

Summary
During division of Gram-negative bacteria, invagination of the cytoplasmic membrane and inward growth of the peptidoglycan (PG) are followed by the cleavage of connective septal PG to allow cell separation. This PG splitting process requires temporal and spatial regulation of cell wall hydrolases. In Escherichia coli, LytM factors play an important role in PG splitting. Here we identify and characterize a member of this family (DipM) in Caulobacter crescentus. Unlike its E. coli counterparts, DipM is essential for viability under fast-growth conditions. Under slow-growth conditions, the ΔdipM mutant displays severe defects in cell division and FtsZ constriction. Consistent with its function in division, DipM colocalizes with the FtsZ ring during the cell cycle. Mutagenesis suggests that the LytM domain of DipM is essential for protein function, despite being non-canonical. DipM also carries two tandems of the PG-binding LysM domain that are sufficient for FtsZ-ring localization. Localization and fluorescence recovery after photobleaching microscopy experiments suggest that DipM localization is mediated, at least in part, by the ability of the LysM tandems to distinguish septal, multilayered PG from non-septal, monolayered PG.
Keywords: LytM, endopeptidase, peptidoglycan, localization, division, LysM
Introduction
The peptidoglycan (PG) withstands the internal osmotic pressure of bacterial cells and maintains their shape (Vollmer et al., 2008a). This cell wall structure is essential for bacterial viability under most conditions, and a wide array of antibiotics target its biosynthetic pathway. In Gram-negative bacteria, the PG is thought to largely consist of a monolayer of glycan strands held together by covalently linked peptide bridges (Vollmer and Holtje, 2004). Evidence suggests that the glycan chains are oriented parallel to the cytoplasmic membrane and roughly perpendicular to the long axis of the cell (Verwer et al., 1978; Vollmer and Holtje, 2004; Gan et al., 2008). This covalently-linked network (also known as sacculus) forms a strong, elastic fabric that surrounds the cytoplasmic membrane and protects it from osmotic bursting. Peptide bridges must be cleaved to accommodate insertion of new glycan strands while preserving the integrity of the PG. This task is particularly challenging in the single layered PG of Gram-negative bacteria. Hence, PG growth requires exquisite temporal and spatial coordination between cell wall synthase and hydrolase activities.
During cell constriction, the PG grows inward concurrently with membrane invagination (Woldringh, 1976; Poindexter and Hagenzieker, 1981; Judd et al., 2005), transiently forming a multilayered wall (Holtje, 1998). This process is governed by the membrane-spanning cell wall machinery that is associated with the cytokinetic FtsZ ring (den Blaauwen et al., 2008; Vollmer and Bertsche, 2008). The repetition of cell wall material insertion and the pulling force generated through FtsZ ring constriction (Osawa et al., 2008; Osawa et al., 2009) result in inward PG growth, which is followed by degradation and release of digested PG. This key degradation process is often referred to as septal PG splitting as it cleaves the PG that links the two future daughter cells, thereby allowing outer membrane invagination and cell separation. Indeed, about 30% of the new septal PG is released during constriction (Uehara and Park, 2008).
Several cell wall enzymes such as amidases and endopeptidases have been shown to play important roles in PG splitting in Escherichia coli (Rodolakis et al., 1973; Starka et al., 1974; Heidrich et al., 2001; Hara et al., 2002; Heidrich et al., 2002; Priyadarshini et al., 2006; Priyadarshini et al., 2007; Uehara et al., 2009). These PG hydrolases often have redundant functions, and in most cases, single deletions of their corresponding genes have little effect on growth and division (Vollmer et al., 2008b). However, inactivation of the genes encoding all three N-acetylmuramyl-L-alanine amidases AmiA, AmiB and AmiC results in long cell chains that are held together by connective septal cell wall (Heidrich et al., 2001; Priyadarshini et al., 2007). Other major players in septal PG splitting in E. coli include the LytM factors EnvC and NlpD. An ΔenvC mutant forms short cell chains with regularly spaced constrictions (Rodolakis et al., 1973; Hara et al., 2002; Ichimura et al., 2002; Bernhardt and de Boer, 2004), and this cell chaining phenotype is dramatically exacerbated when the ΔenvC mutation is combined with a ΔnlpD deletion (Uehara et al., 2009). While endopeptidase activity has been demonstrated for some LytM factors of Gram-positive bacteria (Browder et al., 1965; Firczuk et al., 2005; Cohen et al., 2009; Reste de Roca et al., 2010), the LytM domains of NlpD and EnvC lack some or all zinc-coordinating residues involved in catalysis. Recent evidence suggests that NlpD and EnvC do not hydrolyze the PG themselves, but rather stimulate the activity of PG amidases (Uehara et al., 2010). AmiB, AmiC, EnvC and NlpD have all been shown to accumulate at the septum during E. coli constriction (Bernhardt and de Boer, 2003; Bernhardt and de Boer, 2004; Uehara et al., 2009), consistent with an involvement in septal PG splitting. The mechanism of septal localization is, however, not well understood and is generally assumed to involve protein-protein interaction with the cell division protein complex. In the case of the amidase AmiC, this is supported with experimental evidence showing that septal localization is dependent on the late cell division protein FtsN (Bernhardt and de Boer, 2003).
Our knowledge of cell wall hydrolysis in Gram-negative bacteria is primarily limited to studies in a single model organism, E. coli. In this study, we identify and characterize a periplasmic LytM factor in the Gram-negative bacterium Caulobacter crescentus. We describe similarities to the E. coli paradigm as well as important differences. Additionally, we present evidence of a protein localization mechanism that relies on differential recognition of monolayered and multilayered PG.
Results
The LysM and LytM domain-containing protein DipM is involved in cell division
Since proteins belonging to the LytM family have been identified to play roles in the cell division process of E. coli (Bernhardt and de Boer, 2004; Uehara et al., 2009), we searched the C. crescentus genome for proteins that contain this LytM domain. Multiple predicted proteins were identified, but one protein (CC1996) caught our attention because it contained four LysM PG-binding motifs in addition to a LytM domain (see Fig. 4A for a schematic representation of its domain organization). Further analysis of its primary sequence identified a signal peptide with a cleavage site for a signal peptidase at the N-terminus and no transmembrane-spanning segments, suggesting that CC1996 is a soluble periplasmic protein.
Fig. 4.

Determination of the DipM region involved in protein localization. (A) Schematic of the DipM domain organization and representation of mCherry. (B) Fluorescent micrographs of mCherry fused to different fragments of DipM: (i) DipM-mCherry (CJW3124); (ii) DipMΔ501-609-mCherry (CJW3439); (iii) DipMΔ297-609-mCherry (CJW3121); (iv) DipM1–296Δ121–167-mCherry (CJW3526); (v) DipM1–296Δ175–223-mCherry (CJW3528); (vi) mCherry-DipMΔ1-30 (CJW3116); (vii) mCherry-DipMΔ1-236 (CJW3117); (viii) DipMΔ54-609-mCherry (CJW2959). All inducible mCherry fusions were expressed from the chromosome by adding vanillic acid to a final concentration of 250 μM about 5 to 6 h prior to microscopy. In the case of the N-terminal mCherry fusions, the signal peptide is encoded by the vector. Note that wild-type DipM is also produced in these backgrounds. The white bar corresponds to 1 μm.
To characterize the function of CC1996, we created a mutant strain in which the full cc_1996 coding sequence was substituted by an Ω spectinomycin-resistance cassette. The resulting mutant strain grew at a slower rate than the wild-type parent on PYE plates at 30°C, taking approximately twice as long to produce visible colonies. Cells from these colonies exhibited division defects (data not shown). From here on, CC1996 will be called DipM (Division Involved Protein with LysM domains). The severity of the defects associated with the ΔdipM mutation was dependent on growth conditions. In minimal M2G liquid medium at 30°C or below, the ΔdipM mutant was viable, but it was unable to propagate at 37°C in M2G liquid medium or in rich PYE liquid medium at a temperature of 30°C or above (Fig. 1A). Supplementing M2G medium with increasing amounts of rich PYE medium slowed the growth of the ΔdipM strain in liquid cultures (Fig. 1A). Even at 22°C, increasing concentrations of PYE in liquid M2G medium were accompanied by a dramatic enhancement of cell filamentation in the population (Fig. 1B). The morphology of wild-type cells was unaffected by such switching of growth conditions (data not shown). Collectively, our data indicate that conditions that normally increase growth rate adversely affect the ability of ΔdipM cells to grow and divide.
