limited DNA amounts.
SNP arrays
Microarrays have been developed to interrogate over 906,000 SNP loci and this platform can also be utilized for analysis of DNA methylation. The SNP-chip methodology is based on hybridization differences between perfectly matched and single mismatch probes representing the different SNPs. DNA is digested sequentially with one or more MRE, such as HpaII and then with StyI or NspI, which is used to ligate linkers for PCR amplification. Digestion with MREs can result in an experiment-induced loss of heterozygosity as the allele without methylation will be cleaved and therefore not amplified by PCR, resulting in decreased or absence of signal for that allele. Thus, the SNP-array platform allows allele specific methylation to be assessed at heterozygous loci. The SNP-array platform is not specifically designed for the purpose of identifying allele-specific methylation, and can only do so if two criteria are satisfied. First, the MRE site must occur within a StyI or NspI fragment. Second, the MRE site itself cannot be polymorphic. The SNP-array approach has allowed identification of dense gene-body methylation on the active X chromosome, and reduced gene-body methylation on the inactive X chromosomes, as well as allele specific methylation in cell lines [46–48].
Microarrays & methyl-sensitive restriction enzymes
Methyl-sensitive restriction enzymes remain one of the most precise and useful tools for studying DNA methylation. The initial discovery of CGIs came from the analysis of abundant small DNA fragments generated by digestion with the MRE HpaII (HpaII tiny fragments), uncovering an unmethylated compartment of the genome with a distinctly CG rich sequence composition [49]. The HpaII tiny fragment enrichment by ligation-mediated PCR, or HpaII tiny fragment enrichment by ligation-mediated PCR (HELP) assay, uses HpaII along with its methylation-insensitive isoschizomer MspI to identify unmethylated CpG sites within the sequence 5′-CCGG-3′ [50]. Genomic DNA digested separately with each enzyme is size-selected to capture small DNA fragments. Custom adaptors complementary to digest ends are ligated and the adaptor-ligated molecules are PCR-amplified (LM-PCR). The amplification products can be analyzed using a variety of platforms. For example, Khulan et al. applied HELP with a custom microarray to mouse sperm and brain, competitively hybridizing HpaII and MspI fragments [50]. Data validation in this study included bisulfite pyrosequencing of four loci. The HELP data confirmed that most of the mouse genome is contiguously methylated, punctuated by hypomethylated clusters that often overlapped with transcription start sites. Approximately 30% of CGIs were methylated, and there were many tissue-specific differentially methylated regions that occurred both within and outside of annotated promoters. An advantage of MRE-based assays such as HELP is the positive display of hypomethylated loci. Limitations of HELP are that it interrogates HpaII sites only and is relatively low resolution when applied to arrays. To increase the number of CpGs assessed, MRE and array-based assays have incorporated multiple parallel MRE digests [51]. MRE-based methods have also been used in combination with the next-generation sequencing platforms [52–54].
Microarrays & McrBC
The methylation-dependent restriction enzyme McrBC recognizes methylated DNA and cuts near but not at its recognition sequence. McrBC recognizes RmC(N)55–103RmC, and cuts once between each pair of half-sites, cutting close to one half-site or the other. Thus, the cuts can be distributed over several base pairs and approximately 30 base pairs distant from the methylated base, generating a distribution of DNA ends rather than precisely defined DNA ends. McrBC is useful to size-separate methylated DNA from unmethylated DNA, since the unmethylated DNA remains high-molecular weight after digestion. McrBC digested DNA can be labeled and competitively hybridized on microarrays with DNA that has not been digested by McrBC. Lippman et al. used McrBC depletion of DNA methylation with a custom DNA microarray to analyze a 1.5-Mb portion of the interstitial hk4s heterochromatic knob in A. Thaliana [55]. DNA methylation was not distributed uniformly over this region, but coincided with transposable elements and related repeat elements. McrBC depletion was also used by Nouzova et al. to study DNA methylation in the acute promyelocytic leukemia cell line NB4 before and after differentiation with all-trans-retinoic acid (ATRA) and normal peripheral blood mononuclear cells [56]. ATRA relieves histone deacetylase (HDAC)-mediated gene repression resulting from PML/RARα translocation in acute promyelocytic leukemia. In this study, McrBC digestion was used with a 6800 element CGI microarray, detecting hypermethylation of approximately 70 CGIs near transcription start sites in NB4 cells without ATRA treatment. ATRA differentiation of NB4 cells did not change the methylation status of any assayed CGIs, although significant changes in histone acetylation associated with gene activation were observed.
