

Abstract
Disulphide formation within the endoplasmic reticulum (ER) requires the activity of the ER oxidase Ero1, and as a consequence, generates hydrogen peroxide. The production of hydrogen peroxide is thought to lead to oxidative stress that ultimately results in apoptosis. Here, we show that mammalian peroxiredoxin IV (PrxIV) metabolises hydrogen peroxide produced by Ero1. We demonstrate that the presence of PrxIV within the ER provides a cytoprotective effect against stresses known to enhance Ero1 activity and perturb ER redox balance. Increased Ero1 activity and production of hydrogen peroxide led to preferential hyperoxidation of PrxIV relative to peroxiredoxins in other cellular compartments. The hyperoxidation was increased by the upregulation of Ero1 and by the expression of a hyperactive Ero1. These findings provide the first evidence for an enzymatic mechanism that facilitates peroxide removal from the ER, and show that the oxidation status of PrxIV acts as a marker for ER oxidative stress.
Keywords: Disulphide formation, Ero1, Oxidative stress, Peroxiredoxin
Introduction
Cell stress and cell death caused by protein misfolding have been suggested to be prevalent in tissues with a heavy secretion load and in cells expressing mutant proteins (Schroder and Kaufman, 2005). One of the predicted causes of cell stress under these conditions is the build-up of reactive oxygen species (ROS) formed during disulphide formation or as a consequence of the unfolded protein response (UPR) (Harding et al., 2003; Malhotra et al., 2008; Marciniak et al., 2004). Formation of disulphide bonds involves a series of disulphide-exchange reactions catalysed by members of the protein disulphide isomerase (PDI) family (Ellgaard, 2004; Ellgaard and Ruddock, 2005). The ER oxidase Ero1 (Ero1p in yeast, Ero1α or Ero1β in mammals) catalyses oxidation of PDI (Frand and Kaiser, 1999) by coupling the introduction of a disulphide with reduction of oxygen to generate hydrogen peroxide (Gross et al., 2006). One molecule of hydrogen peroxide is produced for every disulphide introduced into a protein, therefore significant quantities of ROS can be produced within the ER (Harding et al., 2003; Haynes et al., 2004).
Despite the production of ROS during protein folding and disulphide formation, oxidative stress is not induced under normal physiological conditions. In conditions of induced stress, however, such as during the UPR, Ero1 is upregulated (Harding et al., 2003), giving rise to increased ROS production and oxidative stress (Malhotra et al., 2008; Marciniak et al., 2004). At steady state, Ero1 activity is feedback-regulated by formation of non-catalytic disulphides that inactivate the enzyme (Appenzeller-Herzog et al., 2008; Baker et al., 2008; Sevier et al., 2007). Although tightly controlled, Ero1 will be active under such conditions and thus generate hydrogen peroxide. It is therefore likely that mechanisms exist within the ER to prevent ROS accumulation and oxidative stress during normal oxidative protein folding.
The removal of hydrogen peroxide in the cell occurs via several routes, including breakdown by catalase or glutathione peroxidases, or reaction with non-enzymatic oxidants including vitamins and glutathione (GSH) (Valko et al., 2007). In addition, a family of six enzymes (peroxiredoxin I to peroxiredoxin VI) with various cellular locations can act as both peroxidases and peroxide sensors within cells (Phalen et al., 2006; Wood et al., 2003b). We have recently shown that one family member, peroxiredoxin IV (PrxIV, Prx4) forms a characteristic decamer, contains a cleavable signal sequence and is localised to the human ER (Tavender et al., 2008). It is therefore a prime candidate for a peroxidase involved in the removal of ER-generated hydrogen peroxide.
PrxIV is a typical, so-called 2-Cys, peroxiredoxin, which means that it contains a redox-active cysteine residue, which attacks peroxides forming a cysteine sulfenic acid (SOH) in the process. The sulfenic cysteine can then be resolved by reaction with a second free thiol group in an adjacent molecule, forming an interchain disulphide-bonded homodimer and water. Within the cytosol, members of the 2-Cys peroxiredoxins can then be recycled by reduction of the disulphide by either glutathione or thioredoxin. Alternatively, the sulfenic cysteine can react further with hydrogen peroxide to form sulphinic acid (SO2H) and even sulphonic acid (SO3H). These hyperoxidised forms of cysteine inactivate the enzyme; however, an additional enzyme called sulfiredoxin can reduce the sulphinic acid group back to sulfenic acid (Jeong et al., 2006). This mechanism of inactivation and reactivation enables the peroxiredoxins to act as efficient peroxidases at low concentrations of peroxide but once the concentration increases, the enzyme is inactivated, allowing the hydrogen peroxide to be stabilised and initiate signalling events (Phalen et al., 2006).
We tested the hypothesis that PrxIV metabolises the hydrogen peroxide produced by Ero1 by first determining the ability of the enzyme to protect cells from ER stress. We also determined whether PrxIV can act as a peroxidase in isolation and within cells. Our results clearly show that PrxIV has a cytoprotective effect during ER stress and that this protection is due to the metabolism of hydrogen peroxide produced by Ero1. In addition, cellular PrxIV becomes hyperoxidised after excessive oxidative stress, providing a sensor for hyperoxidising conditions in the ER lumen.
Results
PrvIV has a cytoprotective effect against ER stress
To determine whether PrxIV has a role in preventing cell death during ER stress, we initially compared cell viability during stress induction of three HT1080 cell lines; one with normal levels of PrxIV, a second in which PrxIV had been knocked down using shRNA (Tavender et al., 2008) and the third treated with non-targeted shRNA to control for pleiotropic effects. Cells containing reduced amounts of PrxIV were significantly more susceptible to cell death following treatment with tunicamycin or DTT, both known inducers of ER stress (Merksamer et al., 2008) (Fig. 1A,B). Interestingly, at higher DTT concentrations (>2 mM) there was no significant effect of PrxIV knockdown on viability, suggesting that PrxIV is inactivated at these concentrations. To confirm that the effect of PrxIV knockdown was due to an ER-specific process, we induced apoptosis in each cell line using etoposide. Etoposide causes apoptosis through inhibition of topoisomerase II (Barry et al., 1993) and is therefore independent of ER stress. There was no significant difference in viability of the various cell lines tested following treatment with this reagent (Fig. 1C). Hence PrxIV knockdown compromises the cellular tolerance towards stress generated within the ER.
