

Summary
Caulobacter crescentus attachment is mediated by the holdfast, a complex of polysaccharide anchored to the cell by HfaA, HfaB and HfaD. We show that all three proteins are surface-exposed outer membrane (OM) proteins. HfaA is similar to fimbrial proteins and assembles into a high molecular weight (HMW) form requiring HfaD, but not holdfast polysaccharide. The HfaD HMW form is dependent on HfaA but not on holdfast polysaccharide. We show that HfaA and HfaD form homomultimers and that they require HfaB for stability and OM translocation. All three proteins localize to the late predivisional flagellar pole, remain at this pole in swarmer cells, and localize at the stalk tip after the stalk is synthesized at the same pole. Hfa protein localization requires the holdfast polysaccharide secretion proteins and the polar localization factor PodJ. A hfaB mutant is much more severely deficient in adherence and holdfast attachment than hfaA and hfaD mutants. A hfaA, hfaD double mutant phenocopies either single mutant, suggesting that HfaB is involved in holdfast attachment beyond secretion of HfaA and HfaD. We hypothesize HfaB secretes HfaA and HfaD across the outer membrane, and the three proteins form a complex anchoring the holdfast to the stalk.
Keywords: attachment, Caulobacter crescentus, Hfa, holdfast
Introduction
Adherence is a key step for bacteria to establish interactions with a variety of biotic and abiotic surfaces and marks the initiation of the infection process. Once initial attachment occurs, many bacteria go on to form biofilms, which are multicellular communities embedded within a matrix that adopt a variety of structures, thereby offering protection from environmental changes from antibiotics (Stoodley 2002). Many cell surface structures play important roles in the initiation and maintenance of adherence and biofilm formation, including flagella, pili, curli, and polysaccharides (Bodenmiller et al., 2004, Van Houdt & Michiels, 2005, Stoodley et al., 2002). For example, Staphylococcus sp. and Escherichia coli produce several polysaccharides important for biofilm growth (Van Houdt & Michiels, 2005). A polysaccharide adhesin made entirely of N acetylglucosamine (NAG) was identified in Staphylococcus sp. (PIA/PNAG)(Mack et al., 1994, Mack et al., 1996, Cramton et al., 1999) and was also identified in various other bacterial species (Lasa, 2006). The E. coli pga locus encodes proteins that produce a β-1, 6 linked NAG polysaccharide required for biofilm formation (Wang et al., 2004). In addition, β-1, 4 linked NAG is a polysaccharide component of the C. crescentus polar holdfast adhesin as shown by wheat germ agglutinin (WGA) binding (Merker & Smit, 1988). The exact composition and structure of the holdfast is unknown, but in addition to NAG it contains the Hfa proteins that are the subject of this paper. C. crescentus is a ubiquitous aquatic Gram-negative bacterium with a dimorphic lifecycle producing both motile swarmer cells and sessile stalked cells at every cell division. Caulobacter sp. form biofilms both in vitro and in the environment (Corpe, 1972, Entcheva-Dimitrov & Spormann, 2004, Smit et al., 2000) and the holdfast is essential for biofilm formation (Corpe, 1972, Entcheva-Dimitrov & Spormann, 2004, Smit et al., 2000, Merker & Smit, 1988).
Adhesion is a developmentally regulated process in C. crescentus; motility, flagella and pili of the swarmer cell are all important for the initial stages of adherence to surfaces (Bodenmiller et al., 2004, Levi & Jenal, 2006). Strong permanent attachment of C. crescentus to surfaces requires the holdfast, which is localized to the tip of the stalk, a polar cell envelope extension (Smith et al., 2003, Bodenmiller et al., 2004, Cole et al., 2003, Mitchell & Smit, 1990). C. crescentus cells can also adhere to each other via their holdfasts in groups of 2 to more than 100 cells, called a rosette (Poindexter, 1964). Digestion of the holdfast with lysozyme, which cleaves oligomers of β-1, 4 linked NAG, weakens the force of adhesion but cells remain surface associated suggesting that additional components of the holdfast are needed for surface attachment (Li et al., 2005, Tsang et al., 2006). Two major genetic loci have been identified that are directly associated with the function and biosynthesis of holdfast polysaccharide, the hfs and hfa loci. The hfs, or holdfast synthesis, locus encodes proteins involved in the biosynthesis and secretion of the holdfast polysaccharide (Smith et al., 2003, Toh et al., 2008). HfsE, HfsG, and HfsH are predicted to synthesize the holdfast polysaccharide repeat units, HfsF is predicted to translocate them across the inner membrane, and the paralogs HfsC and HfsI are thought to polymerize them in the periplasm (Toh et al., 2008). HfsD, HfsA and HfsB have amino acid similarity to the type I capsule translocation machinery of E. coli and are predicted to secrete holdfast polysaccharide (Whitfield & Naismith, 2008, Whitfield & Paiment, 2003, Smith et al., 2003).
The hfa locus is important for attaching the holdfast polysaccharide to C. crescentus cells (Cole et al., 2003, Mitchell & Smit, 1990, Kurtz & Smit, 1992, Kurtz & Smit, 1994). The hfa locus is a single operon comprised of hfaA, hfaB and hfaD that is cell cycle regulated by CtrA (Laub et al., 2002, Laub et al., 2000, Janakiraman & Brun, 1999). Insertion mutations within the hfa genes result in varying degrees of decreased adherence and holdfast shedding from the cell body (Ong et al., 1990, Kurtz & Smit, 1992, Cole et al., 2003). Earlier characterization of the hfa locus included hfaC, encoding an ABC-transporter-like protein (Kurtz & Smit, 1994), but analysis of insertion mutants and experiments presented in this paper demonstrate that HfaC is not required for holdfast anchoring (Cole et al., 2003).
Both HfaB and HfaD are associated with the membrane fraction of the cell and are enriched in purified stalks, consistent with a localized anchor function (Cole et al., 2003). HfaB is a lipoprotein with 42% similarity and 31% identity over a 143 amino acid region to CsgG (Cole et al., 2003), which is required for secretion of curlin monomers (CsgA) across the outer membrane (OM) (Robinson et al., 2006). Curli are E. coli fimbrial structures important for attachment and biofilm formation (Kikuchi et al., 2005). HfaA is similar to fimbrial family proteins and HfaD has weak similarity to a variety of adhesins and other surface proteins (Kurtz & Smit, 1994, Cole et al., 2003).
In this paper, we show that hfaA, hfaB and hfaD play distinct roles in holdfast attachment to the cell. We demonstrate that HfaA is part of a high molecular weight (HMW) complex that is resistant to heat and SDS but sensitive to formic acid, a property shared with amyloid proteins such as CsgA. The ability of HfaA to form HMW complexes is dependent on HfaD, but not the presence of holdfast polysaccharide. HfaD also forms a HMW complex that is not dependent on holdfast or HfaA, but overall stability of HfaD requires HfaA. We establish that HfaA, HfaB and HfaD are associated with the OM and are polarly localized; this polar localization is dependent on podJ and hfsDAB. Additionally, HfaB is important for the stability and proper OM targeting of HfaA and HfaD suggesting a role for HfaB in secretion or as a chaperone for HfaA and HfaD. Yeast two-hybrid (Y2H) and co-immunoprecipitation (CoIP) analyses demonstrate HfaA-HfaA and HfaD-HfaD interactions. Taken together, these data support a model where HfaB secretes HfaA and HfaD at the cell pole where they form a multiprotein structure involved in anchoring the holdfast.
Results
Deletion of hfaA, hfaB or hfaD causes holdfast shedding and a decrease of holdfast function
Plasmid insertions in each of the hfa genes were previously used to determine that HfaA, HfaB and HfaD, but not HfaC, were important for holdfast anchoring to the cell (Cole et al., 2003). Because insertion mutations can have polar or dominant negative effects, we made in-frame, non-polar deletions of each gene to specifically examine the role of each Hfa protein in holdfast anchoring. Microscopic examination of cells and holdfasts bound to microscope cover-slips or plastic multi-well plates for 45 min was used to examine the role of hfa genes in holdfast anchoring and in the adherence of single cells. Long-term (24–72 h) attachment assays in plastic multi-well plates were used to assess the role of hfa genes in biofilm formation. Finally, WGA-FITC, which binds specifically to NAG oligomers was used to quantify the percentage of predivisional (PD) cells with holdfasts, because these cells are past the cell differentiation stage and should therefore have a holdfast.
In the cover-slip assay, deletion of hfaA resulted in a significant amount of holdfast shedding and a decrease in cell adherence relative to the wild-type C. crescentus strain CB15 (Fig.1 panels A, B, E and F). In the short-term binding assay, the hfaA mutant had 69.5% adherence (Table 1), whereas the hfaA mutant had drastically reduced biofilm formation at all time points (Fig. 2). Only 18.6% of hfaA mutant PD cells had a holdfast as compared to 53.7% for CB15 (Table 1). Finally, the hfaA mutant had reduced rosette formation when grown in liquid cultures as compared to CB15 (Table 1). The hfaD deletion mutant essentially phenocopied the hfaA mutant for holdfast shedding (Fig. 1 panels M and N), short-term binding, lectin labeling, and rosette formation (Table 1). The hfaD mutant was strongly deficient in biofilm formation and lagged behind the hfaA mutant at all time points (Fig. 2).
Figure 1.
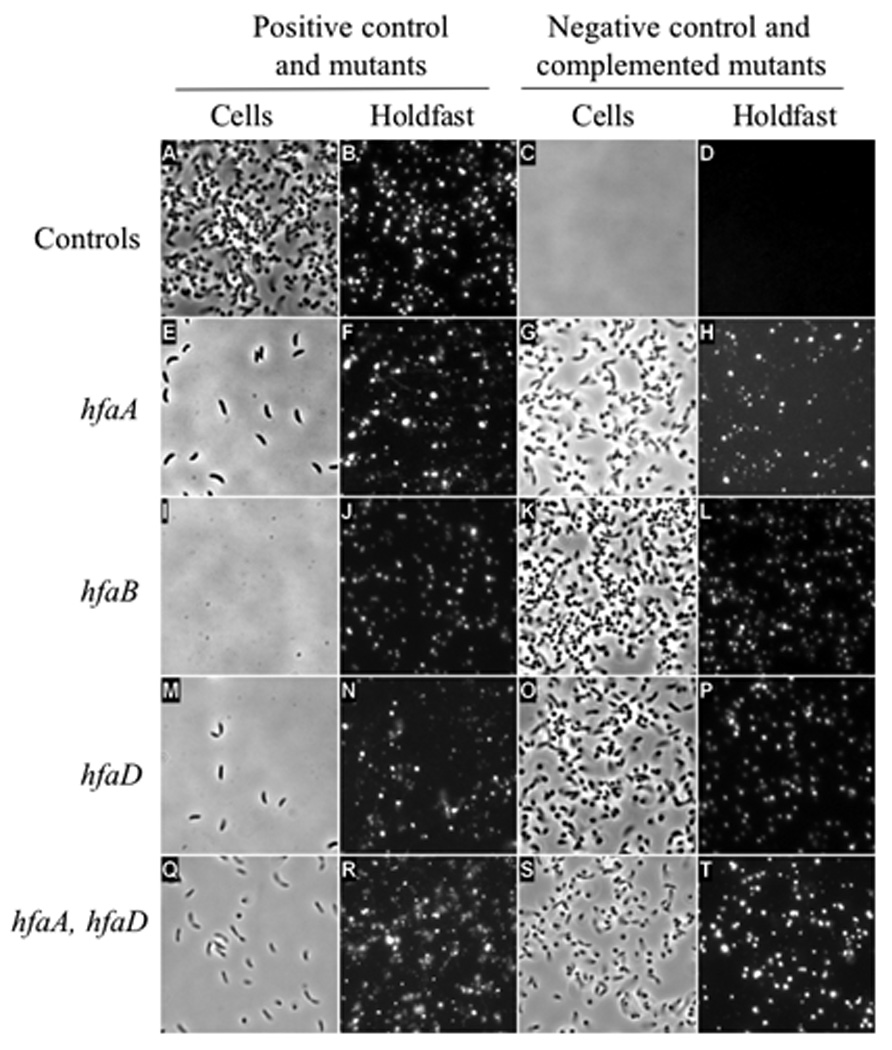
Cover-slip binding assay and lectin labeling showing holdfast shedding in hfa deletion mutants and complemented mutants. Panels A, C, E, G, I, K, M, O, Q and S are phase contrast images of cells. Panels B, D, F, H, J, L, N, P, R and T are fluorescence images of holdfast stained with lectin. All images are representative areas of the glass cover-slip that was submerged in a culture of each strain washed, stained, and imaged. (A, B) CB15/pMR20; (C, D) NA1000/pMR20; (E, F) CB15 ΔhfaA/pMR20; (G, H) CB15 ΔhfaA/pMR20hfaA; (I, J) CB15 ΔhfaB/pMR20; (K, L) CB15 ΔhfaB/pMR20hfaB; (M, N) CB15 ΔhfaD/pMR20; (O, P) CB15 ΔhfaD/pMR20hfaD; (Q, R) CB15 ΔhfaA, ΔhfaD/pMR20; (S, T) CB15 ΔhfaA, ΔhfaD/pMR20hfaA,hfaD.
TABLE 1.
Presence of holdfast and adherence of hfa deletion mutants.
| Strain | Lectin Bindinga (mean±SE) |
Short term Bindingb (mean±SE) |
Rosette formationc (Percent/total cells) |
|---|---|---|---|
| CB15/pMR20 | 53.7±4.4 | 100.0±1.9 | 61.5/326 |
| NA1000/pMR20 | 0.7±0.5 | 3.1±0.0 | 0.0/449 |
| CB15 ΔhfaA/pMR20 | 18.6±2.6 | 69.5±1.6 | 38.3/264 |
| CB15 ΔhfaB/pMR20 | 2.8±1.0 | 34.6±7.4 | 0.8/259 |
| CB15 ΔhfaD/pMR20 | 17.8±3.3 | 67.6±1.6 | 33.1/414 |
| CB15 ΔhfaA ΔhfaD/pMR20 | 19.3±7.9 | 57.5±5.7 | 27.6/298 |
| CB15 ΔhfaA/pMR20Phfa-hfaA | 69.2±4.9 | 89.6±2.4 | ND |
| CB15 ΔhfaB/(pMR20Phfa-hfaB) | 66.6±11.5 | 90.5±4.0 | ND |
| CB15 ΔhfaD/(pMR20Phfa-hfaD) | 77.5±10.9 | 85.4±3.2 | ND |
| CB15 ΔhfaAΔhfaD/pMR20Phfa-hfaA, hfaD | 66.9±13.1 | 89.7±6.0 | ND |
Percent of PD cells with polar WGA-lectin binding
Cell adherence to polystyrene measured by crystal violet assay after 45 min.