The comprehensive high-throughput arrays for relative methylation (CHARM) method by Irizarry et al. is another array-based method for methylation profiling using McrBC [57]. By optimizing probe location and CpG density on custom arrays, optimal specificity and sensitivity were achieved. Because neighboring CpG sites tend to have a highly correlated methylation status, neighboring probe signals are averaged to reduce background noise without loss of sensitivity or specificity, though modestly reducing resolution. By comparing CHARM to methylated DNA immunoprecipitation (MeDIP) or HpaII on arrays, Irizarry et al. suggested that McrBC yields better methylome coverage than HpaII and less of a bias for CpG density than MeDIP. Using CHARM, aberrant DNA methylation was found in colon cancer at sequences up to 2-kb flanking CGIs, called CGI shores [58]. A proportion of the CGI shores exhibit conserved tissue-specific methylation that correlates with expression of the corresponding gene. These data demonstrate the utility of McrBC-based methylation detection, and the specific new insights afforded by the CHARM method.
Methylated DNA immunoprecipitation-chip analysis
In addition to MREs and McrBC, methylation can be assessed by immunoprecipitation of methylated DNA with a monoclonal antibody against 5-methylcytosine (MeDIP) [17]. A major advantage of MeDIP-based detection is that it is not limited to a specific restriction site and theoretically any fragment with a methylated CpG is immunoprecipitated. MeDIP enrichment of methylated DNA has been applied to various platforms for assessing DNA methylation. One approach involves the coupling of MeDIP with DNA microarrays to obtain relative methylation levels at the loci represented on the array. This MeDIP-chip has been used in the analysis of cancer cell lines [17,59], stem cells [41,42] and the methylome of A. Thaliana [37,38], among others. Defining a region as methylated from the MeDIP-chip data depends on several factors including the sequence context of the loci (CpG poor regions versus CpG rich regions), the number of probes in a region and their hybridization values. DNA methylation status is expressed relative to input DNA or to another sample. There are three main issues to consider when performing MeDIP-Chip assay. First, the use of sonication results in DNA fragment sizes of approximately 200 bp that typically have multiple CpGs. Thus, methylation detected by MeDIP is assigned to discrete regions rather than individual CpG sites. Second, the differing hybridization characteristics of different DNA regions have a significant effect on their specific signal intensities. Third, the determination of methylation from the array signal(s) should take into consideration the local CpG density [17,43]. It is also important to be familiar with the software packaged with commercial arrays, as many of these were originally designed to analyze chromatin immunoprecipation data, which has noticeably different characteristics than methylation data. Overall, MeDIP-chip is a cost-effective way to assess relative levels of methylation at tens of thousands of loci in a particular cell line or tissue.
Newer algorithms have been specifically designed for analyzing MeDIP experiments and are freely available. Modeling experimental data with MeDIP enrichment is a combination of analytical and experimental methodologies that improve the interpretation of MeDIP-chip data [60]. Since MeDIP enrichment of DNA fragments is dependent not only on methylation state but also the density of CpGs within a given fragment, there is not necessarily a linear relationship between enrichment and true methylation values. Using MeDIP enrichment, estimates of relative and absolute DNA methylation were highly correlated with methylation values derived from the gold-standard bisulfite sequencing.
Methylated CpG island recovery assay
The Methylated CpG Island Recovery Assay (MIRA) is an alternative to MeDIP for selecting/enriching for methylated DNA, particularly at CGIs [18,19]. MIRA involves size fractionation of DNA, either by sonication or with MseI, which recognizes 5′-TTAA, a site that is typically found outside of CGIs. After digestion, adaptors are ligated to the DNA followed by selective binding of methylated fragments on a column with full-length MBD2b and MBD3L1 proteins. MBD2b is a methyl-binding protein that exhibits a high affinity for methylated DNA while also being able to differentiate between methylated and unmethylated DNA [18]. MBD3L1, however, lacks the methyl-CpG binding domain but can interact with MBD2b and results in improved enrichment of methylated DNA [18]. The methylated DNA eluted from the column is then amplified by PCR, fluorescently labeled and hybridized to a microarray. This approach has been used to identify HOX genes as potential targets for DNA hypermethylation in cancer cell lines and early-stage lung cancer [19], as well as provide a methylation map of human B cells using a tiling array with median probe spacing of 100 bp [20]. The advantages of the MIRA technique are that it does not require a particular DNA sequence other than methylated CpGs nor does it require DNA to be denatured to single-strands as for MeDIP. In addition, DNA fragments with a low CpG density may also be enriched by MIRA [20].