Fig. 1.
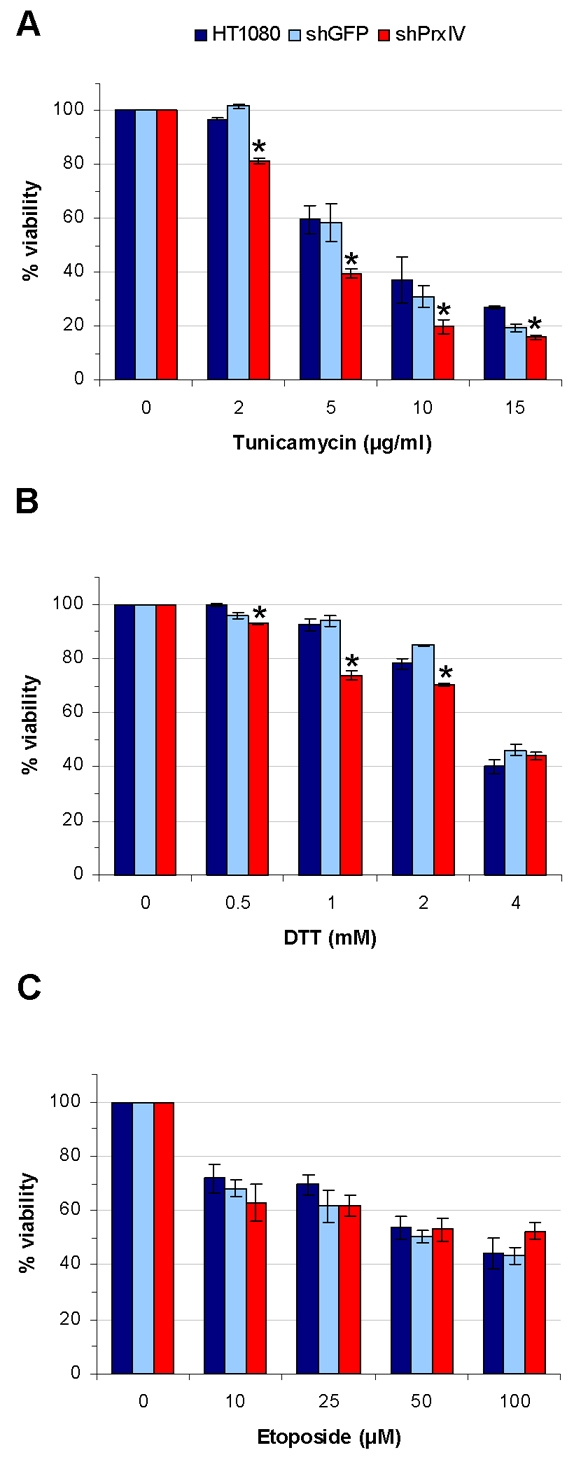
PrxIV protects against ER-stress-induced cell death. (A-C) Crystal Violet viability assays comparing survival of HT1080 cells (dark blue bars) with HT1080 cells expressing shRNA against GFP (shGFP, pale blue bars) or PrxIV (shPrxIV, red bars) following treatment with increasing concentrations of tunicamycin for 48 hours (A), DTT for 24 hours (B) or etoposide for 24 hours (C). Data comprise means ± s.d. for three independent replicates. Asterisks (*) indicate where shPrxIV survival differs significantly (P<0.05) from both HT1080 as well as shGFP, as judged by unpaired, two-tailed t-tests assuming unequal variance.
Given the cytoprotective effect of PrxIV following DTT treatment we postulate that this enzyme metabolises peroxides generated as a consequence of DTT treatment, thus protecting cells during protein misfolding. These peroxides could be produced as a result of Ero1 oxidising DTT directly (Gross et al., 2006) or oxidising thiols generated by DTT treatment. Likewise, the protection of cells from apoptosis following tunicamycin treatment might reflect the increased misfolding and non-native disulphide formation, which would ultimately lead to increased Ero1 activity and production of ROS (Harding et al., 2003). Alternatively, the induction of the UPR itself might lead to production of ROS via an as yet unknown mechanism (Malhotra et al., 2008). Regardless of the source of ROS, our results clearly demonstrate that the presence of PrxIV protects mammalian cells from apoptosis caused as a result of induction of the UPR.
PrxIV has peroxidase activity both in isolation and within cells
The ability of PrxIV to protect cells from the consequences of the UPR is likely to reside in its enzymatic activity. To characterize this activity and to demonstrate that PrxIV can act as a peroxidase, we prepared recombinant protein by expression in E. coli. Using a standard peroxide-turnover assay (Kim et al., 2005), we showed that human PrxIV exhibits peroxidase activity in vitro (Fig. 2A). PrxIV is a typical 2-Cys peroxiredoxin, and consequently forms a homodecamer (Tavender et al., 2008). Each subunit contains a redox-active cysteine residue (the peroxidatic cysteine), which attacks peroxides to form cysteine sulfenic acid (SOH) in the process. The sulfenic cysteine can then be resolved by reaction with a second free thiol (the resolving cysteine) in an adjacent molecule, forming a disulphide-bonded dimer; hence formation of a disulphide-liked dimer is integral to peroxiredoxin enzymatic activity (Wood et al., 2003b). However, human PrxIV also forms a disulphide via a third cysteine residue (Cys51) (Tavender et al., 2008), which connects subunits within the decamer (Fig. 2B). When our recombinant protein was separated by SDS-PAGE under non-reducing conditions, several high molecular weight products were evident, suggesting that both the Cys51 and the active-site cysteines had formed interchain disulphide bonds (Fig. 2C, lane 1). To distinguish between PrxIV interchain disulphides, we mutated the peroxidatic and resolving cysteines (DM) or Cys51 to alanine (C51A), as well as all three together (DM51). The C51A and DM recombinant proteins both now ran as interchain disulphide-bonded dimers, whereas the DM51 mutant ran under denaturing conditions as a monomer (Fig. 2C). The presence of only disulphide-linked dimers with the C51A mutant indicates that PrxIV might well have the same mechanism of action as other peroxiredoxins (Wood et al., 2003b).