Fig. 1.
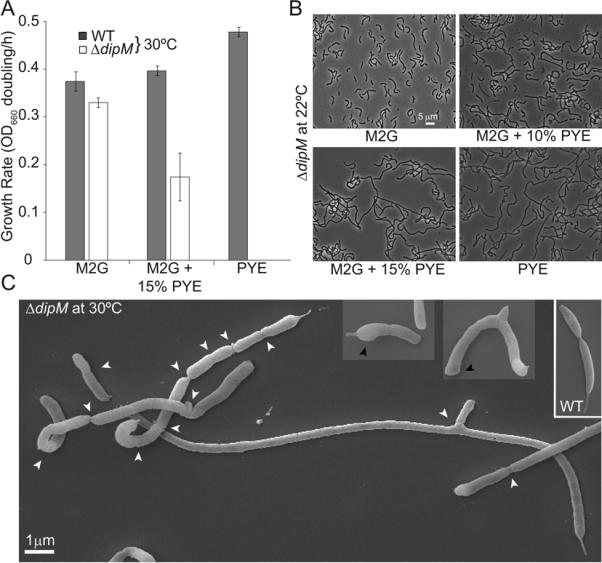
Phenotypic characterization of the ΔdipM mutant. (A) Growth rates (in doublings/h) of wild-type and ΔdipM (CJW3137) CB15N strains grown at 30°C in liquid cultures containing M2G minimal medium, M2G medium supplemented with 15% of rich PYE medium, or PYE medium. Average values and standard deviations for three experiments are shown. (B) Phase contrast images of ΔdipM cells grown at 22°C in different media. ΔdipM cells from overnight M2G cultures at 30°C were used to inoculate culture tubes containing M2G medium, PYE medium or M2G supplemented with 10 or 15% of PYE medium, which were then incubated at 22°C for an additional 24 h prior to imaging. At the time of imaging, the cultures were at an OD660 ≤ 0.5. (C) Scanning EM images of ΔdipM mutant cells grown at 30°C in liquid M2G medium. White and black arrows indicate constriction sites and bulged poles, respectively. For comparison, the inset shows scanning EM images of a wild-type predivisional cell grown under the same conditions.
For all the following experiments, the ΔdipM strain was grown in M2G at 30°C. Besides the filamentation phenotype, ΔdipM cells tended to have short or no stalks, suggesting a potential stalk growth defect. Additionally, time-lapse microscopy of ΔdipM cells revealed frequent release of vesicles from the division sites (Movie S1), suggesting instability of the outer membrane at these sites. This phenotype is common among cell wall peptidase (or amidase) mutants in which the coordination between outer membrane invagination and inward PG growth has been perturbed (Rodolakis et al., 1973; Heidrich et al., 2001; Hara et al., 2002; Heidrich et al., 2002; Bernhardt and de Boer, 2004; Priyadarshini et al., 2006).
Mutations in division proteins involved in cell-wall synthesis can cause altered cell pole morphologies (Taschner et al., 1988; Priyadarshini et al., 2006). Similarly, the poles of ΔdipM cells often displayed an aberrant round morphology and were frequently the widest region of the cells (Fig. 1C, black arrowheads). This was particularly striking since wild-type C. crescentus cells have pointed poles (Aaron et al., 2007) (Fig. 1C, inset). Time-lapse microscopy of growing ΔdipM cells showed that the poles grow wider as they age (Movie S2), occasionally resulting in cell branching (Fig. 1C, Movie S3). These findings indicate that DipM plays an important role in cell morphogenesis.
Filamentation of ΔdipM cells is associated with altered FtsZ dynamics
In E. coli, the absence of the two main LytM factors EnvC and NlpD produces chains of cells kept together by regular septa, indicating a defect in splitting of the PG generated during cell division (Uehara et al., 2009). The ΔdipM cell filaments were considerably different; they often displayed several constriction sites but typically at irregular intervals (Fig. 1C, white arrow heads). To determine the degree of cytoplasmic compartmentalization in the ΔdipM filaments, we carried out fluorescence loss in photobleaching (FLIP) microscopy experiments. Small regions of ΔdipM cells producing cytoplasmic GFP were subjected to a series of photobleaching laser pulses (Figure 2A, yellow dotted regions). The extent of GFP signal loss inside the cell filaments was used as an indication of the size of the cytoplasmic compartment. We found that ΔdipM cell filaments had cytoplasmic compartments of sizes varying from the expected size for a normal cell to the size of an entire cell filament (Fig. 2A). Out of 104 FLIP events, 70 of them (67%) generated bleached cytoplasmic spaces much larger than normal cell size, indicating a frequent defect in cytoplasmic compartmentalization.
Fig. 2.

Characterization of the cell filamentation phenotype associated with the ΔdipM mutation. (A) FLIP experiment of diffusible cytosolic GFP expressed in ΔdipM cell filament (strain CJW3449) to assess the extent of cell compartmentalization. Yellow dotted circles indicate the regions targeted for photobleaching. The green dotted lines show the extension of the cytoplasmic space as determined by the extent of bleaching.(B) Transmission electron micrographs of isolated sacculi from wild-type (CB15N), ΔdipM (CJW3137), FtsZ-depleted (YB1585) and FtsA-depleted (CJW3187) cells. Depletion was achieved by growing cells in the absence of xylose (inducer) for 5 h (FtsZ) or 8 h (FtsA). In the wild-type panel, the arrow points to a PHB (polyhydroxybutyrate) granule trapped in the sacculus. In the ΔdipM panel, the arrowheads and arrows indicate PG-rings and SP-rings, respectively. (C) Selected frames from a time-lapse microscopy sequence of a ΔdipM cell filament (CJW3448) expressing FtsZ-YFP (green) and MipZ-CFP (red). ftsZ-yfp expression was induced by the addition of 0.3% xylose for 3 h prior to the start of the time-lapse experiment. In all panels, the scale bars correspond to 1 μm.
To better understand the ΔdipM filamentation phenotype, we examined FtsZ localization dynamics in growing cells by time-lapse microscopy. The FtsZ rings (labeled with FtsZ-YFP) in ΔdipM filaments (strain CJW3430) displayed several abnormal behaviors. Some were unstable and changed their position rapidly before completing constriction (Movie S4, arrow). Others appeared to stall or to constrict at very slow rates (Movie S4; Fig. S1, arrows). The average lifetime of FtsZ rings that eventually resulted in division was 80 ± 8 min and 140 ± 28 min for the wild-type and ΔdipM mutant strains (n = 20 in both cases), respectively. Occasionally, an aggregation of FtsZ-YFP signal was visible for extended periods of time (Fig. S1, line) before condensing into a ring (Fig. S1, arrowhead). Some FtsZ rings were able to complete their constriction and reassemble at other locations well before cell separation occurred (Movie S5). A possible explanation for this disconnection is that the splitting of the PG synthesized by the divisome was considerably delayed and occurred after the FtsZ ring had finished constricting and had reformed elsewhere. Close coordination between constriction of the FtsZ ring and constriction of the cell was only observed in rare cases (data not shown) and even then, the process took an unusual long time.
In E. coli, mutants of proteins involved in PG splitting tend to accumulate PG material at potential division sites (Priyadarshini et al., 2007; Uehara et al., 2009). These PG accumulations can be observed in uranyl acetate-stained EM preparations of purified sacculi and are referred to as PG rings (PeptidoGlycan) when no constriction is evident or SP rings (Septum Peptidoglycan) when constriction is visible (Priyadarshini et al., 2007). Consistent with DipM playing a role in PG hydrolysis at division, both types of rings were observed in purified ΔdipM sacculi (Fig. 2B, arrowheads and arrows indicate PG and SP rings, respectively). No such rings were observed in sacculi of dividing wild-type cells or in sacculi of filamentous FtsZ or FtsA-depleted cells (Fig. 2B). These results suggest that DipM is involved in splitting the PG during division.
