

Abstract
A major goal of population genomics is to reconstruct the history of natural populations and to infer the neutral and selective scenarios that can explain the present-day polymorphism patterns. However, the separation between neutral and selective hypotheses has proven hard, mainly because both may predict similar patterns in the genome. This study focuses on the development of methods that can be used to distinguish neutral from selective hypotheses in equilibrium and nonequilibrium populations. These methods utilize a combination of statistics on the basis of the site frequency spectrum (SFS) and linkage disequilibrium (LD). We investigate the patterns of genetic variation along recombining chromosomes using a multitude of comparisons between neutral and selective hypotheses, such as selection or neutrality in equilibrium and nonequilibrium populations and recurrent selection models. We perform hypothesis testing using the classical P-value approach, but we also introduce methods from the machine-learning field. We demonstrate that the combination of SFS- and LD-based statistics increases the power to detect recent positive selection in populations that have experienced past demographic changes.
GENOMES contain information related to the history of natural populations. Past neutral and selective processes may have left footprints in the genome. Recent advances in population genetics aim to understand the patterns of genetic diversity and identify events that have led to genetic adaptations. Among them, positive selection has been a focus of many recent studies (Harr et al. 2002; Kim and Stephan 2002; Glinka et al. 2003; Akey et al. 2004; Orengo and Aguadé 2004). Their goal is to (i) provide evidence of positive selection, (ii) estimate the strength and the rate of selection, and (iii) localize the targets of selection. These objectives form the basis of a long-term pursuit, which is the understanding of the molecular basis of adaptation of populations in a changing environment.
Positive selection can cause genetic hitchhiking when a beneficial mutation spreads in the population (Maynard Smith and Haigh 1974). When a strongly beneficial mutation occurs and spreads in a population, linked neutral or slightly deleterious variants hitchhike with it, and their frequency increases. According to Maynard Smith and Haigh's model, three patterns are generated locally around the position of the beneficial mutation. First, the level of variability will be reduced since standing variation of the population that is not linked to the beneficial allele vanishes, and tightly linked polymorphisms may fix (Kaplan et al. 1989; Stephan et al. 1992). Second, the site frequency spectrum (SFS), which describes the frequency of allelic variants, shifts from its neutral expectation toward rare and high-frequency derived variants (Braverman et al. 1995; Fay and Wu 2000). The third signature describes the emergence of specific linkage disequilibrium (LD) patterns around the target of positive selection, such as an elevated level of LD in the early phase of the fixation process of the beneficial mutation and a decay of LD across the selected site at the end of the selective phase (Kim and Nielsen 2004; Stephan et al. 2006).
The availability of genome-wide SNP data has made possible the scanning of genomes and the identification of loci that may have been targets of recent selective events. Several approaches have been developed within the last years that can detect the molecular signatures of positive selection (Kim and Stephan 2002; Jensen et al. 2005; Nielsen et al. 2005). While the methods of Kim and Stephan (2002) and Jensen et al. (2005) are designed to analyze subgenomic SNP data, the approach of Nielsen et al. (2005) can be applied to both subgenomic and whole-genome data (reviewed in Pavlidis et al. 2008). For this reason we concentrate here on the latter procedure. This method, called SweepFinder, calculates the probability P(x) that a polymorphism of multiplicity x is linked to a beneficial mutation using a simple selective model and the SFS prior to the selective event. Then, for each location in the genome it compares a selective with a neutral model assuming independence between the SNPs, therefore calculating the composite likelihood ratio Λ. Thus, it identifies regions where the likelihood of the selective sweep is greater than that of the neutral model using the maximum value ΛMAX of Λ.
The ω-statistic, developed by Kim and Nielsen (2004), detects specific LD patterns caused by genetic hitchhiking (described above). In the study by Kim and Nielsen (2004) the maximum value of the ω-statistic was used to identify the targets of selective sweeps. Later, Jensen et al. (2007) studied its performance in separating demographic from selective scenarios. An important result by Jensen et al. (2007) is the demonstration that for demographic parameters relevant to nonequilibrium populations (such as the cosmopolitan populations of Drosophila melanogaster) the ω-statistic can distinguish between neutral and selective scenarios. This article further develops SweepFinder and the ω-statistic such that they can eventually be applied to whole-genome SNP data sets that have been collected from nonequilibrium populations. In particular, populations undergoing population-size bottlenecks are of interest as these size changes may confound the patterns of selective sweeps (Barton 1998). For this reason we use the following approach: first, we theoretically analyze the genealogies of bottlenecked populations under neutrality and show to what extent they resemble the genealogies of single hitchhiking (SHH) events. We also point out the importance of high-frequency-derived variants in the identification of selective sweeps. Second, we study the statistical properties of SweepFinder and the ω-statistic separately and in combination. As the main result, we demonstrate that the combination of these two methods (that include both SFS and LD information) increases the power for detecting recent SHH events in nonequilibrium populations, in particular when machine-learning techniques are employed. Third we analyze the performance of SweepFinder and the ω-statistic in the detection of recurrent hitchhiking (RHH) events.
METHODS
Modifications of the ω-statistic and SweepFinder:
The proposed modifications aim at (i) adapting the ω-statistic for the analysis of whole-genome data and (ii) increasing the accuracy of SweepFinder to predict the target of selection. Instead of fixed windows, variable-size windows are used in the ω-statistic, and in the SweepFinder algorithm a fraction of monomorphic sites is incorporated.
The hitchhiking model by Maynard Smith and Haigh (1974) predicts that an excess of LD arises after the completion of the selective sweep within each of the two regions flanking the selected site, but does not extend across the two regions (Stephan et al. 2006; McVean 2007; Pfaffelhuber et al. 2008). This is due to the assumption that any observed polymorphism around the sweep has been introduced in the population prior the selective sweep and entered the beneficial genetic background through recombination. Since independent recombination events are necessary to explain polymorphisms on both sides of the selective sweep, the LD vanishes across the site of the beneficial mutation, but not within each side. This genomic footprint may be captured using the ω-statistic (Kim and Nielsen 2004). Assume a genomic window with S segregating sites that is split into a left and right subregion with l and S – l segregating sites, respectively. The ω-statistic (Equation 1) quantifies to what extent average LD is elevated on each side of the selective sweep (see the numerator of Equation 1) but not across the selected site (see the denominator of Equation 1):
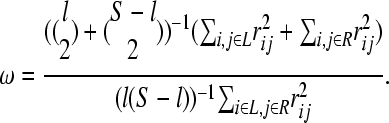 |
(1) |
The ω-statistic considers the space between the left and right subregions as the center of the selective sweep. Thus, a genomic region may be scanned and scores are reported for each position. Then, using simulations, a significance threshold is determined. The maximum value ωMAX predicts the target of recent positive selection. In the original version of the ω-statistic, the borders of the left and right subregions are assumed constant (Kim and Nielsen 2004; Jensen et al. 2007). This may be valid for a subgenomic analysis, when the recombination rate ρ and mutation rate θ do not fluctuate much or a single selective event may have occurred. However, in a whole-genome study these parameters that affect the extent of LD may vary dramatically. Additionally, the polymorphism patterns may have been shaped by recurrent selective sweeps. Thus, the constant-border approach implemented by Kim and Nielsen (2004) may be limited. If the subregions are large, then ωMAX tends to decrease and the signal disappears. On the other hand, short subregions might contain no SNPs and the ω-statistic cannot be calculated.
We have implemented a variable-window size ω-statistic. The borders of the left and right subregions vary and the configuration that maximizes ω is reported. This approach overcomes the aforementioned problems inherent in the constant-border approach of Kim and Nielsen (2004). Thus, it may be suitable for scanning large genomic regions or whole chromosomes characterized by variable ρ or θ parameters and shaped by recurrent adaptive substitutions.
A naive implementation of the ω-statistic scanning algorithm would recalculate the LD of the positions as the center of the sweep moves along the chromosome. This is particularly critical for the variable-window size approach since the number of calculations increases. Our implementation, as illustrated in supporting information, Table S1, guarantees a single calculation between any two sites that may participate in the ω calculation. Thus, it results in an algorithm that is efficient when the number of polymorphisms is large. Calculations are performed using a matrix Z (Table S1), which stores the unweighted ZnS (Kelly 1997) values (not divided by the number of comparisons) for all possible windows. For a pair (i, i + 1), Zi,i+1 equals the correlation coefficient between these two positions. This value is then added to all cells Zj,i+1, with j < i to form the ZnS for the region [j, i + 1]. With this method all possible numerators of the ω-statistic are formed. When the left and right subregions are defined by [i, k] and [k + 1, j], respectively, then the denominator is simply a weighted version of Zi,j − Zi,k − Zk+1,j.