Fig. 2.
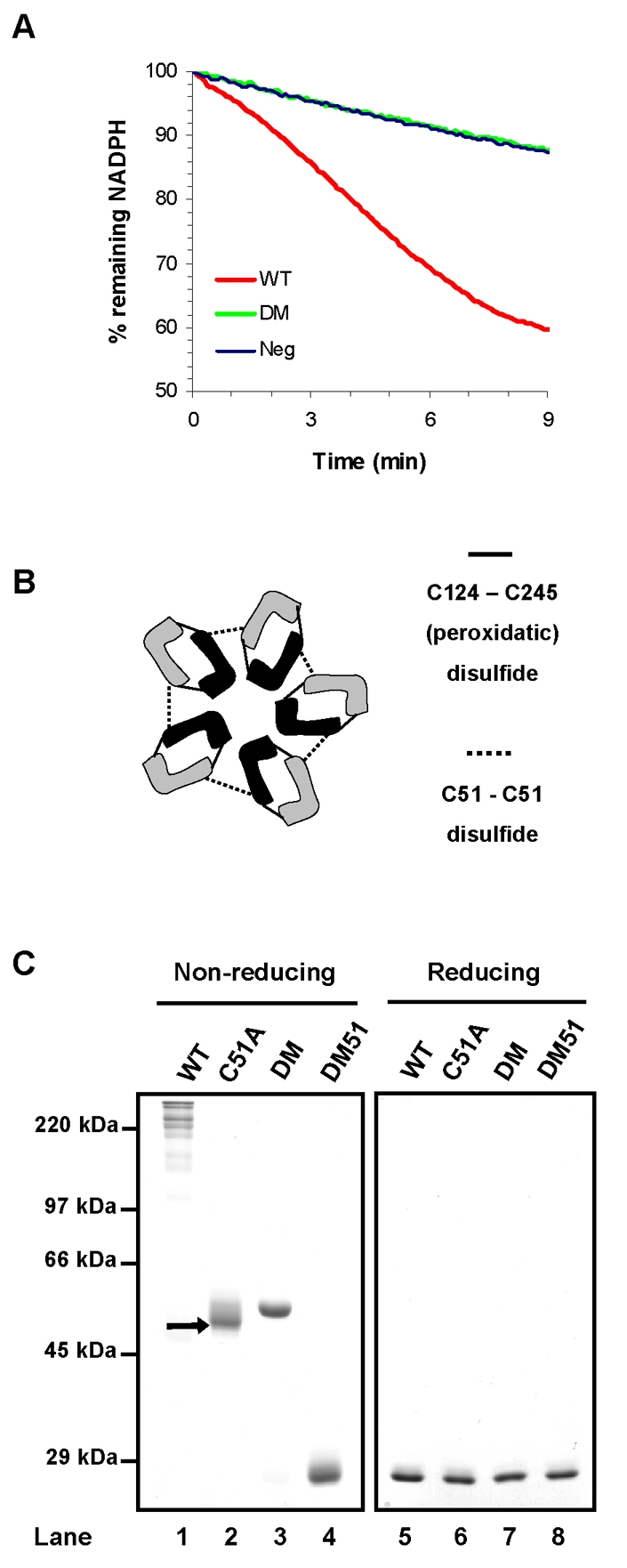
PrxIV has peroxidase activity in vitro. (A) Purified PrxIV activity (WT, red line) in an assay coupling hydrogen peroxide reduction with NADPH oxidation via thioredoxin and thioredoxin reductase. PrxIV DM (green line) displays no activity beyond background NADPH oxidation observed when no PrxIV is added (Neg, blue line). (B) Depiction of interchain disulphides formed within the PrxIV decamer. For clarity, individual PrxIV molecules within each dimeric unit are represented in different shades. (C) Non-reducing SDS-PAGE analysis and Coomassie Blue staining (left panel) showing disulphide-bonded species for purified PrxIV (WT) and cysteine mutants (C51A, DM, DM51). Samples were treated with 40 mM NEM to maintain thiol-disulphide status and 3 μg protein was loaded per lane. Arrow indicates the faster-migrating C51A dimer. All interchain disulphides disappear under reducing SDS-PAGE conditions (right panel).
A striking feature of PrxIV C51A mutant protein is the presence of a faster-migrating disulphide-linked dimer, which is absent with PrxIV DM (Fig. 2C, arrow). We hypothesised that this more-compact molecule contains two inter-molecular disulphides (depicted Fig. 3A) between the two pairs of peroxidatic and resolving cysteines in adjacent subunits. Incubation of PrxIV with thioredoxin in vitro (Fig. 3B, lanes 4-6) caused a step-wise change in mobility of C51A from high-mobility dimer to low-mobility dimer to monomer, consistent with reduction from 2 to 1 to 0 inter-molecular disulphides. Complete, rapid reduction of the Cys51 disulphide was observed for DM, which cannot form peroxidatic disulphides (Fig. 3B, lanes 7-9). These effects combined in the wild-type enzyme to give a reduction from high molecular weight structures to dimers reminiscent of C51A (Fig. 3B, lanes 1-3). Thus, catalytically active PrxIV displays disulphide-linked dimeric species that are distinct from the dimers formed by non-catalytically active PrxIV.
Fig. 3.