Percentage of the total cells counted that are bound in rosettes
Figure 2.

Biofilm formation of hfa mutants. (A) Graph of relative biofilm formation for 24 h, 48 h, and 72 h. The assay was performed in triplicate from three independent cultures grown on different days. The mean±SE is given at each timepoint (B) Coverslips from a representative assay at the 24 h timepoint stained with crystal violet.
Deletion of hfaB had the most severe phenotype of the hfa deletion mutants. Very few cells of the hfaB mutant remained bound to the surface in coverslip assays and it had the most severe holdfast shedding phenotype (Fig. 1 panels I and J). The hfaB mutant had only 34.6% binding when examined with the short-term binding assay (Table 1) and biofilm formation was similar to the negative control NA1000 (Fig. 2). Finally, only 2.8% of the PD cells had WGA-labeled holdfast (Table 1). The hfaB mutant rarely made rosettes (Table 1). Each hfa deletion mutant was complemented with the deleted gene expressed from the hfa promoter on the low-copy replicating plasmid pMR20 to levels comparable to CB15 (Fig. 1. compare panels G, H, K, O, P, S and T to panels A and B and Table 1).
Deletion of hfaC produced in a wild-type holdfast phenotype. There was no difference between the hfaC mutant and CB15 in holdfast synthesis, short-term surface binding and biofilm formation (Fig. S1)
Together, these data indicate that the hfaA, hfaB and hfaD genes are important for proper holdfast anchoring and surface attachment of C. crescentus and confirm that hfaC plays no role adhesion.
Hfa proteins are localized to the OM
HfaB and HfaD are found in the total membrane fraction of the cell (Cole et al., 2003). We used fractionation experiments to determine the envelope fraction to which Hfa proteins localize. CB15 ΔrsaA was used for the isolation of OMs as the separation of the inner membrane (IM) and OM is facilitated by the absence of the surface array protein, RsaA (Kurtz, 2004). We separated sarkosyl-insoluble OMs and sarkosyl-soluble IMs from CB15 ΔrsaA expressing each of the Hfa proteins as functional translational fusions (Fig. S2 and Table S1) to a C-terminal M2 tag. HfaA, HfaB and HfaD were found exclusively in the OM (Fig. 3). To confirm IM and OM separation, we assayed for proteins known to be associated with each membrane. FlgH, a component of the flagellar outer ring (Jenal et al., 1994), was found exclusively in the OM (Fig. 3) and McpA, methyl-accepting chemotaxis protein A (Krikos et al., 1983, Alley et al., 1992), fractionated only with the IM, as expected (Fig. 3). These data indicate that the HfaA, HfaB and HfaD reside in the OM of the cell.
Figure 3.
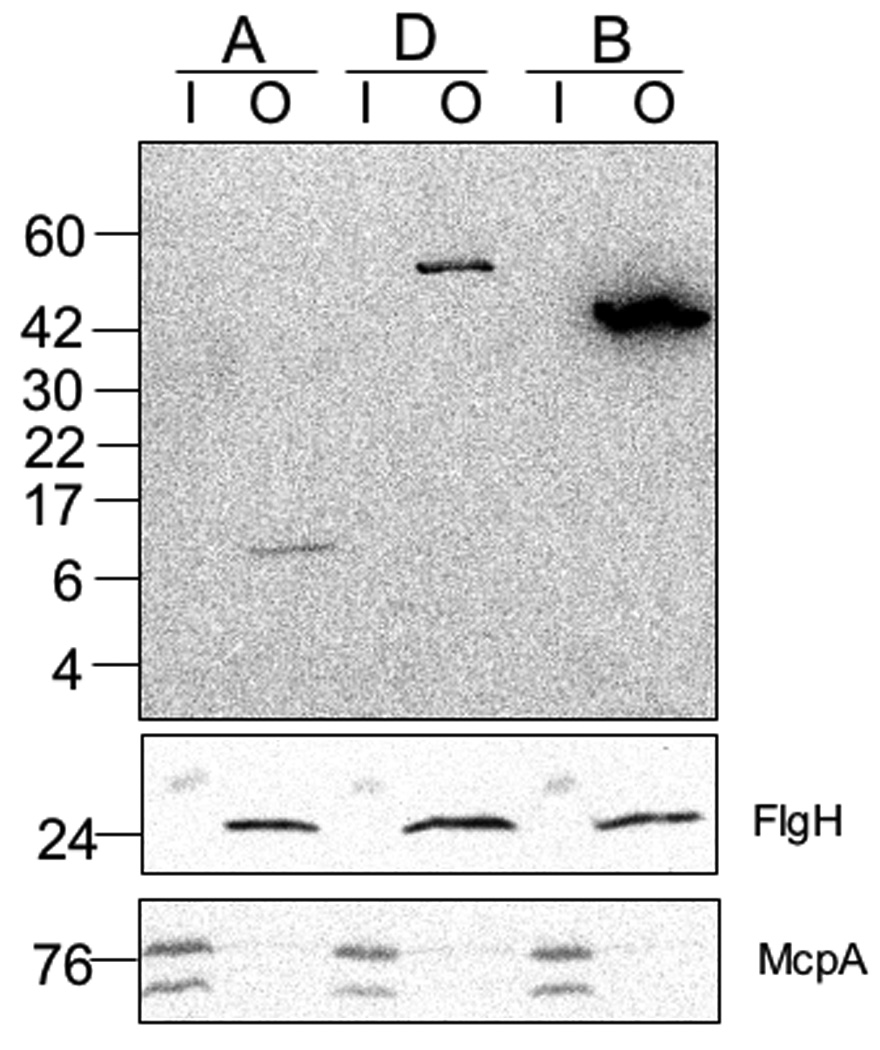
Hfa proteins reside in the OM. Samples are as follows: A) HfaA-M2 (CB15 ΔrsaA hfaA::pJM21hfaA); D) HfaD-M2 (CB15 ΔrsaA hfaD::pJM23hfaD); B) HfaB-M2 (CB15 ΔrsaA hfaB::pJM21hfaB). There are 30 µg per lane of total protein derived from either inner or OMs for HfaB and HfaD and 60 µg per lane of total protein for HfaA samples prior to formic acid treatment. IM proteins (I) and OM proteins (O). Western Blot of HfaA-M2, HfaB-M2 and HfaD-M2 containing IMs and OMs probed with α-M2-HRP. Western Blot of McpA and FlgH control proteins in IMs and OMs were probed with α-McpA or α-FlgH. For the McpA and FlgH control Western 20 µg of protein per lane was loaded.
The Hfa proteins are polarly localized
Because HfaA and HfaD are important for holdfast anchoring, we determined whether they colocalize with the holdfast. The Hfa proteins were detected in cells using immunofluorescence microscopy (IFM) of functional M2 fusions in CB15. HfaA-M2 and HfaD-M2 exhibited a low frequency of random localization and both were polarly localized in the vast majority of cells in which they could be detected (Table 2). HfaA-M2 localized at a pole in 17.3% of cells and HfaD-M2 in 14.3% (Table 2A). We analyzed localization in cells with visible stalks and PD cells, which have all proceeded past the initiation of stalk synthesis. HfaA and HfaD localized to the stalk tip in 80% and 84.2% of stalked and PD cells with a detectable signal (Fig. 4, panels F and H, Table 2A). HfaA and HfaD localized to the flagellar pole before completion of cell division in 20% and 15.8% of PD cells, respectively (Fig. 4, panels F and H and Table 2A). A CB15 negative control with no M2 epitope tag exhibited weak and diffuse M2 antibody staining (Fig. 4, panel B). Flagellar pole localization of HfaA and HfaD in PD cells was mostly observed in deeply constricted cells, suggesting that this localization occurs at the end of the cell cycle, consistent with the timing of hfaABD transcription (Janakiraman & Brun, 1999). Therefore, we hypothesize that HfaA and HfaD first localize at the flagellar pole of the PD cell where they are first expressed (Janakiraman & Brun, 1999), are retained at that pole in swarmer cells, and persist at that pole as it develops into the stalk pole during cell differentiation (Fig. 4I). This model is consistent with the localization of HfaBmCherry described below.
TABLE 2.
Localization of HfaA and HfaD in permeabilized and unpermeabilized cells.
| All cellsa | Predivisionalb | ||||
|---|---|---|---|---|---|
| Treatment | Protein (cells counted) | HfaA (915) | HfaD (1221) |
HfaA (273) | HfaD (428) |
| A) Permeabilizedc | Polard | 17.3% | 14.3% | 20.4% | 22.2% |
| Stalked polee | - | - | 80% | 84.2% | |
| Flagellar polee | - | - | 20% | 15.8% | |
| Non-polar | 4.8% | 3.1% | - | - | |
| Protein (cells counted) | HfaA (937) | HfaD (830) | HfaA (311) | HfaD (252) | |
| B) Unpermeabilizedf | Polard | 31.5% | 27.8% | 34% | 29.8% |
| Stalked polee | - | - | 82.2% | 82.6% | |
| Flagellar polee | - | - | 17.8% | 17.3% | |
| Non-polar | 0.65% | 1.7% | - | - | |
Percentage determined as proportion of all cells.
Percentage determined as proportion of predivisional cells only.
Permeabilized cells were fixed with 2.5% formaldehyde and then permeabilized with Triton X-100, EDTA and lysozyme.
Percentage of either all cells or just predivisional cells with polar localization
Percentage of stalked and flagellar pole localization is determined from the percentage of PD cells that have polar localization.
Unpermeabilized cells were fixed only with 2.5%.
Figure 4.
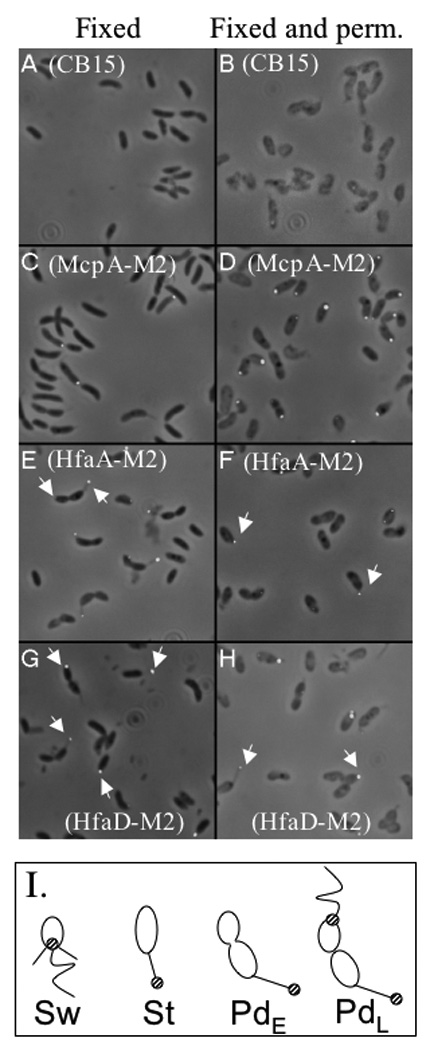
Localization of HfaA-M2 and HfaD-M2 using immunofluorescence microscopy. Panels A, C, E, and G fixed cells. Panels B, D, F, and H fixed and permeabilized cells. Panels A and B, CB15; Panels C and D, NA1000 mcpA::pRCM22; Panels E and F, CB15 hfaA::pJM23hfaA; Panels G and H, CB15 hfaD::pJM23hfaD. White arrows in panels E thru H indicate polar localization for HfaA and HfaD in swarmer, PD and stalked cells. (I) Cartoon of C. crescentus indicating the localization of HfaA and HfaD (striped circle) in swarmer (Sw), stalked (St), early PD (PdE) and late-PD (PdL) cells.
We were unable to detect HfaB by IFM using M2 fusions in either permeabilized or unpermeabilized cells. This could result from the M2 tag being inaccessible to the antibody as it is likely buried within the folded protein or the membrane. As an alternative, we used an HfaB C-terminal mCherry fusion and found that HfaBmCherry had a localization pattern similar to HfaA and HfaD (Fig. S2C, 5A panel 1, and Table 3). We used time-lapse microscopy to determine the localization of HfaBmCherry during the cell cycle using (Fig. 5B and movie S1). HfaBmCherry was first seen at the flagellar pole of late PD cells (Fig. 5B, panel 1). After cell division, HfaBmCherry remained at the flagellar pole throughout the swarmer to stalked cell transition (Fig. 5B, panels 2 and 3). During stalk formation HfaBmCherry was pushed out with the stalk tip and then remained at the tip of the stalk in early PD cells (Fig. 5B, panel 4). During the late PD stage, new HfaBmCherry appeared at the flagellar pole and HfaBmCherry was localized to both the stalk tip and the flagellar pole (Fig. 5B, panels 5, and 6 and Table 3).
Figure 5.