SweepFinder detects the shift of the SFS as a signature of hitchhiking. Demographic effects are incorporated through the neutral SFS, which is either provided by the user or calculated from the data itself. Monomorphic sites are generally excluded from the analysis (Nielsen et al. 2005; Svetec et al. 2009) since tests that include them may be more sensitive to assumptions regarding the mutation rate (Nielsen et al. 2005). Additionally, for realistic mutation rates, the majority of the sites remain monomorphic. Thus, by including invariant sites, the data set and the computational time required for the analysis increase dramatically. On the other hand, the decrease of diversity represented by the monomorphic sites constitutes a well-known signature of the hitchhiking effect. Omitting them may decrease the power of the tests (Nielsen et al. 2005) and lead to inaccurate predictions about the target of selection. Inaccuracies mainly emerge due to changes in the input site density when only polymorphic sites are included. We incorporate a fraction of the monomorphic sites into the analysis in a way that (i) generates a uniform input site density and (ii) preserves the signature of low diversity in regions of depleted variation. Additionally, since only a small fraction of monomorphic sites are used, the computational time is only increased slightly. Given a genomic region with S polymorphic sites we include Sq monomorphic sites, where 0 < q < 1. In this study q = 0.1, so that the number of monomorphic sites are in the same order as the polymorphic sites. We proceed as follows. In the first step, there are S − 1 intervals between the S polymorphic sites. A monomorphic site is included at a random location within the largest interval. In the second step there are S + 1 sites and S intervals and the process is repeated. The cutoff value is defined by treating the neutral simulations in the same way. With this process the SNP density differences are reduced and monomorphic sites are embedded in regions of depleted variation.
Quantifying the effects of population bottlenecks on neutral genealogies:
The ω-statistic and SweepFinder can scan genomes from natural populations that have experienced demographic changes and detect targets of selection. We investigated whether the neutral demographic scenarios inferred by Li and Stephan (2006) and Thornton and Andolfatto (2006) to describe the demography of a European population of D. melanogaster can result in patterns along a recombining chromosome that resemble selective sweeps. In particular, we examined which effects of population bottlenecks are responsible for the polymorphism patterns that mimic the effects of selective sweeps. We focused on the properties of genealogies that are generated by those two demographic models because genealogies reflect demographic properties more comprehensively than summary statistics.
A way to measure the effect of a bottleneck on the genealogies of a recombining genome is through the ratio 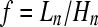 of the total length to the height of the coalescent. Short, star-like genealogies have large ratios and
of the total length to the height of the coalescent. Short, star-like genealogies have large ratios and  is obtained for a n-furcated star-like tree. On the other hand, for genealogies with long internal branches the ratio takes small values and
is obtained for a n-furcated star-like tree. On the other hand, for genealogies with long internal branches the ratio takes small values and 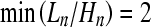 is obtained when the genealogy is dominated by two very long internal branches. Using simulations we first calculate the percentage of n-furcated star-like genealogies (with large f values) in a region of 50 kb. Then, for each simulated instance we relate the percentage of n-furcated star-like genealogies with the resemblance to a selective sweep as this is measured using SweepFinder (see theoretical analyses).
is obtained when the genealogy is dominated by two very long internal branches. Using simulations we first calculate the percentage of n-furcated star-like genealogies (with large f values) in a region of 50 kb. Then, for each simulated instance we relate the percentage of n-furcated star-like genealogies with the resemblance to a selective sweep as this is measured using SweepFinder (see theoretical analyses).
The joint effects of population bottlenecks and selective sweeps on high-frequency derived alleles:
A hallmark of selective sweeps in constant populations is the excess of high-frequency-derived variants around the target of positive selection. High-frequency-derived variants consist of mutations that were present in the population prior to the selective sweep, hitchhike with the beneficial allele, and, due to recombination, appear as polymorphisms. This signature forms the basis of a multitude of neutrality tests that are based on the SFS (Fay and Wu 2000; Kim and Stephan 2002; Nielsen et al. 2005) and contributes to the precise detection of the target of selection. However, in natural populations positive selection may occur simultaneously with demographic changes. Using simulations from the demographic models that were inferred by Li and Stephan (2006) and Thornton and Andolfatto (2006), we examine whether high-frequency-derived alleles occur when demographic changes occur simultaneously with positive selection.
Measuring the precision of the inferred selective sweep position:
An objective of the genome-scanning studies is the precise prediction of the selective sweep locations. Usually, every position or a subset of them is scored for a given statistic (for example, the ω-statistic or the SweepFinder). Thus, peaks and valleys are formed along the genomic region. Then, some of the peaks may survive a cutoff value delimiting the potential targets of selection. As illustrated in Figure S1, we determine the distance between a peak on the landscape of the statistic and the closest location where a selective sweep has occurred given a user-defined threshold. In Figure S1, two selective sweeps have occurred recently in the history of the population. The positions of the sweeps are illustrated as vertical green lines. A peak is defined as the highest point in an isolated region by the cutoff value. Thus, five peaks (a to e) have been formed in the example of Figure S1. D measures the distance between a peak and the closest selective sweep location. On the basis of this approach we can measure the accuracy of the different methods. Furthermore, we implemented a simple randomization of the peaks to evaluate the quality of the predictions. This is necessary because finite genomic regions are simulated, and therefore the distance between any location and the target of selection is bounded.
Supervised-learning techniques:
We introduce supervised-learning approaches from the field of machine learning that can be useful for the classification of a genomic region as either neutral or selected. In a classification problem, the goal is to separate these classes using a function, which is inferred from the available data. Such a process is called “learning from the data” or “supervised learning” and is related to finding the optimal hyperplane that distinguishes the two classes. Typically, in a supervised-learning problem, data consist of pairs of input and output objects. Input consists of a vector of multiple entries that summarize the data and are called features. Inputs can be set arbitrarily depending on the specific problem. However, the efficiency of the algorithm increases when they are independent and capture the whole information of the data. Output can be binary, denoting the class that the object belongs to. In supervised learning the goal is to use the input to predict the value of the output, and the problem can be formulated as teaching the computer the combinations of feature values that are associated with either of the classes. In the specific problem we examine here, the output is coded as neutrality/selection. Then, using simulations of the neutral demographic model and the model with selection we train the algorithm to separate these two classes. As input for the machine-learning approach we use ωMAX, ΛMAX (from the original algorithms), and combinations of ω and Λ, such as the distance between the genomic positions of ωMAX and ΛMAX and the correlation coefficient between ω and Λ. The reasoning for this choice of inputs is as follows. First, ΛMAX and ωMAX capture different aspects of the data. ΛMAX is affected mostly by the SFS, whereas ωMAX is affected by LD. Even if SFS and LD can be correlated (Kim and Nielsen 2004), it is expected that this correlation is lower than that from using statistics that are based exclusively on the SFS or LD. Second, previous studies have shown that ΛMAX and ωMAX are relatively robust to demographic changes (but see Orengo and Aguadé 2010). Third, it seems intuitively obvious that the peaks of ω and Λ profiles should point to the same genomic location if a selective sweep has occurred. Thus, using the distance between the peaks or the correlation of the profiles should increase the classification performance of the algorithm. In this study, both the distance between the peaks and the correlation between the profiles are used.
For each demographic scenario that was simulated in this study, we used a subset of simulations for training and the remaining for testing the performance. The supervised-learning approach can be employed to classify a certain genomic region as either neutral or selected. However, within a region the specific target of selection cannot be specified by the method itself. To achieve this, the features of the method (i.e., the ω and Λ profiles) should be inspected. Tables 1–4 provide information about the accuracy of the features under various demographic scenarios.
TABLE 1.
Equilibrium neutrality vs. selection in equilibrium populations
| Model parameter | Performance | SF | SF* | ω | ω* | SVM |
|---|---|---|---|---|---|---|
| α = 500 | TP (FP = 0.03) | 0.85 | 0.97 | 0.13 | 0.14 | 0.9 |
| α = 500 | Median distance (bp) from target (SD) | 1728 (5597) | 754 (1333) | 528 (480) | 540 (525) | — |
| α = 2500 | TP (FP = 0) | 0.97 | 0.99 | 0.82 | 0.85 | 0.98 |
| α = 2500 | Median distance (bp) from target (SD) | 5383 (4509) | 4582 (3905) | 789 (657) | 794 (680) | — |
Using the SVM approach a false positive rate (FP) is estimated. For this FP rate, the true positive rates (TP) of the various neutrality tests are compared. The median distance and the standard deviation (SD) are also shown. SF, original SweepFinder; SF*, modified SweepFinder; ω, ω algorithm with constant-size windows; ω*, ω algorithm with variable-size windows.
TABLE 2.
Nonequilibrium neutrality vs. selection in equilibrium populations
| Model parameter | Performance | SF | SF* | ω | ω* | SVM |
|---|---|---|---|---|---|---|
| α = 500 | TP (FP = 0.26) | 0.1 | 0.41 | 0.04 | 0.03 | 0.75 |
| α = 500 | Median distance (bp) from target (SD) | 899 (878) | 522.982 (824) | 423 (428) | 603 (513) | — |
| α = 2500 | TP (FP = 0.18) | 0.73 | 0.93 | 0.72 | 0.74 | 0.84 |
| α = 2500 | Median distance (bp) from target (SD) | 3065 (3209) | 2074 (3361) | 917 (1653) | 956 (1629) | — |
TABLE 3.