An intriguing observation in the ΔdipM cell filaments was the frequent relocation of the FtsZ ring before completion of constriction (Movies S4 and S5). In C. crescentus, the positioning of the FtsZ ring is determined by bipolar gradients of the FtsZ assembly inhibitor MipZ that form after segregation of the chromosome partitioning complex (Thanbichler and Shapiro, 2006). We hypothesized that the impairment in FtsZ ring constriction in ΔdipM cells may allow time for the next round of replication and segregation of the chromosomal partitioning complex. This would result in MipZ localizing at these slowly constricting or halted FtsZ rings, causing their disassembly and relocation to another division site. This idea was supported by time-lapse microscopy of FtsZ-YFP and MipZ-CFP in ΔdipM cells, which showed MipZ-CFP moving to the location of FtsZ ring and causing their disappearance (Fig. 2C).
Together, our observations show that in the absence of DipM, the ability of the FtsZ rings to constrict is frequently impeded, probably by an alteration in PG splitting. These stalled and slowly constricting FtsZ rings then become susceptible to disassembly by MipZ bound to partitioning complexes before completion of cell constriction, contributing to the observed ΔdipM division phenotype.
DipM localizes near midcell and at the base of the stalk at specific stages of the cell cycle
Next, we examined the localization of a functional DipM-mCherry construct produced in place of DipM under native expression conditions. Time-lapse microscopy of cell cycle-synchronized populations showed early localization of the fusion protein at the old pole where a stalk later formed (Fig. 3A and Movie S6). In the stalked cell stage, DipM-mCherry formed a band near midcell while maintaining an old-pole accumulation. DipM-mCherry retained the medial localization throughout the cell constriction process until cell separation occurred. This pattern is consistent with the localization of CC1996 (now DipM) in asynchronous populations as reported in a recent proteome-wide study (Werner et al., 2009). Interestingly, the old-pole accumulation of DipM-mCherry disappeared during the late cell cycle stages (Figure 3A) and did not reappear during the cell cycle of the stalked progeny (Movie S6) despite continued growth of the stalk, raising the possibility of an early role for DipM in stalk generation. A summary of DipM localization during the cell cycle is depicted in Fig. 3B.
Fig. 3.

Localization of DipM during the cell cycle and its dependence on other division proteins. (A) Time-course microscopy of synchronized cells (CJW3124) expressing dipM-mCherry as the only copy of dipM from the native chromosomal promoter and progressing through the cell cycle following synchronyzation. Time 0 corresponds to purified swarmer cells resuspended in M2G medium at 30°C. (B) Schematic of the localization pattern of DipM determined from time-lapse experiments using synchronized cell cycle populations as in (A) and Movie S6. (C) The percentage of cells with DipM-mCherry and FtsZ-YFP localization near midcell and with discernible cell constriction were plotted over time. The data were obtained from two independent time-course experiments (137 and 196 cells) starting with synchronized swarmer cells (CJW3436) that were followed through a cell cycle while growing on a M2G-agarose pad. (D) Localization of DipM-mCherry in FtsA-depleted cells. Cultures of CJW3447 cells carrying ftsA under xylose-inducible expression were grown to an OD660 of 0.1 in M2G medium supplemented with 0.3% xylose (inducer), after which the cells were washed and resuspended in M2G medium lacking xylose. After about 4 h of FtsA depletion and cell filamentation, fluorescent images of FtsZ-YFP and DipM-mCherry were acquired. ftsZ-yfp expression was induced with vanillic acid (250 μM) for 3 h prior to imaging. (E) DipM-mCherry localization in the mreBQ26P mutant (strain CJW3437). (F) DipM-mCherry localization in FtsZ-depleted cells. Cultures of CJW3438 cells carrying ftsZ under xylose-inducible expression and dipM-mCherry under the native promoter were grown to an OD660 of 0.1 in M2G medium supplemented with 0.3% xylose (inducer), after which the cells were washed, resuspended in M2G medium lacking xylose and spotted in an agarose pad of the same media. Depletion was followed by microscopic observation during which fluorescent images of DipM-mCherry were acquired. The image corresponds to about 4 h of FtsZ depletion and cell filamentation. The white scale bars correspond to 1 μm.
Co-visualization of DipM-mCherry and FtsZ-YFP showed that DipM localized to the FtsZ ring position about 15 to 20 min after FtsZ ring formation and 20 min before cell constriction could be detected (Fig. 3C; under these conditions, the cell cycle is about 120 min), indicating an early localization to the FtsZ ring. In E. coli, FtsA and ZipA are the first known proteins to be recruited to the FtsZ ring and are required for the recruitment of other division proteins (Adams and Errington, 2009). No ZipA homolog has yet been described for C. crescentus but FtsA has been shown to localize early to the FtsZ ring and to be essential for division (Ohta et al., 1997; Martin et al., 2004). Time-lapse microscopy of DipM-mCherry and FtsZ-YFP in FtsA-depleted cell filaments revealed that DipM-mCherry colocalized with the non-constricting FtsZ-YFP rings and followed FtsZ-mYFP after disassembly and reassembly at other locations (Fig. 3D), suggesting that DipM localization to FtsZ rings is independent of FtsA. DipM-mCherry also formed bands (rings) in an ftsA temperature-sensitive strain at the restrictive temperature (data not shown). In C. crescentus, MreB also displays an early recruitment to the FtsZ ring and this recruitment is prevented by a Q26P substitution in MreB (Aaron et al., 2007). In an mreBQ26P background, DipM-mCherry localization near midcell was retained (Fig. 3E), suggesting that DipM localization to the FtsZ ring is also MreB-independent. Not surprisingly, however, DipM localization was dependent on FtsZ as DipM-mCherry failed to form bands in FtsZ-depleted cells (Fig. 3F).
The N-proximal tandem of LysM domains is necessary and largely sufficient for DipM localization
In order to determine the region(s) of DipM required for its localization, we generated a series of protein truncations fused to mCherry. These fusions (with the exception of the one shown in Fig. 4B, i, which was produced from the native promoter) were produced from the chromosome under the control of the vanillate-inducible promoter (vanAp) in an otherwise wild-type dipM background. We verified the size and integrity of the fusions by Western blot analysis (Fig. S2)
The DipM protein can be divided into three regions based on its domain composition (Fig. 4A): an N-terminal region carrying two neighboring PG-binding LysM motifs, a central region carrying two more adjacent LysMs and a C-terminal region containing the predicted LytM peptidase domain. The region containing the LytM domain was dispensable for localization (see localization of DipMΔ501-609-mCherry; Fig. 4B, ii). The absence of the N-terminal region carrying the first LysM tandem abolished almost all localization (mCherry-DipMΔ1-236; Fig.4B, vii). Conversely, the N-terminal region with the first LysM tandem alone displayed a slightly weaker, but otherwise normal localization pattern even in the absence of the second LysM tandem and LytM domain (DipMΔ297-609-mCherry; Fig. 4B, iii). The presence of two LysMs in tandem was critical for localization as an in-frame deletion of either LysM motif within the tandem (DipM1–296Δ121–167-mCherry and DipM1–296Δ175–223-mCherry) resulted in dispersed localization in the periplasm (Fig. 4, iv and v, respectively). Since the first and second proline-rich linkers were preserved in these constructs, these results indicate that these linkers are not important for localization; instead they argue that the N-proximal tandem of LysM domains is the major cis-acting determinant of localization.
The LysM domains of DipM can differentiate between two types of peptidoglycan
The LysM domain is a widespread protein module that binds PG (Buist et al., 2008) and a recent atomic force microscopy study has demonstrated that the LysM-PG interaction occurs with high affinity and specificity (Andre et al., 2008). Many LysM-containing proteins are cell-wall hydrolases and by binding to PG, the LysM domains are thought to increase the local concentration of the enzyme, bring the catalytic domain closer to its substrate, and/or help the enzyme slide along the polymeric PG (Steen et al., 2005). DipM has a predicted LytM peptidase domain at its C-terminus. This LytM domain, while dispensable for localization (Fig. 4B, ii), was required for DipM function. This was shown by producing the localization-proficient DipMΔ501-609-mCherry construct (carrying the two LysM tandems but lacking the LytM domain) in cells in which wild-type dipM is under vanillate-controlled expression (strain CJW3122). Cells had a normal morphology in the presence of vanillic acid, but repression of wild-type dipM expression after removal of vanillic acid from the culture medium resulted in cell filamentation characteristic of the DipM- phenotype (Fig. 5A). Interestingly, in these DipM-depleted cells, the localization-proficient DipMΔ501-609-mCherry fusion not only accumulated in bands, as we would expect for filamentous cells that form FtsZ rings (e.g., see DipM localization in FtsA-depleted cells; Fig. 3D), but also accumulated over wide regions along the cell filaments (Fig. 5A). These accumulations were not the result of an uneven periplasmic volume along the cell filaments, as demonstrated by the largely dispersed distribution of freely diffusible mCherry in the periplasm of ΔdipM cells (Fig. S3). Moreover, freely diffusible, periplasmic mCherry molecules were clearly present in the outer membrane vesicles shed by the ΔdipM mutant, unlike the DipMΔ501-609-mCherry construct, consistent with the later binding to the PG through its LysM domains.