Indirect methylation detection with demethylating agents and expression arrays
Although not a direct assay of DNA methylation, blocking DNA methylation through siRNA of DNMT1, treatment of cell lines with demethylating agents such as 5-aza-2′deoxycytidine (5-aza) alone, or 5-aza in combination with histone deacetylase inhibitors followed by expression-array analysis can identify genes that may have been suppressed by DNA methylation [61–65]. 5-aza is a cytidine analog that is incorporated into DNA and can then covalently bind and inhibit DNA methyl-transferases, resulting in passive demethylation. 5-aza treatment results in activation of some genes that were silenced by DNA methylation, provided that the appropriate transcription factors are present. However, interpretation of this indirect assessment of methylation is complicated by the fact that genes apparently without promoter methylation may also exhibit an increase in expression following 5-aza treatment [66]. Presumably, this results from demethylation at other loci within the same gene or in genes upstream that are required for its expression, though direct effects on nonmethylated sites cannot be ruled out. Furthermore, this approach is best applied to cells grown in culture, such as cell lines or early passage primary cells [67], as 5-aza requires replication to induce passive demethylation. The application of this approach to primary tumor cells is particularly useful in that it addresses epigenetic artifacts seen in longer-term cell cultures, though non-transformed cells may arrest growth following 5-aza exposure.
Methylation-sensitive restriction enzyme tags & Sanger sequencing
DNA methylation has also been assessed through traditional Sanger sequencing combined with MRE in digital karyotyping [15,16]. Using a combination of MRE that recognize 6–8 bp sites and methylation-insensitive restriction enzymes, a library of short sequence tags is generated. The number of tags sequenced reflects the level of methylation at each recognition site, with lower tag counts representing greater methylation levels. In this method, the number of sites analyzed depends on the MRE used – use of AscI for example can generate over 5000 unique tags that correspond to more than 4000 genes. Approximately 1 μg of starting DNA is sufficient for this procedure.
Next-generation sequencing
Massively parallel sequencing platforms from Roche (Basel, Switzerland)/454, Illumina (CA, USA)/Solexa and Applied Biosystems (CA, USA)/SOLiD™ have transformed genomic and epigenomic research [68]. After applying a DNA methylation detection reagent, tens of millions of DNA fragments are sequenced in parallel. Sequencing has several advantages over array platforms, particularly that sequencing-based methods allow assessment of DNA methylation in interspersed repeat sequences that are inaccessible using microarrays.
Methyl-seq, HELP-seq & methyl sensitive cut counting
The reliability of MREs enables their straightforward application to next-generation sequencing (MRE-seq) to dramatically increase the number of CpGs that can be analyzed. There are many ways to optimally deploy MRE-seq methods that depend on the number of enzymes used and the fragment sizes analyzed. To date, three variations for combining MREs with next-generation sequencing have been described.
The first method, termed Methyl-seq, used the methylation-sensitive enzyme, HpaII, and its methylation-insensitive isoschizomer, MspI, in combination with Illumina sequencing [54]. Separate aliquots of the same DNA sample are digested with HpaII or MspI, ligated to adaptors, and subjected to Illumina sequencing. A total of 255,266 MspI sites were assayed, and these sites were then grouped into 90,612 regions based on proximity. Using these parameters, approximately 65% of the CGIs in the human genome were assayed. Although this method is biased towards CGIs (which constitute 1–2% of the genome), non-CGI sites account for approximately 61% of regions assayed, including a variety of genomic sequences such as promoters, exons, introns and intergenic regions.
The HELP assay has also been applied to next-generation sequencing in HELP-seq [52]. Similar to methyl-seq, this method also used the HpaII/MspI isoschizomer pair and included ligation of HpaII-complementary adaptors and ligation-mediated PCR prior to Illumina library construction. HELP-seq was generally concordant with HELP-array data, and identified additional unmethylated loci, such as a non-CGI alternative promoter of KCNQI, which was hypomethylated in erythroid progenitor cells. There are several advantages to this method, including the option of using as little as 10 ng of DNA. Furthermore, the MspI library sequencing allows copy-number variation to be determined, since MspI is not affected by methylation status. These advantages make HELP-seq potentially useful for limited or archival samples, provided that the DNA is not significantly sheared or degraded.