Neutrality vs. selection in nonequilibrium populations (deep bottlenecks)
| Model Parameter | Performance | SF | SF* | ω | ω* | SVM |
|---|---|---|---|---|---|---|
| Sweep in phase 1 | TP (FP = 0.51) | 0.64 | 0.66 | 0.39 | 0.49 | 0.71 |
| Median distance (bp) from target (SD) | 10813 (6768) | 10497 (6832) | 11986 (6595) | 10239 (6186) | — | |
| Random target distance (SD) | 11053 (6827) | 11308 (6803) | 11575 (6645) | 11944 (6945) | — | |
| Sweep in phase 2 | TP (FP = 0.20) | 0.62 | 0.64 | 0.36 | 0.44 | 0.73 |
| Median distance (bp) from target (SD) | 9666 (6531) | 10828 (6896) | 11854 (6500) | 10469 (6123) | — | |
| Random target distance (SD) | 11508 (6885) | 11397 (6808) | 11877 (6750) | 11555 (6804) | — | |
| Sweep in phase 2* | TP (FP = 0.08) | 0.72 | 0.78 | 0.63 | 0.12 | 0.97 |
| Median distance (bp) from target (SD) | 9512 (6659) | 10986 (6977) | 10905 (6482) | 11328 (6487) | — | |
| Random target distance (SD) | 12067 (6983) | 12265 (6920) | 11647 (6950) | 13236 (7213) | — | |
| Sweep in phase 3 | TP (FP = 0.56) | 0.53 | 0.55 | 0.48 | 0.46 | 0.63 |
| Median distance (bp) from target (SD) | 10377 (6831) | 10845 (6833) | 11342 (6662) | 10624 (6541) | — | |
| Random target distance (SD) | 12202 (6908) | 11641 (6860) | 12151 (6920) | 12220 (6824) | — |
 |
TABLE 4.
Neutrality vs. selection in nonequilibrium populations (shallow bottlenecks)
| Model Parameter | Performance | SF | SF* | ω | ω* | SVM |
|---|---|---|---|---|---|---|
| Sweep in phase 1 | TP (FP = 0.27) | 0.46 | 0.49 | 0.22 | 0.25 | 0.5 |
| Median distance (bp) from target (SD) | 10116 (6872) | 10691 (7001) | 10268 (6658) | 10868 (6670) | — | |
| Random target distance (SD) | 11604 (6862) | 11452 (6835) | 10744 (6895) | 11192 (7115) | — | |
| Sweep in phase 2 | TP (FP = 0.22) | 0.58 | 0.56 | 0.27 | 0.32 | 0.6 |
| Median distance (bp) from target (SD) | 10233 (6866) | 11059 (6807) | 11659 (6721) | 11531 (6643) | — | |
| Random target distance (SD) | 11725 (6889) | 11375 (6855) | 10846 (6829) | 11245 (6882) | — | |
| Sweep in phase 2* | TP (FP = 0.35) | 0.67 | 0.74 | 0.65 | 0.4 | 0.67 |
| Median distance (bp) from target (SD) | 9610 (6814) | 10148 (6962) | 11356 (6683) | 10680 (6539) | — | |
| Random target distance (SD) | 11906 (6889) | 12102 (6846) | 12432 (6894) | 11583 (7079) | — | |
| Sweep in phase 3 | TP (FP = 0.25) | 0.4 | 0.38 | 0.23 | 0.27 | 0.46 |
| Median distance (bp) from target (SD) | 10232 (6710) | 10447 (6744) | 11693 (6965) | 10829 (6625) | — | |
| Random target distance (SD) | 11372 (6906) | 11574 (6857) | 11666 (6817) | 13068 (6914) | — |
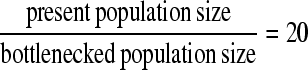 |
Traditionally, when neutrality tests are employed to detect targets of positive selection, neutral simulations are performed and the 5% percentile is used as a threshold. This methodology assumes that neutrality tests produce significantly larger values in data with selection. This may be the case when the population size remains constant. However, in nonequilibrium models the values of the neutrality tests may overlap significantly between neutral models and models with selection, and therefore their performance decreases. Combining different statistics that capture different aspects of the data may contribute to increasing the classification performance.
Several methods have been developed for data classification. For example, Bayesian classifiers, rule-based classifiers, k-nearest neighbors, and linear discriminant analysis are some of the approaches that have been applied to supervised-learning problems (Duda et al. 2000; Han and Kamber 2000; Hastie et al. 2001). Here, we demonstrate the use of SVMs with a radial basis kernel, which is the most widespread kernel. In general, SVM uses a nonlinear mapping to transform the original training data into a higher-dimensional space and to search for an optimal linear hyperplane in this space. A great advantage of SVMs is that they are highly accurate and less prone to overfitting; i.e., they have desirable generalization properties (Han and Kamber 2000).
Implementation and code availability:
The C++ source code is available from http://www.bio.lmu.de/∼pavlidis. For the ω-statistic, the user is able to choose between constant- or variable-window-size scanning modes. Additionally, besides r2 various other measurements of LD, such as abs(D) and abs(Dω) (Langley and Crow 1974), may be used in Equation 1. There are no specific library dependencies and the software can be installed on any Linux machine that runs the g++ compiler. Also, the modified version of SweepFinder that has been used here to analyze data with monomorphic sites is provided. In this version the likelihood curve of monomorphic sites has been modified so that the probability to observe a monomorphic site is high in the proximity of the sweep position but becomes negligible as distance increases (the rate of decrease is larger than in the original version). The original version of SweepFinder is provided by the website of Rasmus Nielsen (http://people.binf.ku.dk/rasmus/webpage/sf.html). Furthermore, perl scripts that have been used in the analysis are available from http://www.bio.lmu.de/∼pavlidis or upon request from the authors.
THEORETICAL ANALYSES
The genealogies of bottlenecked populations may resemble those of SHH in constant-size populations:
Past demographic changes such as bottlenecks may confound the patterns of a selective sweep (Barton 1998). Similarly to a selective sweep, a bottleneck scenario may result in coalescent trees dominated by either external or internal branches. Short coalescent trees with long external branches are obtained when, due to a rapid, recent, and severe decrease of population size, the time of the most recent common ancestor of the sample is found within the bottleneck period. On the other hand, if some of the lineages escape the bottleneck, then long internal branches will be created. In recombining genomic regions, short and long trees may alternate, creating sweep-like patterns in the SFS (Barton 1998).
We illustrate the effect of bottlenecks on genealogies using the demographic scenarios that have been inferred by Li and Stephan (2006) and Thornton and Andolfatto (2006) to describe the history of the European population of D. melanogaster. Scaling the time in units of 4N generations (where N is the present effective population size) the Li and Stephan (2006) model describes a four-epoch scenario. Backward in time, the population experiences a bottleneck from 0.0367 time units until 0.0375 time units. Within this bottleneck period Nb = 0.002N, where Nb denotes the effective population size in the bottleneck. Then, instantly, the size of the population size changes to 7.5N, and eventually at the time 0.1395 it becomes 1.5N. The bottleneck phase models the founding of the European population from the ancestral population, whereas the transition from 7.5N to 1.5N models a (forward-in-time) expansion of the ancestral population. The demographic scenario inferred by Thornton and Andolfatto (2006) implements a three-epoch model. The values of the parameters depend on the ratio ρ/θ and here we use the results obtained when ρ/θ = 10. The present population size N is estimated to be 2.4 × 106, and backward in time at 0.0042 it contracts to 0.029N. Finally, the population reaches instantly the present-day level at time 0.022.
The demographic model of Li and Stephan (2006) produces both star-like and long genealogies in the same genomic region of a recombining chromosome (Figure 1). The length of these trees is on average shorter than that of the standard neutral trees, thus reducing variation. The effect of the Thornton and Andolfatto (2006) demographic model is similar, but milder. On average, it creates shorter genealogies and effectively reduces the nucleotide polymorphism. However, it does not result in extreme star-like coalescent trees as often as the Li and Stephan (2006) model (Figure 1). This is because the population-size changes are milder, the bottleneck period is longer, and starts (backward in time) very recently in the usual coalescent time scale, allowing for a series of coalescent events.
Figure 1.—

Histogram of the ratio 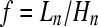 for the following demographic scenarios. (A) a single realization of the bottleneck scenario inferred by Li and Stephan (2006). Long coalescent trees that escape the bottleneck tend to produce small ratios (<4). On the other hand, genealogies that coalesce within the bottleneck period produce star-like trees because of the recent, rapid, and severe contraction of the population. (B) A realization of the bottleneck scenario inferred by Thornton and Andolfatto (2006). In contrast to Li and Stephan (2006), coalescent events occur continuously. (C) The standard neutral model. For the Li and Stephan (2006), Thornton and Andolfatto (2006), and the neutral scenario, 12 chromosomes of 50 kb have been simulated. The recombination rate is ρ = 0.05/bp and the mutation rate θ = 0.004/bp. The parameter values for the Li and Stephan (2006) and Thornton and Andolfatto (2006) scenarios are described in the main text.
for the following demographic scenarios. (A) a single realization of the bottleneck scenario inferred by Li and Stephan (2006). Long coalescent trees that escape the bottleneck tend to produce small ratios (<4). On the other hand, genealogies that coalesce within the bottleneck period produce star-like trees because of the recent, rapid, and severe contraction of the population. (B) A realization of the bottleneck scenario inferred by Thornton and Andolfatto (2006). In contrast to Li and Stephan (2006), coalescent events occur continuously. (C) The standard neutral model. For the Li and Stephan (2006), Thornton and Andolfatto (2006), and the neutral scenario, 12 chromosomes of 50 kb have been simulated. The recombination rate is ρ = 0.05/bp and the mutation rate θ = 0.004/bp. The parameter values for the Li and Stephan (2006) and Thornton and Andolfatto (2006) scenarios are described in the main text.