PrxIV forms distinct disulphide-bonded dimers. (A) Representation of interchain disulphides formed by PrxIV mutants. (B) Non-reducing SDS-PAGE analysis of purified PrxIV species following thioredoxin addition, in the presence of thioredoxin reductase (*) and NADPH, under aerobic conditions. High-mobility dimers (2 × S-S), low-mobility dimers (1 × S-S) and redox monomers (0 × S-S) are indicated. (C) Anti-PrxIV western blotting of lysates from HT1080 cells overexpressing PrxIV variants. Arrow indicates faster-migrating dimers under non-reducing conditions. A fraction of each sample was also analysed under reducing conditions. (D) Similar analysis was performed using semi-permeabilised cells prepared from HT1080 cells expressing PrxIV C51A and incubated for 30 minutes at 37°C with indicated GSH concentrations. In each experiment, further thiol reactivity was blocked by addition of 40 mM NEM.
The characterisation of PrxIV recombinant protein allowed us to interpret the redox forms of PrxIV seen in cell lines expressing the wild type, C51A or DM mutant. Analysis by non-reducing SDS-PAGE showed that the high molecular weight species seen with the wild-type protein in vitro do not predominate within the cell (Fig. 3C, lane 4), with the distribution instead balanced in favour of lower-order species. This is at least partly due to variable formation of the Cys51 disulphide in vivo, as evident in the cell line expressing PrxIV DM (Fig. 3C, lane 6). Similarly, the C51A mutant expressed in cells displayed both monomeric and dimeric redox species compared with exclusively disulphide-bonded dimers in vitro (compare Fig. 3C, lane 5 with Fig. 2C, lane 2). In addition, a faster-mobility dimer was again seen (indicated with an arrow). Interestingly, when the cytosol was removed from cells expressing the C51A mutant by preparing semi-permeabilised cells and the redox state of PrxIV analysed, a pattern more similar to the recombinant material was seen (Fig. 3D, lane 1). No monomeric species was evident and a higher-mobility dimer prevailed. The addition of GSH to these semi-permeabilised cells to levels normally found in the cytosol (Schafer and Buettner, 2001) restored the PrxIV redox profile found in intact cells (Fig. 3D, lane 3), and also directly reduced recombinant PrxIV C51A in vitro (supplementary material Fig. S1). Hence, it would appear that within the intact ER, the peroxidatic and resolving thiols exist in equilibrium between reduced and oxidised forms. In addition, it is clear that GSH, either directly or indirectly, maintains the redox balance of PrxIV by reducing the peroxidatic disulphide. Furthermore, purified recombinant PrxIV is clearly very sensitive to oxidation, presumably by oxygen radicals present in the buffers used for purification. In this respect PrxIV mirrors the behaviour of recombinant PrxII, which is exquisitely sensitive to oxidation (Peskin et al., 2007).
Identification of distinct PrxIV dimeric species created an opportunity to visualise the peroxidatic process in vivo (depicted in Fig. 4A). Formation of a disulphide between the peroxidatic and resolving cysteine is a consequence of peroxide reactivity, so it follows that modulating PrxIV activity should result in visible transitions between the redox states. Indeed, supply of exogenous hydrogen peroxide to cells overexpressing PrxIV C51A caused an increase in the higher-mobility dimer relative to untreated cells (Fig. 4B upper panel, lanes 1 and 2, arrow). As peroxide concentration increased, this shift disappeared, correlating with elevated detection of hyperoxidised PrxIV using an antibody that recognises the hyperoxidised active site common to PrxI-PrxIV (Fig. 4B, middle panel). Hyperoxidation of the peroxidatic cysteine generates cysteine-sulfinic acid (Cys-SO2H) or sulfonic acid (Cys-SO3H) derivatives, which inhibit the formation of a disulphide with the resolving cysteine (see Fig. 4A).
Fig. 4.

PrxIV redox states demonstrate peroxidase activity in vivo. (A) Schematic representing changes in PrxIV oxidation state accompanying increasing peroxide reactivity. Peroxidatic (SHPer) and resolving (SHRes) thiols are indicated for each PrxIV molecule. (B) Western blot analysis of PrxIV C51A redox state in cells treated for 10 minutes at 37°C with indicated hydrogen peroxide concentrations, then 40 mM NEM. Analyses were performed under non-reducing or reducing conditions as indicated, with tubulin serving as a loading control.
PrxIV metabolises the hydrogen peroxide produced by Ero1α
Having demonstrated that PrxIV is active and can be hyperoxidised by addition of hydrogen peroxide to cells, we next asked whether the hydrogen peroxide produced by Ero1 is a substrate for endogenous PrxIV. It is known that DTT is a substrate for yeast Ero1p (Gross et al., 2006) and human Ero1α (supplementary material Fig. S2) and that cells overexpressing Ero1α have an increased resistance to this reducing agent (Chakravarthi and Bulleid, 2004; Mezghrani et al., 2001). To determine whether hydrogen peroxide was produced by Ero1 following addition of DTT to cells, we looked for hyperoxidation of PrxIV in HT1080 cells treated with increasing concentrations of DTT for 5 minutes. Such a brief treatment with DTT would lead to turnover of DTT by Ero1, but is too short to induce gene expression in the time course of the experiment. Following DTT treatment, there was a marked increase in the level of hyperoxidised PrxIV (Fig. 5A and supplementary material Fig. S3). The antibody used also recognises the hyperoxidised versions of PrxI-PrxIII, and in particular, a slightly faster-migrating protein than PrxIV (Fig. 5A). To confirm the identities of the hyperoxidised proteins, we compared their mobility with those of PrxI-PrxIV (Fig. 5A,B). The upper band clearly migrates with PrxIV, whereas the predominant lower band migrates with PrxIII. In addition, when PrxIV was overexpressed we saw a similar hyderoxidation of the overexpressed protein in response to DTT treatment, further verifying that the increase in signal is due to the specific hyperoxidation of PrxIV (supplementary material Fig. S4). In contrast to PrxIV, there is no increase in hyperoxidation of PrxI-PrxIII. Hence DTT treatment leads to hyperoxidation specifically of PrxIV. The most likely explanation for this result is that Ero1 oxidises the added DTT or thiols generated during DTT treatment, dramatically increasing hydrogen peroxide concentration in the ER lumen and therefore PrxIV hyperoxidation.
Fig. 5.