Ball et al. reported a third variation of MRE-seq, using HpaII/MspI digestion with Illumina sequencing to analyze DNA methylation in the PGP1 Epstein–Barr virus-transformed B-lymphocyte cell line [53]. This approach, termed methyl-sensitive cut counting (MSCC), assayed approximately 1.4 million unique HpaII sites. Using MSCC and a complementary method, bisulfite padlock probe sequencing to assay the methylation status of approximately 10,000 CpGs, highly expressed genes were found to be associated with high gene-body methylation and low promoter methylation. MSCC read counts were linearly related to bisulfite padlock probe sequencing percent methylation at 381 CpG sites that were assayed with both methods, suggesting that MSCC allows relative quantitation of methylation levels.
Methyl-seq, HELP-seq and MCSS demonstrate the utility of MRE for analysis of DNA methylation. The single CpG resolution and ability to assay a more significant portion of the methylome including most CGIs makes MRE and next-generation sequencing a relatively simple and accurate method to assay DNA methylation in the near future. When used alone, the MRE-seq methods assess relative rather than absolute methylation levels.
MeDIP-seq
As detailed in a previous section, MeDIP involves immunoprecipitation of methylated DNA, which then can be analyzed by a variety of methods. An important advantage of MeDIP over restriction enzymes is that it is not biased for a particular nucleotide sequence other than CpG. Two recent publications describe MeDIP combined with Illumina sequencing (MeDIP-seq). Down et al. performed MeDIP-chip and MeDIP-seq on human sperm cells [69]. DNA sonicated to less than 800 bp was end-repaired, ligated to adaptors, immunoprecipitated and then amplified with adapter primers. Libraries were size selected and insert sizes of 85–160 bp were subjected to sequencing on an Illumina Genome Analyzer. An analytical method for MeDIP-chip and MeDIP-seq data, called Bayesian tool for methylation analysis (BATMAN) was developed to allow quantification of methylation levels across a range of CpG densities. After performing MeDIP-seq on a human sperm DNA sample, Batman and a smoothing function were used to extend each read to 500 bp surrounding its mapping site, thereby achieving coverage of approximately 60% of all CpGs in the human genome, including approximately 90% of sites within CGIs. Comparison of MeDIP-seq with MeDIP-chip and bisulfite sequencing data revealed good concordance [69,70].
Pomraning et al. performed MeDIP-chip and MeDIP-seq to examine the Neurosporacrassa methylome [71]. This protocol differed from Down et al. in that they performed MeDIP before end repair and adapter ligation. Since the MeDIP immunoprecipitation step is performed on ssDNA, this success of the downstream Illumina library construction depends on reannealing of immunoprecipitated DNA after MeDIP, the efficiency and accuracy of which is not fully understood. The authors provide several useful experimental and analytical tips:
To prevent cross-reaction of the antimethyl-cytosine antibody with RNA, it is necessary to include an RNase step during DNA extraction and a gel-purification step after sonication to remove all RNA;
Alternative and less expensive commercial sources of adaptors and enzymes for library generation are available;
Alternative alignment programs, such as CashX and reference-guided assembly, might be useful to map methylation in repetitive sequences.
One inherent limitation of MeDIP in its current form is the lower resolution compared with MRE or bisulfite-based methods. On the other hand, MeDIP-seq provides comprehensive methylome coverage at a fraction of the cost of shotgun bisulfite sequencing. Experimental and analytic advances will undoubtedly improve this method in the near future and make it more widely useful for methylome analysis.
Reduced representation bisulfite sequencing
The gold standard for DNA methylation analysis of individual genes is bisulfite treatment to convert unmethylated cytosines to uracil followed by cloning and Sanger sequencing. This method provides quantitative, allelic, contiguous and base-pair resolution of CpG methylation. The bisulfite approach, however, has been difficult to apply on a genome-wide scale for mammals for several reasons. First, DNA methylation is concentrated at repetitive elements, and short sequence reads corresponding to repeats are more challenging to assemble uniquely onto a bisulfite-converted genome sequence. Recently, two groups have used bisulfite treatment followed by whole-genome sequencing to annotate the DNA methylome of A. Thaliana at single base-pair resolution [70,72] and one group has done the same with human embryonic stem cells [7]. However, the Arabidopsis genome is much smaller than the mammalian genome and contains far fewer repetitive elements. Second, sequencing the methylome of larger genomes is cost-prohibitive. Third, the conversion of unmethylated cytosine to uracil/thymidine during bisulfite treatment reduces genome complexity and can make aligning reads from single-copy regions more difficult.