Next we used simulations to examine the relationship between the percentage of star-like genealogies, the number of segregating sites, and ΛMAX of SweepFinder, which can be considered a proxy for the resemblance of polymorphism patterns (based on the SFS) to a signature of a selective sweep. A 50-kb genomic region was simulated using ms (Hudson 2002) for a sample of 12 chromosomes. The recombination rate ρ = 0.05/bp and the mutation rate θ = 0.004/bp. The demographic model describes a recent population bottleneck (as inferred by Li and Stephan (2006)). As illustrated in Figure 2, a small number of star-like trees create a large number of segregating sites and small ΛMAX values. Similarly, when a genomic region is dominated by short, star-like genealogies, the number of segregating sites and ΛMAX decrease. Even if this constitutes a polymorphism valley, the pattern does not look like a sweep because of a lack of the high-frequency derived variants (Kim and Stephan 2002). On the other hand, the simultaneous presence of star-like and long genealogies creates sweep-like patterns. For intermediate frequencies of star-like genealogies, ΛMAX assumes large values. Since neighboring genealogies are not independent, star-like genealogies form clusters and effectively create valleys of reduced polymorphism resembling a selective sweep. These results help to interpret some of our findings below.
Figure 2.—

The relation between (A) ΛMAX and (B) the percentage of star-like genealogies and the number of segregating sites in the Li and Stephan (2006) demographic scenario. We have performed neutral simulations for 12 recombining chromosomes, assuming a length of 50 kb. The recombination rate ρ = 0.05/bp and the mutation rate θ = 0.005/bp. The parameter values for the demographic model inferred by Li and Stephan (2006) are described in the main text. The number of short genealogies in the Li and Stephan (2006) scenario determines both the number of segregating sites and the sweep resemblance (measured by the SweepFinder statistic). When a genomic region is dominated by short star-like genealogies only a few segregating sites are present. Even if this constitutes a polymorphism valley, the pattern does not look like a single sweep because of a lack of the high-frequency derived variants (Kim and Stephan 2002). Similarly, when the star-like trees are absent ΛMAX is small. On the other hand, the simultaneous presence of star-like and long genealogies creates sweep-like patterns. This is because star-like trees tend to cluster together along the recombining chromosome, creating valleys within polymorphism islands.
Selective sweeps in nonequilibrium populations may result in a loss of high-frequency-derived variants and violate the assumptions of SweepFinder and the ω-statistic:
We examined the effects of selective sweeps on polymorphisms, when they occur within demographic bottlenecks. A 50-kb genomic fragment was simulated under the bottleneck model inferred by Thornton and Andolfatto (2006), and a selective sweep (α = 2500) was assumed to take place within the bottleneck period (Thornton and Jensen 2007). First, we show that the combined action of selective sweeps and bottlenecks results in SFS that differ considerably from those generated by selective sweeps in equilibrium populations. Figure 3 compares the modifications of the average SFS around the target of selection in a constant-size demographic scenario with the model inferred by Thornton and Andolfatto (2006). It is apparent that in equilibrium demographic models there is a dramatic increase of the class n − 1 in the proximity of the selective sweeps (Figure 3A). Neutrality tests based on the SFS can detect the increase of the high-frequency-derived variants and therefore the accurate prediction of the target of selection is possible. In nonequilibrium scenarios, when population contraction and selective sweeps co-occur, the n − 1 class vanishes in a large genomic region around the target of selection (Figure 3B). The joint effect of selection and population contraction increases the probability of coalescences, resulting in short genealogies where the most recent common ancestor is located within the bottleneck phase. Consequently, the frequency of the n − 1 class vanishes in the present-day sample. Furthermore, the part of the genealogy that is older than the selective sweep/bottleneck phase is eliminated. Therefore the vast majority of the present-day polymorphisms are younger than the selective sweep. This violates the assumptions of SweepFinder and the ω-statistic and may result in imprecise prediction of the target of selection.
Figure 3.—

A selective sweep causes a spatial modification of the SFS. The mean and the variance of the frequency are modified when a selective sweep has occurred in the middle of a 50-kb genomic fragment. The 50-kb region is split in 2-kb nonoverlapping windows and in each one the average mean (fi) (A and C) and the variance var(fi) (B and D) of the frequency fi of the polymorphism class i is calculated. In A the plots refer to a selective event in equilibrium populations (α = 2500) that has been completed recently, whereas in C, the plots refer to the nonequilibrium model of Thornton and Andolfatto (2006) (α = 2500). The solid lines refer to the singletons, the dashed lines to the class 11, and the gray lines to the classes 2–10. The dramatic change of the high-frequency derived alleles in A contributes to the precise localization of the selective event. On the contrary, in C the high-frequency-derived SNPs are absent even in the proximity of the selective sweep. This is because the length of the branches of the coalescent tree that may generate high-frequency-derived variants are very small due to the simultaneous action of the sweep and the bottleneck. Therefore, the observed polymorphisms (mostly singletons) are younger than the selective event and spread over the whole genomic region, obscuring the location of the selective sweep.
STATISTICAL PERFORMANCE OF THE TESTS IN THE DETECTION OF SHH
In this section, the discrimination capacity of SweepFinder and the ω-statistic is scrutinized, and the distance between the predicted and the true target of selection is evaluated for single sweeps under the scenarios (i) selection vs. neutrality in equilibrium populations (i.e., standard neutral populations), (ii) selection in equilibrium populations vs. neutrality in nonequilibrium populations (i.e., populations that have experienced past demographic changes), and (iii) selection vs. neutrality in nonequilibrium populations. The performance is assessed as follows. First, the false positive (FP) rate of the SVM is estimated. Using this false positive rate we compare the true positive (TP) rates of each test. Thus, all comparisons refer to the same false positive rate. Second, for the evaluation of the distance between the true and predicted targets we use only simulated results that survive the threshold defined by the false positive rate. Finally, for the nonequilibrium models with selection we implement a simple randomization process to assess the quality of results (see methods).
SHH vs. neutrality in equilibrium populations:
We simulate a single selective sweep in the middle of a 50-kb genomic region using the ssw software (Kim and Stephan 2002). The parameter values have been chosen for their relevance to natural populations of D. melanogaster. Specifically, the parameter α = 2Ns, where s is the selection coefficient of the beneficial mutation, assumes the values 500, 2500, and 5000 that are realistic for D. melanogaster (Beisswanger and Stephan 2008). For all data sets the mutation rate θ = 0.005/bp, similar to the estimation of θ for the European population of D. melanogaster by Li and Stephan (2006). The scaled recombination rate ρ = 0.05/bp, so that the ratio ρ/θ =10 (Thornton and Andolfatto 2006). The standard neutral simulations were performed using the same value of ρ. We used a sample size of 12 for all simulations.
Each realization of the selective sweep was compared with those of the standard neutral model that are obtained using 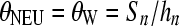 . θNEU denotes the θ value used in standard neutral simulations, θW is Watterson's (1975) estimator of θ obtained using the number of segregating sites Sn of the selective sweep realization, and
. θNEU denotes the θ value used in standard neutral simulations, θW is Watterson's (1975) estimator of θ obtained using the number of segregating sites Sn of the selective sweep realization, and 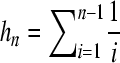 . Thus, a selective sweep is compared with the standard neutral realizations that on average create the observed number of polymorphic sites [F θ procedure (Ramos-Onsins et al. 2007)]. Alternative approaches to calculating the threshold value may use the observed number of segregating sites Sn or take into account the uncertainty on θ by considering a prior distribution of θ. In neutral equilibrium populations these approaches result in the same threshold values for the models tested in this study (Figure S2). Here, for the calculation of thresholds we use the Fθ approach. Since, the null model is represented by an equilibrium standard neutral model, θ can be estimated using the estimator θW. Figure S2 shows that the cutoff value of the ω-statistic decreases as Sn increases and the opposite tendency is seen for the SweepFinder statistic.
. Thus, a selective sweep is compared with the standard neutral realizations that on average create the observed number of polymorphic sites [F θ procedure (Ramos-Onsins et al. 2007)]. Alternative approaches to calculating the threshold value may use the observed number of segregating sites Sn or take into account the uncertainty on θ by considering a prior distribution of θ. In neutral equilibrium populations these approaches result in the same threshold values for the models tested in this study (Figure S2). Here, for the calculation of thresholds we use the Fθ approach. Since, the null model is represented by an equilibrium standard neutral model, θ can be estimated using the estimator θW. Figure S2 shows that the cutoff value of the ω-statistic decreases as Sn increases and the opposite tendency is seen for the SweepFinder statistic.
Consistent with previous studies (Jensen et al. 2007) a selective sweep is discriminated easily from the standard neutral model. Indeed as illustrated in Figure 4A, the ωMAX and ΛMAX are distributed to a large extent distinctly even for relatively small values of α (e.g., 500). Results are summarized in Table 1. Next, the distance between the true target of selection and the predicted target of selection is estimated (Table 1). The ω-statistic is more accurate than the SweepFinder and the median distance from the target of selection is about 0.5 kb. However, the performance of SweepFinder in discriminating the two scenarios is higher. Combining SweepFinder with the ω-statistic increases the classification performance (last column in Table 1).