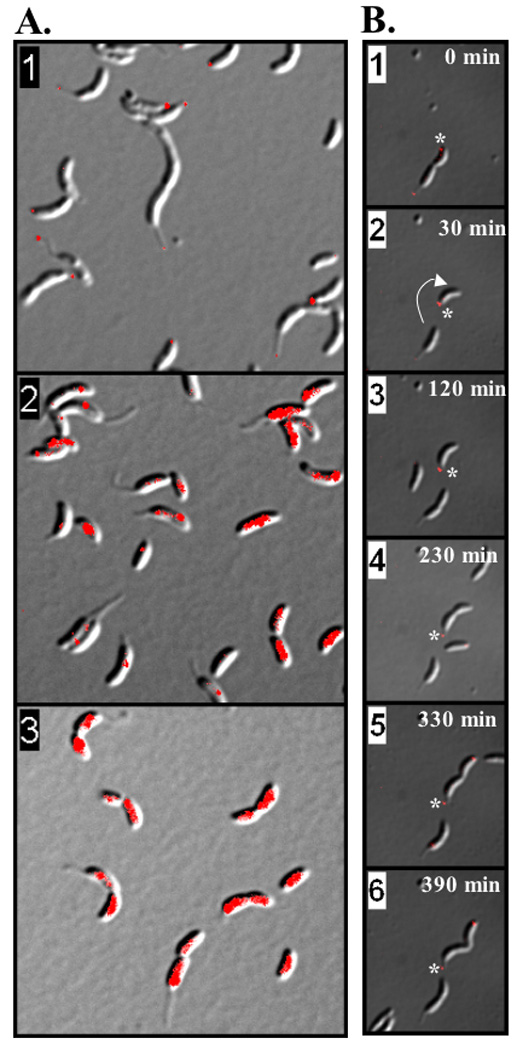
HfaB is polarly localized and its localization is dependent on both PodJ and HfsDAB. (A) Localization of HfaBmCherry using differential interference contrast (DIC) and epifluorescence microscopy. White arrows indicate polar localization of HfaB in PD, stalked and swarmer cells. Panel 1 is CB15 hfaB::pCHYChfaAB; Panel 2, CB15 ΔpodJ hfaB::pCHYChfaAB; Panel 3, CB15 ΔhfsDAB hfaB::pCHYChfaAB. (B) Cell cycle localization of HfaBmCherry. Each frame is a single time point within the time-lapse experiment. Time stamps are shown for each frame. The white asterisks marks the swarmer pole of a PD cell that releases the new swarmer cell that is followed through the cell cycle progression. The white arrow shows the 180° rotation of the swarmer cell just after cell division. The time-lapse movie from which these frames were taken can be found in supplementary information (Movie S1)
TABLE 3.
Localization of HfaBmCherry.
| Localization Pattern | Cell type (cells counted) | |
|---|---|---|
| Percent of All cells (979) | Percent of Predivisional Cells (306) | |
| Polara | 38.2% | 44.4% |
| Stalk Tipb | - | 98.5%c |
| Flagellar Poleb | - | 1.5%c |
| Non-Polara | 5.5% | 2.6% |
Percentage determined as proportion of all cells.
Percentage determined as proportion of PD cells only.
Percentage of stalked and flagellar pole localization is determined from the percentage of PD cells that have polar localization.
Our combined fluorescence microscopy results suggest that the Hfa proteins localize to the new flagellar pole late in the predivisional stage as they are expressed in the swarmer cell compartment; in the subsequent cell cycle remain localized at this pole, which then develops into the new stalked pole (Fig. 4, panel I and 5B).
Polar localization of Hfa proteins requires PodJ and the HfsDAB holdfast polysaccharide translocation apparatus
Numerous regulators of polar development have been identified in Caulobacter and some of them are required for holdfast synthesis (Brown et al., 2009). The PodJ protein is required for optimal motility in swarm agar, pili synthesis, holdfast synthesis, flagellum ejection, and serves as a localization factor at the flagellar pole of the predivisional cell for a number of regulatory proteins (Crymes et al., 1999, Hinz et al., 2003, Viollier et al., 2002b, Viollier et al., 2002a, Lawler et al., 2006). Since Hfa proteins first localize at the flagellar pole of predivisional cells (Fig. 5A and 5B, panel 1), their localization was examined in a ΔpodJ mutant. HfaBmCherry did not localize to the pole, but was found dispersed around the cell in the ΔpodJ mutant (Fig. 5A, panel 3), indicating that the polar localization of HfaB requires PodJ. IFM localization of HfaA and HfaD also showed a marked reduction of polar localization in the podJ mutant (Fig. S4). Because the biosynthesis and secretion of holdfast polysaccharide and creation of the anchor are likely to be coordinated, we examined localization of the Hfa proteins in an hfsDAB mutant. HfsDAB is the putative holdfast polysaccharide translocation apparatus and deletion of their genes abolishes holdfast synthesis (Smith et al., 2003). HfaBmCherry was delocalized in the hfsDAB mutant (Fig. 5A, panel 2). IFM localization of HfaA and HfaD also indicated a marked reduction of polar localization of these proteins in the hfsDAB mutant (Fig. S4). HfaBmCherry was present at similar levels in CB15 and in the hfsDAB and podJ mutants (Fig. S3A). HfaA and HfaD levels were decreased both podJ and hfsDAB mutants (Fig. S5) so the lack of HfaA and HfaD polar localization in the hfsDAB and podJ mutants may be a reflection of their instability. Localization of HfaBmCherry was not affected in an hfaA mutant (Fig. S3C) and HfaBmCherry had a similar abundance in both CB15 and the hfaA mutant (Fig. S3B).
HfaA has sequence and biochemical properties of amyloid proteins
In previous studies, we were unable to detect HfaA by SDS-PAGE or Western Blot using standard conditions (Cole et al., 2003). The similarity of HfaB to CsgG led us to investigate the possibility that HfaA could be an amyloid protein. HfaA has similarity to fimbrial family proteins based on PROPSEARCH, including E. coli curlin CsgA, Salmonella enteritidis fimbrial protein precursor SEF14, E. coli CsgB, Pisolithus tinctorius class I hydrophobin, E. coli F fimbrial precursor, Salmonella typhimurium CsgB, Klebsiella pneumoniae fimbrial subunit type I precursor (Fig. S6), but HfaA does not have any notable repeat domains similar to those of CsgA.
An alignment of HfaA sequences from the prosthecate bacteria using CLUSTAL W (Thompson et al., 1994, Larkin et al., 2007) indicated a low level of overall sequence conservation with two highly conserved regions of 18 and 33 amino acids. Because it is often difficult to identify amyloid proteins by their primary amino acid sequence, we analyzed CB15 HfaA using two different programs, AGGRESCAN and TANGO, which identify amino acid sequences important for promoting protein aggregation or amyloid fiber formation (Conchillo-Sole et al., 2007) (Fernandez-Escamilla et al., 2004). Both programs identified three common aggregation domains in HfaA and all three sequences are within the two highly conserved regions of HfaA (Fig. S7). AGGRESCAN identified a fourth possible aggregation domain that is conserved among the Caulobacter and Asticcacaulis genera but not in Brevundimonas diminuta, Maricaulis maris, and Oceanocaulis alexandrii. In addition, we examined the secondary structure of HfaA using PSIPRED (Jones, 1999). HfaA is predominantly composed of predicted β-strands with the exception of the predicted signal sequence, which is consistent with other amyloid proteins (Figs S8).
Curlins and hydrophobins have properties of amyloid proteins, including heat- and SDS-resistance (Wang et al., 2007, Collinson et al., 1999). CsgA and other fimbrial adhesins require treatment with formic acid or hexafluoroisopropanol (HFIP) to completely depolymerize the protein (Collinson et al., 1991, St. Geme & Cutter, 2000). To determine if HfaA has similar properties, we performed Western Blot analysis of OM fractions containing a functional HfaA-M2 fusion expressed in CB15 (Fig. S2, Table S1). We compared samples treated with both heat and SDS to those treated with formic acid or HFIP and found that HfaA migrated at its predicted molecular size of 12 kDa only when treated with formic acid or HFIP. Heat/SDS-treated HfaA migrated as a high molecular weight (HMW) complex that barely entered the stacking gel (Fig. 6A). These data demonstrate that HfaA is part of a HMW complex that is resistant to heat and SDS and is solubilized by formic acid or HFIP. These results and the sequence analysis presented above suggest that HfaA may be a polymerizing amyloid-like protein.
Figure 6.
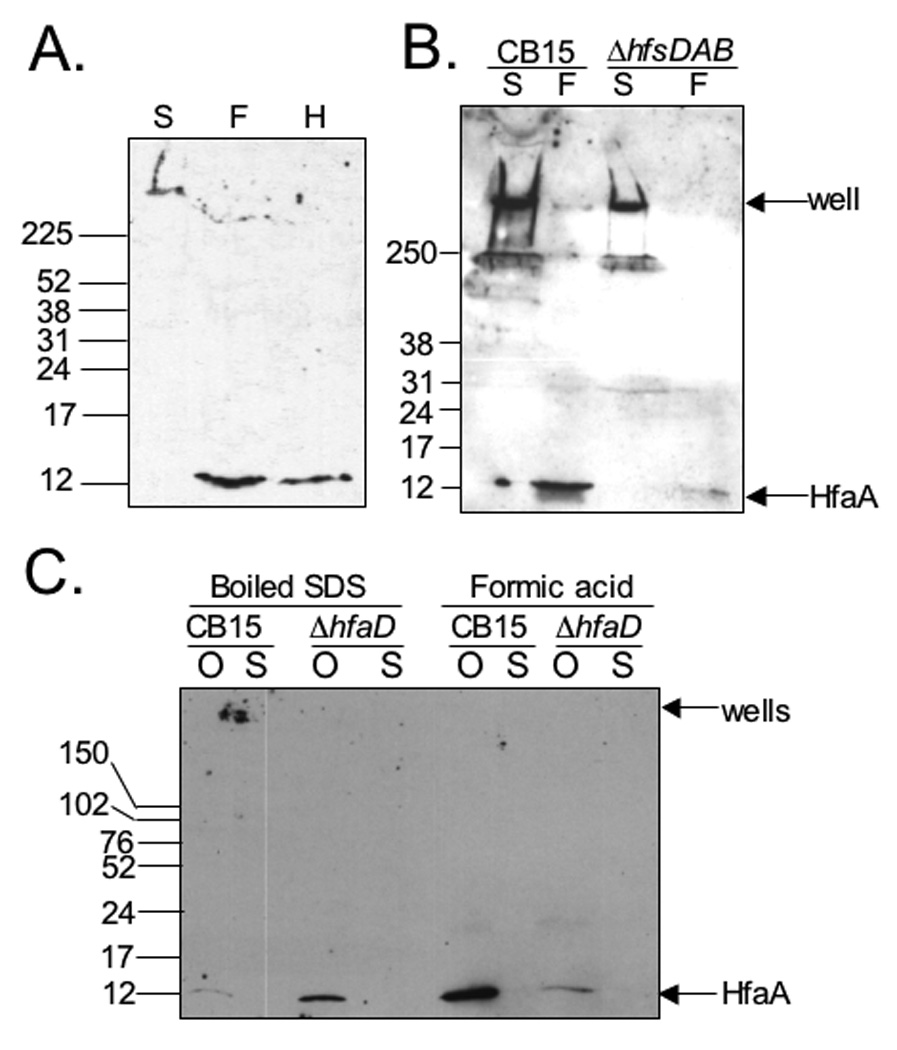
The heat and SDS resistance of HfaA is dependent on HfaD, but not HfsDAB. (A) HfaA is resistant to heat and SDS. Western Blot of OM proteins probed with M2-specific polyclonal antibody. (S) CB15hfaA::pJM23hfaA treated with heating and SDS. (F) CB15hfaA::pJM23hfaA treated with 90% formic acid, heating and SDS. (H) CB15 hfaA::pJM23hfaA treated with HFIP (B) HfaA is not dependent on the holdfast polysaccharide for heat and SDS resistance. Western Blot of OMPs probed with M2-specific antibody. Arrows indicate the wells of the gel and the HfaA monomer size of 12 kDa. CB15 hfaA::pJM23hfaAM2 and CB15 ΔhfsDAB hfaA::pJM23hfaA (S) Boiled in SDS (F) Treated with formic acid and then boiled with SDS. (C) HfaA is dependent on HfaD for heat and SDS resistance. Western blot of OM proteins probed with M2-specific antibody. Samples were either boiled in SDS or first treated with formic acid and then boiled in SDS. CB15 hfaA::pJM23hfaAM2 and CB15 ΔhfaD hfaA::pJM23hfaAM2 (O) OMP (S) culture supernatant.
HfaD, but not holdfast polysaccharide, is required for HfaA aggregate formation
To determine whether HfaA aggregate formation is dependent on the presence of holdfast polysaccharide, we examined HfaA aggregate formation in OM fractions of a ΔhfsDAB mutant. In both CB15 and CB15 ΔhfsDAB, HfaA was resistant to heat and SDS and could only be observed as a 12-kDa monomer when cells were treated with formic acid prior to boiling in SDS sample buffer indicating that the presence of the holdfast polysaccharide is not required for formation of the HMW species of HfaA (Fig. 6B). There was a decrease in the steady-state level of HfaA in the hfsDAB mutant (Fig. S5), but transcription from the hfa promoter was the same in the hfsDAB mutant and CB15 (Fig. S9).
In the curlin system, CsgB is a nucleator for CsgA fibril formation (Bian & Normark, 1997, Hammer et al., 2007). Since HfaA has properties of bacterial amyloid proteins, HfaD could serve as a nucleator for HfaA fiber formation. While HfaD is not similar to CsgB it does have amino acid similarity to other bacterial adhesins based on PROPSEARCH. To determine if HfaD has an effect on the formation of the HMW species of HfaA, HfaA-M2 was examined from OM fractions of an hfaD mutant. In contrast to wild-type cells where HfaA-M2 remained in a HMW form when treated only with heat and SDS, HfaA-M2 migrated as a 12-kDa monomer in the ΔhfaD mutant even without formic acid treatment (Fig. 6C). Although there was a decrease in the steady-state level of HfaA-M2 in the hfaD mutant, transcription from the hfa promoter in each of the hfa mutants was similar to wild-type (Fig S9). We conclude that HfaD is necessary for the formation of HfaA HMW complexes.
HfaD forms a HMW complex whose formation requires HfaA and HfaB but not holdfast polysaccharide
Since HfaD is required for the formation of HfaA HMW complexes, we asked whether HfaD also forms HMW complexes. In wild-type cells whose extracts were treated with heat and SDS, a HMW form, a 45-kDa monomeric form and a low abundance 28-kDa cleavage product of HfaD-M2 were detected (Fig. 7A, lane 1). The HfaD-M2 HMW species was eliminated when the sample was treated with formic acid or HFIP and lyophilized, with a concomitant increase in abundance of the monomeric form and 24-kDa cleavage product (Fig. 7A lanes 3 and 5). The 28-kDa cleavage product was caused by incubation at room temperature and lyophilization as shown by in samples in which formic acid and HFIP were not added (Fig 7A, lanes 2 and 4). The HMW form of HfaD was absent in hfaA and hfaB mutants (Fig 7A, lanes 6 and 7) but was present in the hfsDAB mutant (Fig 7A, lane 8). The level of HfaD-M2 was decreased similarly in the hfsDAB, hfaA and hfaB mutants, indicating that the failure of detecting the HMW form of HfaD-M2 in the hfaA and hfaB mutants was not due to low to the lower level of the protein (Fig 7A, lanes 6 to 8). Transcription from the hfa promoter was not affected in any of hfa mutants (Fig. S9). Therefore, the stability of HfaD-M2 requires HfaA and HfaB and at least one of the HfsDAB translocation proteins.