PrxIV hyperoxidation indicates peroxide production during Ero1 activity in vivo. (A) Western blot analysis of Prx hyperoxidation following treatment of HT1080 cells with increasing concentrations of DTT for 5 minutes at 37°C. PrxIV is indicated (*) and tubulin provides a loading control. (B) Included for comparison are blots for untreated HT1080 lysate showing the mobility of each human typical 2-Cys Prx. (C) Anti-PrxSO3 and anti-tubulin western blotting of cell lysates from untreated HEK TREX Ero1α-V5 (UT) and cells treated with 500 μM H2O2 or 2 mM DTT for 5 minutes (asterisk indicates PrxIV). (D) Similar analysis to that shown in A was performed for HEK TREX Ero1α-V5 cells following incubation with or without doxycyclin for 16 hours. Samples were also probed using anti-PrxIV to ascertain expression following Ero1α induction. (E) Western blot analysis of lysates from HEK cells expressing Ero1α-V5 (WT) or a deregulated variant (C104,131A). Lysates were prepared following incubation for 16 hours in the presence or absence of doxycyclin and probed using anti-V5 (top panel), anti-tubulin (middle) and anti-PrxSO3 (bottom). Included for comparison is lysate from uninduced WT cells treated for 5 minutes with 1 mM DTT (WT + DTT). In all experiments, free thiols were alkylated using 40 mM NEM following treatment.
To demonstrate that the hydrogen peroxide produced was from the activity of Ero1α we determined the effect of overexpressing Ero1α on the level of hyperoxidised PrxIV. We predicted that cells overexpressing this enzyme would exhibit more-pronounced hyperoxidation of PrxIV following addition of DTT. Consequently, we constructed a HEK293 cell line where expression of Ero1α could be induced (supplementary material Fig. S5). It has been shown previously that the induction of expression of Ero1α or of deregulated versions of Ero1α does not lead to a UPR, making this system a good tool to analyse specifically the effect of overexpression of Ero1α (Appenzeller-Herzog et al., 2008). We also did not see any UPR following the induction of Ero1α expression using doxycyclin, as judged by a lack of induction of BiP (supplementary material Fig. S5).
To assess the sensitivity of endogenous PrxIV to hyperoxidation in the HEK293 cell background, we first determined the effect of addition of hydrogen peroxide or DTT to the HEK293 TREX Ero1α cell line without induction of Ero1α (Fig. 5C). Preferential PrxIV hyperoxidation was again observed following treatment of these cells with DTT for 5 minutes (Fig. 5C, lane 3), a striking contrast to the widespread peroxiredoxin hyperoxidation stimulated by addition of hydrogen peroxide (Fig. 5C, lane 2). This result again highlights the fact that addition of DTT results specifically in the production of hydrogen peroxide in the ER, leading to PrxIV hyperoxidation. When Ero1α expression was induced by the addition of doxycyclin, a marked enhancement in PrxIV hyperoxidation occurred in response to the incubation with DTT when compared with non-induced cells (Fig. 5D, compare lanes 3 and 5 with 4 and 6; supplementary material Fig. S6). The increase in the level of hyperoxidised PrxIV could not be explained by an increased expression of PrxIV because there was no increase in the total amount of PrxIV following induction of Ero1α (Fig. 5D, lanes 7 and 8). These results clearly demonstrate that hydrogen peroxide produced by Ero1α in the presence of DTT caused specific hyperoxidation of PrxIV. As hyperoxidation inactivates the peroxidatic cysteine, these results go some way to explain the loss of PrxIV protection from apoptosis seen at increased DTT concentrations (Fig. 1B). Inactivation of PrxIV is not seen after exposure of cells to tunicamycin, probably because tunicamycin treatment results in protein misfolding and non-native disulphide formation, but does not cause an acute need for rapid disulphide formation in a manner that DTT does. Hence the consequence of DTT treatment will be a sudden surge of peroxide, whereas tunicamycin treatment causes a UPR followed by oxidative stress (Marciniak et al., 2004).
To demonstrate that the hyperoxidation of PrxIV can occur in the absence of added DTT we constructed an additional HEK293 cell line that allowed induction of a deregulated version of Ero1α. It has recently been shown that both yeast Ero1p and human Ero1α are regulated by the formation of disulphide bonds, in the case of Ero1 occurring between catalytic and non-catalytic cysteines (Appenzeller-Herzog et al., 2008; Baker et al., 2008; Sevier et al., 2007). Hence, Ero1α is in an inactive state and requires activation by reduction of these regulatory disulphides. Mutation of the non-catalytic cysteines involved in the formation of the regulatory disulphides leads to the formation of a deregulated Ero1α, which when expressed in cells causes increased ER oxidation (Appenzeller-Herzog et al., 2008). When expression of such a deregulated version of Ero1α was induced there was a higher level of hyperoxidised PrxIV in the cell line expressing the deregulated Ero1α compared with the cell line expressing wild-type Ero1α (Fig. 5E, compare lanes 4 and 5; supplementary material Fig. S7). Hence the ability of PrxIV to metabolise the hydrogen peroxide produced by Ero1α is not dependent upon using DTT as a substrate and is probably due to an increased oxidation of the physiological substrates of Ero1α. Indeed, in an equivalent cell line, the levels of oxidised PDI increased following induction of Ero1α expression (Appenzeller-Herzog et al., 2008). Taken together, these results clearly demonstrate that PrxIV can metabolise the hydrogen peroxide produced by Ero1α in the ER of mammalian cells.