Fig. 5.

Localization and mobility of the DipM LysM tandems in FtsZ- and DipM-depleted cells. (A) Localization of the localization-proficient, inactive DipMΔ501-609-mCherry fusion in CJW3446 cells inducing the synthesis of wild-type DipM (growth in the presence of 250 μM of the vanillic acid inducer), or repressing the synthesis of wild-type DipM (growing without vanillic acid inducer). (B) FRAP experiments of DipMΔ501-609-mCherry (carrying two LysM tandems) in DipM- or FtsZ-depleted CJW3446 cells (n=21 and 20, respectively). Depletion of DipM or FtsZ was achieved by removing vanillic acid or xylose from the cultures for about 24 and 5 h, respectively. Regions within cell filaments were photobleached with a 1s laser pulse series. Top, examples of fluorescent images of DipMΔ501-609-mCherry in DipM- and FtsZ-depleted cells before and after photobleaching. The photobleached regions are indicated by yellow dotted circles. The scale bar corresponds to 1 μm. Bottom, the percentage of fluorescence recovery was plotted as a function of time following photobleaching. The average values and the best fit (see Material and methods) are shown in blue and red, respectively. (C) FRAP experiments of DipMΔ297-609-mCherry (carrying a single LysM tandem) in DipM- or FtsZ-depleted cells (CJW3445 and CJW3530, respectively). Regions of cell filaments were photobleached with a 300ms laser pulse series. Quantification was performed as in panel (B).
We considered the possibility that the widespread accumulations of DipMΔ501-609-mCherry in DipM- cells might be caused by the presence of specific PG material that is temporarily left behind in the absence of DipM activity, to which the LysM domains of DipM have a higher binding affinity. To test this idea, we carried out fluorescence recovery after photobleaching (FRAP) experiments. As expected for freely diffusing molecules, a periplasmic mCherry fusion displayed fast kinetics of fluorescence recovery after photobleaching that were not significantly different in DipM- and FtsZ-depleted cells (data not shown). In contrast, we observed no detectable recovery of DipMΔ501-609-mCherry fluorescence over 10 min within the photobleached regions (Fig. 5B; n=21 cells). This is consistent with the LysM tandems of DipM strongly binding to the PG of ΔdipM cells, virtually eliminating protein mobility. On the other hand, in FtsZ-depleted cells where no septal PG is present and DipM has an uniform spatial distribution, recovery of DipMΔ501-609-mCherry signal within photobleached regions occurred at a detectable rate (Fig. 5B, n=20 cells) with a recovery half-time of 13.6 s (±4.6 s). In the absence of fluorescence recovery in the DipM-depleted cells, we could not calculate a recovery half-time value for DipMΔ501-609-mCherry for quantitative comparison. Therefore, to reduce binding to the PG and thereby increase mobility in the periplasm, we performed FRAP experiments with a mCherry reporter construct that contains only one LysM tandem (instead of two) in DipM-depleted and FtsZ-depleted cells. Mobility of this construct (DipMΔ297-609-mCherry) was increased in both backgrounds (Fig. 5C), consistent with our interpretation that PG binding through the LysM domains largely accounts for the restricted mobility. Perhaps more importantly, fluorescence recovery was much faster in the FtsZ-depleted cells, with estimated recovery half-times of 1.1 s (±0.271 s) and 17.6 s (±1.7 s) in FtsZ-depleted and DipM-depleted cells, respectively (Fig. 5C). Thus, there is about a 16-fold difference in mobility for a construct with a single LysM tandem. Although the second LysM tandem cannot localize by itself, the observation that in conjunction with the first tandem, protein localization is enhanced (Fig. 4B, ii versus iii) suggests that the difference in binding selectivity could be even larger when two LysM tandems are present.
Taken together, these results suggest that localization of DipM is mediated by the discriminating specificity of LysM tandems for different types of PG.
The non-canonical LytM domain of DipM is required for protein function
The C-terminal region carrying the LytM domain of DipM is dispensable for localization (Fig. 4B, ii) but is essential for DipM function (Fig. 5A), consistent with a role in PG splitting during constriction. LytM domains have been shown to have metallo-endopeptidase activity in Gram-positive bacteria (Ramadurai et al., 1999; Horsburgh et al., 2003). The crystal structure of Staphylococcus aureus LytM identifies three conserved amino acids that directly coordinate zinc (Odintsov et al., 2004; Firczuk et al., 2005). Only one of these residues is conserved in DipM (H585; Fig. 6A). The other two amino acids are substituted by asparagines (N503 and N507, Fig. 6A), which, at least in principle, can coordinate zinc (Harding, 2004; Passerini et al., 2007).
Fig. 6.
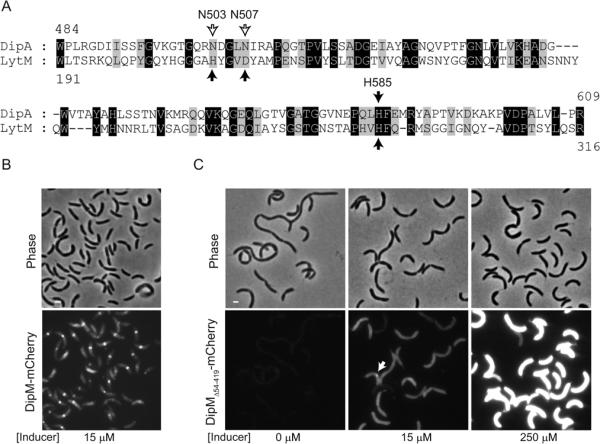
The non-canonical LytM domain of DipM can support function. (A) Alignment between S. aureus LytM catalytic domain and the non-canonical LytM domain of C. crescentus DipM. Black arrows indicate the conserved residues that are directly involved in metal coordination in the crystal structure of S. aureus LytM. The empty arrows indicate the non-conserved residues in DipM at positions of metal coordination. (B) Images of ΔdipM cells expressing wild-type dipM fused to mCherry (strain CJW3440). The cells were grown in the presence of 15 μM vanillic acid (inducer) for 5 h in M2G medium at 30°C to induce the synthesis of the mCherry fusions. Under these conditions, the level of expression of wild-type dipM allele was sufficient to fully suppress the cell filamentation phenotype caused by the ΔdipM mutation. (C) Images of ΔdipM cells carrying the non-canonical LytM domain of DipM (and no LysM domains) fused to mCherry (DipMΔ54–419-mCherry; strain CJW3444). Different levels in DipMΔ54–419-mCherry proteins were obtained by growing the cells in the presence of different concentrations of the vanillic acid inducer. The arrow indicates an example of the infrequent accumulation of DipMΔ54-419-mCherry signal at constriction sites.
To examine whether the LytM domain alone can support protein function, we generated two additional mCherry fusions, one to the full-length DipM protein (DipM-mCherry) and the other to the LytM domain alone (DipMΔ54-419-mCherry). The synthesis of these fusions was then induced from the vanillic acid-inducible promoter on a low-copy copy plasmid (pRVCHYC-2) in a ΔdipM background. The minimal concentration of inducer (vanillic acid) needed to obtain full complementation of the ΔdipM filamentation phenotype with the wild-type DipM-mCherry construct was 15 μM (Fig. 6B). Consistent with the notion that the non-canonical LytM domain is required for function, expression of DipMΔ54-419-mCherry partially suppressed the ΔdipM filamentation phenotype (Fig. 6C), especially when overexpressed (i.e., in the presence of 250 μM of inducer).
As expected, this DipMΔ54-419-mCherry fusion lacking the LysM tandems displayed a largely uniform fluorescent signal, except for a slightly brighter signal at deeply constricted sites on rare occasions (Fig. 6C, arrow). These small accumulations of signal are possibly caused by a local increase in periplasmic volume due to outer membrane instability, as observed for the freely diffusing periplasmic mCherry (Fig. S3) and as suggested previously for the envC mutant (Bernhardt and de Boer, 2004). The defect in localization was not due to mCherry being cleaved off from the fusion as verified by Western blot (Fig. S2). Importantly, the suppression of the ΔdipM filamentation phenotype by the LytM domain alone remained only partial even when it was overproduced (in the presence of 250 μM of inducer; Fig. 6C). This demonstrates the importance of positioning the LytM domain activity at the correct cellular locations.
