

Abstract
Chlorite dismutase (Cld) is a heme enzyme capable of rapidly and selectively decomposing chlorite (ClO2−) to Cl− and O2. The ability of Cld to promote O2 formation from ClO2− is unusual. Heme enzymes generally utilize ClO2− as an oxidant for reactions such as oxygen atom transfer to, or halogenation of, a second substrate. The X-ray crystal structure of Dechloromonas aromatica Cld co-crystallized with the substrate analogue nitrite (NO2−) was determined to investigate features responsible for this novel reactivity. The enzyme active site contains a single b-type heme coordinated by a proximal histidine residue. Structural analysis identified a glutamate residue hydrogen-bonded to the heme proximal histidine that may stabilize reactive heme species. A solvent-exposed arginine residue likely gates substrate entry to a tightly confined distal pocket. On the basis of the proposed mechanism of Cld, initial reaction of ClO2− within the distal pocket generates hypochlorite (ClO−) and a compound I intermediate. The sterically restrictive distal pocket probably facilitates the rapid rebound of ClO− with compound I forming the Cl− and O2 products. Common to other heme enzymes, Cld is inactivated after a finite number of turnovers, potentially via the observed formation of an off-pathway tryptophanyl radical species through electron migration to compound I. Three tryptophan residues of Cld have been identified as candidates for this off-pathway radical. Finally, a juxtaposition of hydrophobic residues between the distal pocket and the enzyme surface suggests O2 may have a preferential direction for exiting the active site.
Keywords: Heme, Chlorite dismutase, Crystal structure
Introduction
Anthropogenic deposition of pollutant compounds has led to the proliferation of microbes capable of degrading a wide range of compounds and utilizing a variety of terminal electron acceptors [1–3]. Examples include organisms capable of using AsO4− [4–6] and SeO4− [7, 8] from industrial or agricultural output. Dechloromonas aromatica RCB is a beta-proteobacterium that has been able to exploit the primarily man-made oxidants perchlorate (ClO4−) and chlorate (ClO3−) as terminal respiratory acceptors to yield innocuous chloride ion and molecular oxygen [9–11].
The pathway involves two enzymes [12]. The first is a molybdopterin-dependent oxotransferase, perchlorate reductase [13]. Perchlorate reductase enzymes, which are highly homologous to dissimilatory nitrate reductases, reductively convert ClO4− or ClO3− to chlorite (ClO2−) [13, 14]. The buildup of ClO2− is toxic to the organism and is removed via the action of a second enzyme, chlorite dismutase (Cld) [15]. Cld is a protophorphyrin IX (b-type) heme enzyme responsible for decomposition of chlorite into Cl− and O2. Whereas other heme enzymes are known to react with chlorite, the highly efficient generation of O2 from this substrate is unique to Cld [16–21]. Previous kinetic studies have confirmed the stoichiometric conversion of ClO2− to Cl− and O2 [21]. Oxygen-18 labeling studies have demonstrated ClO2− as the sole source of product O2, thereby requiring the formation of a new O–O bond during the course of catalysis [21, 22]. This makes Cld the only known enzymatic system outside photosystem II that has evolved to efficiently catalyze O–O bond formation. The recent crystal structure of Cld from Azospira oryzae has a β-sheet-dominated fold that is novel among heme enzymes [23]. Furthermore, annotated cld genes are widespread in non-perchlorate-metabolizing bacteria and archaea, constituting their own independent cluster of orthologs [9]. Clds therefore appear to occupy a unique, largely undescribed niche in heme enzyme chemistry.
The central intermediate for many heme enzymes is a ferryl porphyrin radical species known as compound I [(Por•+)Fe(IV)=O]. Chlorite is a thermodynamically robust oxidant [Em,pH 7.0, 25 °C (ClO2−/Cl−) = +1,175 mV], and should be capable of compound I formation. The postulated mechanism of Cld involves heterolytic cleavage of the Cl–O bond to form compound I [21, 22]. The hypochlorite (ClO−) generated is proposed to rebound in a nucleophilic attack on the oxo group of compound I, generating Cl− and O2 and returning the enzyme to its resting state (Scheme 1). However, a concerted mechanism, involving iron and bidentate chlorite in an energetically strained four-membered ring in the transition state, has not been definitively ruled out.
Scheme 1.