Discussion
Previously, we have shown that PrxIV is an ER-localised enzyme (Tavender et al., 2008). A partial knockdown of the enzyme rendered cells more susceptible to cell death induced by the addition of hydrogen peroxide, although we could not demonstrate any effect of this knockdown on the turnover of exogenously added peroxide. Our inability to detect a gross effect of PrxIV knockdown probably reflects the partial nature of the knockdown, the nature of the assays used and the fact that other cellular peroxidases might well mask any specific consequence of PrxIV knockdown. In particular, the peroxide assays were carried out with semi-permeabilised cells and we now know from work presented in this paper that the recycling of PrxIV is compromised in semi-permeabilised cells. Importantly, we have now shown that the enzyme can act to turnover hydrogen peroxide in vitro and that it provides a cytoprotective effect against stresses likely to raise the levels of hydrogen peroxide in the ER lumen. In addition, we show that increasing the activity of Ero1 leads to hyperoxidation of PrxIV, an event that could only occur if Ero1 produces hydrogen peroxide. This result is the first indication that hydrogen peroxide is produced by Ero1 in cells.
Our results show that the activity of PrxIV in the ER is linked to hydrogen peroxide production by Ero1, providing a mechanism by which the cell is protected from a potentially harmful consequence of disulphide formation. We suggest a model (Fig. 6) where the oxidation of PDI by Ero1 is initially controlled by the presence of regulatory disulphides within Ero1 (Appenzeller-Herzog et al., 2008; Baker et al., 2008). Once these disulphides are broken, Ero1 oxidises PDI and in the process generates hydrogen peroxide. The hydrogen peroxide is then metabolised by peroxiredoxin IV, which in the process becomes oxidised at the peroxidatic cysteine to form sulfenic acid. This can be resolved by the formation of a disulphide between the peroxidatic and resolving cysteine residues. Once oxidised, the PrxIV needs to be recycled to regenerate the peroxidatic cysteine. The reduction of the active-site disulphide might be carried out by a member of the thioredoxin-domain-containing PDI family of proteins, several of which are reduced in vivo and hence competent for reduction of PrxIV disulphides (Jessop and Bulleid, 2004). Such a role for a PDI family member would mirror the role of thioredoxin in the cytosol in resolving the active-site disulphide in PrxI and PrxII. Alternatively, glutathione might directly reduce PrxIV, as suggested by its ability to reduce the protein in vitro. Once the peroxidatic cysteine becomes hyperoxidised to sulfinic acid, as happens when cells are treated with DTT, then it could potentially be returned to a sulfenic acid form by the action of a sulfiredoxin (Woo et al., 2003). However, no such enzyme has been identified in the ER, so it remains to be determined whether such an activity exists.
Fig. 6.

PrxIV metabolises hydrogen peroxide produced during formation of disulphide bonds in the human ER. Schematic illustrating the role of PrxIV in peroxide elimination following oxidation of PDI by Ero1. Oxidation of PDI facilitates the introduction of a disulphide into a client protein during PDI-catalysed oxidative folding (depicted top left). As Ero1 is re-oxidised by molecular oxygen the hydrogen peroxide produced is catabolised by PrxIV, resulting in changes in PrxIV redox state (SHPer, peroxidatic cysteine; SHRes, resolving cysteine). Continued peroxidase activity requires a thiol-dependent recycling pathway (represented in light blue on bottom right) which clearly exists for PrxIV. This might involve a thioredoxin-domain-containing PDI family member, but its nature remains unclear.
Such a model for peroxide removal will depend in part upon the ability of PrxIV to rapidly metabolise any hydrogen peroxide produced. Hydrogen peroxide will also react with cysteine residues in other proteins and might well result in disulphide formation (Karala et al., 2009). However, the reactivity of hydrogen peroxide towards cysteines in protein is highly selective, with the peroxidatic cysteine in peroxiredoxins being particularly sensitive to oxidation (Wood et al., 2003a). The cytosolic peroxiredoxins have been shown to be highly reactive towards hydrogen peroxide with an apparent second-order rate constant of 1.3×107 M−1 second−1 for PrxII (Peskin et al., 2007). Such high reactivity would result in hydrogen peroxide preferentially reacting with the peroxidatic cysteine within PrxIV rather than cysteines in other ER proteins. In fact a recent proteome-wide analysis of the disulphide proteome showed that protein thiols do not become oxidised randomly after treatment with hydrogen peroxide (Le Moan et al., 2006).
PrxIV is an abundant and ubiquitous protein found throughout the metazoan kingdom. However, it is absent from fungi, so in yeast an alternative mechanism to remove hydrogen peroxide must exist. Indeed, even in mammals, other ER-resident glutathione peroxidase homologues have been identified (Raykhel et al., 2007), although they have not been characterised in any detail. Although the pathway described here is clearly active and is required to protect cells from oxidative stress, there might be alternative mechanisms that contribute to peroxide removal. In support of this idea, a Prdx4-knockout mouse has recently been reported, which has been characterised in terms of the effect of PrxIV removal on spermatogenesis (Iuchi et al., 2009). Interestingly, the absence of PrxIV leads to elevated spermatogenic cell death, which occurs after oxidative stress. However, the animals are still fertile, so the absence of PrxIV must be compensated by the upregulation of alternative pathways to remove the peroxide formed during disulphide formation. These pathways are unlikely to involve the non-enzymatic removal of peroxide because low molecular weight thiols such as glutathione have been shown to have poor reactivity towards ROS (Winterbourn and Metodiewa, 1999).
The formation of hyperoxidised forms of the peroxiredoxins adds an additional layer of regulation to the enzyme, which at least in the cytosol, can facilitate an acute response to hydrogen peroxide. This response is exemplified by the inactivation of PrxII as a result of oxidative insult, resulting in hydrogenperoxide-dependent cell-cycle arrest (Phalen et al., 2006). It might be that PrxIV has a similar role in response to hydrogen peroxide levels in the ER lumen. Low levels of hydrogen peroxide are tolerated because of the action of PrxIV, but if levels of hydrogen peroxide increase dramatically, for example during UPR, then PrxIV becomes hyperoxidised, resulting in its inactivation and an acute rise in the levels of ER-generated peroxide. The burst of peroxide could then lead to cell-cycle arrest and eventually apoptosis. The fact that PrxIV is hyperoxidised at high concentrations of DTT and that this modification negates any hypersensitivity of the PrxIV-knockdown cell line to DTT supports this possibility. It is interesting to note that PrxIV is upregulated dramatically in line with ER oxidoreductases as the ER expands during B-cell differentiation to ensure efficient synthesis of immunoglobulins (van Anken et al., 2003). However, PrxIV does not seem to be induced during the UPR (Tavender et al., 2008). It could be that during differentiation, levels of PrxIV need to increase to accommodate the increased load of disulphide formation, but during UPR, levels need to remain constant to allow signalling of oxidative stress. Clearly, more work is needed to clarify this point.