Figure 4.—

The joint distributions of ΛMAX and ωMAX in scenarios with and without selection. (A) We compare the joint distribution of ΛMAX and ωMAX between a model with selection (α = 500) in a constant population and a standard neutral model. The overlap between the distributions is limited and the scenarios can be discriminated by the SweepFinder (y-axis) and to a lesser extent by the ω-statistic (x-axis). (B) We compare a model with selection (α = 500) with a neutral model that has experienced a bottleneck as it has been inferred by Li and Stephan (2006). Neither of the statistics can discriminate accurately the two scenarios (see also Table 2). Note that the scales of the statistics are different in A and B.
SHH in equilibrium populations vs. neutrality in nonequilibrium populations:
Using simulations, selective sweeps have been generated as described above. For realizing past bottleneck events we used the Li and Stephan (2006) demographic history for the European population of D. melanogaster. We follow a similar approach as described in the previous section to assess the cutoff value. However, since the null hypothesis is not represented by the standard neutral model, θW is not an appropriate estimator of θ. Instead, we use the generalized unbiased estimator 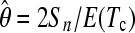 , where E(Tc) is the expected total length of the coalescent of n sequences (Zivkovic and Wiehe 2008). E(Tc) depends only on the demographic history of the population.
, where E(Tc) is the expected total length of the coalescent of n sequences (Zivkovic and Wiehe 2008). E(Tc) depends only on the demographic history of the population.
For large values of α (α = 2500) the true positive rate of the statistics ωMAX and ΛMAX is greater than 70% when the false positive rate is 18% (Table 2). For the same false positive rate, the true positive rate of the modified version of SweepFinder is above 90%. However, when smaller selection coefficients (e.g., α = 500) define the hitchhiking effect, the selective sweep may be inseparable from bottleneck scenarios similar to that inferred by Li and Stephan (2006), using the original version of SweepFinder or the ω-statistic (TP rates < 10%, Table 2 and Figure 4B). The modified version of SweepFinder has a larger discrimination performance (true positive rate ∼40%). The low discrimination performance is indicated by the resemblance of genealogies between bottleneck models and selective sweeps in constant populations (see also theoretical analyses). The distributions of ωMAX and ΛMAX are largely overlapping as illustrated in Figure 4B. The SVM approach performs considerably better than any of the tests alone. The true positive rate is 75% when the false positive is 26% (Table 2). The main reason for the superior performance of the SVM approach is that it uses information about the distance of the peaks. In the scenarios with selection the target can be predicted accurately (Table 2); therefore, the distance between the peaks is considerably smaller than in the neutral scenarios.
SHH vs. neutrality in nonequilibrium populations:
In this section we examine the statistical performance of the neutrality tests to detect selection in a genomic region and assess the distance between the true and the predicted targets of selection. We focus on two bottleneck scenarios. The first one describes a deep and short-lasting bottleneck (model A), whereas the second scenario describes a shallow and long-lasting bottleneck (model B). In both cases the severity (i.e., the product depth × length) is the same (=0.375 in units of 4N), and the bottleneck begins (backward in time) at 0.01. The present effective population size is assumed 106, and the simulated region 50 kb. The recombination rate ρ for the whole region is set to 500. In the deep bottleneck scenario, the depth
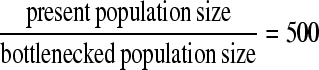 |
and the length of the bottleneck is 0.00075.
In the shallow bottleneck scenario, the depth equals 20 and the length 0.01875.
Neutral simulations have been performed using Hudson's ms (Hudson 2002) and simulations with selection using the mbs algorithm (Teshima and Innan 2009). The design of simulations is as follows. In both cases we fix the number of polymorphic sites (=50) by employing broad uniform priors on θ and accepting only those instances that result in 50 segregating sites. This is justified by the dependence of the ω-statistic and SweepFinder on the number of segregating sites (Figure S2 and Figure S3) and the large variance on segregating sites that neutral bottleneck scenarios generate. Furthermore, the rejection process guarantees that the total length of the tree, the posterior θ values, and the number of segregating sites are coupled. The 25th and 75th quantiles of the posterior distribution of θ are 32 and 52, respectively, for the deep-bottleneck scenario and 32 and 48 for the shallow scenario; therefore, the ratio ρ/θ is close to 10. In the simulations with selection, we examine scenarios of selective sweeps occurring recently (between the present and the bottleneck, sweep in phase 1), within the bottleneck (sweep in phase 2), and after the bottleneck (backward in time, sweep in phase 3). The parameters of the models with selection are described in Table 3 and Table 4 for the deep and shallow models, respectively. Similar to the neutral cases, a broad uniform prior on θ has been used, and we condition on observing 50 segregating sites. The posterior range of θ depends on the timing of the selective sweep; therefore, the ratio ρ/θ is close to 10 when the sweep is either recent or old, but it decreases when the selective sweep occurs within the bottleneck phase.
First, we examined the performance of the ω-statistic and SweepFinder to detect whether a genomic region has been shaped by positive selection. Results are presented in Table 3 and Table 4. For all comparisons, we used the false positive rate that is reported by the SVM. Then, we compare the TP rates between the various tests; the performance of a test is better when the TP rate is higher. The combination of SweepFinder and ω-statistic performs better than each test (SVM column in Table 3 and Table 4). Also, SweepFinder outperforms the ω-statistic. In model A (deep bottleneck), when the sweep is either recent or old, the discrimination between neutral and selective models becomes problematic; when the false positive rate is about 50%, the true positive is as low as 70 and 63%, respectively, for the SVM approach. For the separate tests, the performance is even lower. This result suggests that recent or old selection in populations that have experienced deep bottlenecks cannot be discriminated from neutrality. However, when selection has occurred within the bottleneck phase, the false positive rate decreases to 20% and the true positive rate is 73% for the SVM and about 10% lower for the SweepFinder (Table 3, sweep phase 2). Higher discrimination performance is achieved when the sweep completes within the bottleneck (Table 3, sweep phase 2*), but this requires unrealistically high values of s.
In model B (shallow bottleneck), the discrimination performance is slightly better than that of model A. However, again the most challenging scenarios are either recent or old sweeps and the performance increases when the sweep occurs within the bottleneck phase (Table 4). Finally, the distances between the true target and the predicted target of selection are estimated. For both models A and B the distance is large and close to random expectations (Table 4).
Distinguishing RHH from neutrality in equilibrium populations:
In contrast to single selective sweep (SHH) models, recurrent selected substitutions occur randomly along a chromosome according to a time-homogeneous Poisson process at a rate v per generation (Kaplan et al. 1989; Wiehe and Stephan 1993; Stephan 1995). Well-known patterns of SHH models are modified under RHH. As an example, the SFS is skewed toward the rare variants; however, the excess of high-frequency-derived alleles decreases (Kim 2006; Jensen et al. 2008). Previously, Jensen et al. (2007) have shown that it is difficult to separate RHH models from neutrality on the basis of ωMAX-values or site frequency spectrum statistics. We explore the same problem with our new versions of the ω-statistic and the SweepFinder algorithm. Using the software developed by Jensen et al. (2008) we simulated 100-kb genomic regions for a given reduction of heterozygosity (Wiehe and Stephan 1993), namely 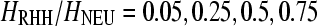 , or 0.95.
, or 0.95. 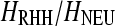 denotes the ratio of heterozygosity in the RHH model to the heterozygosity in the absence of selective sweeps. The selection coefficient s = 0.0001 or 0.01. The null hypothesis is represented by the standard neutral model.
denotes the ratio of heterozygosity in the RHH model to the heterozygosity in the absence of selective sweeps. The selection coefficient s = 0.0001 or 0.01. The null hypothesis is represented by the standard neutral model.
The null model used for the SweepFinder calculations and represented by the SFS of the population prior to the selective sweep in the SHH cases (n-SFS) cannot be described precisely by the standard neutral model. The population size is assumed to be constant. However, since adaptive mutations occur according to a time-homogeneous Poisson process it remains obscure what the “prior to the sweep” SFS should be. Here, we follow two approaches. First, we assume that the n-SFS is derived from the standard neutral model and second that the n-SFS is obtained from the genomic region itself. Clearly, both approaches are approximations. On one hand, using the standard neutral model we increase the sensitivity of the SweepFinder. On the other hand, the nucleotide polymorphism patterns of the genomic region under investigation have been shaped by selective sweeps, so the n-SFS forms a conservative null model with small sensitivity. However, if real data are consistent with the RHH model, the standard neutral model cannot be supported as a null model since the whole genome will be affected by recurrent sweeps.
When the n-SFS is derived from the data itself then the power of the SweepFinder is greater for small values (e.g., 0.0001) than for large values (e.g., 0.01) of the selection coefficient s (Figure S4). Even if this appears to be counterintuitive, it is reasonable because when s is small the footprints of the selective sweep are local, and a large fraction of the genome remains neutral. On the other hand, for large values of s almost the entire genomic region may be affected by RHH, contradicting the assumption of the SweepFinder test that only a small fraction of the genome has been shaped by positive selection (Figure S4).