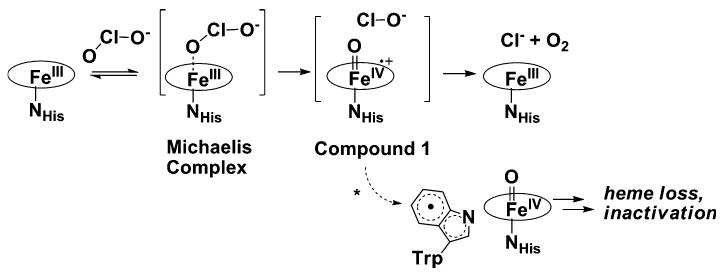
Proposed mechanism of action of chlorite dismutase. The asterisk denotes the off-pathway formation of the tryptophanyl radical
Heme enzymes have evolved to catalyze a wide variety of reactions via the compound I intermediate [24, 25]. These include one-electron oxidation of diverse cosubstrates ranging from small aliphatic to large aromatic compounds (peroxidases) [24, 25], the two-electron oxidation of peroxide leading to O2 (catalase), hydroxylation (peroxygenases, cytochromes P450), and chlorination (chloroperoxidases) [19, 26–34]. It should be noted that catalase does not catalyze O–O bond formation as this bond is already present in the peroxide substrate. Many of these enzymes can also catalyze the oxidation, albeit less efficiently, of more than one cosubstrate [24, 35, 36]. However, the proposed chlorite-derived compound I is surprisingly faithful to chlorite decomposition [22]. When the Cld reaction is run in the presence of a variety of cosubstrates that could potentially undergo oxidation, oxygenation, or chlorination, Cld-catalyzed chlorite decomposition is overwhelmingly favored [22, 35]. This acute specificity of Cld for a single substrate is unusual.
Here we report the crystal structure of D. aromatica RCB Cld bound with the substrate analog and enzyme inhibitor, nitrite (NO2−). The current structure offers new insight into structural features responsible for the rapid and highly selective removal of chlorite through the catalytic action of Cld.
Materials and methods
Protein expression and purification
Protein expression and purification were carried out as previously described [21]. Briefly, the gene encoding Cld from D. aromatica with its twin arginine leader peptide removed was expressed from the pET41a vector in Tuner(DE3) Escherichia coli cells (Novagen). A single colony from a freshly streaked plate was used to grow an overnight 5-mL culture, which was inoculated 1:100 into 50 mL of terrific broth supplemented with kanamycin (50 mg mL−1) and grown on a shaker–incubator at 37 °C (New Brunswick, 250 rpm). The freshly saturated (6 h) culture was inoculated 1:100 into 1 L of terrific broth/kanamycin and the culture was grown (30 °C) to the mid-logarithmic phase (optical density at 600 nm of 0.4–0.5). Hemin, FeSO4, and isopropyl β-d-thiogalactopyranoside were then added to final concentrations of 1, 1.6, and 0.1 mM, respectively, and the temperature was reduced to 20 °C. After 16 h, the culture was centrifuged and the resulting cell pellet resuspended in 5 mL g−1 of wet cell paste of 100 mM phosphate buffer (pH 6.8). The suspension was lysed by sonication and clarified by centrifugation (41,000g for 45 min at 4 °C). The deep-red supernatant was dialyzed against 20 mM tris(hydroxymethyl)amino-methane–Cl pH 8.6 (4 °C) and loaded onto (diethyl-amino)ethyl resin (GE Biosciences) equilibrated with the same buffer. The flow-through was collected and concentrated in a nitrogen gas pressurized stirred cell to 5–15 mL. The sample was then loaded onto a 500-mL Sephacryl S-200 HR gel filtration column and run at 0.4 mL min−1 with 100 mM phosphate buffer (pH 6.8). The eluted proteins were screened by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and the pure fractions were pooled. Protein was concentrated by centrifugation at 4,000g and 4 °C using 10-kDa molecular mass cutoff Amicon concentrators. Protein aliquots were snap-frozen in liquid nitrogen and stored at −20 °C at 5–20 mg mL−1 as determined by the Bradford assay. Heme stoichiometry was determined via the pyridine hemochrome assay.
Measurement of binding constants
For each titration, an 8.0 μM enzyme sample was prepared in 150 μL of the appropriate buffer in a quartz cuvette and its spectrum was recorded. Solutions of the ligands to be titrated (nitrite and acetate) were prepared in the same buffers at 3–2,000 mM and added in 5-μL aliquots. Spectra were repeatedly measured after each addition until the samples equilibrated. When the titration appeared complete, a ligand stock of tenfold higher concentration was added in 5-μL aliquots to ensure that a clear end point had been reached. Difference spectra were generated from spectra that had been corrected for sample dilution. The wavelength of maximal absorption change was used to construct a plot of the change in absorbance versus the total added ligand concentration and this was fit by least-squares regression (Kaleidagraph) to an equilibrium isotherm of the form:
Here, [L]T is the total added ligand concentration irrespective of the ligand protonation state, KD is the apparent dissociation constant, and ΔAbs∞ is the asymptote of the rectangular hyperbola, which describes the change in absorbance in the presence of an infinite amount of added ligand. Each calculated KD from the maximal absorbance change was checked against other absorption changes, ensuring consistent determinations of KD regardless of the wavelength monitored. Since nitrite has an absorbance at 354 nm (εmax = 22.7 M−1 cm−1) [37], the changing absorbance of the Soret band owing to the conversion of unligated enzyme to ligated enzyme (386 nm) was not used because of overlap with the nitrite absorption band.