Figure 7.
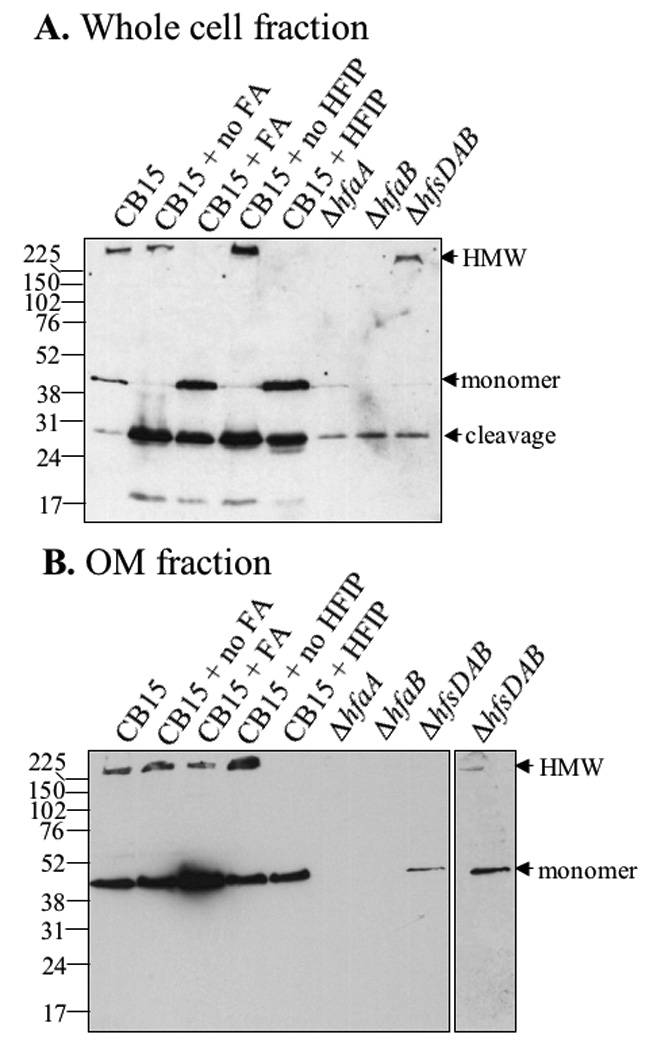
HfaD forms a HMW complex whose formation requires HfaA and HfaB, but not holdfast polysaccharide. Blots are probed with M2-specific antibody. Lane 1) CB15 HfaD-M2; 2) CB15 HfaD-M2 incubated and lyophilized, but not treated with formic acid; 3) CB15 HfaD-M2 treated with formic acid; 4) CB15 HfaD-M2 lyophilized, but not treated with HFIP; 5) CB15 HfaD-M2 treated with HFIP; 6) CB15 ΔhfaA HfaD-M2; 7) CB15 ΔhfaB HfaD-M2; 8) CB15 ΔhfsDAB HfaD-M2. A) Western Blot of whole cell extracts B) Western Blot of 10 µg OM fractions.
One possible explanation for the lack of the HMW form of HfaD-M2 and its relatively low level in hfaA and hfaB mutants is that HfaA and/or HfaB are required for the assembly of HfaD in the OM and unassembled HfaD is unstable. This model is supported by the fact that the vast majority of HfaD-M2 detected in these mutants is degraded. We therefore tested OMs for the presence of HfaD. In wild-type cells, HfaD-M2 from OMs had essentially the same behavior as from whole cells, except that the degradation products, which would not be expected to be assembled in the OM, were not detectable (Fig. 7B). Formic acid still disassembled the HMW form, although not to completion (Fig. 7B, lane 3), whereas disassembly by HFIP was complete (Fig. 7B, lane 5). HfaD was not detectable as a HMW or monomeric species in the OM fraction of either the hfaA or the hfaB mutants (Fig. 7B, lanes 6 and 7). The steady-state level of HfaD was greatly reduced in the hfsDAB mutant, but the HMW species was still present (Fig. 7B, lanes 8 and overexposure of lane 8 shown as lane 9).
These results indicate that HfaD forms a HMW species associated with the OM and that the formation of this HMW species requires HfaA and HfaB but not holdfast polysaccharide or the holdfast polysaccharide secretion machinery. They further suggest that HfaD requires HfaA and HfaB for assembly in the OM (see below).
HfaA-HfaA and HfaD-HfaD interaction
Since HfaA and HfaD form HMW complexes that depend on each other, we tested their interaction using Y2H analysis. We also tested their interaction with HfaB. HfaA interacted strongly with itself, but no significant interactions with HfaB or HfaD were found. There were no significant interactions of HfaB with itself or the other Hfa proteins. While expression of HfaB was detected in yeast (data not shown), HfaB may not interact with HfaA or HfaD in yeast because HfaB is an OM lipoprotein (Cole et al., 2003) that may not fold correctly in the yeast cytoplasm. When HfaD was expressed from the bait vector (pGBKT7) in the presence of the empty prey vector (pGADT7), there was vigorous growth on selective media (Table 4). Therefore, interactions of the HfaD bait could not be interpreted (Table 4).
TABLE 4.
Yeast two-hybrid analysis of Hfa protein interactions.a
| Vectors (Bait + Prey)b |
Weak Interaction (-His/-Leu/-Trp) |
Strong Interaction (-Ade/-His/-Leu/-Trp) |
|---|---|---|
| Control Interactions | ||
| Lam + T-antigen (negative) | − | − |
| p53 + T-antigen (positive) | + | + |
| HfaA + pGADT7 | +/− | − |
| HfaB + pGADT7 | − | − |
| HfaD + pGADT7 | + | + |
| Lam + HfaA | − | − |
| Lam + HfaB | − | − |
| Lam + HfaD | − | − |
| Hfa Protein Interaction Data (Bait + Prey) |
||
| HfaA + HfaA | + | + |
| HfaA + HfaB | +/− | − |
| HfaA + HfaD | +/− | − |
| HfaB + HfaA | − | − |
| HfaB + HfaB | − | − |
| HfaB + HfaD | − | − |
| HfaD + HfaA | + | + |
| HfaD + HfaB | + | + |
| HfaD + HfaD | + | + |
“+” indicates a strong growth similar to positive control; “+/− “ indicates less growth than the positive but more than the negative control and “− “ indicates little or no growth like the negative control.
pGBKT7 and pGADT7 are the bait and prey vectors
In vitro transcription/translation and CoIP were performed to verify protein-protein interactions detected with the Y2H system and Western blots. HfaD interacted with itself (Fig. 8A, lane 1) as compared to the non-specific binding of HfaD to the lamin C negative control bait protein (Fig. 8A, lane 3). Similarly, HfaA interacted with itself (Fig. 8B, lane 1) as compared to the negative control (Fig. 8B, lane 3). We were unable to detect a significant interaction between HfaA and HfaD using CoIP (data not shown).
Figure 8.
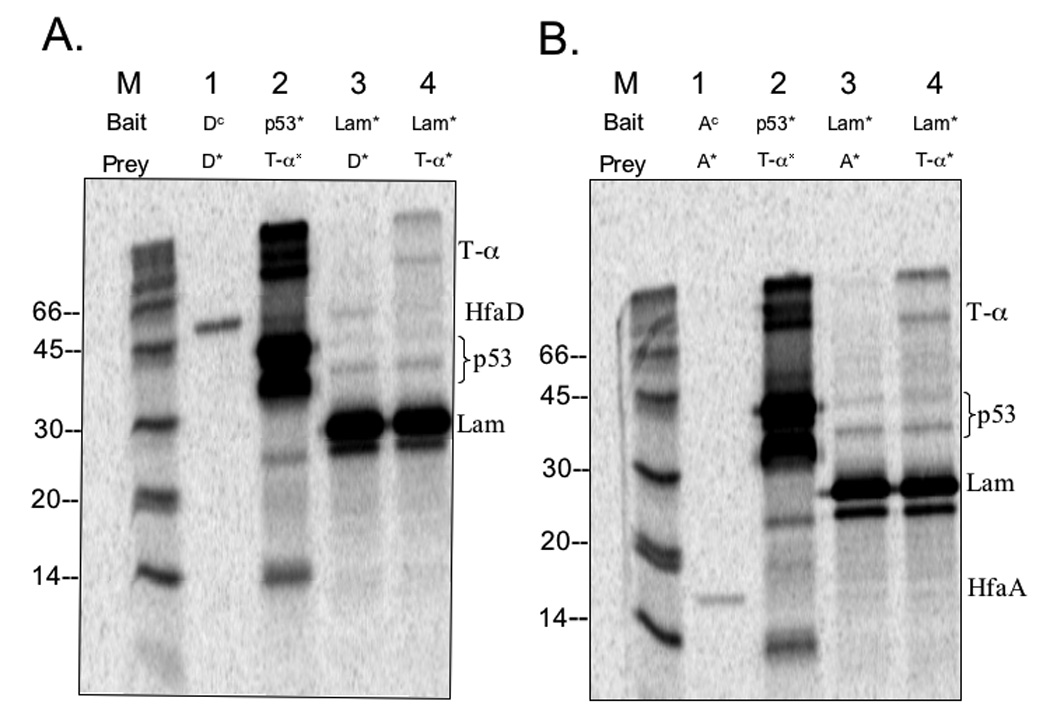
CoIP gels showing Hfa protein interactions. Autoradiograph of SDS-PAGE gel that contains CoIP reactions of proteins expressed in vitro. Immunoprecipitation of c-myc tagged bait proteins with anti-cmyc was used to pulldown any prey proteins that interacted with the bait proteins. Non-radiolabeled proteins are indicated with a superscript c (c) for “cold”; proteins with an asterisk (*) indicate 35S-methionine radiolabeled protein. The positive control for CoIP is p53* bait + T-α* prey. The negative control for CoIP is Lam* bait + T-α* prey. The background level for Hfa protein non-specific binding is established with Lam* bait + Hfa* prey. Protein molecular mass: T-α = 65-kDa; HfaD = 50-kDa; p53 = 35-kDa; Lam = 28-kDa; HfaA = 16-kDa. (A) HfaD CoIP and controls. Lane M: 14C marker; Lane 1: HfaDc bait + HfaD* prey; Lane 2: p53* bait + T-α* prey; Lane 3: Lam* bait + HfaD* prey; Lane 4: Lam* bait + T-α* prey. (B) HfaA CoIP and controls. Lane M: 14C marker; Lane 1: HfaAc bait + HfaA* prey; Lane 2: p53* bait + T-α* prey; Lane 3: Lam* bait + HfaA* prey; Lane 4: Lam* bait + T-α* prey.
The Hfa proteins are exposed on the cell surface
A role in holdfast anchoring for the Hfa proteins suggests that they may be exposed on the cell surface. To test this, we examined HfaA and HfaD localization in cells that were fixed with formaldehyde but not permeabilized by IFM. As a control, we used the polarly localized inner membrane protein McpA with an M2 tag (Alley et al., 1992, Krikos et al., 1983), which should only be detected in permeabilized cells. McpA-M2 was polarly localized in 35.9% of the total cell population in permeabilized cells, but only 4.6% in fixed and non-permeabilized cells (Fig. 4, compare panels C and D). Both HfaA and HfaD had the same localization pattern in cells that were fixed compared to cells that were fixed and permeabilized (Table 2) (Fig. 4, compare panels E and G to panels F and H). In the absence of permeabilization, a higher proportion of cells had polarly localized HfaA-M2 and HfaD-M2 (Table 2B), suggesting that the permeabilization procedure affected their conformation, stability, or the accessibility of the M2 epitope. The ability to detect HfaA and HfaD in unpermeabilized cells indicates that HfaA and HfaD are exposed on the surface of the cell, consistent with a role in holdfast polysaccharide anchoring.
We used a protease sensitivity assay to confirm the exposure of Hfa proteins on the cell surface. Whole cells expressing HfaA-M2, HfaB-M2 or HfaD-M2 were probed with 25 µg/ml proteinase K for up to 30 min. McpA was used as an inner membrane protein control for determining the integrity of the cells and the detergent Triton X-100 was used as an additional control to permeabilized the cells. HfaD was sensitive to proteinase K with or without detergent (Fig. 9A). McpA was not digested with intact whole cells, but was degraded in the presence of detergent (Fig. 9A). HfaB was partially sensitive to proteinase K in whole cells, but was completely digested in the presence of detergent (Fig. 9B). Because HfaA is expressed at very low levels, HfaA-M2 cannot be detected in whole cell extracts. Therefore, we enriched for OM proteins (OMPs) after the protease susceptibility assay. HfaA was sensitive to proteinase K both with and without detergent (Fig. 9C). The inner membrane protein control McpA was only degraded in the presence of the detergent whereas the flagellar OM protein FlgH was partially sensitive to proteinase K both with and without detergent.
Figure 9.