Discussion
Cell wall proteins including the LytM factors have been proposed to play a crucial role in cell division by separating or promoting the separation of the interlinked PG of daughter cells (Rodolakis et al., 1973; Starka et al., 1974; Lange and Hengge-Aronis, 1994; Heidrich et al., 2001; Hara et al., 2002; Bernhardt and de Boer, 2004; Garcia and Dillard, 2006; Priyadarshini et al., 2006; Uehara et al., 2009; Uehara et al., 2010). In this study, we identify DipM, a protein with a non-canonical LytM domain that has a similar role in PG splitting in C. crescentus. Consistent with this function, DipM is a periplasmic protein that localizes to the future site of division in C. crescentus (Fig. 3A-C) and while its function is dispensable for viability under slow growth conditions (e.g., low temperature or low-nutrient medium), it is essential for viability under fast growing conditions (Fig. 1A). This indicates that although some functional redundancy exists, this redundancy is not sufficient to sustain rapid cell multiplication. Even under slow-growth conditions, DipM function is very important as ΔdipM cells display severe cell division defects with a large fraction of the population forming long filaments that occasionally divide. These ΔdipM cell filaments are constricted at irregular intervals (Fig. 1C), unlike the chain-forming mutants in E. coli (Heidrich et al., 2001; Uehara et al., 2009). This cell filamentation phenotype is in part due to a de-synchronization between the progression of cell division and segregation of the partition complex, causing the disassembly of FtsZ rings by the partitioning complex-associated protein MipZ (Fig. 2C). A similar effect is observed under conditions in which recruitment of septal proteins (such as MurG and PBP3) to the FtsZ ring is considerably impaired (Y. Tsuge, G. Ebersbach and C. Jacobs-Wagner, unpublished results). Desynchronization between cytokinesis and segregation of the DNA partition complex in ΔdipM cells seems to be caused by an apparent stalling or slower constriction of the FtsZ ring (Movies S4 and S5; Fig. S1). A defect in FtsZ ring constriction also appears to occur in E. coli when the LytM factor EnvC is absent (Hara et al., 2002). Interestingly, a mutation in the envC gene causes a synthetic sick phenotype when combined with a deletion of the minCDE operon (Bernhardt and de Boer, 2004), and MinD is a functional homolog of MipZ.
How could DipM affect FtsZ ring constriction? Cell division in Gram-negative bacteria involves the invagination of three cell layers: the cytoplasmic membrane, the PG and the outer membrane. The cytoplasmic membrane maintains a close association with the outer membrane and the growing PG through the cell division protein complex (MacAlister et al., 1987; Gerding et al., 2007). A possible reason for the slowdown in FtsZ ring constriction in ΔdipM cells may involve the loss of contact between the cytoplasmic and outer membranes, as coordinated invagination of the cytoplasmic and outer membranes (Poindexter and Hagenzieker, 1981; Judd et al., 2005) requires proper splitting of the growing PG. The shedding of vesicles at division sites in the ΔdipM mutant (Movie S1) is consistent with loss of interaction between the outer membrane and the other two layers (Knox et al., 1966; Deatherage et al., 2009). Another, perhaps more attractive possibility by which DipM affects FtsZ ring constriction relates more directly to its function in splitting FtsZ-ring-directed peptidoglycan. In E. coli, cleavage of septal PG during constriction is accompanied by the release of PG fragments (Uehara and Park, 2008). In the absence of DipM, other hydrolysis-inducing activities can partially take over at least under slow-growth rate conditions, but their activity may not be as selective and they may also cause the cleavage of peptide bonds involved in inward PG growth, causing it to stall or even rescind. While FtsZ rings can generate force by themselves in tubular liposomes (Osawa et al., 2008), recent theoretical calculations suggest that inward PG growth can help the progressivity of FtsZ ring constriction (Lan et al., 2009). The frequent stalling (Fig. 1S) and occasional rescinding (Movie S1) of FtsZ ring constriction in ΔdipM cells are consistent with this notion.
Similarly to DipM, the two main LytM factors involved in septal PG splitting in E. coli, EnvC and NlpD (Uehara et al., 2009), have non-conserved amino acids at the metal-coordinating positions (Ichimura et al., 2002). It was recently shown that theses two proteins do not hydrolyze the PG but instead acts as activators of PG amidases (Uehara et al., 2010). It is possible that DipM has a similar regulatory function, although the LytM domain of DipM is essential for function and is sufficient to partially suppress the ΔdipM phenotype when overexpressed (Fig. 6). Future work will be required to determine the specific role of DipM LytM domain in PG splitting.
In C. crescentus, the FtsZ ring, which forms well before initiation of cell constriction, directs PG synthesis, not only during division, but also during a large portion of the cell elongation phase (Aaron et al., 2007). DipM is recruited relatively early to the FtsZ ring location well before cell constriction becomes discernible (Fig. 3C), suggesting a possible early role in FtsZ-ring-directed PG synthesis. Our data suggest that localization occurs, at least in part, through the preferential binding of the LysM tandems of DipM to the FtsZ ring-directed PG. In the absence of DipM activity, a DipM construct carrying the LysM tandems (but not the LytM domain) accumulates not only in bands at presumed FtsZ ring locations, but also in large adjacent regions (Fig. 5A). FRAP experiments suggest that these regions contain PG for which the DipM LysM domains have higher binding affinity (Fig. 5B-C). We envision that these regions correspond to multilayered PG synthesized by the FtsZ-associated cell wall enzymes. FtsZ-directed PG growth temporarily generates multilayered PG that is progressively removed by cell wall hydrolases. Our findings suggest that DipM greatly contributes to such activity by mediating –directly or indirectly– the selective cleavage of the crosslinks most distal from the cytoplasmic membrane and that DipM accumulates where it is needed through preferential binding of its LysM tandems to multilayered PG rather than monolayered PG. This idea predicts that the absence of DipM would result in accumulation of multiple layers of glycan strands at existing and former FtsZ ring positions, which is consistent with the formation of PG- and SP-rings in ΔdipM cell filaments (Fig. 2B). Similarly, the frequent instability of the FtsZ ring in the ΔdipM background (Fig. S1, lines and Movie S4) would cause the accumulation of glycan strands over wide regions, thereby generating large patches of multilayered PG throughout the length of ΔdipM cell filaments. Consistent with this, cryo-electron tomography of ΔdipM sacculi reveals notable thickening of PG throughout the mutant sacculi (Goley et al., in press), in good agreement with the localization pattern and the FRAP analysis of the inactive, localization-proficient LysM-containing constructs in the ΔdipM mutant (Fig. 5).
How would the LysM domains of DipM recognize multilayered PG over the more common monolayered form? Structural and biochemical studies reveal that LysM domains bind to the N-acetylglucosamine moiety of the PG (Buist et al., 2008). DipM contains two LysM tandems (Fig. 4A) and the N-proximal LysM tandem is sufficient to mediate septal localization (Fig. 4iii), although not as strongly as when it is connected to the second LysM tandem (Fig. 4ii). It was recently shown in vitro that a single LysM domain of Pteris ryukyuensis Chitinase A binds to glycan chains of four N-acetylglucosamine residues in a 1:1 ratio, whereas a tandem of this LysM domain binds to these glycan chains in a 1:2 ratio (Ohnuma et al., 2008). These stoichiometries suggest the possibility that each LysM tandem of DipM binds to two adjacent glycan strands. In the regular monolayered PG, adjacent glycan strands are connected by peptide bridges that are stretched by the turgor pressure (Oldmixon et al., 1974; Koch, 2000). In contrast, in the multilayered, septal PG, the underlying glycan strands are connected with each other and with the stress-bearing layer by peptide bridges that are in a relaxed state. Accordingly, the glycan strands are closer to each other when the peptide cross-links are relaxed, perhaps allowing the simultaneous binding of the two LysM domains in the tandem to adjacent glycan strands. Conversely, adjacent glycan strands connected with stretched peptides (as in the stress-bearing, monolayered PG) may be too far apart, allowing only one of the LysM domains of the tandem to bind at a time, reducing the binding strength, which would be consistent with our FRAP measurements (Fig. 5B-C). Turgor pressure is also thought to produce a conformational change in the stress-bearing glycan chains (from straight to a tessera conformation) by changing the relative orientation of the disaccharides (Koch, 1998; Ursinus et al., 2004; Vollmer and Seligman, 2010). Therefore, an alternative hypothesis for the septal localization of DipM may involve the relaxation state of the glycan strands rather than that of the peptide cross-links. It is formally possible that the LysM domains in tandem bind to N-acetylglucosamine residues of the same glycan strand and that the spacing and arrangement between N-acetylglucosamines might be more favorable for the binding of both LysM motifs of each tandem when the glycan strands are in a relaxed state. In either scenario, DipM would preferentially bind to the underlying, unstressed glycan strands and thereby localize to the multilayered FtsZ-directed PG by distinguishing a physical difference in PG. We also cannot rule out the possibility that the multilayered PG presents a chemical difference for which the LysM tandem has a higher affinity. This would be reminiscent of the proposed localization mechanism for FtsN and other SPOR-containing proteins (Ursinus et al., 2004; Yang et al., 2004; Mishima et al., 2005; Gerding et al., 2009; Lutkenhaus, 2009). Localization of these proteins to the septum is mediated by their SPOR domain, which preferentially binds to PG glycan strands stripped of peptide chains. Differential recognition of PG forms by PG-binding domains may thus represent an effective strategy for spatial and temporal regulation of cell wall activities.