Crystallization and X-ray data collection
Cld crystals were grown in the presence of the substrate analog nitrite (NO2−) using 16–18% poly(ethylene glycol 8000), 0.2 M calcium acetate, and 0.1 M 2-(N-morpholino)ethanesulfonate (MES) buffer at pH 6.5 as previously reported [38]. Cld crystals were also grown at higher pH using 16–18% poly(ethylene glycol 8000), 0.2 M calcium acetate, and 0.1 M bicine buffer at pH 9.0.
Before X-ray data collection, Cld crystals were soaked in a solution consisting of the appropriate pH mother liquor containing 20% poly(ethylene glycol 600). X-ray data sets were collected from single crystals cooled to −173 °C at the Advanced Photon Source, Argonne National Laboratory (beamline 19-ID, Structural Biology Consortium–Collaborative Access Team) [38]. Both the pH 6.5 and 9.0 crystals had similar diffraction quality and were isomorphous (space group P212121).
Structure determination and refinement
The structure of D. aromatica Cld was solved by molecular replacement into the pH 9.0 data set. The search model consisted of monomers derived from the recent crystal structure of Azospira oryzae Cld (solved as a hexameric oligomer) [23], which is 97% sequence identical to D. aromatica Cld, inserted into a pentamer manifold derived from Thermoplasma acidophilum Cld [Protein Data Bank (PDB) entries 2vdx and 3dtz, respectively]. The program PHASER, part of the CCP4 program suite, successfully placed three pentamers within the crystallographic asymmetric unit (ASU) [39]. During refinement a fourth pentamer was located in the electron density and placed to complete the ASU. Model building was carried out using 15-fold (for the initial three pentamers identified) then 20-fold-averaged maps generated using COOT [40]. Refinement of the model was performed using REFMAC5.5 imposing 15-fold then 20-fold noncrystallographic symmetry (NCS) restraints on the peptide backbone and amino acid side chains in conjunction with TLS parameters [41, 42]. Tight NCS restraints were imposed during the early stages of model building and refinement. The NCS restraints were relaxed as model building progressed, with the final refinement cycle imposing medium NCS restraints on peptide backbone residues and loose restraints on amino acid side chains. In addition to the NCS restraints, the Fe–O bond distance between the heme iron and nitrite ion (NO2−) was restrained to 2.0 Å during refinement. Model building and refinement were deemed to be complete when all interpretable areas of the 2Fo − Fc and Fo − Fc maps were explained. Good stereochemistry and minimized R factors were used as criteria to confirm the quality of the finished model.
The pH 6.5 structure was solved by difference Fourier, and refined using the same parameters and restraints in REFMAC5.5 as those used in the final rounds of refinement for the pH 9.0 structure. The same criteria as for the pH 9.0 structure were used to decide when the pH 6.5 structure was finished.
Coordinates and structure factors have been deposited in the PDB with accession numbers 3m2q (pH 6.5) and 3m2s (pH 9.0).
Results
X-ray data collection and crystal structure overview
Diffraction data in the space group P212121 were recorded to 3.0-Å resolution for Cld/nitrite co-crystals grown at pH 9.0 (Table 1) or pH 6.5 [38]. The pH 6.5 diffraction data statistics have been published previously [38]. The data collected at either pH have similar statistics, with an overall Rmerge of 8.7–10.5%, where all shells display between 97 and 100% completeness with multiplicity between 6.7 and 8.3. Cld/nitrite co-crystals at both pH 9.0 and 6.5 provided a final structure to 3.0-Å resolution (Table 1) with four pentamers in the ASU. The crystal structures of three hemeless Cld homologs from different organisms have been determined through structural genomics initiatives (PDB codes 3dtz, 1vdh, and 1t0t) and are pentameric [43]. These homologous proteins are not expected to have Cld activity, as the organisms lack perchlorate reductase and are not known to metabolize chlorite; however, their biological function is currently unknown. De Geus et al. [23] have recently solved the crystal structure to 2.1-Å resolution of a hexameric form of a bone fide Cld from A. oryzae (PDB code 2vdx) that contained heme, but demonstrated by native mass spectrometry that the physiological oligomer in solution is indeed pentameric. In the D. aromatica Cld crystal structure reported here the oligomeric state is the physiological pentamer (Fig. 1a, Fig. S1).
Table 1.