To retain the advantages of methylation detection by bisulfite while avoiding the prohibitive cost of shotgun bisulfite sequencing, Meissner et al. developed a technique that interrogates DNA fragments from a small proportion, or reduced representation, of the bisulfite-treated genome [73–75]. The genome reduction comes from DNA digestion with methylation-insensitive restriction enzyme MspI and fragment size selection. After digestion, the ends of the DNA are filled-in with dGTP and methylated dCTP, followed by the addition of an A overhang to enable adaptor ligation. The adaptors used for this assay are methylated at cytosine residues to prevent conversion during the bisulfite treatment step. The adaptor-ligated DNA is then size selected on a gel and two fractions are excised – the sizes of which depend on the organism. For mouse DNA, approximately 300,000 MspI fragments that span 40–220 bp are analyzed, which corresponds to nearly 1.4 million CpG sites analyzed at the nucleotide level [74]. These fragments are then bisulfite treated, PCR amplified and size selected once again to generate a sequencing library. Several factors must be considered with this approach. First, it is important to be aware of how the choice of a restriction enzyme to fractionate the DNA will bias the portion of the genome that is represented. A second consideration is the process of mapping reads of bisulfite converted DNA to the genome. Several mapping algorithms for bisulfite genomes have been developed, and vary in their performance [70,72,74,76]. Compared with other sequencing methods, reduced representation bisulfite sequencing (RRBS) provides an efficient way to generate absolute quantification of methylation of more than 1 million CpG sites at single base-pair resolution. Methylation at non-CpG cytosines can also be assessed by RRBS.
Shotgun sequencing methodology is a powerful method for the re-sequencing of whole human genomes and the de novo assembly of new but less complex genomes. Shotgun sequencing of bisulfite-treated DNA has been successfully applied to the approximately 135-Mb genome of A. Thaliana [70,72] and to human embryonic stem cells [7]. Many regions of the mammalian genome do not contain the CpG dinucleotide and, thus, a large number of sequence reads will be uninformative. Prior selection of sequences, for example through sequence capture methodology, enrichment of methylated DNA or enrichment of unmethylated DNA, followed by shotgun sequencing, should increase the efficiency and decrease the cost of this approach. Currently, however, shotgun bisulfite sequencing that first employs selective reduction of the genome (e.g., RRBS) is a more accessible option to a greater number of laboratories.
Reduced representation bisulfite sequencing and shotgun bisulfite sequencing require algorithms that are tailored to mapping the sequence reads from bisulfite treated DNA back onto the genome. Four different algorithms have been developed for this computationally intensive problem [70,72,74,76]. The reduction in base complexity from the bisulfite conversion and the fact that a CpG can be methylated or unmethylated are addressable, through complex issues when aligning bisulfite reads. Owing to the bisulfite conversion process, the forward and reverse strands of DNA are no longer complementary and the sequence reads therefore are compared with four different bisulfite-converted genomes (forward BS, forward BS reverse complement, reverse BS, reverse BS reverse complement) for methylated as well as the unmethylated genome. Thus, for this mapping there is increased search space along with a reduction of sequence complexity, requiring significant computation time for the read mapping steps. Newer versions of alignment algorithms are likely to reduce compute time significantly.
Future perspective
In the short term, it will be challenging to complete single-nucleotide maps for more than a handful of the hundreds of methylomes in a single individual. However, the breathtaking pace of advances in next-generation sequencing methodology, including notable increases in sequence reads per lane and read length, along with development of paired-end sequencing have created optimism for the future of sequencing-based methylome mapping. Parallel technology developments in genome sequencing such as direct single-molecule sequencing have significant potential for further advancing methylation mapping. Similarly, the direct detection of 5-methylcytosine in DNA via inexpensively produced nanopores, if they become amenable to high throughput, could be technologically transformative [77].
In addition to 5-methycytosine, another nucleotide base, 5-hydroxymethylcytosine (HMC), has been noted in genomic DNA from mouse brain and human embryonic stem cells, but inexplicably absent from a human cancer cell line [8,9]. Early research in T2, T4 and T6 bacteriophage demonstrated they contained HMC in their genome [78]. In mammals, the enzyme TET1 can convert 5-methylcytosine to 5-hydroxymethylcytosine [9]. It may be that HMC can lead to genomic demethylation, either through a passive mechanism via the inability of DNMT1 to bind [79], or through an active mechanism acting as an intermediate for a glycosylase [80]. Reagents and platforms for distinguishing HMC from 5-methylcytosine patterns across the genome would be most useful.
Executive summary.