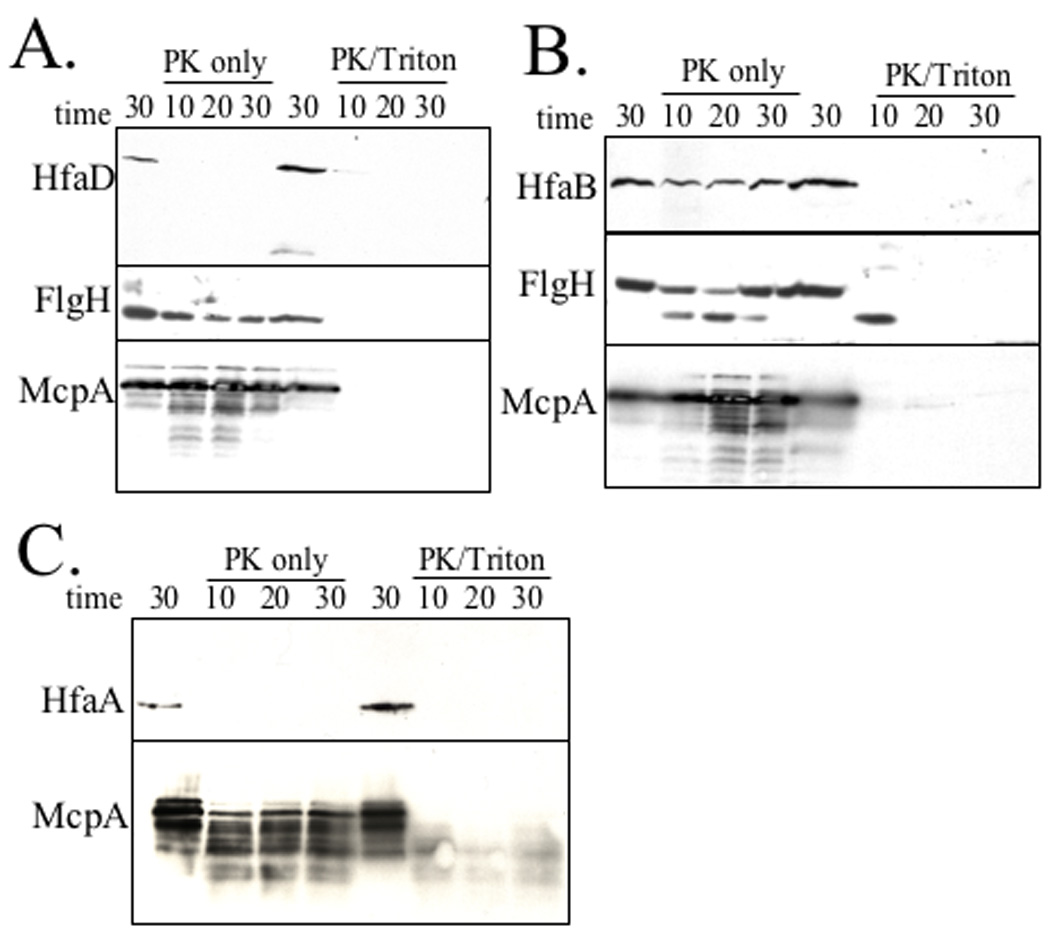
Protease susceptibility of HfaA, HfaB, and HfaD. Western blots of OM or whole cells incubated at 37°C with 25 µg/ml proteinase K (PK) for 10 min, 20 min or 30 min. McpA and FlgH were used as IMP and OMP controls, respectively. The buffer only control was incubated for 30 min. Western blots were incubated with α-M2-HRP, α-FlgH or α-McpA. Lanes 1–4 contain proteinase K only (PK only). Lanes 5–8 contain proteinase K and 0.1% Triton X-100 (PK +Triton). Lanes 1 and 5, no proteinase; Lane 2 and 6, 10 min; Lane 3 and 7, 20 min; Lane 4 and 8, 30 min. (A) All lanes contain CB15 HfaD-M2 (YB2579). (B) All lanes contain CB15 HfaB-M2 (YB2580). (C) All lanes contain CB15 HfaA-M2 (YB 2578).
These data, together with the results of IFM localization in non-permeabilized cells, indicate that Hfa proteins are surface-exposed. The partial resistance of HfaB and FlgH to proteinase K may reflect the presence of two different forms of these proteins, perhaps because of their participation in macromolecular complexes. Alternatively, HfaB and FlgH could be more resistant to protease because they are integral membrane proteins, which could limit access by the protease.
HfaB is required for the export of HfaA and HfaD to the OM
HfaB is localized in the OM and has similarity to CsgG, the E. coli OM secretion protein for the CsgA and CsgB curlin proteins (Robinson et al., 2006, Loferer et al., 1997), and has a similar predicted secondary structure to CsgG (Figs. S10 and S11). We hypothesized that HfaB may be an OM pore that secretes HfaA and HfaD. In addition, experiments presented above suggested that HfaD-M2 targeting to the OM requires HfaA and HfaB. To investigate this possibility, the effect of an hfaB deletion on the OM targeting of HfaA and HfaD was examined. HfaA-M2 and HfaD-M2 could not be detected in the periplasm or the OM of a ΔhfaB mutant, possibly due to rapid degradation of the proteins (data not shown). Therefore, HfaA-M2 and HfaD-M2 were overexpressed in the ΔhfaB mutant from a xylose inducible promoter on a high copy plasmid. When their expression was induced, HfaA-M2 and HfaD-M2 were both detected in the periplasm but not the OM of the ΔhfaB mutant (Fig. 10A and 10B), suggesting that HfaB is important for localization and/or secretion of HfaA and HfaD to the OM.
Figure 10.
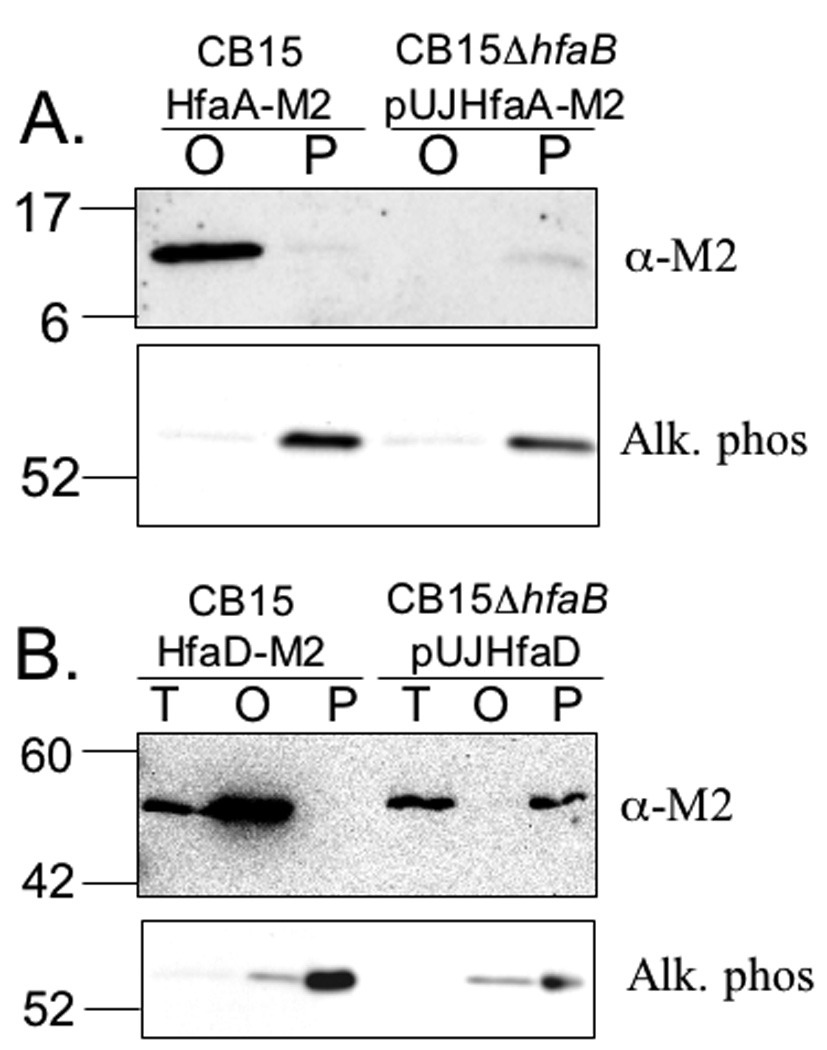
HfaB is necessary for stability and proper cellular partitioning of HfaA and HfaD. Equal amounts of OMs (O), periplasms (P) or total membranes (T) were loaded onto SDS-PAGE gels and transferred to nitrocellulose. Alkaline phosphatase was used as a marker for periplasmic proteins and a zymogram of alkaline phosphatase for each sample is shown below the Western blot in each panel. Western blots were probed with anti-M2 conjugated to HRP (A) OM and periplasmic fractions were extracted from YB2578 (CB15 HfaA-M2) and YB4270 (CB15 ΔhfaB/pUJhfaA-M2). HfaA-M2 expression was induced in YB4270 with 0.3% w/v xylose for 3 h. (B). Total membrane, OM and periplasmic fractions were extracted from YB2579 (CB15 HfaD-M2) and YB4271 (CB15 ΔhfaB/pUJhfaD-M2). HfaD-M2 expression was induced in YB4271 with 0.3% xylose w/v for 5 h.
In order to determine if the sole function of HfaB is to secrete and localize HfaA and HfaD, we analyzed the phenotype of an hfaA hfaD double mutant. The hfaA, hfaD double mutant phenocopied the single hfaA and hfaD mutants for all measured phenotypes and was significantly less deficient than the hfaB mutant (Fig. 1 and 2 and Table 1). These results indicate that HfaB performs functions important for holdfast anchoring and adherence beyond the export and localization of HfaA and HfaD. In addition, these data suggest that HfaA and HfaD contribute equally to anchoring the holdfast and are part of the same pathway.
Discussion
Polysaccharides can be anchored to the cell by different mechanisms in bacteria. In E. coli, Wzi, an OM protein, is thought to be involved in anchoring the K30 type I capsule to the cell, but the anchoring mechanism has not been elucidated (Rhan et al., 2003, Power et al., 2006). The S. pneumoniae capsular polysaccharide can either be anchored via the cell wall or a phospholipid anchor (Cartee et al., 2005, Sørensen et al., 1990). Alternatively, polysaccharides can be anchored to the cell surface by glycosylation of proteins. Neisseria meningitidis pili have O-linked glycosylation that is believed to occur via a pathway similar to wzy-dependent O-antigen synthesis (Power et al., 2006, Power et al., 2000). Camplylobacter jejeuni has both N-linked and O-linked protein glycosylation systems (Szymanski et al., 2003). Extensive O-linked glycosylation of C. jejeuni flagella results in carbohydrate representing 10% of the mass of the flagellin (Thibault et al., 2001). The HfaABD proteins provide an extremely strong anchor for the holdfast polysaccharide to the cell envelope of C. crescentus that is able to resist forces in the µ N range (Tsang et al., 2006).
A number of the findings presented in this paper indicate that the Hfa holdfast anchor system shares some properties with the curli system. We have shown that HfaA is resistant to heat and SDS and requires treatment with formic acid or HFIP for dissociation into a monomeric 12-kDa form. These properties are similar to other fibrillar adhesins, such as Hia and Hsf, or bacterial amyloid proteins such as curlins (Cotter et al., 2005, St. Geme & Cutter, 2000, Wang et al., 2007, Elliot et al., 2003), suggesting that HfaA may form a fibril-like structure involved in attachment of the holdfast polysaccharide to the cell. Indeed, HfaA has some amino acid similarity to fimbrial family proteins and to the curlin monomers AgfA and CsgA. Programs that identify aggregation domains typical of amyloid proteins identified three such domains and all three are in the most conserved regions of a protein that is not highly conserved overall. Alternatively, HfaA is a fibrillar surface protein similar to the Hsf and Hia adhesins of H. influenzae, which require denaturation with formic acid to reduce them to their monomeric form, but do not have other properties of amyloid proteins (St Geme et al., 1998). The hypothesis that HfaA is an amyloid protein is supported by the fact that HfaB, which is required for HfaA and HfaD assembly in the OM, is homologous to CsgG, the OM secretion channel for both CsgA and CsgB (Robinson et al., 2006). Furthermore, the level of HfaA and HfaD is greatly reduced in an hfaB mutant without the hfaB mutation affecting their transcription, suggesting the possibility that they are destabilized. Destabilization of the curlin CsgA and its nucleator CsgB has been observed in mutants of the curlin secretion channel csgG gene (Loferer et al., 1997) and for the. H. influenzae HMW adhesins in mutants lacking HMWB secretion protein (Grass et al 2002). Interestingly, HfaA and HfaD appear to be destabilized in an hfsDAB mutant. Destabilization of HfaA and HfaD could be due to the absence of a putative Hfa-Hfs complex when the holdfast translocation machinery is absent. Alternatively, if glycosylation of Hfa proteins is involved in holdfast attachment, loss of glycosylation could be the cause of their destabilization in an hfsDAB mutant. Other proteins have been shown to become unstable when they are no longer glycosylated (e.g. HMW adhesins) (Grass et al., 2003).
Despite sequence similarity between some of the holdfast anchor proteins and some of the proteins belonging to the curli system, these two systems differ in several respects, suggesting that the Hfa proteins may have unique properties. The curli monomer CsgA begins polymerization extracellularly and is associated with the cell via CsgB, a nucleator protein similar in sequence and size to CsgA (Hammar et al., 1996, Hammer et al., 2007). HfaD may play the role of nucleator for HfaA since it is necessary for the formation of the HMW species of HfaA. Alternatively, HfaD may be an integral part of the anchor structure with HfaA. However, HfaD is not similar to either CsgB or HfaA and is significantly larger than HfaA (415 vs 146 amino acids). There are no other proteins in the C. crescentus genome with significant similarity to CsgA or HfaA. Also encoded in the curli operon are CsgE and CsgF, which both interact with CsgG and are believed to be periplasmic chaperones required for proper folding of CsgA and CsgB, respectively (Robinson et al., 2006, Chapman et al., 2002). No homologs of CsgE and CsgF are found in the C. crescentus genome.
The HMW forms of HfaA and HfaD could indicate that they polymerize and/or that they are part of a heterogeneous macromolecular complex. The Y2H and CoIP data shows that HfaA interacts strongly with itself supporting the idea that HfaA forms a multimeric structure in C. crescentus. HfaD was also found to interact with itself, suggestive of a multimeric HfaD structure as well. Although we did not observe a significant interaction between HfaA and HfaD in the CoIP analysis, HfaA and HfaD may require post-translational modification to fold and interact properly, which would not occur when the proteins are generated in vitro. For example, some bacterial flagellins cannot assemble properly when they are not glycosylated (Ewing et al., 2009) (Twine et al., 2009). Alternatively, HfaA and HfaD could require another protein(s) to interact strongly or to facilitate fiber formation, such as HfaB. Finally, HfaA and HfaD may also require integration into the membrane for proper interaction, which would not occur in either the CoIP or Y2H experiments.
The fact that treatment with formic acid destroys the HMW form of HfaA could be due to depolymerization of a curlin-like polymer. Alternatively, the HMW form of HfaA could be due to its glycosylation by the holdfast polysaccharide, which should be hydrolyzed by formic acid. The ability of HfaA to maintain its HMW form in the absence of the holdfast polysaccharide implies that the holdfast polysaccharide is not responsible for the HMW form of HfaA. Similarly, the HMW form of HfaD does not require holdfast polysaccharide.