Experimental procedures
Strains and culture conditions
All C. crescentus cultures were grown in liquid M2G medium or on solid PYE-agar medium at 30°C (Ely, 1991), unless otherwise indicated. E. coli strains were grown in LB broth. Antibiotics for C. crescentus were used at the following final concentrations (in μg/ml) for liquid and solid media correspondingly: Gentamycin, 2 and 5; kanamycin, 5 and 20; oxytetracycline, 1 and 2; spectinomycin, 25 and 100; apramycin 8. For E. coli, the following concentrations (in μg/ml) were used for liquid and solid media: Ampicillin, 150 and 200; gentamycin, 10 and 20; kanamycin, 50; oxytetracycline, 20; spectinomycin, 50. The strains and plasmids used in this study are listed in Tables 1 and 2, respectively. Their construction is described in the supplementary text.
Table 1.
List of strains used in this study.
| Strain | Genotype | Source |
|---|---|---|
| C. crescentus | ||
| CB15N | synchronizable derivative of C. crescentus CB15 (also known as NA1000) | (Evinger and Agabian, 1977) |
| CJW1715 | CB15N mreBQ26P | (Aaron et al., 2007) |
| CJW2959 | CB15N vanA::pVsigpepCHYN-4 | (Takacs et al., 2010) |
| CJW3116 | CB15N dipM::pVspCHYdipMΔ1-30 | This study |
| CJW3117 | CB15N dipM::pVspCHYdipMΔ1-236 | This study |
| CJW3121 | CB15N vanA::pVdipMΔ297-609CHY | This study |
| CJW3122 | CB15N dipM::pVdipMΔ501-609CHY | This study |
| CJW3124 | CB15N dipM::pCHYCdipM | This study |
| CJW3137 | CB15N ΔdipM::Ω–Spc | This study |
| CJW3187 | CB15N ftsA::pXMCS2ftsA | This study |
| CJW3430 | CB15N ΔdipM::Ω–Spc vanA::pMT383 | This study |
| CJW3436 | CB15N dipM::pCHYCdipM vanA::pMT383 | This study |
| CJW3437 | CB15N mreBQ26P dipM::pdipMCHY | This study |
| CJW3438 | CB15N ftsZ::pBJM dipM::pCdipMCHY | This study |
| CJW3439 | CB15N vanA::pVdipMΔ501-609CHY | This study |
| CJW3440 | CB15N ΔdipM::ΩSpc/pRVdipMCHY | This study |
| CJW3444 | CB15N ΔdipM::ΩSpc /pRVCHYCdipMΔ54-419 | This study |
| CJW3445 | CB15N dipM::pVdipMΔ297-609CHY | This study |
| CJW3446 | CB15N dipM::pVdipMΔ501-609CHY ftsZ::pBJM | This study |
| CJW3447 | CB15N ftsA::pXMCS2ftsA vanA::pMT383 dipM::pdipMCHY | This study |
| CJW3448 | CB15N ΔdipM::Ω–Spc vanA::pMT383 mipZ-cfp | This study |
| CJW3449 | CB15N ΔdipM::Ω–Spc xylX::pXGFPMCS-2 | This study |
| CJW3455 | CB15N vanA::pMT383 mipZ-cfp | W. Schofield |
| CJW3530 | CB15N vanA:: pVdipMΔ297-609CHY ftsZ:: pBJM | This study |
| CJW3526 | CB15N vanA::pVLysM1A | This study |
| CJW3528 | CB15N vanA::pVLysM1B | This study |
| CJW3530 | CB15N ftsZ:pBJM pVdipMΔ297-609CHY | This study |
| CJW3550 | vanA::pVsigpepMCHYN-4 ftsZ::pBJM | This study |
| CJW3551 | ΔdipM::Ω–Spc vanA::pVsigpepMCHYN-4 | This study |
| CJW3591 | CB15 dipM::pdipMCHY divE309 (ftsAts) Rifr | This study |
| MT196 | CB15N vanA::pMT383 | (Thanbichler and Shapiro, 2006) |
| PC8848 | CB15 divE309 (ftsAts) Rifr | (Ohta et al, 1997) |
| YB1585 | CB15N ftsZ::pBJM | (Wang et al., 2001) |
| E. coli | ||
| DH5a | Cloning strain | Invitrogen |
| TOP10 | Cloning strain | Invitrogen |
Table 2.
List of plasmids used in this study.
| Plasmid | Characteristic | Source |
|---|---|---|
| pCHYC-4 | pMB1 replicon with oriT and mCherry | (Thanbichler et al., 2007) |
| pCR2.1-TOPO | Cloning vector | Invitrogen |
| pdipMCHY | pCHYC-4 carrying dipM421-609 | This study |
| pNPTS138 | pLitmus38 derivative carrying oriT and sacB | MRK Alley |
| pNPTSΔdipM::Ω | pNPTS138 carrying ΔdipM::Ω-Spc | This study |
| pRVCHYC1 | Vector with oriT, oriV, vanAp and mCherry | (Thanbichler et al., 2007) |
| pRVdipMCHY | pRVCHYC-1 carrying dipM | This study |
| pRVdipMΔ54-419CHY | pRVCHYC-1 carrying dipMΔ54-419 | This study |
| pTOPOdipM | pCR2.1-TOPO carrying dipM | This study |
| pVCHYC-4 | pMB1 replicon with oriT, vanAp and mCherry | (Thanbichler et al., 2007) |
| pVCHYN-4 | pMB1 replicon with oriT, vanAp and mCherry | (Thanbichler et al., 2007) |
| pVdipMΔ297-609CHY | pVCHYC-4 carrying dipMΔ297-609 | This study |
| pVdipMΔ501-609CHY | pVCHYC-4 carrying dipMΔ501-609 | This study |
| pVspCHYdipMΔ1-30 | pVsigpepCHYN-4 carrying dipMΔ1-30 | This study |
| pVspCHYdipMΔ1-236 | pVsigpepCHYN-4 carrying dipMΔ1-236 | This study |
| pVsigpepCHYN-4 | pVCHYN-4 carrying dipMΔ54-609 | (Takacs et al., 2010) |
| pVLysM1ACHY | pVCHYC-4 carrying dipM1–296Δ121–167 | This study |
| pVLysM1BCHY | pVCHYC-4 carrying dipM1–296Δ175–223 | This study |
| pXMCS-2 | pMB1 replicon with oriT and xylXp promoter | (Thanbichler et al., 2007) |
| pXMCS2ftsA | pXMCS-2 with ftsA under xylXp control | This study |
Microscopy and analysis
Light microscopy images were obtained using a Nikon E1000 or NIKON E80i equipped with a Hamamatsu ORCA ER camera or a NIKON E80i equipped with an Andor iXonEM+ CCD camera. Cells were grown to an OD660 between 0.2 and 0.3, and immobilized on agarose-padded slides containing M2G medium for imaging at room temperature. Images were taken and processed with Metamorph 7.1.4 software or ImageJ. Cell size measurements were performed as described (Angelastro et al., 2009). For the FLIP experiments, a Photonic Instrument Micropoint Laser system was used with a 481-nm Laser Dye, set at 15 pulses with a power of 2 at the attenuator plate and 20 for the internal attenuator. For the FRAP experiments, a 552-nm Laser Dye was used set at 8 or 2 pulses with a power of 1 and 40 for the internal attenuator. For both FLIP and FRAP experiments the laser intensity was cut by 70% through a beam splitter, and attenuated 4X by a neutral density filter. The fluorescence recovery data was processed in Microsoft Excel. Fitting was achieved using the MATLAB “cftool” application and function F(t)=A(1-e-bt) where F, t, A, and b are fluorescence intensity, time, maximal recovery fluorescence, and recovery rate, respectively.