Reagents
Methylation-sensitive restriction enzymes are capable of distinguishing methylated versus unmethylated DNA, but only at CpGs within their specific recognition sites.
Affinity-based methods include an antibody against 5-methylcytosine and columns that contain methylated DNA binding proteins.
Chemical-based treatment of DNA converts unmethylated cytosines while leaving methylated cytosines intact, allowing for single base pair resolution of methylation.
Platforms
BeadArray™ (Illumina) is used with bisulfite treated DNA to analyze a high number of samples at a moderate number of CpGs across the genome.
Microarrays can be used with restriction enzyme or affinity-based enrichment methods and provide relative levels of methylation across the genome.
Next-generation sequencing-based methods can assess tens of millions of DNA fragments, allowing for detection of DNA methylation across the entire genome, including interspersed repeat sequences that are inaccessible using microarrays.
Future perspective
Limitations owing to cost and detection level need to be addressed to allow for complete single nucleotide maps of all methylomes of an individual.
Direct sequencing of 5-methylcytosine could provide more complete methylome maps.
Reagents to distinguish 5-hydroxymethylcytosine from 5-methylcytosine would be very useful to assess the genomic distribution and potential function of 5-hydroxymethylcytosine.
Footnotes
Financial & competing interests disclosure: The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Trasler JM, Trasler DG, Bestor TH, Li E, Ghibu F. DNA methyltransferase in normal and Dnmtn/Dnmtn mouse embryos. Dev Dyn. 1996;206:239–247. doi: 10.1002/(SICI)1097-0177(199607)206:3<239::AID-AJA2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 2.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 3.Maraschio P, Zuffardi O, Dalla Fior T, Tiepolo L. Immunodeficiency, centromeric heterochromatin instability of chromosomes 1, 9, and 16, and facial anomalies: the ICF syndrome. J Med Genet. 1988;25:173–180. doi: 10.1136/jmg.25.3.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu GL, Bestor TH, Bourc'his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 5.Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nat Genet. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- 6.Ramsahoye BH, Biniszkiewicz D, Lyko F, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ The first whole-genome shotgun bisulfite sequencing reported for human cells. Non-CpG methylation was found to be prevalent in embryonic stem cells, and was reduced upon differentiation and restored in induced pluripotent stem cells.
- 8.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Describes the presence of 5-hydroxymethylcytosine in mammals and suggests enzymatic regulation. Future epigenome studies will need to consider this new base when analyzing epigenetic patterns and new reagents are needed for its study.
- 9.Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Describes the presence of 5-hydroxymethylcytosine in mammals, and suggest enzymatic regulation. Future epigenome studies will need to consider this new base when analyzing epigenetic patterns and new reagents are needed for its study.
- 10.Rollins RA, Haghighi F, Edwards JR, et al. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–163. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das R, Dimitrova N, Xuan Z, et al. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci USA. 2006;103:10713–10716. doi: 10.1073/pnas.0602949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanagan JM, Wild L. An epigenetic role for noncoding RNAs and intragenic DNA methylation. Genome Biol. 2007;8:307. doi: 10.1186/gb-2007-8-6-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakyan VK, Blewitt ME, Druker R, Preis JI, Whitelaw E. Metastable epialleles in mammals. Trends Genet. 2002;18:348–351. doi: 10.1016/s0168-9525(02)02709-9. [DOI] [PubMed] [Google Scholar]
- 14.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu M, Yao J, Cai L, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 16.Bloushtain-Qimron N, Yao J, Snyder EL, et al. Cell type-specific DNA methylation patterns in the human breast. Proc Natl Acad Sci USA. 2008;105:14076–14081. doi: 10.1073/pnas.0805206105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber M, Davies JJ, Wittig D, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]; ▪▪ First paper to describe the use of the methylcytosine antibody to immunoprecipitated DNA, and provides a protocol used by subsequent publications.
- 18.Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]; ▪ Describes the use of methyl-binding column to enrich for methylated DNA sequences that are hybridized to DNA microarrays.