Finally, the hyperoxidation of PrxIV demarcates PrxIV-SO2 and PrxIV-SO3 as novel indicators of hyperoxidising conditions within the ER. Unlike disulphide formation, this modification directly reports on the levels of hydrogen peroxide in the ER. Similarly to other peroxiredoxins, PrxIV therefore provides an effective monitor of the oxidative burden within its cellular compartment.
Materials and Methods
Chemicals, reagents and antibodies
All reagents were acquired from Sigma and enzymes from Promega (Southampton, UK) unless otherwise stated. Rabbit polyclonal anti-PrxI, anti-PrxII, anti-PrxIII, anti-PrxIV and anti-PrxSO3 were purchased from Ab Frontier (Seoul, Korea), a mouse monoclonal antibody recognising α-tubulin was a generous gift from Keith Gull (University of Oxford, Oxford, UK) and rabbit polyclonal anti-BiP was kindly supplied by Richard Zimmerman (Universität des Saarlandes, Saarbrücken, Germany). Rabbit polyclonal anti-Ero1α was obtained from Cell Signaling Technology (Danvers, MA) and mouse monoclonal anti-V5 from Invitrogen (Paisley, UK).
Cell-based analyses
Crystal Violet viability assays, site-directed mutagenesis and creation of cell lines stably underexpressing or overexpressing PrxIV have been previously described (Tavender et al., 2008). All primer sequences used are available on request. HT PrxIV C51A, DM and HT shGFP cell lines were constructed in an identical fashion. V5-tagged human Ero1α was amplified from cDNA by PCR and inserted into first pcDNA5/FRT/V5-His-TOPO and from there into pcDNA5/FRT/TO-TOPO (Invitrogen) by TA cloning in accordance with the manufacturer's instructions. The deregulated mutant was generated from this by site-directed mutagenesis. HEK293 cells expressing each Ero1α-V5 construct were prepared using the Flp-In™ system (Invitrogen). All cells were cultured at 37°C using Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM L-glutamine and 10% foetal bovine serum. Where appropriate, doxycyclin-mediated expression was induced as indicated in the text.
Samples for western blot analysis of cells following treatment with H2O2 or DTT was performed by first preparing cell suspensions from subconfluent cultured cells. Cells were trypsinised, washed with PBS and resuspended in serum-free DMEM at a density of 107 cells per ml. Samples were treated as indicated in the text then free thiols alkylated using 40 mM N-ethylmaleimide (NEM) on ice for 5 minutes. Cells were subsequently isolated by centrifugation, washed with PBS and lysed by boiling for 5 minutes in SDS-PAGE sample buffer (31.25 mM Tris-HCl, pH 6.8, 2% w/v SDS, 5% v/v glycerol, 0.01% w/v Bromophenol Blue) added to give an equivalent concentration of 104 cells per μl.
For analysis of lysates from cells at steady state, or following doxycyclin-induced expression but no further treatment, NEM was added directly to culture medium of cells growing in subconfluent monolayer. Both attached and floating cells were harvested by trypsinisation and/or centrifugation, washed with PBS and lysed as above by addition of SDS-PAGE sample buffer. Semi-permeabilised cells were prepared as described previously (Wilson et al., 1995) and treated as for above cell suspensions except treatments were performed in KHM buffer rather than DMEM.
In vitro analyses
His-tagged PrxIV variants were cloned into pRSFDuet-1 (Novagen, Nottingham, UK) for E. coli BL21-DE3 expression. Previously created human expression constructs (Tavender et al., 2008) were used as cDNA templates along with primers designed to remove the 37 residue N-terminal signal peptide and incorporate an N-terminal thrombin cleavage site. E. coli expression was induced for 3 hours with 0.5 mM IPTG and standard Ni2+-agarose (Qiagen, Crawley, UK) batch purification performed. His-tags were cleaved with 10 U thrombin protease (GE Healthcare, Amersham, UK) per mg purified protein for 16 hours at 4° C and PrxIV decamers then separated to homogeneity using Superdex 200™ column chromatography (GE Healthcare). PrxIV activity assays and step-wise reduction by thioredoxin were performed using previously established reaction conditions (Kim et al., 2005). Final concentrations were 4.5 μM PrxIV, 3 μM thioredoxin, 1.5 μM thioredoxin reductase, 200 μM NADPH and 100 μM hydrogen peroxide. Assay buffer was 50 mM HEPES, pH 7.0. Ero1α was assayed for activity in the presence of 12.5 mM DTT using an oxygen-consumption assay, as described previously (Baker et al., 2008).
Electrophoresis and western blotting
Samples for SDS-PAGE were resuspended in SDS sample buffer and heated to 100°C for 5 minutes. For reducing conditions, DTT was added to 50 mM, for non-reducing conditions DTT was omitted. Gels were stained for 30 minutes using Coomassie Blue (45% methanol, 9% acetic acid, 0.1% w/v Coomassie brilliant blue) followed by overnight destain (15% methanol, 10% acetic acid) or western blotting was performed exactly as described previously (Tavender et al., 2008).