Data collection and refinement statistics
| Data collectiona | pH 9.0 | |
|---|---|---|
| Detector type | Area Detector Systems Corporation CCD | |
| Source | Advanced Photon Source | |
| Space group | P212121 | |
| Unit cell (Å3) | 123 × 203 × 247 | |
| Wavelength (Å) | 0.9784 | |
| Resolution (Å)b | 50.00–3.00 (3.10–3.00) | |
| Measured reflections | 882,314 | |
| Unique reflections | 120,425 | |
| Completeness (%)b | 99.6 (97.2) | |
| Rmerge (%)b,c | 10.5 (34.8) | |
| I/σIb | 17.1 (4.7) | |
| Redundancyb | 7.6 (6.7) | |
| Refinement | pH 9.0 | pH 6.5 |
| Resolution (Å)b | 48.31–3.00 (3.08–3.00) | 50.0–3.04 (3.12-3.04) |
| Number of reflections; working set/test setb,d | 116,905/6,174 (8,411/445) | 110,582/5,834 (6,222/323) |
| R factorb,e | 18.8 (26.2) | 18.1 (23.5) |
| Rfreeb,d,e | 23.5 (33.5) | 21.9 (28.5) |
| Protein atoms | 39,420 | 39,420 |
| Other atoms | 100 | 180 |
| Ramachandran statisticsf | ||
| Allowed (%) | 99.5 | 99.5 |
| Disallowed (%) | 0.5 | 0.5 |
| Root mean square deviation | ||
| Bond lengths (Å) | 0.016 | 0.016 |
| Bond angles (°) | 1.6 | 1.5 |
| Average B factor (Å2) | 38.9 | 32.4 |
| DPI (Å)g | 0.4243 | 0.4451 |
| Protein Data Bank code | 3m2s | 3m2q |
Data collection statistics for pH 6.5 data can be found in Goblirsch et al. [38]
Numbers in parentheses represent values in the highest-resolution shell
Rmerge = Σi |Ihkl,i − 〈Ihkl〉|/Σhkl Σi Ihkl,i, where I is the observed intensity and 〈I〉 is the average intensity of multiple measurements
Rfree, R factor based on 5% of the data (test set) excluded from refinement. The data selected were the same for the pH 6.5 and 9.0 data sets
R factor = Σ| |Fo| − |Fc| |/Σ|Fo|, where |Fo| is the observed structure factor amplitude and |Fc| is the calculated structure factor amplitude
Based on values obtained from PROCHECK [66]
Diffraction-component precision indicator based on the R factor [67]
Fig. 1.
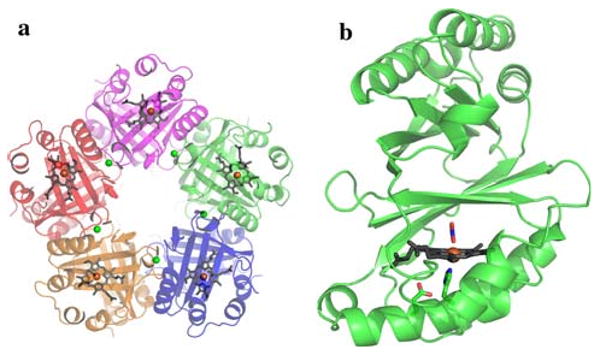
Overview of the chlorite dismutase (Cld) crystal structure. a The pentamer structure. b Cld monomer. Monomers shown as a cartoon with heme drawn in gray sticks, and the iron center represented as an orange sphere. Calcium ions are shown as green spheres. Nitrite and relevant residues are colored by atom (carbon, green). This figure was generated using PyMOL (http://www.pymol.org/)
A cation is bound at the monomer/monomer interface. As 200 mM calcium acetate was used for the crystallization, this has been assigned as a calcium ion (Fig. S2). Sequence alignment of over 300 Clds and their homologs identified Asp-192, one of the Ca2+ ligands, as highly conserved, possibly indicating a structural role for this residue. However Ca2+ is not required for pentamer formation, as the previously published structural genomics models do not have Ca2+ bound and all are pentameric in the crystal structure. Each monomer contains a single protoporphyrin IX or b-type heme coordinated by a proximal His-170, hydrogen-bonded to Glu-220 (Fig. 1b).
Structure analysis and model building
Comparing 20-fold-averaged 2Fo − Fc maps between the pH 6.5 and pH 9.0 structures reveals a region of density exclusive to the pH 6.5 model (Fig. S3). Subsequent rounds of model building followed by refinement revealed this density as a single molecule of the anionic buffer MES hydrogen-bonded to Arg-183 (Fig. S3). Outside the bound MES, the superimposed pH 6.5 and 9.0 structures are highly similar (root mean square deviation per monomer of 0.148 Å). The figures, unless noted otherwise, were generated using the pH 6.5 structure, as pH 6.5 is closer to the optimal pH for Cld activity (pH 5.2) [44].
Both unaveraged and 20-fold-averaged maps clearly identified the binding of a ligand to the distal face of the heme iron. This ligand was assigned as a nitrite ion (NO2−) (Fig. 2a). Formation of a Cld/nitrite complex leads to a redshift in the UV/vis spectrum of the Soret band (Fig. S4), and nitrite binds Cld with a KD of 1,120 ± 50 μM at pH 6.5 (Fig. S5a). Two different binding modes of nitrite have been crystallographically characterized in other heme proteins; N binding and O-monodentate binding [45–47]. Owing to the medium resolution of the D. aromatica Cld crystal structure, both N binding and O-monodentate binding as well as O,O-bidentate nitrite binding modes were modeled and subsequently refined against the Cld data. Only the O-monodentate binding model (nitrito ligation) adequately explained the heme distal electron density, and resulted in no steric clashes with the nearby Arg-183 (Fig. 2a). Acetate, a component of the crystal growth medium, is also capable of binding to Cld (Fig. S4). However, acetate binding (KD = 36,000 ± 2,000 μM, pH 6.5, Fig. S5b) under the crystal growth conditions (7.8-fold excess of acetate over nitrite) is unlikely to be a major component of the distally bound species, and this is consistent with the triatomic shape of the electron density.