- 19.Rauch T, Wang Z, Zhang X, et al. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci USA. 2007;104:5527–5532. doi: 10.1073/pnas.0701059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. A human B cell methylome at 100-base pair resolution. Proc Natl Acad Sci USA. 2009;106:671–678. doi: 10.1073/pnas.0812399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frommer M, McDonald LE, Millar DS, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pfeifer GP, Riggs AD. Genomic sequencing by ligation-mediated PCR. Mol Biotechnol. 1996;5:281–288. doi: 10.1007/BF02900367. [DOI] [PubMed] [Google Scholar]
- 23.Clark SJ, Statham A, Stirzaker C, Molloy PL, Frommer M. DNA methylation: bisulphite modification and analysis. Nat Protoc. 2006;1:2353–2364. doi: 10.1038/nprot.2006.324. [DOI] [PubMed] [Google Scholar]
- 24.Gitan RS, Shi H, Chen CM, Yan PS, Huang TH. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Res. 2002;12:158–164. doi: 10.1101/gr.202801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatada I, Hayashizaki Y, Hirotsune S, Komatsubara H, Mukai T. A genomic scanning method for higher organisms using restriction sites as landmarks. Proc Natl Acad Sci USA. 1991;88:9523–9527. doi: 10.1073/pnas.88.21.9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costello JF, Hong C, Plass C, Smiraglia DJ. Restriction landmark genomic scanning: analysis of CpG islands in genomes by 2D gel electrophoresis. Methods Mol Biol. 2009;507:131–148. doi: 10.1007/978-1-59745-522-0_11. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalgo ML, Liang G, Spruck CH, 3rd, Zingg JM, Rideout WM, 3rd, Jones PA. Identification and characterization of differentially methylated regions of genomic DNA by methylation-sensitive arbitrarily primed PCR. Cancer Res. 1997;57:594–599. [PubMed] [Google Scholar]
- 28.Huang TH, Laux DE, Hamlin BC, Tran P, Tran H, Lubahn DB. Identification of DNA methylation markers for human breast carcinomas using the methylation-sensitive restriction fingerprinting technique. Cancer Res. 1997;57:1030–1034. [PubMed] [Google Scholar]
- 29.Toyota M, Ahuja N, Suzuki H, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res. 1999;59:5438–5442. [PubMed] [Google Scholar]
- 30.Huang TH, Perry MR, Laux DE. Methylation profiling of CpG islands in human breast cancer cells. Hum Mol Genet. 1999;8:459–470. doi: 10.1093/hmg/8.3.459. [DOI] [PubMed] [Google Scholar]
- 31.Bibikova M, Fan JB. GoldenGate assay for DNA methylation profiling. Methods Mol Biol. 2009;507:149–163. doi: 10.1007/978-1-59745-522-0_12. [DOI] [PubMed] [Google Scholar]
- 32.Ladd-Acosta C, Pevsner J, Sabunciyan S, et al. DNA methylation signatures within the human brain. Am J Hum Genet. 2007;81:1304–1315. doi: 10.1086/524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci USA. 2005;102:15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raval A, Tanner SM, Byrd JC, et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129:879–890. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan PS, Chen CM, Shi H, et al. Dissecting complex epigenetic alterations in breast cancer using CpG island microarrays. Cancer Res. 2001;61:8375–8380. [PubMed] [Google Scholar]
- 36.Ching TT, Maunakea AK, Jun P, et al. Epigenome analyses using BAC microarrays identify evolutionary conservation of tissue-specific methylation of SHANK3. Nat Genet. 2005;37:645–651. doi: 10.1038/ng1563. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Yazaki J, Sundaresan A, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell. 2006;126:1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]; ▪▪ First high-resolution mapping of eukaryotic methylome using methylated DNA immunoprecipitation (MeDIP) coupled with tiling microarray analysis.