While HfaA does not require holdfast polysaccharide for formation of the HMW polymer, it does require HfaD. This result supports a role for HfaD as a nucleator for HfaA polymerization or its participation in a complex with HfaA. The fact that a hfaA hfaD double mutant phenocopies either single mutant is consistent with HfaA and HfaD working together in a complex or a pathway for holdfast attachment. The precise mechanism by which HfaA and HfaD attach the holdfast to the cell remains to be determined. However, the localization of Hfa proteins in the OM at the stalk tip and their exposure to the cell surface strongly suggest that they are directly involved in holdfast attachment.
Examining the localization pattern of proteins in the cell can shed light on their regulation and function. The Hfa proteins first localize at the flagellar pole of late predivisional cells, coincident with transcription of the hfaABD operon in the swarmer compartment of these cells (Janakiraman & Brun, 1999). They remain at the incipient stalked pole during the swarmer to stalk transition and are localized at the tip of the stalk as it is formed and elongated. The IFM and HfaBmCherry data indicate that the developmental regulator PodJ is needed for polar localization of the Hfa proteins. PodJ is a localization factor that first localizes to the pole opposite the stalk in stalked cells and is required for the localization of other developmental regulators and polar morphogenesis proteins to that pole (Crymes et al., 1999, Hinz et al., 2003, Viollier et al., 2002b, Viollier et al., 2002a, Lawler et al., 2006). PodJ may act as a direct target for the polar localization of Hfa proteins. Alternatively, the role of PodJ in polar localization of the Hfa proteins may be related to its requirement for holdfast polysaccharide synthesis since HfsDAB proteins are required for the polar localization of Hfa proteins.
Both the OM fractionation and IFM data suggest that the Hfa proteins are exposed to the cell surface. The protease susceptibility analysis of HfaA, HfaB and HfaD confirm that HfaA and HfaD are on the cell surface because they are protease sensitive in intact cells. HfaB was partially sensitive to protease in intact cells, suggesting that it is also surface exposed; however a significant fraction of HfaB was protease-resistant under our experimental conditions. In contrast to HfaB, its homolog CsgG is completely resistant to protease unless cells are permeabilized (Loferer et al., 1997). The partial protease-resistance of HfaB may be due to its assembly in a hypothesized secretion channel for HfaA and HfaD, or HfaB may be part of a macromolecular complex with other proteins, for example HfaA and HfaD, the proteins of the holdfast secretion machinery, or of the pili or flagellum machineries, which are localized at the same pole during part of the cell cycle. Finally, one caveat of our protease sensitivity experiments is that Hfa proteins were detected with M2 epitope tags. Even if these tagged fusions are functional, the Hfa proteins themselves may be more resistant to proteases than our results suggest because once the tag has been removed the remainder of the protein cannot be detected. Nonetheless, these results clearly establish that the Hfa proteins are exposed on the cell surface.
Deletion analyses demonstrate that each of the Hfa proteins is necessary for holdfast attachment to the cell. Based on all our assays, HfaA and HfaD appear to contribute equally to holdfast attachment and are dependent on each other for proper function based on the lack of HfaD in an hfaA mutant and on the lack of the HMW form of HfaA in an hfaD mutant. Loss of HfaB results in the most severe phenotype with an almost complete loss of surface adhesion and a strong holdfast shedding phenotype. Since HfaB is required for the export of both HfaA and HfaD through the OM, part of the phenotype of a hfaB mutant is probably due to the combined loss of HfaA and HfaD from the OM. Interestingly, the hfaA hfaD double mutant has a weaker shedding phenotype than the hfaB mutant, indicating that HfaB may be directly involved in holdfast anchoring or that HfaB secretes an additional factor involved in holdfast anchoring that has not been identified.
Many bacterial adhesin molecules work as multimeric protein structures (Cotter et al., 2006, Jerzejas et al., 2000). Based on the predicted function of each of the Hfa proteins and current data, we suggest a model for the holdfast anchor. We propose that HfaB forms an OM channel or serves as a chaperone for the secretion of HfaA and HfaD across the OM. HfaD and HfaA could form an anchor in one of several possible combinations: 1) HfaD is a nucleator protein and HfaA is the fibrillar anchor; 2) HfaD forms a fibrillar structure with HfaA at its tip, as in the case for type I pili, or 3) HfaA and HfaD form a heterogeneous polymer. HfaA and HfaD may stay associated with HfaB to form this structure or HfaB could secrete additional components involved in the holdfast anchor.
Experimental methods
Bacteria strains, plasmids, and growth conditions
The bacterial and yeast strains and plasmids used in these studies are described in Table S2. E. coli strains were grown at 37°C in Luria-Bertani (LB) broth or on LB agar (Sambrook et al., 1989). C. crescentus strains were grown at 30°C in peptone-yeast extract (PYE) broth or on PYE agar (Poindexter, 1964) or M2 minimal medium (Johnson & Ely, 1977) supplemented with 0.2% glucose (M2G). For strains containing xylose inducible constructs, PYE was supplemented with either 0.2% glucose for repression of expression or 0.3% xylose for induction of expression. Antibiotics (Amresco, Solon, Ohio) were used at the following concentrations (µg/ml liquid/agar in C. crescentus; µg/ml liquid/agar in E.coli): kanamycin (1.25/5; 25/25), tetracycline (1/2; 12/12.5), chloramphenicol (0.5/1; 20/30), streptomycin (5/5; 30/30). Naladixic acid was used at 20 µg/ml in PYE agar. Yeast strains were grown at 30°C in yeast extract-peptone-dextrose (YPD) or YPD agar (Guthrie & Fink, 1991). To test interactions, yeast strains were plated on minimal synthetic medium (0.67% yeast nitrogen base, 10 % amino acid drop out solution) (SD)
Recombinant DNA methods
DNA ligation, restriction digests and PCR were performed according to manufacturers’ recommendations. Primers (Operon Technologies, Huntsville, AL) used in these studies are listed in Supporting Information Table S3. Restriction enzymes and T4 DNA ligase were obtained from New England Biolabs, Beverly, MA. Plasmids were purified from E. coli using Qiagen Midi or Qiaprep Spin columns (Qiagen, Valencia, CA). Plasmids were introduced into E. coli by either chemical transformation or electroporation (Inoue et al., 1990, Dower et al., 1988). Plasmids were introduced into C. crescentus by conjugation (Ely, 1979, Ely, 1991). Sequencing was performed by the Indiana Molecular Biology Institute, Indiana University, Bloomington, IN using dideoxynucleotide chain termination method (Sanger et al., 1977) and ABI BigDye (Applied Biosystems, Foster, CA) with double stranded plasmid or PCR templates, which were purified with Qiaquick gel extraction columns (Qiagen).
Chromosomal DNA was isolated using Promega Magic MiniPrep DNA purification system as previously described (Promega, Madison, WI) (Janakiraman & Brun, 1997).
Deletion mutations in C. crescentus CB15 were generated in two steps using homologous recombination and sacB selection with pNPTS138 as previously described [see Supporting information (Gonin et al., 2000)].
Bioinformatic analysis
The PROPSEARCH (http://abcis.cbs.cnrs.fr/propsearch/) program was used to identify proteins with amino acid similarity to the Hfa proteins (Hobohm & Sander, 1995). CLUSTALW (http://www.ebi.ac.uk/Tools/clustalw2/index.html) was used to perform multiple protein sequence alignments (Larkin et al., 2007, Higgins, 1994). TANGO (http://tango.crg.es/) and AGGRESCAN (http://bioinf.uab.es/aggrescan/) programs were used to identify aggregation domains within the HfaA protein sequence (Rousseau et al., 2006) (Conchillo-Sole et al., 2007). The PSIpred program (http://bioinf4.cs.ucl.ac.uk:3000/psipred/) was used to predict the secondary structure of HfaA (Bryson et al., 2005, Jones, 1999).
Cell fractionation and protein analysis
Whole-cell lysates were prepared by resuspending bacterial pellets from 1 ml of bacterial culture into 50 µl of 10 mM Tris, pH 8 and 50 µl of 2× SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) buffer. Samples were boiled for 5 min and loaded on the gel.
OM proteins were isolated by sarkosyl insolubility (Spinola et al., 1986, Thanassi et al., 2002, Nikaido, 1994). Briefly, 100 ml of cells were grown to an OD600=0.6 to 0.7 in PYE, centrifuged at 5,000 × g at 4°C for 15 min, suspended in 20 ml 10 mM HEPES, pH 7.4, and lysed by two passes through a French press at 16,000 psi. Unbroken cells were removed by centrifugation at 10,000 × g at 4°C for 15 min. The supernatant was removed and centrifuged at 100,000 × g at 4°C for 1 h. The pellet was suspended in 20 ml 10 mM HEPES pH 7.4 and 1% sodium lauryl sarcosine, rocked at room temperature (RT) for 45 to 60 min and centrifuged at 100,000 × g for 1 h. The pellet, which contains the OM, was washed briefly with 10 mM Tris, pH 7.8 and then suspended in 500 µl 10 mM Tris, pH 7.8 and stored at –80°C. The sarkosyl soluble inner membrane fraction, which was the supernatant, was isolated by ethanol precipitation by adding ethanol to a final concentration of 85%, incubating overnight at –20°C and centrifuging at 12,000 × g for 15 min at 4°C. Protein pellets were suspended in 1 ml of 10 mM Tris, pH 7.8 and stored at –80-°C. A rapid micro procedure for isolation of OMs was performed as described (Carlone et al., 1986). Periplasmic extracts were isolated from 3 ml of an exponentially grown culture by chloroform extraction as previously described (Ames et al., 1984). Extracts were stored at −20°C.
Culture supernatants were isolated from 5 ml of an exponentially grown culture by centrifugation at 10,000 × g for 15 min at 4°C. The supernatant was passed through a 0.2 µM filter and centrifuged at 100,000 × g for 1 h at 4°C to remove any residual cells and outer membrane blebs. Supernatant proteins were precipitated on ice for 20 min with 10% trichloroacetic acid (TCA) and centrifuged for 15 min at 10,000 × g at 4°C. The TCA protein pellet was washed with 10 ml ice-cold 100% acetone and centrifuged again. The acetone was decanted and the pellet air-dried and suspended in 200 µl 1 M Tris, pH 9.
Formic acid and HFIP treatment of HfaA samples
Protein samples that contained HfaA were treated with either formic acid as described (Collinson et al., 1999) or HFIP (Sigma, St. Louis, MO) prior to analysis by SDS-PAGE, unless otherwise indicated. Both periplasmic extracts and culture supernatants were lyophilized overnight. Whole cell pellets from 1 ml of culture, periplasmic extracts and culture supernatants were incubated at RT in the dark for 2 h with 400 µl of 90% formic acid or 95% HFIP for 10 min. (Sigma). Isolated OMs or total membranes (60 µg) or the final membrane pellet obtained from a rapid micro membrane protocol (Carlone et al., 1986), were incubated with 200 µl of formic acid or HFIP. After incubation with formic acid or HFIP, all samples were lyophilized until dry. Two volumes of deionized water were added to each formic acid sample that was lyophilized again to remove any traces of formic acid. All samples were suspended in equal volumes of 1 M Tris, pH 9 and 2× SDS-PAGE sample buffer (0.125 M Tris, 4% SDS, 25% glycerol, 4% dithiothreitol, 10% β-mercaptoethanol, and 0.2% bromophenol blue) and boiled for 5 min prior to electrophoresis.
Protein quantification was performed using the SDS-modified Lowry assay (Lowry, 1951, Hartree, 1972).
Antibodies and Western Blot analysis
Proteins were suspended in an equal volume of 2X Laemelli sample buffer (Laemmli, 1970) and boiled for 5 min prior to loading and electrophoresis on an SDS-PAGE gel. Proteins were transferred to nitrocellulose and incubated with antibody overnight at RT or 4°C. Goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to horseradish peroxidase (HRP) (Biorad, Hercules, CA) were preabsorbed with acetone powder of C. crescentus NA1000 and used at a dilution of 1:20,000 and 1:10,000, respectively. HRP conjugated M2 antibody (Sigma) was used at 1:1250. dsRed Living Colors polyclonal antibody (Clontech) was used at 1:1000. FlgH (Jenal et al., 1994) and McpA (Alley et al., 1992) antibodies were used at 1:1000 and 1:10,000, respectively, and were supplied by Dr. Lucy Shapiro, Department of Developmental Biology, Stanford University, Stanford, CA. Western blots were developed with Supersignal Dura or Pico chemiluminescent substrate (Pierce, Rockford, IL). Western blots of yeast whole-cell lysates (TCA method, Clontech protocol #PT3024-1) were probed with either c-myc monoclonal antibody (Clontech) at 1:300 or HA monoclonal antibody (Santa Cruz Biotechnology) at 1:100.
Short-term binding assays
To determine the efficiency of single cell attachment to surfaces, cultures were grown in plastic multiwell plates for 45 min, the plates were washed vigorously and crystal violet (CV) was used to stain total proteins of the bound cells. This CV binding assay provides a short-term quantitative measure of cells bound tightly to the plastic surface and was performed as previously described with the following modifications (Bodenmiller et al., 2004). Plates were incubated with shaking at RT for 45 min instead of 15 min. Wells were washed three times with 1 ml PYE, 750 µl of 1% (wt/vol) CV solution was added and plates were incubated without shaking for 20 min instead of 15 min. All assays were performed in triplicate and each strain was examined using three independent cultures. To obtain the final percent binding, the PYE blank was subtracted from all samples and then each sample was normalized to C. crescentus CB15 binding, which was set at 100%. All samples are shown as the mean ± standard error (SE).
Biofilm assays
To assess efficiency of biofilm formation, cultures were grown overnight at 30°C in M2G. Cultures were diluted in M2G to an OD600= 0.05 and 3 ml was inoculated in triplicate into 12-well plates for each time point. Plastic coverslips were placed upright in each well. CB15 was used as a positive control and NA1000 and M2G medium were used as negative controls. Plates were incubated at 30°C without shaking for 24 h, 48 h and 60 h. At each timepoint, coverslips were removed, washed for 10 to 20 sec. with a water bottle and stained in 0.1% CV for 20 min. After staining, the coverslips were washed again with a water bottle to remove excess stain. For the three replicates, one coverslip was kept as a visual staining sample and the other two coverslips were washed in 1 ml of 10% acetic acid to remove and quantify the CV staining at A600. The OD600 of each well was also determined. To quantify the amount of long-term binding, the CV A600 of M2G was subtracted from CV A600 of each sample. Then, the CV As600 value of each well was divided by culture OD600 to normalize for cell growth.