Fig. 2.
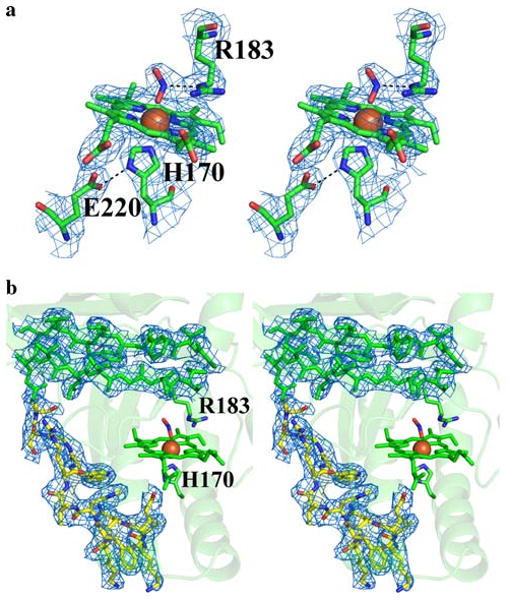
Stereo images of Cld 2Fo − Fc electron density. The blue mesh represents the 20-fold-averaged 2Fo − Fc map contoured at 3.0σ. a Cld active site at pH 6.5. Nitrite is modeled as a direct ligand bound end-on to the heme (nitrito ligation). The model is drawn as sticks and colored by atom (carbon, green). b The newly modeled residues 218–230 drawn as sticks and colored by atom (carbon, yellow). In addition, part of a β-sheet is displayed to show the general quality of the fit between the electron density and the model. The model is drawn as sticks and colored green. Iron is drawn as an orange sphere. The heme porphyrin is drawn as sticks and colored green. Arg-183 and His-170 are drawn as sticks and colored by atom (carbon, green). This figure was generated using PyMOL (http://www.pymol.org/)
The initial 20-fold-averaged 2Fo − Fc maps clearly showed the presence of a 13-residue stretch that was disordered, and thus absent, in the original 2vdx search model (residues 218–230). Subsequent refinement confirmed the presence of the missing fragment (Fig. 2b), which contributes significantly to the formation of the proximal heme pocket. In particular, Glu-220 within this stretch of residues is strictly conserved amongst bone fide Clds, and forms a hydrogen bond with the proximal His-170 ligand to heme iron (Fig. 2a). Hydrogen bonding of Glu-220 to the imidazole of His-170 lends anionic/imidazolate character to the proximal ligand (Scheme 1) [48]. A similar aspartate/histidine motif is widely observed in heme peroxidases [49], and has been shown to stabilize the iron in its reactive five-coordinate high-spin ferric state.
Discussion
The structure of D. aromatica Cld reveals a sequestered distal heme environment promoting the efficient and specific conversion of chlorite to Cl− and O2. Heme-based peroxidases and catalases depend on broadly conserved arginine/histidine or asparagine/histidine residues in their distal pockets for positioning substrates, stabilizing charge, and facilitating bond cleavage [24, 49–51]. The histidine residues have a further role as active-site bases toward H2O2 (pKa > 11). In Cld, a single Arg-183 is the only apparent ionizable residue in the distal pocket, and has one face of the side chain primarily in contact with hydrophobic side chains, whereas the other is solvent-exposed (Figs. 2a, 3). Although the Cld reaction is not expected to require base catalysis (HClO2/ClO2− pKa = 1.82), Arg-183 may fulfill any number of other chemical roles that are analogous to those described for peroxidases, such as the well-studied Arg-38 in horseradish peroxidase isoenzyme C shown to be critical for substrate positioning, polarization of the peroxide O–O bond, and compound I stabilization [52–54]. In addition, it likely acts as a gate controlling the entry of substrate chlorite into the distal pocket, and thus reactivity at the iron center. The Cld crystal structure from A. oryzae has a solvent-accessible heme cofactor, which the authors suggest allows free diffusion of substrate to the enzyme active site (Fig. 4a) [23]. However, the 13-residue stretch that is disordered in the A. oryzae structure, but ordered in the D. aromatica structure (residues 221–228; Fig. 2b), completely sequesters the heme from solvent (Fig. 4b). Examination of D. aromatica Cld suggests access to the heme distal pocket is likely via increased flexibility of the Arg-183 side chain in the absence of a suitable distally bound heme ligand (Fig. 3). Unusually, the Arg-183 side chain makes no hydrogen bonds or salt bridges with surrounding protein residues, only the heme-bound nitrito, and one face of the side chain is directly solvent accessible. As such, Arg-183 is likely to have considerable mobility in the absence of an interaction with a suitable heme iron distal ligand. This may be a factor contributing to the poor diffraction quality of Cld crystals in the absence of bound ligand [38]. Direct and unhindered binding of substrate to the iron is supported by steady-state kinetic data for Cld, which exhibits simple saturation kinetics, with the Michaelis complex being modeled as a collision complex between chlorite and the enzyme [21].