Electron microscopy
A diluted suspension of purified sacculi was spotted for 15 min on Carbon-B coated Formvar 200 mesh copper grids (TedPella) that had been previously air-ionized for 30 s. The grids were then washed with water and stained for 10 min with a 3% uranyl acetate solution, washed again and allowed to dry. Imaging was carried out on a Jeol JEM-1230 transmission electron microscope operating at 80 kV. Digital images were acquired with a Hamamatsu ORCA-HR camera. SEM images were obtained as described previously (Takacs et al. 2010).
Molecular biology techniques and Western blot
For PCR reactions, the TaKaRa PrimeSTAR-HS DNA polymerase was used as directed by the manufacturer. DNA restriction and ligase reactions were carried out with enzymes from New England BioLabs. Immunoblots were done using standard techniques and an anti-RFP antibody.
Supplementary Material
Acknowledgments
The authors wish to thank the Jacobs-Wagner lab members for helpful discussion and critical reading of the manuscript, and L. Shapiro for the anti-RFP antibody. We also thank L. Shapiro, M. Thanbichler and their colleagues for agreeing on the DipM name. This work was supported by the National Institutes of Health (GM076698 and GM065835 to C.J-W.), the European Commission within the EUR-INTAFAR project (to W.V.) and the Howard Hughes Medical Institute (to C.J-W.). S.P. was supported in part by a PEW Latin American fellowship and C.N.T. was supported in part by a Yale College Dean's Office Science, Technology and Research Scholars (STARS) II fellowship.
References
- Aaron M, Charbon G, Lam H, Schwarz H, Vollmer W, Jacobs-Wagner C. The tubulin homologue FtsZ contributes to cell elongation by guiding cell wall precursor synthesis in Caulobacter crescentus. Mol Microbiol. 2007;64:938–952. doi: 10.1111/j.1365-2958.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- Adams DW, Errington J. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol. 2009;7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- Andre G, Leenhouts K, Hols P, Dufrene YF. Detection and localization of single LysM-peptidoglycan interactions. J Bacteriol. 2008;190:7079–7086. doi: 10.1128/JB.00519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelastro PS, Sliusarenko O, Jacobs-Wagner C. Polar localization of the CckA histidine kinase and cell cycle periodicity of the essential master regulator CtrA in Caulobacter crescentus. J Bacteriol. 2009 doi: 10.1128/JB.00985-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol. 2004;52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browder HP, Zygmunt WA, Young JR, Tavormina PA. Lysostaphin: Enzymatic Mode of Action. Biochem Biophys Res Commun. 1965;19:383–389. doi: 10.1016/0006-291x(65)90473-0. [DOI] [PubMed] [Google Scholar]
- Buist G, Steen A, Kok J, Kuipers OP. LysM, a widely distributed protein motif for binding to (peptido)glycans. Mol Microbiol. 2008;68:838–847. doi: 10.1111/j.1365-2958.2008.06211.x. [DOI] [PubMed] [Google Scholar]
- Cohen DN, Sham YY, Haugstad GD, Xiang Y, Rossmann MG, Anderson DL, Popham DL. Shared catalysis in virus entry and bacterial cell wall depolymerization. J Mol Biol. 2009;387:607–618. doi: 10.1016/j.jmb.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage BL, Lara JC, Bergsbaken T, Rassoulian Barrett SL, Lara S, Cookson BT. Biogenesis of bacterial membrane vesicles. Mol Microbiol. 2009;72:1395–1407. doi: 10.1111/j.1365-2958.2009.06731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Blaauwen T, de Pedro MA, Nguyen-Disteche M, Ayala JA. Morphogenesis of rod-shaped sacculi. FEMS Microbiol Rev. 2008;32:321–344. doi: 10.1111/j.1574-6976.2007.00090.x. [DOI] [PubMed] [Google Scholar]
- Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991;204:372–384. doi: 10.1016/0076-6879(91)04019-k. [DOI] [PubMed] [Google Scholar]
- Evinger M, Agabian N. Envelope-associated nucleoid from Caulobacter crescentus stalked and swarmer cells. J Bacteriol. 1977;132:294–301. doi: 10.1128/jb.132.1.294-301.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firczuk M, Mucha A, Bochtler M. Crystal structures of active LytM. J Mol Biol. 2005;354:578–590. doi: 10.1016/j.jmb.2005.09.082. [DOI] [PubMed] [Google Scholar]
- Gan L, Chen S, Jensen GJ. Molecular organization of Gram-negative peptidoglycan. Proc Natl Acad Sci U S A. 2008;105:18953–18957. doi: 10.1073/pnas.0808035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DL, Dillard JP. AmiC functions as an N-acetylmuramyl-l-alanine amidase necessary for cell separation and can promote autolysis in Neisseria gonorrhoeae. J Bacteriol. 2006;188:7211–7221. doi: 10.1128/JB.00724-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. Self-enhanced accumulation of FtsN at division sites, and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J Bacteriol. 2009 doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Narita S, Karibian D, Park JT, Yamamoto Y, Nishimura Y. Identification and characterization of the Escherichia coli envC gene encoding a periplasmic coiled-coil protein with putative peptidase activity. FEMS Microbiol Lett. 2002;212:229–236. doi: 10.1111/j.1574-6968.2002.tb11271.x. [DOI] [PubMed] [Google Scholar]
- Harding MM. The architecture of metal coordination groups in proteins. Acta Crystallogr D Biol Crystallogr. 2004;60:849–859. doi: 10.1107/S0907444904004081. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Holtje JV. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Holtje JV. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J Bacteriol. 2002;184:6093–6099. doi: 10.1128/JB.184.22.6093-6099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh GJ, Atrih A, Foster SJ. Characterization of LytH, a differentiation-associated peptidoglycan hydrolase of Bacillus subtilis involved in endospore cortex maturation. J Bacteriol. 2003;185:3813–3820. doi: 10.1128/JB.185.13.3813-3820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Yamazoe M, Maeda M, Wada C, Hiraga S. Proteolytic activity of YibP protein in Escherichia coli. J Bacteriol. 2002;184:2595–2602. doi: 10.1128/JB.184.10.2595-2602.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd EM, Comolli LR, Chen JC, Downing KH, Moerner WE, McAdams HH. Distinct constrictive processes, separated in time and space, divide Caulobacter inner and outer membranes. J Bacteriol. 2005;187:6874–6882. doi: 10.1128/JB.187.20.6874-6882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox KW, Vesk M, Work E. Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J Bacteriol. 1966;92:1206–1217. doi: 10.1128/jb.92.4.1206-1217.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AL. Orientation of the peptidoglycan chains in the sacculus of Escherichia coli. Res Microbiol. 1998;149:689–701. doi: 10.1016/s0923-2508(99)80016-3. [DOI] [PubMed] [Google Scholar]
- Koch AL. Simulation of the conformation of the murein fabric: the oligoglycan, pentamuropeptide, and cross-linked nona-muropeptide. Arch Microbiol. 2000;174:429–439. doi: 10.1007/s002030000227. [DOI] [PubMed] [Google Scholar]
- Lan G, Daniels BR, Dobrowsky TM, Wirtz D, Sun SX. Condensation of FtsZ filaments can drive bacterial cell division. Proc Natl Acad Sci U S A. 2009;106:121–126. doi: 10.1073/pnas.0807963106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange R, Hengge-Aronis R. The nlpD gene is located in an operon with rpoS on the Escherichia coli chromosome and encodes a novel lipoprotein with a potential function in cell wall formation. Mol Microbiol. 1994;13:733–743. doi: 10.1111/j.1365-2958.1994.tb00466.x. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J. FtsN - trigger for septation. J Bacteriol. 2009 doi: 10.1128/JB.01100-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlister TJ, Cook WR, Weigand R, Rothfield LI. Membrane-murein attachment at the leading edge of the division septum: a second membrane-murein structure associated with morphogenesis of the gram-negative bacterial division septum. J Bacteriol. 1987;169:3945–3951. doi: 10.1128/jb.169.9.3945-3951.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ME, Trimble MJ, Brun YV. Cell cycle-dependent abundance, stability and localization of FtsA and FtsQ in Caulobacter crescentus. Mol Microbiol. 2004;54:60–74. doi: 10.1111/j.1365-2958.2004.04251.x. [DOI] [PubMed] [Google Scholar]