Under RHH models selective sweeps occur in different genomic locations during the evolution of the population following a time-homogeneous Poisson process (Wiehe and Stephan 1993). When subgenomic data are analyzed it is possible that the target of selection is either inside or outside of the sequenced genomic region. Furthermore, since selective events occur with a certain probability per generation (Wiehe and Stephan 1993), patterns of polymorphism are shaped by both old and new selective events. However, the ω-statistic and SweepFinder are based on the assumption that a single selective sweep has just been completed. Thus, it is important to test whether the algorithms are able to predict the correct position of the adaptive events.
Incorporating a fraction of monomorphic sites into SweepFinder analysis increases the precision of the algorithm (Figure S5). Similarly, the variable-size sliding window approach appears more accurate than the constant-size sliding window method for high cutoff values. When 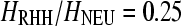 , SweepFinder and the ω-statistic predict that a target of selection is within a 5-kb distance from a true selective sweep position in about 40% of the cases. However, this fraction becomes smaller for higher values of
, SweepFinder and the ω-statistic predict that a target of selection is within a 5-kb distance from a true selective sweep position in about 40% of the cases. However, this fraction becomes smaller for higher values of  (Figure S5).
(Figure S5).
DISCUSSION
The demography of natural populations:
A major challenge of population genomics studies is to identify the loci that driven by positive selection contribute to the adaptation of natural populations and to localize the beneficial mutation accurately (Kim and Stephan 2002; Sabeti et al. 2002; Jensen et al. 2005; Nielsen et al. 2005; Akey 2009; Nielsen et al. 2009; Pickrell et al. 2009). To address these questions, it is important to consider the demographic history of the population, as this neutral nonequilibrium model represents the null (Li and Stephan 2006; Thornton and Andolfatto 2006). Since the standard neutral model does not reflect accurately the demography of most natural populations, neutrality tests should not be performed using the standard neutral scenario as the null model. In this study, we examined two bottleneck scenarios that are relevant to the demographic history of the European population of D. melanogaster (Li and Stephan 2006; Thornton and Andolfatto 2006). The properties of the coalescent trees that underlie these demographic models differ considerably. In a recombining genomic region, the model inferred by Li and Stephan (2006) produces both star-like short coalescent trees and genealogies with long internal branches. Star-like genealogies are generated less frequently by the Thornton and Andolfatto (2006) model (Figures 1 and 2). As a consequence, the null distributions of the neutrality statistics may differ. Thus, inferring the demographic history of a population is a prerequisite for performing genomic scans for selective sweeps, which has been shown to be a challenging task (Myers et al. 2008).
Separating single selective sweeps from neutral models:
When the value of the selection intensity α is large, the joint distribution of Λ and ω overlaps only partially between a model of selection in an equilibrium population and the bottleneck model inferred by Li and Stephan (2006). However, for smaller values of α the two distributions overlap greatly. A useful approach for classifying an observation as either a neutral or selective model is by combining the Λ and ω profiles. Here, we use the distance between the peaks and the correlation of ω and Λ. These features can be used in a classifier (e.g., SVM). Training requires that there are known instances of both neutral and selective models. For simple selective and neutral models this is currently possible, using coalescent-based programs. However, it remains challenging for more complicated scenarios. Forward simulations provide greater flexibility when selective events occur in nonequilibrium populations and they can be used efficiently when the population size is relatively small (i.e., on the order of thousands) or diffusion scaling applies (Hoggart et al. 2007; Chadeau-Hyam et al. 2008; Hernandez 2008).
The rationale for employing combinations of Λ and ω is that under a selective model the two statistics assume high values close to the target of selection. This implies that the target of selection can be localized accurately. Under selection models in equilibrium populations this assumption is met even for small α values. Modifying SweepFinder to include a fraction of nonpolymorphic sites in the analysis increased the accuracy of the algorithm and the performance in separating neutral scenarios from scenarios with selection. Furthermore, both versions of the ω-statistic, the constant- ad the variable-size sliding window approach, are very accurate for selection models in equilibrium populations.
However, in severe nonequilibrium scenarios (e.g., the estimated bottlenecks of Li and Stephan 2006 and Thornton and Andolfatto 2006), when selection and past demographic changes occur within the same model, the target of selection cannot be predicted, neither by SweepFinder nor by the ω-statistic. The accuracy of the target prediction when a selective sweep has occurred within the bottleneck period is comparable to that of randomized experiments. The reason is that polymorphism valleys and short coalescent trees may extend over large genomic regions, and the often used sweep signature of an excess of high-frequency-derived alleles vanishes. This result should be taken into account when regions of strong and recent positive selection are identified in genome scans. Since natural populations can be described by equilibrium demographic models only rarely, the true target of selection may be tens of kilobases away from the predicted target.
In the case of a severe bottleneck, such as model A, recombinants (carrying the selected mutation and the derived neutral allele) are most likely formed in the early period of the selective phase (forward in time), but they will be lost with high probability due to drift after the population size crashes. Therefore, high-frequency-derived variants may not be observed. In contrast, the frequency of rare variants (singletons) will dramatically increase. Therefore, on the basis of site frequency spectrum it is possible to discriminate, to some extent, neutral from nonneutral scenarios (Table 3).
The analysis of the likelihood curves of SweepFinder can provide further insights into the technical reasons that, in the cases of selection in nonequilibrium populations, make the prediction of the target of selection challenging. SweepFinder implements a model of selective sweep, which assumes that each observed SNP existed prior to the sweep. It uses the compound parameter  (named α in Nielsen et al. 2005) and the position x where the selective event occurred. (Here r denotes the recombination rate per basepair.) As Figure S6 illustrates, low- and high-frequency SNPs affect the likelihood in a similar way by contributing high values in the proximity of the sweep. Examining how the SFS changes over a genomic region under an equilibrium demographic model with selection and the Thornton and Andolfatto (2006) model with selection (α = 2500), it is apparent that there is a dramatic increase of the class n − 1 in the proximity of the selective sweep in the equilibrium model (Figure 3), but a very slight change of singletons in the nonequilibrium model. In the equilibrium-model case the precise localization of the sweep is possible, due to the spatial patterns of the rare and high-frequency-derived variants. However, in the Thornton and Andolfatto (2006) model with selection this pattern vanishes, the high-frequency-derived variants disappear and the singletons spread over the whole genomic region. Thus, the target of selection cannot be estimated accurately.
(named α in Nielsen et al. 2005) and the position x where the selective event occurred. (Here r denotes the recombination rate per basepair.) As Figure S6 illustrates, low- and high-frequency SNPs affect the likelihood in a similar way by contributing high values in the proximity of the sweep. Examining how the SFS changes over a genomic region under an equilibrium demographic model with selection and the Thornton and Andolfatto (2006) model with selection (α = 2500), it is apparent that there is a dramatic increase of the class n − 1 in the proximity of the selective sweep in the equilibrium model (Figure 3), but a very slight change of singletons in the nonequilibrium model. In the equilibrium-model case the precise localization of the sweep is possible, due to the spatial patterns of the rare and high-frequency-derived variants. However, in the Thornton and Andolfatto (2006) model with selection this pattern vanishes, the high-frequency-derived variants disappear and the singletons spread over the whole genomic region. Thus, the target of selection cannot be estimated accurately.
It should be noted, however, that the poor performance of SweepFinder and the ω-statistic under the nonequilibrium models (bottlenecked populations with selection) does not imply that the performance of the tests is poor under any nonequilibrium model with selection. These models represent extreme cases that violate major assumptions of the algorithms. The slightly improved performance of the machine-learning approach is due to the fact that it uses information from the sweep scenarios and, furthermore, it combines information from both the ω-statistic and SweepFinder.
Studying a scenario where a selective event took place in a bottleneck period is of great biological importance. Often, population bottlenecks are associated with a major migration event. For example, the bottleneck inferred by Li and Stephan (2006) for the European population of D. melanogaster describes the colonization of Europe from the African ancestral population. Therefore, positive selection may have occurred in the new habitat that contributed to the adaptation of flies to the environmental conditions of Europe. As Tables 3 and 4 show, the performance of the tests (especially the SVM, and to a lesser extent, the SweepFinder) is high when the sweep occurs within the bottleneck. This suggests that the approaches tested in this study can be used for the detection of selective sweeps in populations that have recently migrated to new environments. Furthermore, Tables 3 and 4 suggest that the power of SFS-based tests is higher than LD-based tests.
A difficulty that arises from using simulations with selection to train the algorithms is that the parameters of the scenarios with selection are unknown, i.e., the selection intensity α, the position of the sweep x, and the time at which the sweep occurred. In the models that we presented it was assumed that these parameters are known. However, when real data are analyzed these parameters are generally unknown, and moreover no methods that can estimate them in scenarios with past demographic changes are available. Thus, heuristic approaches have to be used. First, the position x can be assumed to be in the center of the fragment. Then, in real-data analysis overlapping windows should be used so that windows where x is located near their center will exist. The time of the sweep should be recent (<0.1N). In the classical approach this parameter is also implicitly specified by assuming that the sweep has just been complete. Finally, the selection intensity can be drawn from a prior uniform distribution. In this case the training set is composed of a mixture of models with various selection intensities.