Fig. 3.
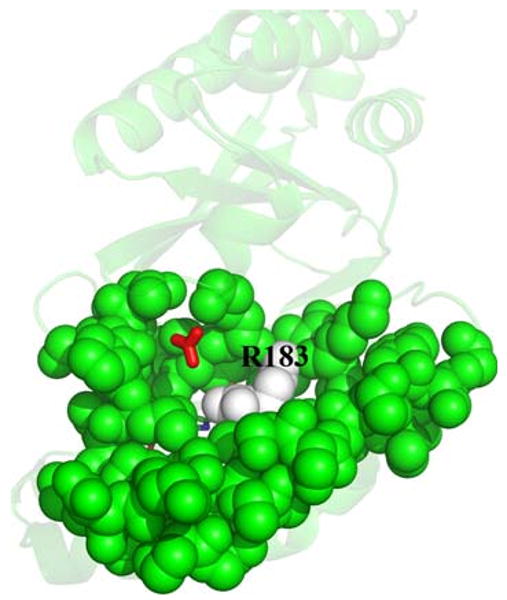
Proposed Arg-183 gate. Surface residues close to the heme active site are drawn as solvent-accessible green spheres except for Arg-183, which is drawn as white spheres. The heme cofactor and bound nitrite are drawn as sticks colored by atom, and lie directly behind Arg-183. A hydrogen carbonate molecule was superimposed from the Azospira oryzae Cld model (Protein Data Bank entry 2vxh) [23] onto the Dechloromonas aromatica Cld model using COOT [40] and is drawn as red sticks. This figure was generated using PyMOL (http://www.pymol.org/)
Fig. 4.
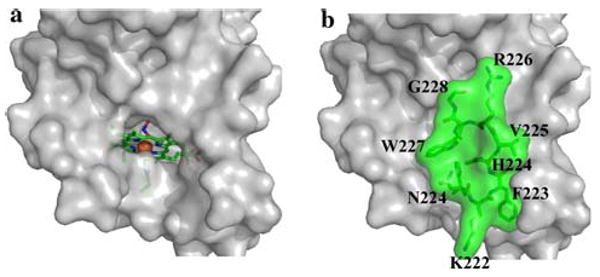
Solvent-sequestered Cld active site. a Solvent-accessible surface of Cld without residues 221–228. b Solvent-accessible surface of the D. aromatica Cld crystal structure, including residues 221–228 (green) that are defined in D. aromatica Cld. The orientation for a and b is identical. Residues surrounding the active site are shown as a gray surface, except residues 221–228, explicitly drawn as green sticks with a green surface. The heme proximal ligand, His-170, and distally bound nitrito ligand are shown as sticks, and the iron center of the heme is shown as an orange sphere. This figure was generated using PyMOL (http://www.pymol.org/)
Once a ligand is coordinated to the heme and Arg-183 mobility is restricted, the distal pocket becomes sequestered from solvent (Fig. 5). In the crystal structure presented here, nitrite, an analogue of substrate chlorite (ClO2−), is modeled as binding to the iron via a single oxygen atom (nitrito ligation), with the nitrogen atom hydrogen-bonded to Arg-183 (Fig. 2a). An analogous coordination mode for chlorite would appear ideal for polarizing and thereby promoting heterolytic cleavage of the O–Cl bond to generate compound I and hypochlorite. Owing to the nature of chlorite, the preferred binding geometry for the substrate could involve rotation around the O–Fe coordination bond, for example by chlorite 180° compared to that observed for nitrite (Fig. 2a). Modeling of chlorite in this alternative geometry into the Cld structure is sterically compatible, and enables hydrogen bonding of the second oxygen atom with Arg-183. Interestingly, the pH 6.5 structure shows Arg-183 hydrogen-bonded to both the nitrito ligand and an anionic buffer molecule poised at the threshold to the active site (Fig. S3). At pH 9, Arg-183 interacts solely with the nitrito ligand. These observations are consistent with an extensive pH-dependent spectroscopic and reactivity study of Cld which showed an enzyme-associated pKa near pH 6.5 [44]. This was attributed to loss of a proton and hence positive charge by Arg-183. The protonated, more electropositive Arg-183 might be expected to be able to accommodate hydrogen-bonding interactions with both anions. This form of the enzyme was also more catalytically active.
Fig. 5.