For quantification of western blots, multiple exposures were performed and the intensities of the most- and least-intense bands were calculated for each time. Based on this, exposures were selected for analysis at which all samples exhibited a linear response to the chemiluminescent substrate. Intensity of each sample was quantified using AIDA 2D densitometry, drawing identical-sized boxes for each band within a given blot and also subtracting a local background for each individual sample.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Wellcome Trust (ref. 74081). In addition we wish to acknowledge the generosity of Andrew Gilmore, Chris Grant and Stephen High (all University of Manchester, UK), as well as Keith Gull (University of Oxford, UK) and Richard Zimmerman (Universität des Saarlandes, Germany) for their contributions of reagents, plasmids, cell lines and antibodies. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/123/15/2672/DC1
References
- Appenzeller-Herzog C., Riemer J., Christensen B., Sorensen E. S., Ellgaard L. (2008). A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 27, 2977-2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K. M., Chakravarthi S., Langton K. P., Sheppard A. M., Lu H., Bulleid N. J. (2008). Low reduction potential of Ero1alpha regulatory disulphides ensures tight control of substrate oxidation. EMBO J. 27, 2988-2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry M. A., Reynolds J. E., Eastman A. (1993). Etoposide-induced apoptosis in human HL-60 cells is associated with intracellular acidification. Cancer Res. 53, 2349-2357 [PubMed] [Google Scholar]
- Chakravarthi S., Bulleid N. J. (2004). Glutathione is required to regulate the formation of native disulfide bonds within proteins entering the secretory pathway. J. Biol. Chem. 279, 39872-39879 [DOI] [PubMed] [Google Scholar]
- Ellgaard L. (2004). Catalysis of disulphide bond formation in the endoplasmic reticulum. Biochem. Soc. Trans. 32, 663-667 [DOI] [PubMed] [Google Scholar]
- Ellgaard L., Ruddock L. W. (2005). The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frand A. R., Kaiser C. A. (1999). Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Mol. Cell 4, 469-477 [DOI] [PubMed] [Google Scholar]
- Gross E., Sevier C. S., Heldman N., Vitu E., Bentzur M., Kaiser C. A., Thorpe C., Fass D. (2006). Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc. Natl. Acad. Sci. USA 103, 299-304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619-633 [DOI] [PubMed] [Google Scholar]
- Haynes C. M., Titus E. A., Cooper A. A. (2004). Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15, 767-776 [DOI] [PubMed] [Google Scholar]
- Iuchi Y., Okada F., Tsunoda S., Kibe N., Shirasawa N., Ikawa M., Okabe M., Ikeda Y., Fujii J. (2009). Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 419, 149-158 [DOI] [PubMed] [Google Scholar]
- Jeong W., Park S. J., Chang T. S., Lee D. Y., Rhee S. G. (2006). Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J. Biol. Chem. 281, 14400-14407 [DOI] [PubMed] [Google Scholar]
- Jessop C. E., Bulleid N. J. (2004). Glutathione directly reduces an oxidoreductase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 279, 55341-55347 [DOI] [PubMed] [Google Scholar]
- Karala A. R., Lappi A. K., Saaranen M. J., Ruddock L. W. (2009). Efficient peroxide-mediated oxidative refolding of a protein at physiological pH and implications for oxidative folding in the endoplasmic reticulum. Antioxid. Redox. Signal. 11, 963-970 [DOI] [PubMed] [Google Scholar]
- Kim J. A., Park S., Kim K., Rhee S. G., Kang S. W. (2005). Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Anal. Biochem. 338, 216-223 [DOI] [PubMed] [Google Scholar]
- Le Moan N., Clement G., Le Maout S., Tacnet F., Toledano M. B. (2006). The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J. Biol. Chem. 281, 10420-10430 [DOI] [PubMed] [Google Scholar]
- Malhotra J. D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S. W., Kaufman R. J. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 105, 18525-18530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., Ron D. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066-3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merksamer P. I., Trusina A., Papa F. R. (2008). Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell 135, 933-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezghrani A., Fassio A., Benham A., Simmen T., Braakman I., Sitia R. (2001). Manipulation of oxidative protein folding and PDI redox state in mammalian cells. EMBO J. 20, 6288-6296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., Winterbourn C. C. (2007). The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885-11892 [DOI] [PubMed] [Google Scholar]
- Phalen T. J., Weirather K., Deming P. B., Anathy V., Howe A. K., van der Vliet A., Jonsson T. J., Poole L. B., Heintz N. H. (2006). Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 175, 779-789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raykhel I., Alanen H., Salo K., Jurvansuu J., Nguyen V. D., Latva-Ranta M., Ruddock L. (2007). A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 179, 1193-1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer F. Q., Buettner G. R. (2001). Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 30, 1191-1212 [DOI] [PubMed] [Google Scholar]
- Schroder M., Kaufman R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739-789 [DOI] [PubMed] [Google Scholar]
- Sevier C. S., Qu H., Heldman N., Gross E., Fass D., Kaiser C. A. (2007). Modulation of cellular disulfide-bond formation and the ER redox environment by feedback regulation of Ero1. Cell 129, 333-344 [DOI] [PubMed] [Google Scholar]
- Tavender T. J., Sheppard A. M., Bulleid N. J. (2008). Peroxiredoxin IV is an endoplasmic reticulum-localized enzyme forming oligomeric complexes in human cells. Biochem. J. 411, 191-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M., Leibfritz D., Moncol J., Cronin M. T., Mazur M., Telser J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44-84 [DOI] [PubMed] [Google Scholar]
- van Anken E., Romijn E. P., Maggioni C., Mezghrani A., Sitia R., Braakman I., Heck A. J. (2003). Sequential waves of functionally related proteins are expressed when B cells prepare for antibody secretion. Immunity 18, 243-253 [DOI] [PubMed] [Google Scholar]
- Wilson R., Allen A. J., Oliver J., Brookman J. L., High S., Bulleid N. J. (1995). The translocation, folding, assembly and redox-dependent degradation of secretory and membrane proteins in semi-permeabilized mammalian cells. Biochem. J. 307, 679-687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn C. C., Metodiewa D. (1999). Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322-328 [DOI] [PubMed] [Google Scholar]
- Woo H. A., Chae H. Z., Hwang S. C., Yang K. S., Kang S. W., Kim K., Rhee S. G. (2003). Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science 300, 653-656 [DOI] [PubMed] [Google Scholar]
- Wood Z. A., Poole L. B., Karplus P. A. (2003a). Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650-653 [DOI] [PubMed] [Google Scholar]
- Wood Z. A., Schroder E., Robin Harris J., Poole L. B. (2003b). Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 28, 32-40 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}