Recurrent selective sweep analysis:
Recurrent selective sweeps invalidate the assumption that a single hitchhiking event has just been completed. In agreement with Jensen et al. (2007), we find that for greater rates v of selective events per generation the power of the tests increases for a given  . One possible explanation is that for smaller v a few strong selective sweeps that affect a large portion of the genome and shift the SFS of large genomic regions have occurred. Thus, the local characteristic of the signature of a selective event is lost. Another possible explanation is that for smaller v the selective events are old on average and the signature of selective sweep has faded away (Jensen et al. 2007).
. One possible explanation is that for smaller v a few strong selective sweeps that affect a large portion of the genome and shift the SFS of large genomic regions have occurred. Thus, the local characteristic of the signature of a selective event is lost. Another possible explanation is that for smaller v the selective events are old on average and the signature of selective sweep has faded away (Jensen et al. 2007).
The variable-size sliding window approach increases the accuracy of the ω-statistic to predict the target of selection. However, the performance is still poor. In ∼20% of the peaks above a certain threshold found in a scan of a given genomic region, the real position of the sweep is located within a 5-kb distance. The performance of the constant-size sliding window is about half that of the variable-size approach and comparable to the randomization experiments. A similar improvement has been achieved with the modified SweepFinder algorithm. RHH models imply that adaptive substitutions occur at a time-homogeneous rate, i.e., uniformly in the history of the population. This assumption may be violated in domesticated populations or in populations that experienced environmental changes. Thus, an increase of the performance of the tests (lower false positive rate, greater accuracy in target prediction) may result when RHH models are incorporated within the SweepFinder or the ω-statistic algorithms.
Recurrent selective sweep parameters such as the rate v of adaptive substitutions and the decrease of heterozygosity have been estimated recently. Jensen et al. (2008) and Li and Stephan (2006) have estimated that heterozygosity has decreased in genomic regions of normal recombination by 50% whereas the estimate of Macpherson et al. (2007) and Andolfatto (2007) is about 20% (i.e.,  = 0.8). We examined the performance of the SweepFinder and the ω-statistic for various levels of heterozygosity reduction,
= 0.8). We examined the performance of the SweepFinder and the ω-statistic for various levels of heterozygosity reduction,  = 0.25, 0.5, 0.75, and 0.95, and selection coefficients s = 10−2 and 10−4 (Figure 5). The power of SweepFinder is greater for the Li and Stephan (2006) and Jensen et al. (2008) estimations than for that of Macpherson et al. (2007) and Andolfatto (2007), given that selection is strong (s = 10−2). For s = 10−4 the differences in the performance of SweepFinder for various levels of
= 0.25, 0.5, 0.75, and 0.95, and selection coefficients s = 10−2 and 10−4 (Figure 5). The power of SweepFinder is greater for the Li and Stephan (2006) and Jensen et al. (2008) estimations than for that of Macpherson et al. (2007) and Andolfatto (2007), given that selection is strong (s = 10−2). For s = 10−4 the differences in the performance of SweepFinder for various levels of 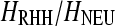 are small. The reason is that for s = 10−4 the diversity is similar for values of
are small. The reason is that for s = 10−4 the diversity is similar for values of  between 0.05 and 0.95. This may be due to inaccuracies of the RHH theory when s is small or due to the stochastic trajectory of the beneficial mutation (Coop and Griffiths 2004; Spencer and Coop 2004).
between 0.05 and 0.95. This may be due to inaccuracies of the RHH theory when s is small or due to the stochastic trajectory of the beneficial mutation (Coop and Griffiths 2004; Spencer and Coop 2004).
Figure 5.—

The distributions of ΛMAX for various levels of the decrease of heterozygosity and s = 10−2. Each distribution is discrete and the size of each bin has been set to 6. (A) For 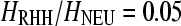 , 0.5, and 0.95 the cutoff values (95th percentile) are 5.7, 9.7, and 11.9, respectively, and the sensitivities of the test (percentage of true positives) given the cutoff values are 0.74, 0.48, and 0.07. The power of SweepFinder is greater for the Li and Stephan (2006) and Jensen et al. (2008) estimations than those of Macpherson et al. (2007) and Andolfatto (2007) because selection is strong (s = 10−2). (B) When s = 10−4 the amount of diversity is similar for
, 0.5, and 0.95 the cutoff values (95th percentile) are 5.7, 9.7, and 11.9, respectively, and the sensitivities of the test (percentage of true positives) given the cutoff values are 0.74, 0.48, and 0.07. The power of SweepFinder is greater for the Li and Stephan (2006) and Jensen et al. (2008) estimations than those of Macpherson et al. (2007) and Andolfatto (2007) because selection is strong (s = 10−2). (B) When s = 10−4 the amount of diversity is similar for  , 0.5, and 0.95. Therefore, the performance of SweepFinder is relatively independent of the
, 0.5, and 0.95. Therefore, the performance of SweepFinder is relatively independent of the 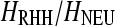 .
.
Time of the selective sweep:
For SHH models (in demographic equilibrium) we assume that the selected mutation has reached fixation very recently. The selective model that underlies the SweepFinder algorithm assumes a recent and strong selective sweep. Therefore, the power of SweepFinder is expected to be higher for recently completed hitchhiking effects. Indeed, simulations have shown that the power decreases exponentially after the selective sweep (P. Pavlidis, unpublished results). It should be mentioned that the demographic scenario that follows the selective sweep (i.e., between the time of completion of the selective sweep and the time of sampling) affects the performance of SweepFinder. Simulations have shown that if the completion of a selective sweep is followed by population expansion, the performance of the likelihood ratio test implemented in SweepFinder remains high even after the completion of the selective sweep (P. Pavlidis, unpublished results). The rationale behind this is that a population expansion decreases the coalescent rate; therefore, the return to the equilibrium SFS is slower and the signature of the selective sweep is preserved for a longer period. In contrast to SweepFinder, which is based on the low- and high-derived variants, the ω-statistic is more sensitive to the time since the completion of the selective sweep. Indeed, the LD pattern captured by the ω-statistic vanishes rapidly (Jensen et al. 2007), comparable to the fixation rate of the high-frequency-derived alleles (Kim and Stephan 2000; Przeworski 2002; Jensen et al. 2007).
Overlapping selective sweeps:
In this study we focused on nonoverlapping selective sweeps. The RHH model we have used describes successive and nonoverlapping selective events. Chevin et al. (2008) have shown that two interfering selective sweeps may modify the pattern of linked neutral variation. A related process, when the targets of selection are located closely to each other in the genome, causes trafficking (Kirby and Stephan 1996; Kim and Stephan 2003). A most extreme scenario, which describes the appearance of beneficial mutations at the same site, is described as “soft” sweep (Hermisson and Pennings 2005). Soft sweeps may emerge during the evolution of organisms (e.g., Plasmodium) with high mutation rates (Nair et al. 2007). Conversely, they may be of limited importance in the evolution of D. melanogaster or Homo sapiens, for instance. The patterns of neutral variation under these selective scenarios are different from those of single selective events. For example, the skew of Tajima's D toward negative values vanishes in the interference scenarios described by Chevin et al. (2008) and can even be positive between the selected sites. In general, SFS-based approaches may not work under overlapping selective sweeps because the frequency of the class of polymorphisms in intermediate frequency may be quite large. In such cases, LD-based statistics can be useful because a multitude of extended haplotypes may exist on the left and right sides of the selected region (Sabeti et al. 2002; Voight et al. 2006; Tang et al. 2007).
Machine-learning approaches in population genetics:
Machine-learning approaches are widely used in a variety of applications from image processing to classification of microarrays. Here, we are interested in the subfield of machine learning that is related to supervised learning or classification. Typically, in a classification problem a training set teaches the algorithm to predict the class label of an input object (Duda et al. 2000; Hastie et al. 2001). The goal is to decide between a selective and a neutral model. However, classifying a data set as either neutral or selective is challenging because the parameters of the neutral and selective models are unknown. Therefore, parameter estimation is required prior to the classification. In the cases that an equilibrium model with selection is employed, the selection intensity α can be estimated using the clsw software (Kim and Stephan 2002) or the SweepFinder algorithm (given that ρ is known). To our knowledge, currently the only method able to estimate α given a nonequilibrium (stepwise) model with selection has been developed by Li and Stephan (2006). On the other hand, several approaches exist for the estimation of parameters in a neutral demographic model (Nielsen 2000; Excoffier et al. 2005; Li and Stephan 2006; Hey and Nielsen 2007). Usually, these approaches require multiple loci to infer the demographic parameters of a population. The next step in a classification problem is feature selection, which aims at using a subset of the features available from the data. Here, ΛMAX, ωMAX, and their combinations (distance between peaks and correlation of ω and Λ) have been used. Combining ω and Λ is powerful in comparisons between equilibrium models with selection and neutral nonequilibrium models when the selection intensity is small (Table 2). Alternatively, various summary statistics, such as Tajima (1989)'s D, Fay and Wu (2000)'s H, or ZnS (Kelly 1997) can be used. Our choice is based on the fact that SweepFinder uses SFS information whereas the ω-statistic is based on LD. The choice of the classification technique is important and depends on the problem and the nature of the data. Here, we demonstrate an application using the SVM classifier (with the radial kernel), as it is implemented in the e1071 package of the R-project. To our knowledge, there are no studies on separating neutral from selective scenarios that use supervised-learning approaches. Future work will provide insight into the feature selection problem and will also evaluate the performance of the supervised-learning approaches.