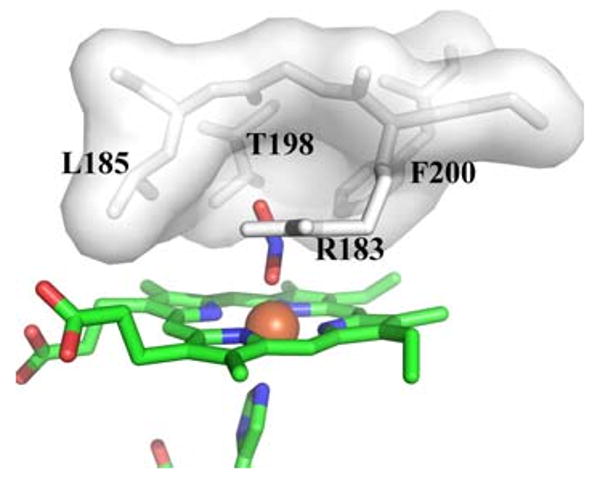
Distal pocket of Cld. Residues lining the distal pocket are shown as white sticks with a transparent surface, except for Arg-183. The heme and ligands are drawn as sticks colored by atom (carbon, green), with the iron as an orange sphere. This figure was generated using PyMOL (http://www.pymol.org/)
After generation of compound I and ClO−, the Cld mechanism proposed by Streit and DuBois [21] requires rebound of ClO− to the compound I intermediate (Scheme 1). The tight distal pocket provided by Leu-185, Thr-198, Phe-200, and Arg-183 would be expected to trap hypochlorite in proximity to the highly reactive compound I center. Such a structural motif is consistent with the specificity of Cld for its small substrate, and the lack of detectable hypochlorite escape during catalytic turnover [21]. Thermal fluctuations allowing ClO− to sample alternative orientations within this confined rebound space likely ensure rapid O–O bond formation once hypochlorite is correctly positioned for nucleophilic attack (Scheme 1). Furthermore, side chains from Leu-185, Thr-198, and Phe-200 also make the tight distal pocket relatively hydrophobic (the hydroxyl group of the Thr-198 side chain points away from the distal pocket and is hydrogen-bonded to a neighboring residue). As hypochlorite has an appreciable dipole moment (2.27 D) about its Cl–O bond [55], orientation of the chlorine atom toward this hydrophobic portion of the distal pocket may be energetically favored. This would further help reposition the hypochlorite oxygen atom for nucleophilic attack on the oxo group of compound I. The structure of A. oryzae Cld contained hydrogen carbonate bound to the surface of the protein close to Arg-183 [23]. This led De Geus et al. [23] to suggest this surface position as a possible rebound pocket for hypochlorite. However, an overlay of the hydrogen carbonate position on the current Cld structure shows the binding site is well removed from the reactive iron center (approximately 11 Å), and would require protein motion to enable transfer from the sequestered distal pocket to this surface site (Fig. 3). Such a remote and solvent-exposed rebound pocket for hypochlorite appears unlikely given the expected efficient rebound with compound I and the evidence for lack of hypochlorite escape. Keeping the hypochlorite within the tight confines of the heme distal pocket would be more consistent with the experimental data regarding Cld catalysis.
The small sequestered Cld distal pocket would appear to be a key feature in explaining both the rapid turnover and the substrate specificity of Cld (kcat/Km = 3.5 × 107 M−1 s−1, 4 °C, pH 6.8) compared with other heme enzymes [21]. The larger distal pockets of peroxygenases and some catalases permit access of a second cosubstrate molecule to the compound I oxidant [56–58]. Access to compound I in peroxidases is sterically restricted, causing substrates to bind and react near the δ-heme (or in a few cases γ-heme) edge [59]. When Cld reacts with chlorite in the presence of potential cosubstrates, the decomposition of chlorite is overwhelmingly favored, with little or no oxidation of the second molecule [22]. Unlike a peroxidase, peroxygenase, or catalase, all of which manage both the generation of an oxidizing species and its reaction with a cosubstrate, Cld generates compound I and hypochlorite in tandem. Tight linkage between compound I formation and hypochlorite rebound are likely responsible for the highly faithful specificity of Cld. The proposed rebound of hypochlorite with compound I within the sequestered distal pocket would rapidly complete the reaction (Scheme 1) and prohibit reduction of the high-valent iron intermediate by external molecules. A recent attempt to generate iron–porphyrin model complexes capable of efficiently producing O2 from chlorite had limited success [60]. The major reason for the poor yield of O2 was attributed to hypochlorite escape after compound I formation allowing comproportionation of compound I with the unoxidized iron–porphyrin complexes to create a new catalytic population incapable of O2 production.