- Mishima M, Shida T, Yabuki K, Kato K, Sekiguchi J, Kojima C. Solution structure of the peptidoglycan binding domain of Bacillus subtilis cell wall lytic enzyme CwlC: characterization of the sporulation-related repeats by NMR. Biochemistry. 2005;44:10153–10163. doi: 10.1021/bi050624n. [DOI] [PubMed] [Google Scholar]
- Odintsov SG, Sabala I, Marcyjaniak M, Bochtler M. Latent LytM at 1.3A resolution. J Mol Biol. 2004;335:775–785. doi: 10.1016/j.jmb.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Ohnuma T, Onaga S, Murata K, Taira T, Katoh E. LysM domains from Pteris ryukyuensis chitinase-A: a stability study and characterization of the chitin-binding site. J Biol Chem. 2008;283:5178–5187. doi: 10.1074/jbc.M707156200. [DOI] [PubMed] [Google Scholar]
- Ohta N, Ninfa AJ, Allaire A, Kulick L, Newton A. Identification, characterization, and chromosomal organization of cell division cycle genes in Caulobacter crescentus. J Bacteriol. 1997;179:2169–2180. doi: 10.1128/jb.179.7.2169-2180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldmixon EH, Glauser S, Higgins ML. Two proposed general configurations for bacterial cell wall peptidoglycans shown by space-filling molecular models. Biopolymers. 1974;13:2037–2060. doi: 10.1002/bip.1974.360131008. [DOI] [PubMed] [Google Scholar]
- Osawa M, Anderson DE, Erickson HP. Reconstitution of contractile FtsZ rings in liposomes. Science. 2008;320:792–794. doi: 10.1126/science.1154520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Anderson DE, Erickson HP. Curved FtsZ protofilaments generate bending forces on liposome membranes. EMBO J. 2009;28:3476–3484. doi: 10.1038/emboj.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini A, Andreini C, Menchetti S, Rosato A, Frasconi P. Predicting zinc binding at the proteome level. BMC Bioinformatics. 2007;8:39. doi: 10.1186/1471-2105-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poindexter JS, Hagenzieker JG. Constriction and septation during cell division in caulobacters. Can J Microbiol. 1981;27:704–719. doi: 10.1139/m81-109. [DOI] [PubMed] [Google Scholar]
- Priyadarshini R, de Pedro MA, Young KD. Role of peptidoglycan amidases in the development and morphology of the division septum in Escherichia coli. J Bacteriol. 2007;189:5334–5347. doi: 10.1128/JB.00415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadarshini R, Popham DL, Young KD. Daughter cell separation by penicillin-binding proteins and peptidoglycan amidases in Escherichia coli. J Bacteriol. 2006;188:5345–5355. doi: 10.1128/JB.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadurai L, Lockwood KJ, Nadakavukaren MJ, Jayaswal RK. Characterization of a chromosomally encoded glycylglycine endopeptidase of Staphylococcus aureus. Microbiology. 1999;145(Pt 4):801–808. doi: 10.1099/13500872-145-4-801. [DOI] [PubMed] [Google Scholar]
- Reste de Roca F, Duche C, Dong S, Rince A, Dubost L, Pritchard DG, Baker JR, Arthur M, Mesnage S. Cleavage Specificity of Enterococcus faecalis EnpA (EF1473), a Peptidoglycan Endopeptidase Related to the LytM/Lysostaphin Family of Metallopeptidases. J Mol Biol. 2010 doi: 10.1016/j.jmb.2010.03.033. [DOI] [PubMed] [Google Scholar]
- Rodolakis A, Thomas P, Starka J. Morphological mutants of Escherichia coli. Isolation and ultrastructure of a chain-forming envC mutant. J Gen Microbiol. 1973;75:409–416. doi: 10.1099/00221287-75-2-409. [DOI] [PubMed] [Google Scholar]
- Starka J, Di Savino D, Michel G, Rodolakis A, Thomas P. Phenotypic expression of an envC-division mutant of Escherichia coli K12. Ann Microbiol (Paris) 1974;125 B:227–232. [PubMed] [Google Scholar]
- Steen A, Buist G, Horsburgh GJ, Venema G, Kuipers OP, Foster SJ, Kok J. AcmA of Lactococcus lactis is an N-acetylglucosaminidase with an optimal number of LysM domains for proper functioning. FEBS J. 2005;272:2854–2868. doi: 10.1111/j.1742-4658.2005.04706.x. [DOI] [PubMed] [Google Scholar]
- Takacs CN, Poggio S, Charbon G, Pucheault M, Vollmer W, Jacobs-Wagner C. MreB drives de novo rod morphogenesis in Caulobacter crescentus via remodeling of the cell wall. J Bacteriol. 2010;192:1671–1684. doi: 10.1128/JB.01311-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschner PE, Ypenburg N, Spratt BG, Woldringh CL. An amino acid substitution in penicillin-binding protein 3 creates pointed polar caps in Escherichia coli. J Bacteriol. 1988;170:4828–4837. doi: 10.1128/jb.170.10.4828-4837.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanbichler M, Iniesta AA, Shapiro L. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 2007;35:e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanbichler M, Shapiro L. MipZ, a spatial regulator coordinating chromosome segregation with cell division in Caulobacter. Cell. 2006;126:147–162. doi: 10.1016/j.cell.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Uehara T, Dinh T, Bernhardt TG. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J Bacteriol. 2009;191:5094–5107. doi: 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Park JT. Growth of Escherichia coli: significance of peptidoglycan degradation during elongation and septation. J Bacteriol. 2008;190:3914–3922. doi: 10.1128/JB.00207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Parzych KR, Dinh T, Bernhardt TG. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. EMBO J. 2010 doi: 10.1038/emboj.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursinus A, van den Ent F, Brechtel S, de Pedro M, Holtje JV, Lowe J, Vollmer W. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J Bacteriol. 2004;186:6728–6737. doi: 10.1128/JB.186.20.6728-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verwer RW, Nanninga N, Keck W, Schwarz U. Arrangement of glycan chains in the sacculus of Escherichia coli. J Bacteriol. 1978;136:723–729. doi: 10.1128/jb.136.2.723-729.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Bertsche U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta. 2008;1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008a;32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Holtje JV. The architecture of the murein (peptidoglycan) in gram-negative bacteria: vertical scaffold or horizontal layer(s)? J Bacteriol. 2004;186:5978–5987. doi: 10.1128/JB.186.18.5978-5987.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Joris B, Charlier P, Foster S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev. 2008b;32:259–286. doi: 10.1111/j.1574-6976.2007.00099.x. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Seligman SJ. Architecture of peptidoglycan: more data and more models. Trends Microbiol. 2010;18:59–66. doi: 10.1016/j.tim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Wang Y, Jones BD, Brun YV. A set of ftsZ mutants blocked at different stages of cell division in Caulobacter. Mol Microbiol. 2001;40:347–360. doi: 10.1046/j.1365-2958.2001.02395.x. [DOI] [PubMed] [Google Scholar]
- Werner JN, Chen EY, Guberman JM, Zippilli AR, Irgon JJ, Gitai Z. Quantitative genome-scale analysis of protein localization in an asymmetric bacterium. Proc Natl Acad Sci U S A. 2009;106:7858–7863. doi: 10.1073/pnas.0901781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldringh CL. Morphological analysis of nuclear separation and cell division during the life cycle of Escherichia coli. J Bacteriol. 1976;125:248–257. doi: 10.1128/jb.125.1.248-257.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Lowe J. Solution structure and domain architecture of the divisome protein FtsN. Mol Microbiol. 2004;52:651–660. doi: 10.1111/j.1365-2958.2004.03991.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.