Fluorescent lectin binding
The WGA binding assay was performed on cells in liquid culture using fluorescein isothiocyanate labeled WGA (FITC-WGA) (Molecular probes, Eugene, OR) at 50 µg/ml final concentration as previously described (Merker & Smit, 1988, Janakiraman & Brun, 1999). This assay provides a quantitative measure of holdfast present on stalks. Only PD cells are counted because the poles of the cell can be unambiguously identified. Quantification of lectin binding was determined by calculating the percentage of PD cells that had fluorescent lectin binding and is shown as the mean ± SE.
Glass coverslip binding
To determine whether holdfasts were shed, each strain was grown with shaking in the presence of a glass cover-slip for 4–5 h, stained with FITC-WGA and observed with epifluorescence microscopy as previously described (Cole et al., 2003).
Rosette formation
Rosette formation was assessed by calculating the number of cells in rosettes per total cells counted. Additionally, the number of cells in each rosette was determined. Cells were grown at 30°C to mid-log phase (OD600=0.3 to 0.5), placed on ice and then examined by light microscopy. Cells were quantified from twenty images and two independent cultures grown on separate days.
Immunofluorescence microscopy
IFM was performed as previously described for permeabilized cells (Quardokus et al., 2001) and for unpermeabilized cells with some modification. Briefly, cells were grown to an OD600= 0.3 to 0.8 and fixed in 2.5% formaldehyde (Ted Pella, Inc., Redding, CA) for 15 min. Cells were spun at 5000 × g for 5 min at 4°C and washed once with phosphate-buffered saline (PBS), pH 7.2. The protocol was then followed as published (Quardokus et al., 2001). M2 polyclonal (Sigma) and Goat anti-rabbit FITC (Biorad) antibodies were used at 1:100. The HfaBmCherry construct was visualized on pads made of 0.6% Seakem agarose (Lonza, Basel, Switzerland) and M2G medium. Timelapse series were done in 10 min intervals on a heated stage at 30°C for up to 15 h.
Epifluorescence microscopy was performed on a Nikon Eclipse 800 or 90i light microscope equipped with a Nikon B-2E FITC filter cube for FITC and a 100× plan Apo oil objective (NA 1.4) or the 83000 quad filter cube and 100× (DIC) oil objective for mCherry. Images were captured using a Princeton Instruments Cooled CCD camera model 1317 or a Photometrics Cascade 1K EMCCD camera and Metamorph Imaging Software package vs. 7.5.4. Assembly and processing of timelapse images was done using ImageJ version 1.42l (http://rsb.info.nih.gov/ij/)(Abramoff et al., 2004).
Protease sensitivity
Cells were grown overnight and diluted into fresh PYE to an OD600=0.15. Cells were grown to mid-log phase and 1 OD600 of cells was prepared for each sample. Cells were centrifuged at 5000 × g for 5 min, washed one time in 50 mM Tris-HCl pH 8 and 5 mM CaCl2 and suspended in 135 µl of 50 mM Tris-HCl pH8, 5 mM CaCl2. Some samples also contained 0.1% Triton X-100 to disrupt cells. Proteinase K was added to a final concentration of 25 µg/ml and the cells were incubated at 37°C for 10, 20 or 30 min. The protease inhibitor phenylmethylsulfonyl fluoride was added to a final concentration of 2 mM and EGTA, a calcium chelator, was added to a final concentration of 5 mM. Cells were centrifuged at 5000 × g for 5 min, suspended in 50 µl of 10 mM Tris-HCl pH 8 and 50 µl 1X SDS-PAGE sample buffer and boiled for 5 min. A buffer only control was used as a protease free control. The IM protein McpA and the OMP FlgH were used as controls to demonstrate whole cell integrity.
The protease sensitivity of HfaA was assessed in a similar manner to HfaB and HfaD with some modification. Five OD600 of cells were prepared for each sample and suspended in a final volume of 600 µl of 50 mM Tris-HCl, pH 8, 5 mM CaCl2 prior to protease digestion as described above. After the addition of protease inhibitor and EGTA, cells were centrifuged and suspended in 1 ml of 10 mM HEPES, pH 7.4 and Complete mini-EDTA free protease inhibitor Roche, Indianapolis, IN) and outer membrane and inner membrane fractions were prepared using sarkosyl solubility (Carlone et al. 1986)
Alkaline phosphatase zymogram
The protocol for alkaline phosphatase zymograms was based on two previously described methods (Michel & Baratti, 1989, Pond et al., 1989). Protein samples were electrophoresed on a 16 × 10 cm SDS-PAGE gel. The gel was washed for 20 min., two times in 0.5 M Tris pH 8.5, 1 mM ZnCl2, 2 mM MgCl2 and 2% Triton X-100. The gel was then washed for 20 min, two times in 0.5 M Tris pH 8.5, 0.1 mM ZnCl2, 1 mM MgCl2 and 2% Triton X-100. To visualize the protein bands with alkaline phosphatase activity, the gel was developed overnight at room temperature in 330 µg/ml 4-Nitro blue tetrazolium chloride and 170 µg/ml 5-Bromo-4-chloro-3-indolyl-phosphate in 0.5 M Tris pH 8.5, 0.1 mM ZnCl2, 1 mM MgCl2 and 2% Triton X-100, washed in warm water three times for 20 min and dried in cellophane.
Yeast two-hybrid analysis
The assay was performed based on manufacturers’ recommendations (Clontech, Mountain View, CA). Individual colonies of yeast co-transformants were suspended in 500 µl of 0.9% NaCl; dilutions of 1:10, 1:100 and 1:1000 of this suspension were plated on SD agar lacking different combinations of amino acids, to determine the strength of the interaction: growth on –his/-leu/-trp SD agar indicates a weak protein-protein interaction; growth on –ade/-his/-leu/-trp SD agar indicates strong interaction. Interactions were scored as positive or negative by comparison of colony sizes and number of colonies relative to the positive (p53 bait + T-α prey) and negative (Lam bait + T-α prey) Y2H controls. In co-transformants showing no interaction using the above method, expression of Hfa proteins was confirmed by Western blot (data not shown).
In vitro transcription/translation of DNA templates and Co-immunoprecipitation
CoIP was performed using the Clontech Matchmaker CoIP kit, protocol # PT3323-1, version # PR32602. Proteins were synthesized by in vitro transcription-translation using the TnT Coupled Reticulocyte Lysate System (Promega). The proteins were expressed using templates that added a polyadenine tail to the mRNA transcripts The DNA templates for in vitro transcription/translation were generated by sub-cloning the hfa genes with epitope tags (and also the Lamin-C control protein and tag) from the Y2H bait and prey vectors to the pSP64 (polyA) vector (Promega) and transforming into E. coli DH5α. Radiolabeling was performed by adding [35S] L-methionine in vitro translation grade (MP Biomedicals, Solon, OH) to the desired protein synthesis reactions. Plasmid DNA used as templates in the protein synthesis reactions was purified from E. coli strains using Qiagen plasmid purification columns with some modification. The purification protocol included phenol-chloroform extraction of the templates to remove RNases. Under RNase-free conditions, the DNA was precipitated with isopropanol, washed twice with 70% ethanol, and suspended in 10 mM Tris, pH 8 with a final DNA concentration of between 700 and 2000 ng/ml. Unlabeled (cold) bait protein was used to pulldown labeled (hot) prey protein; otherwise, the signal from the bait HfaA or HfaD would obscure any signal obtained from CoIP of HfaA or HfaD prey protein. The CoIP reactions were incubated with protein-A beads and c-myc antibody. The bait proteins have a c-myc tag. The prey proteins contain a hemagglutinin epitope tag (YPYDVPDYA) and will only be co-immunoprecipitated with the c-myc antibody if the bait protein and the prey protein can interact with each other. CoIP reactions were analyzed by SDS-PAGE via phosphorimaging scanned with a Typhoon 9200 Variable Mode Imager using ImageQuant 5.2 software (GE Healthcare, Piscataway, NJ).
Supplementary Material
Acknowledgements
We thank the members of the Brun lab for helpful scientific discussion and critical reading of the manuscript. We also thank Miriam Martin for generating strain YB2412 and David Kysela for help with the time-lapse microscopy. Most of this work was supported by grant GM077648 from the National Institutes of Health to Y.V.B. Early stages of this work were supported by grant GM51986 from the National Institutes of Health to Y.V.B. Some of this work was supported by the Indiana METACyt Initiative of Indiana University, funded in part through a major grant from the Lilly Endowment, Inc.
Footnotes
Supporting Information. Supporting information is available at (web site link to be supplied at time of publication).
References
- Abramoff M, Magelhaes P, Ram S. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Alley MRK, Maddock JR, Shapiro L. Polar localization of a bacterial chemoreceptor. Genes Develop. 1992;6:825–836. doi: 10.1101/gad.6.5.825. [DOI] [PubMed] [Google Scholar]
- Ames GF, Prody C, Kustu S. Simple, rapid, and quantitative release of periplasmic proteins by chloroform. J Bacteriol. 1984;160:1181–1183. doi: 10.1128/jb.160.3.1181-1183.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Z, Normark S. Nucleator function of CsgB for the assembly of adhesive surface organelles in Escherichia coli. EMBO J. 1997;16:5827–5836. doi: 10.1093/emboj/16.19.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller D, Toh E, Brun YV. Development of surface adhesion in Caulobacter crescentus. J Bacteriol. 2004;186:1438–1447. doi: 10.1128/JB.186.5.1438-1447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PJ, Hardy GG, Trimble MJ, Brun YV. Complex regulatory pathways coordinate cell-cycle progression and development in Caulobacter crescentus. Adv Microb Physiol. 2009;54:1–101. doi: 10.1016/S0065-2911(08)00001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nuc Acids Res. 2005;33:W36–W38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlone GM, Thomas ML, Rumschlag HS, Sottnek FO. Rapid microprocedure for detergent-insoluble outer membrane proteins from Haemophilus species. J Clin Microbiol. 1986;24:330–332. doi: 10.1128/jcm.24.3.330-332.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartee RT, Forsee WT, Yother J. Initiation and synthesis of the Streptococcus pneumoniae type 3 capsule on a phosphatidylglycerol membrane anchor. J Bacteriol. 2005;187:4470–4479. doi: 10.1128/JB.187.13.4470-4479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, Normark S, Hultgren SJ. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851–855. doi: 10.1126/science.1067484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J, Hardy GG, Bodenmiller D, Toh E, Hinz A, Brun YV. The HfaB and HfaD adhesion proteins of Caulobacter crescentus are localized in the stalk. Mol Microbiol. 2003;49:1671–1683. doi: 10.1046/j.1365-2958.2003.03664.x. [DOI] [PubMed] [Google Scholar]
- Collinson SK, Emody L, Muller KH, Trust TJ, Kay WW. Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis. J Bacteriol. 1991;173:4773–4781. doi: 10.1128/jb.173.15.4773-4781.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinson SK, Parker JM, Hodges RS, Kay WW. Structural predictions of AgfA, the insoluble fimbrial subunit of Salmonella thin aggregative fimbriae. J Mol Biol. 1999;290:741–756. doi: 10.1006/jmbi.1999.2882. [DOI] [PubMed] [Google Scholar]
- Conchillo-Sole O, de Groot NS, Aviles FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC bioinformatics. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpe WA. Microfouling: the role of primary film forming marine bacteria. In: Acker RF, Brown BF, DePalma JR, Iverson WP, editors. Proceedings of the Third Intermational Congress on Marine Corrosion and Fouling. Gaithersburg, MD: Northwestern University Press; 1972. pp. 598–609. [Google Scholar]
- Cotter SE, Surana NK, Grass S, St. Geme JW. Trimeric autotransporters require trimerization of the passenger domain for stability and adhesive activity. J Bacteriol. 2006;188:5400–5407. doi: 10.1128/JB.00164-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter SE, Yeo HJ, Jeuhne T, St. Geme JW. Architecture and adhesive activity of the Haemophilus influenzae Hsf adhesin. J Bacteriol. 2005;187:4656–4664. doi: 10.1128/JB.187.13.4656-4664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun. 1999;67:5427–5433. doi: 10.1128/iai.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crymes WB, Jr, Zhang D, Ely B. Regulation of podJ expression during the Caulobacter crescentus cell cycle. J Bacteriol. 1999;181:3967–3973. doi: 10.1128/jb.181.13.3967-3973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dower WJ, Miller JF, Ragsdale CW. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988;16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot MA, Karoonuthaisiri N, Huang J, Bibb MJ, Cohen SN, Kao CM, Buttner MJ. The chaplins: a family of hydrophobic cell-surface proteins involved in aerial mycelium formation in Streptomyces coelicolor. Genes Dev. 2003;17:1727–1740. doi: 10.1101/gad.264403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely B. Transfer of drug resistance factors to the dimorphic bacterium Caulobacter crescentus. Genetics. 1979;91:371–380. doi: 10.1093/genetics/91.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991;204:372–384. doi: 10.1016/0076-6879(91)04019-k. [DOI] [PubMed] [Google Scholar]
- Entcheva-Dimitrov P, Spormann AM. Dynamics and control of biofilms of the oligotrophic bacterium Caulobacter crescentus. J Bacteriol. 2004;186:8254–8266. doi: 10.1128/JB.186.24.8254-8266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing CP, Andreishcheva E, Guerry P. Functional characterization of flagellin glycosylation in Campylobacter jejuni 81–176. J Bacteriol. 2009;191:7086–7093. doi: 10.1128/JB.00378-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature biotechnology. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Gonin M, Quardokus EM, O'Donnol D, Maddock J, Brun YV. Regulation of stalk elongation by phosphate in Caulobacter crescentus. J Bacteriol. 2000;182:337–347. doi: 10.1128/jb.182.2.337-347.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass S, Buscher AZ, Swords WE, Apicella MA, Barenkamp SJ, Ozchlewski N, St Geme JW., 3rd The Haemophilus influenzae HMW1 adhesin is glycosylated in a process that requires HMW1C and phosphoglucomutase, an enzyme involved in lipooligosaccharide biosynthesis. Mol Microbiol. 2003;48:737–751. doi: 10.1046/j.1365-2958.2003.03450.x. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. San Diego: Academic Press; 1991. p. 933. [Google Scholar]