Acknowledgments
We thank Dirk Metzler, Stefan Laurent, and Stephan Hutter for valuable discussions on data analysis, and Yun Song for the discussion of the usefulness of alternative LD measures. Matthew Stephen's comments and the comments from two anonymous referees improved the manuscript considerably. This research was supported by grants from the Volkswagen-Foundation to P.P. (grant I/824234), National Science Foundation (grant DEB-1002785) and Worcester Foundation to J.D.J., and the German Research Foundation to W.S. (grants Ste 325/7-1 and Ste 325/12-1 of the Research Unit 1078).
Supporting information available online at http://www.genetics.org/cgi/content/full/genetics.110.116459/DC1.
References
- Akey, J. M., 2009. Constructing genomic maps of positive selection in humans: Where do we go from here? Genome Res. 19 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey, J. M., M. A. Eberle, M. J. Rieder, C. S. Carlson, M. D. Shriver et al., 2004. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2 e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto, P., 2007. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 17 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N., 1998. The effect of hitch-hiking on neutral genealogies. Genet. Res. 72 123–133. [Google Scholar]
- Beisswanger, S., and W. Stephan, 2008. Evidence that strong positive selection drives neofunctionalization in the tandemly duplicated polyhomeotic genes in Drosophila. Proc. Natl. Acad. Sci. USA 105 5447–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman, J. M., R. R. Hudson, N. L. Kaplan, C. H. Langley and W. Stephan, 1995. The hitchhiking effect on the site frequency spectrum of DNA polymorphisms. Genetics 140 783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadeau-Hyam, M., C. J. Hoggart, P. F. O'Reilly, J. C. Whittaker, M. D. Iorio et al., 2008. Fregene: simulation of realistic sequence-level data in populations and ascertained samples. BMC Bioinformatics 9 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevin, L.-M., S. Billiard and F. Hospital, 2008. Hitchhiking both ways: effect of two interfering selective sweeps on linked neutral variation. Genetics 180 301–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop, G., and R. C. Griffiths, 2004. Ancestral inference on gene trees under selection. Theor. Popul. Biol. 66 219–232. [DOI] [PubMed] [Google Scholar]
- Duda, R. O., P. E. Hart and D. G. Stork, 2000. Pattern Classification, Ed. 2. Wiley-Interscience, Indianapolis.
- Excoffier, L., A. Estoup and J.-M. Cornuet, 2005. Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay, J. C., and C. I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka, S., L. Ometto, S. Mousset, W. Stephan and D. De Lorenzo, 2003. Demography and natural selection have shaped genetic variation in Drosophila melanogaster: a multi-locus approach. Genetics 165 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J., and M. Kamber, 2000. Data Mining: Concepts and Techniques, The Morgan Kaufmann Series in Data Management Systems. Morgan Kaufmann, San Francisco.
- Harr, B., M. Kauer and C. Schlötterer, 2002. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 99 12949–12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie, T., R. Tibshirani and J. H. Friedman, 2001. The Elements of Statistical Learning. Springer, New York.
- Hermisson, J., and P. S. Pennings, 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169 2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez, R. D., 2008. A flexible forward simulator for populations subject to selection and demography. Bioinformatics 24 2786–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J., and R. Nielsen, 2007. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 104 2785–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart, C. J., M. Chadeau-Hyam, T. G. Clark, R. Lampariello, J. C. Whittaker et al., 2007. Sequence-level population simulations over large genomic regions. Genetics 177 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Jensen, J. D., Y. Kim, V. Bauer DuMont, C. F. Aquadro and C. D. Bustamante, 2005. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics 170 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, J. D., K. R. Thornton, C. D. Bustamante and C. F. Aquadro, 2007. On the utility of linkage disequilibrium as a statistic for identifying targets of positive selection in nonequilibrium populations. Genetics 176 2371–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, J. D., K. R. Thornton and P. Andolfatto, 2008. An approximate Bayesian estimator suggests strong, recurrent selective sweeps in Drosophila. PLoS Genet. 4 e1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, N. L., R. R. Hudson and C. H. Langley, 1989. The hitchhiking effect revisited. Genetics 123 887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, J. K., 1997. A test of neutrality based on interlocus associations. Genetics 146 1197–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., 2006. Allele frequency distribution under recurrent selective sweeps. Genetics 172 1967–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and R. Nielsen, 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2000. Joint effects of genetic hitchhiking and background selection on neutral variation. Genetics 155 1415–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2003. Selective sweeps in the presence of interference among partially linked loci. Genetics 164 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby, D. A., and W. Stephan, 1996. Multi-locus selection and the structure of variation at the white gene of Drosophila melanogaster. Genetics 144 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, C. H., and J. F. Crow, 1974. The direction of linkage disequilibrium. Genetics 78 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., and W. Stephan, 2006. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genet. 2 e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson, J. M., G. Sella, J. C. Davis and D. A. Petrov, 2007. Genomewide spatial correspondence between nonsynonymous divergence and neutral polymorphism reveals extensive adaptation in Drosophila. Genetics 177 2083–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23 23–35. [PubMed] [Google Scholar]
- McVean, G., 2007. The structure of linkage disequilibrium around a selective sweep. Genetics 175 1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, S., C. Fefferman and N. Patterson, 2008. Can one learn history from the allelic spectrum? Theor. Popul. Biol. 73 342–348. [DOI] [PubMed] [Google Scholar]
- Nair, S., D. Nash, D. Sudimack, A. Jaidee, M. Barends et al., 2007. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol. Biol. Evol. 24 562–573. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., 2000. Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., S. Williamson, Y. Kim, M. J. Hubisz, A. G. Clark et al., 2005. Genomic scans for selective sweeps using SNP data. Genome Res. 15 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., M. J. Hubisz, I. Hellmann, D. Torgerson, A. M. Andrés et al., 2009. Darwinian and demographic forces affecting human protein coding genes. Genome Res. 19 838–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, D. J., and M. Aguadé, 2004. Detecting the footprint of positive selection in a European population of Drosophila melanogaster: multilocus pattern of variation and distance to coding regions. Genetics 167 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orengo, D. J., and M. Aguadé, 2010. Uncovering the footprint of positive selection on the X chromosome of Drosophila melanogaster. Mol. Biol. Evol. 27 153–160. [DOI] [PubMed] [Google Scholar]
- Pavlidis, P., S. Hutter and W. Stephan, 2008. A population genomic approach to map recent positive selection in model species. Mol. Ecol. 17 3585–3598. [DOI] [PubMed] [Google Scholar]
- Pfaffelhuber, P., A. Lehnert and W. Stephan, 2008. Linkage disequilibrium under genetic hitchhiking in finite populations. Genetics 179 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell, J. K., G. Coop, J. Novembre, S. Kudaravalli, J. Z. Li et al., 2009. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 19 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Onsins, S. E., S. Mousset, T. Mitchell-Olds and W. Stephan, 2007. Population genetic inference using a fixed number of segregating sites: a reassessment. Genet. Res. 89 231–244. [DOI] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419 832–837. [DOI] [PubMed] [Google Scholar]
- Spencer, C. C. A., and G. Coop, 2004. SelSim: a program to simulate population genetic data with natural selection and recombination. Bioinformatics 20 3673–3675. [DOI] [PubMed] [Google Scholar]
- Stephan, W., 1995. An improved method for estimating the rate of fixation of favorable mutations based on DNA polymorphism data. Mol. Biol. Evol. 12 959–962. [DOI] [PubMed] [Google Scholar]
- Stephan, W., T. H. E. Wiehe and M. W. Lenz, 1992. The effect of strongly selected substitutions on neutral polymorphism: analytical results based on diffusion theory. Theor. Popul. Biol. 41 237–254. [Google Scholar]
- Stephan, W., Y. S. Song and C. H. Langley, 2006. The hitchhiking effect on linkage disequilibrium between linked neutral loci. Genetics 172 2647–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetec, N., P. Pavlidis and W. Stephan, 2009. Recent strong positive selection on Drosophila melanogaster HDAC6, a gene encoding a stress surveillance factor, as revealed by population genomic analysis. Mol. Biol. Evol. 26 1549–1556. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, K., K. R. Thornton and M. Stoneking, 2007. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 5 e171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teshima, K. M., and H. Innan, 2009. mbs: modifying Hudson's ms software to generate samples of DNA sequences with a biallelic site under selection. BMC Bioinformatics 10 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K., and P. Andolfatto, 2006. Approximate Bayesian inference reveals evidence for a recent, severe bottleneck in a Netherlands population of Drosophila melanogaster. Genetics 172 1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K. R., and J. D. Jensen, 2007. Controlling the false-positive rate in multilocus genome scans for selection. Genetics 175 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4 e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]
- Wiehe, T. H. E., and W. Stephan, 1993. Analysis of a genetic hitchhiking model, and its application to DNA polymorphism data from Drosophila melanogaster. Mol. Biol. Evol. 10 842–854. [DOI] [PubMed] [Google Scholar]
- Zivkovic, D., and T. Wiehe, 2008. Second-order moments of segregating sites under variable population size. Genetics 180 341–357. [DOI] [PMC free article] [PubMed] [Google Scholar]