- 38.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis Thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]
- 39.Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nat Genet. 1994;6:236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- 40.Shen Y, Chow J, Wang Z, Fan G. Abnormal CpG island methylation occurs during in vitro differentiation of human embryonic stem cells. Hum Mol Genet. 2006;15:2623–2635. doi: 10.1093/hmg/ddl188. [DOI] [PubMed] [Google Scholar]
- 41.Shen Y, Matsuno Y, Fouse SD, et al. X-inactivation in female human embryonic stem cells is in a nonrandom pattern and prone to epigenetic alterations. Proc Natl Acad Sci USA. 2008;105:4709–4714. doi: 10.1073/pnas.0712018105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fouse SD, Shen Y, Pellegrini M, et al. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 44.Smith AE, Chronis C, Christodoulakis M, et al. Epigenetics of human T cells during the G0-->G1 transition. Genome Res. 2009;19:1325–1337. doi: 10.1101/gr.085530.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu CL, Schreiber SL, Bernstein BE. Development and validation of a T7 based linear amplification for genomic DNA. BMC Genomics. 2003;4:19. doi: 10.1186/1471-2164-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 47.Yuan E, Haghighi F, White S, et al. A single nucleotide polymorphism chip-based method for combined genetic and epigenetic profiling: validation in decitabine therapy and tumor/normal comparisons. Cancer Res. 2006;66:3443–3451. doi: 10.1158/0008-5472.CAN-05-3739. [DOI] [PubMed] [Google Scholar]
- 48.Kerkel K, Spadola A, Yuan E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008;40:904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 49.Bird AP, Taggart MH, Smith BA. Methylated and unmethylated DNA compartments in the sea urchin genome. Cell. 1979;17:889–901. doi: 10.1016/0092-8674(79)90329-5. [DOI] [PubMed] [Google Scholar]
- 50.Khulan B, Thompson RF, Ye K, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16:1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schumacher A, Kapranov P, Kaminsky Z, et al. Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res. 2006;34:528–542. doi: 10.1093/nar/gkj461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oda M, Glass JL, Thompson RF, et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 2009;37:3829–3839. doi: 10.1093/nar/gkp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brunner AL, Johnson DS, Kim SW, et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19:1044–1056. doi: 10.1101/gr.088773.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lippman Z, Gendrel AV, Black M, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 56.Nouzova M, Holtan N, Oshiro MM, et al. Epigenomic changes during leukemia cell differentiation: analysis of histone acetylation and cytosine methylation using CpG island microarrays. J Pharmacol Exp Ther. 2004;311:968–981. doi: 10.1124/jpet.104.072488. [DOI] [PubMed] [Google Scholar]
- 57.Irizarry RA, Ladd-Acosta C, Carvalho B, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–790. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Keshet I, Schlesinger Y, Farkash S, et al. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat Genet. 2006;38:149–153. doi: 10.1038/ng1719. [DOI] [PubMed] [Google Scholar]
- 60.Pelizzola M, Koga Y, Urban AE, et al. MEDME: an experimental and analytical methodology for the estimation of DNA methylation levels based on microarray derived MeDIP-enrichment. Genome Res. 2008;18:1652–1659. doi: 10.1101/gr.080721.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karpf AR, Peterson PW, Rawlins JT, et al. Inhibition of DNA methyltransferase stimulates the expression of signal transducer and activator of transcription 1, 2, and 3 genes in colon tumor cells. Proc Natl Acad Sci USA. 1999;96:14007–14012. doi: 10.1073/pnas.96.24.14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamashita K, Upadhyay S, Osada M, et al. Pharmacologic unmasking of epigenetically silenced tumor suppressor genes in esophageal squamous cell carcinoma. Cancer Cell. 2002;2:485–495. doi: 10.1016/s1535-6108(02)00215-5. [DOI] [PubMed] [Google Scholar]
- 63.Foltz G, Yoon JG, Lee H, et al. DNA methyltransferase-mediated transcriptional silencing in malignant glioma: a combined whole-genome microarray and promoter array analysis. Oncogene. 2009;28(29):2667–2677. doi: 10.1038/onc.2009.122. [DOI] [PubMed] [Google Scholar]
- 64.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 65.Shames DS, Girard L, Gao B, et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006;3:E486. doi: 10.1371/journal.pmed.0030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gius D, Cui H, Bradbury CM, et al. Distinct effects on gene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach. Cancer Cell. 2004;6:361–371. doi: 10.1016/j.ccr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 67.Mueller W, Nutt CL, Ehrich M, et al. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene. 2007;26:583–593. doi: 10.1038/sj.onc.1209805. [DOI] [PubMed] [Google Scholar]
- 68.Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255–264. doi: 10.1016/j.ygeno.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 69.Down TA, Rakyan VK, Turner DJ, et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nat Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lister R, O'Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pomraning KR, Smith KM, Freitag M. Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods. 2009;47:142–150. doi: 10.1016/j.ymeth.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 72.Cokus SJ, Feng S, Zhang X, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ First description of whole-genome bisulfite sequencing.
- 73.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meissner A, Mikkelsen TS, Gu H, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ Reduced representation bisulfite sequencing maps of 12 different mouse cell types.
- 75.Smith ZD, Gu H, Bock C, Gnirke A, Meissner A. High-throughput bisulfite sequencing in mammalian genomes. Methods. 2009;48:226–232. doi: 10.1016/j.ymeth.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 78.Wyatt GR, Cohen SS. The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem J. 1953;55:774–782. doi: 10.1042/bj0550774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valinluck V, Sowers LC. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 2007;67:946–950. doi: 10.1158/0008-5472.CAN-06-3123. [DOI] [PubMed] [Google Scholar]
- 80.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]