- Hammar M, Bian Z, Normark S. Nucleator-dependent intercellular assembly of adhesive curli organelles in Escherichia coli. Proc Natl Acad Sci USA. 1996;93:6562–6566. doi: 10.1073/pnas.93.13.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer ND, Schmidt JC, Chapman MR. The curli nucleator protein CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc Natl Acad Sci USA. 2007;104:12494–12499. doi: 10.1073/pnas.0703310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartree EF. Determination of protein: a modification of the Lowry method that gives a linear photometric response. Analyt Biochem. 1972;48:422–427. doi: 10.1016/0003-2697(72)90094-2. [DOI] [PubMed] [Google Scholar]
- Higgins DG. CLUSTAL V: multiple alignment of DNA and protein sequences. Methods in molecular biology (Clifton, N.J. 1994;25:307–318. doi: 10.1385/0-89603-276-0:307. [DOI] [PubMed] [Google Scholar]
- Hinz AJ, Larson DE, Smith CS, Brun YV. The Caulobacter crescentus polar organelle development protein PodJ is differentially localized and is required for polar targeting of the PleC development regulator. Mol Microbiol. 2003;47:929–941. doi: 10.1046/j.1365-2958.2003.03349.x. [DOI] [PubMed] [Google Scholar]
- Hobohm U, Sander C. A sequence property approach to searching protein databases. J Mol Biol. 1995;251:390–399. doi: 10.1006/jmbi.1995.0442. [DOI] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmid. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Janakiraman RS, Brun YV. Transcriptional and mutational analyses of the rpoN operon in Caulobacter crescentus. J Bacteriol. 1997;179:5138–5147. doi: 10.1128/jb.179.16.5138-5147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janakiraman RS, Brun YV. Cell cycle control of a holdfast attachment gene in Caulobacter crescentus. J Bacteriol. 1999;181:1118–1125. doi: 10.1128/jb.181.4.1118-1125.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal U, White J, Shapiro L. Caulobacter flagellar function, but not assembly, requires FliL, a non-polarly localized membrane protein present in all cell types. J Mol Biol. 1994;243:227–244. doi: 10.1006/jmbi.1994.1650. [DOI] [PubMed] [Google Scholar]
- Jerzejas MJ, Hollingshead SK, Lebowitz J, Chantalat L, Briles DE, Lamani E. Production and characterization fo the functional fragment of pneumococcal surface protein A. Arch Biochem Biophys. 2000;373:116–125. doi: 10.1006/abbi.1999.1544. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Ely B. Isolation of spontaneously derived mutants of Caulobacter crescentus. Genetics. 1977;86:25–32. doi: 10.1093/genetics/86.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Mizunoe Y, Takade A, Naito S, Yoshida S. Curli fibers are required for development of biofilm architecture in Escherichia coli K-12 and enhance bacterial adherence to human uroepithelial cells. Microbiol Immunol. 2005;49:875–884. doi: 10.1111/j.1348-0421.2005.tb03678.x. [DOI] [PubMed] [Google Scholar]
- Krikos A, Mutoh N, Boyd A, Simon MI. Sensory Transducers of E.coli are Composed of Discrete Structural and Functional Domains. Cell. 1983;33:615–622. doi: 10.1016/0092-8674(83)90442-7. [DOI] [PubMed] [Google Scholar]
- Kurtz HD. 2004 personal communication. [Google Scholar]
- Kurtz HD, Jr, Smit J. Analysis of a Caulobacter crescentus gene cluster involved in attachment of the holdfast to the cell. J Bacteriol. 1992;174:687–694. doi: 10.1128/jb.174.3.687-694.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz HD, Jr, Smit J. The Caulobacter crescentus holdfast: Identification of holdfast attachment complex genes. FEMS Microbiol Lett. 1994;116:175–182. doi: 10.1111/j.1574-6968.1994.tb06697.x. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lasa I. Towards the identification of the common features of bacterial biofilm development. Internat Microbiol. 2006;9:21–28. [PubMed] [Google Scholar]
- Laub MT, Chen SL, Shapiro L, McAdams HH. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc Natl Acad Sci USA. 2002;99:4632–4637. doi: 10.1073/pnas.062065699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laub MT, McAdams HH, Feldblyum T, Fraser CM, Shapiro L. Global analysis of the genetic network controlling a bacterial cell cycle. Science. 2000;290:2144–2148. doi: 10.1126/science.290.5499.2144. [DOI] [PubMed] [Google Scholar]
- Lawler ML, Larson DE, Hinz AJ, Klein D, Brun YV. Dissection of functional domains of the polar localization factor PodJ in Caulobacter crescentus. Mol Microbiol. 2006;59:301–316. doi: 10.1111/j.1365-2958.2005.04935.x. [DOI] [PubMed] [Google Scholar]
- Levi A, Jenal U. Holdfast formation in motile swarmer cells optimizes surface attachment during Caulobacter crescentus development. J Bacteriol. 2006;188:5315–5318. doi: 10.1128/JB.01725-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Smith CS, Brun YV, Tang JX. The elastic properties of the Caulobacter crescentus adhesive holdfast are dependent on oligomers of N-acetylglucosamine. J Bacteriol. 2005;187:257–265. doi: 10.1128/JB.187.1.257-265.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loferer H, Hammar M, Normak S. Availability of the fibre subunit CsgA and the nucleator protein CsgB during assembly of fibronectin-binding curli is limited by the intracellular concentration of the novel lipoprotein CsgG. Mol Microbiol. 1997;26:11–23. doi: 10.1046/j.1365-2958.1997.5231883.x. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NR, Farr AL, Randall RJ. Protein measurement using the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mack D, Nedelmann M, Krokotsch A, Schwarzkopf A. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1, 6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol. 1996;178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack D, Nedelmann M, Krokotsch A, Schwarzkopf A, Heesemann J, Laufs R. Characterization of transposon mutants of biofilm-producing Staphylococcus epidermidis impaired in the accumulative phase of biofilm production: genetic identification of a hexosamine-containing polysaccharide intercellular adhesin. Infect Immun. 1994;62:3244–3253. doi: 10.1128/iai.62.8.3244-3253.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker RI, Smit J. Characterization of the adhesive holdfast of marine and freshwater Caulobacters. J Appl Environ Microbiol. 1988;54:2078–2085. doi: 10.1128/aem.54.8.2078-2085.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel GPF, Baratti JC. Phosphate-irrepressible alkaline phosphatase of Zymomonas mobilis. 1989;135:453–460. doi: 10.1016/0378-1097(92)90139-f. [DOI] [PubMed] [Google Scholar]
- Mitchell D, Smit J. Identification of genes affecting production of the adhesion organelle of Caulobacter crescentus CB2. J Bacteriol. 1990;172:5425–5431. doi: 10.1128/jb.172.9.5425-5431.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido H. Isolation of outer membranes. Methods Enzymol. 1994;235:225–234. doi: 10.1016/0076-6879(94)35143-0. [DOI] [PubMed] [Google Scholar]
- Ong CJ, Wong MY, Smit J. Attachment of the adhesive holdfast organelle to the cellular stalk of Caulobacter crescentus. J Bacteriol. 1990;172:1448–1456. doi: 10.1128/jb.172.3.1448-1456.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poindexter JS. Biological properties and classification of the Caulobacter group. Microbiol Mol Biol Revs. 1964;28:231–295. doi: 10.1128/br.28.3.231-295.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond JL, Eddy CK, Mackenzie KF, Conway T, Borecky DJ, Ingram LO. Cloning, sequencing, and characterization of the principal acid phosphatase, the phoC+ product, from Zymomonas mobilis. 1989;171:767–774. doi: 10.1128/jb.171.2.767-774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power PM, Roddam LF, Dieckelmann M, Srikhanta YN, Tan YC, Berrington AW, Jennings MP. Genetic characterization of pilin glycosylation in Neisseria meningitidis. Microbiol. 2000;146:967–979. doi: 10.1099/00221287-146-4-967. [DOI] [PubMed] [Google Scholar]
- Power PM, Seib KL, Jennings MP. Pilin glycosylation in Neisseria meningitidis occurs by a similar pathway to wzy-dependent )-antigen biosynthesis in Escherichia coli. Biochem Biophys Res Comm. 2006;347:904–908. doi: 10.1016/j.bbrc.2006.06.182. [DOI] [PubMed] [Google Scholar]
- Quardokus E, Din N, Brun YV. Cell cycle and positional constraints on FtsZ localization and the initiation of cell division in Caulobacter crescentus. Mol Microbiol. 2001;39:949–959. doi: 10.1046/j.1365-2958.2001.02287.x. [DOI] [PubMed] [Google Scholar]
- Rhan A, Beis K, Naismith JH, Whitfield C. A novel outer membrane protein, Wzi, is involved in surface assembly of the Escherichia coli K30 group 1 capsule. J Bacteriol. 2003;185:5882–5890. doi: 10.1128/JB.185.19.5882-5890.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson LS, Ashman EM, Hultgren SJ, Chapman MR. Secretion of curli fibre subunits is mediated by the outer membrane-localized CsgG protein. Mol Microbiol. 2006;59:870–881. doi: 10.1111/j.1365-2958.2005.04997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: confusion of the kinds? Current opinion in structural biology. 2006;16:118–126. doi: 10.1016/j.sbi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. Cold Spring Harbor: Cold Spring Harbor Press; 1989. p. A.2. [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit J, Sherwood CS, Turner RF. Characterization of high density monolayers of the biofilm bacterium Caulobacter crescentus: evaluating prospects for developing immobilized cell bioreactors. Can J Microbiol. 2000;46:339–349. [PubMed] [Google Scholar]
- Smith CS, Hinz A, Bodenmiller D, Larson DE, Brun YV. Identification of genes required for synthesis of the adhesive holdfast in Caulobacter crescentus. J Bacteriol. 2003;185:1432–1442. doi: 10.1128/JB.185.4.1432-1442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen UBS, Henrichsen J, Chen HC, Szu SC. Covalent linkage between the capsular polysaccharide and the cell wall peptidoglycan of Streptococcus pneumoniae revealed by immunochemical methods. Microb Pathog. 1990;8:325–334. doi: 10.1016/0882-4010(90)90091-4. [DOI] [PubMed] [Google Scholar]
- Spinola SM, Peacock J, Denny FW, Smith DL, Cannon JG. Epidemiology of colonization by nontypable Haemophilus influenzae in children: a longitudinal study. J Infect Dis. 1986;154:100–109. doi: 10.1093/infdis/154.1.100. [DOI] [PubMed] [Google Scholar]
- St Geme JW, 3rd, Kumar VV, Cutter D, Barenkamp SJ. Prevalence and distribution of the hmw and hia genes and the HMW and Hia adhesins among genetically diverse strains of nontypeable Haemophilus influenzae. Infect Immun. 1998;66:364–368. doi: 10.1128/iai.66.1.364-368.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St. Geme JW, Cutter D. The Haemophilus influenzae Hia adhesin is an autotransporter protein that remains uncleaved at the C-terminus and fully cell associated. J Bacteriol. 2000;182:6005–6013. doi: 10.1128/jb.182.21.6005-6013.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P, Sauer K, Davies D, Costerton J. Biofilms as complex differentiated communities. Ann Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- Szymanski CM, Logan SM, Linton D, Wren BW. Campylobacter - a tale of two protein glycosylation systems. Trends Microbiol. 2003;11:233–238. doi: 10.1016/s0966-842x(03)00079-9. [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Stathopoulos C, Dodson K, Geiger D, Hultgren SJ. Bacterial outer membrane ushers contain distinct targeting and assembly domains for pilus biogenesis. J Bacteriol. 2002;184:6260–6269. doi: 10.1128/JB.184.22.6260-6269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault P, Logan SM, Kelly JF, Brisson JR, Ewing CP, Trust TJ, Guerry P. Identification of the carbohydrate moieties and glycosylation motifs in Campylobacter jejuni flagellin. J Biol Chem. 2001;276:34862–34870. doi: 10.1074/jbc.M104529200. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nuc Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh E, Kurtz H, Jr, Brun Y. Characterization of the Caulobacter crescentus holdfast polysaccharide biosynthesis pathway reveals significant redundancy in the initiating glycosyltransferase and polymerase steps. J Bacteriol. 2008;190:7219–7231. doi: 10.1128/JB.01003-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang PH, Li G, Brun YV, Freund LB, Tang JX. Adhesion of single bacterial cells in the micronewton range. Proc Natl Acad Sci USA. 2006;103:5764–5768. doi: 10.1073/pnas.0601705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twine SM, Reid CW, Aubry A, McMullin DR, Fulton KM, Austin J, Logan SM. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol. 2009;191:7050–7062. doi: 10.1128/JB.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houdt R, Michiels CW. Role of bacterial cell surface structures in Escherichia coli. Res Microbiol. 2005;156:626–633. doi: 10.1016/j.resmic.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Viollier PH, Sternheim N, Shapiro L. A dynamically localized histidine kinase controls the asymmetric distribution of polar pili proteins. EMBO J. 2002a;21:4420–4428. doi: 10.1093/emboj/cdf454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viollier PH, Sternheim N, Shapiro L. Identification of a localization factor for the polar positioning of bacterial structural and regulatory proteins. Proc Natl Acad Sci USA. 2002b;99:13831–13836. doi: 10.1073/pnas.182411999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Preston JF, Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Smith DR, Jones JW, Chapman MR. In vitro polymerization of a functional E. coli amyloid protein. J Biol Chem. 2007;282:3713–3719. doi: 10.1074/jbc.M609228200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield C, Naismith JH. Periplasmic export machines for outer membrane assembly. Current opinion in structural biology. 2008;18:466–474. doi: 10.1016/j.sbi.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield C, Paiment A. Biosynthesis and assembly of group 1 capsular polysaccharides in Escherichia coli and related extracellular polysaccharides in other bacteria. Carbohydr Res. 2003;338:2491–2502. doi: 10.1016/j.carres.2003.08.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.