As with other heme enzymes that react with oxidants, Cld is known to lose activity after a certain number of turnovers. EPR studies suggest that Cld inactivation occurs through the initial formation of an off-pathway tryptophanyl radical species, eventually leading to heme degradation [22]. The radical species is produced by the migration of an electron from surrounding tryptophan residue(s) to the compound I cation porphyrin radical. The crystal structure of A. oryzae Cld identified a highly conserved tryptophan residue in Cld homologs that is adjacent to the heme, and is strictly conserved in Clds from perchorate/chlorate respirers [23]. The corresponding Trp-155 in D. aromatica Cld occupies a similar position and has been implicated in forming the observed organic radical [22, 23]. Inspection of the active-site pocket in D. aromatica Cld suggests two other tryptophan residues (Trp-156, Trp-227) as possible candidates (Fig. 6a). Although Trp-156 and Trp-227 are not highly conserved across all Cld homologs, they are conserved across Clds from known perchlorate/chlorate respirers. Trp-156 is juxtaposed to Trp-155, and was also identified as a possible radical site in the A. oryzae Cld crystal structure. The proximity of these two residues could allow for delocalization of the radical species between them. Trp-227, in the proximal heme pocket (Fig. 6a), is part of the 13-residue stretch that was ordered in the D. aromatica Cld structure compared with the A. oryzae Cld structure. This Trp-227 could undergo one-electron oxidation via a hydrogen-bonding network originating at His-170, the proximal ligand to the iron. The 20-fold-averaged Fo − Fc map displayed a strong, spherical peak within the proximal pocket that has been modeled as water (B factor of 22.7 Å2) (Fig. 6b). Refinement of water at this position does not return positive difference electron density, which might suggest the presence of a more-electron-rich molecule or ion. In a 3.0-Å-resolution crystal structure, the ability to model and refine a water molecule suggests high occupancy and little disorder. Thus, the most direct pathway of electron transfer to Trp-227 involves residues His-170, Glu-220, and His-224, and a water bridging His-224 and Glu-220 (Fig. 6b). In cytochrome c peroxidase (PDB entry 16sv) [61], the radical-forming tryptophan residue resides in a hydrogen-bonding network with heme-bound histidine and aspartate residues positioned similarly to residues His-170 and Glu-220 in Cld [62, 63], and these hydrogen bonds are known to be key for tryptophanyl radical formation in cytochrome c peroxidase [64].
Fig. 6.
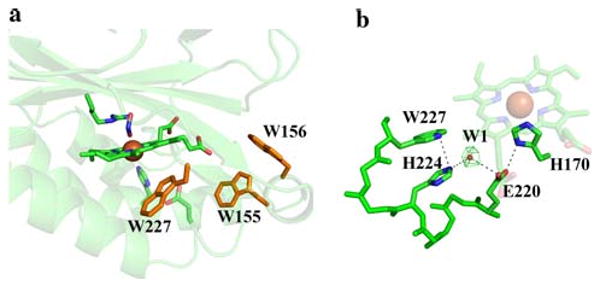
Possible sites for a tryptophan radical. a Three tryptophan residues that might be involved in radical formation are shown as orange sticks. The heme, ligands, and Arg-183 are drawn as sticks colored by atom (carbon, green), with the iron as an orange sphere. b Possible electron relay involving Trp-227. The green mesh represents a 20-fold-averaged Fo − Fc peak at 11σ modeled as water shown as a red sphere. The heme and residues are drawn as sticks colored by atom (carbon, green), with the iron as an orange sphere. This figure was generated using PyMOL (http://www.pymol.org/)
Residues immediately surrounding the active site suggest that following completion of the enzymatic cycle there could be a preferred exit direction for molecular oxygen that is distinct from the proposed entry point of chlorite. The apolar O2 product would be expected to preferentially take a hydrophobic route, but identification of O2 channels in enzymatic systems is challenging. A recent study of copper amine oxidase homologs in complex with xenon (an analog for O2) suggested that multiple dynamic pathways probably exist [65]. These were constantly changing as the enzymes evolved, being restrained only by the common tertiary structure that dictated the hydrophobic interactions between secondary structures. In Cld such an area exists between a β-sheet and an α-helix that leads from the distal pocket to the protein surface (Fig. 7). This area abuts Phe-200 of the distal pocket, and is in a direction orthogonal to the postulated place of substrate entry at Arg-183 (Fig. 7a). The opposite side of the enzyme has a predominance of polar residues between the distal pocket and the solvent, which would be an unfavorable environment for O2 (Fig. 7b). Some directional control of O2 expulsion from Cld may be critical as all perchlorate-respiring bacteria are either facultatively anaerobic or microaerophilic, and perchlorate respiration is downregulated in the presence of O2 [9]. However, the ultimate fate of Cld-generated O2 in these organisms is unknown.
Fig. 7.
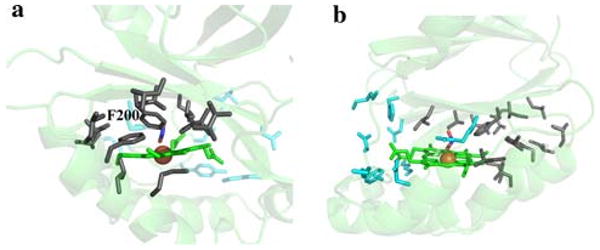
Putative O2 exit direction. a Polar residues are drawn in cyan sticks and nonpolar residues are drawn as gray sticks. The heme is colored green, the nitrito ligand is in atom colors, and the iron is shown as an orange sphere. b A 90° rotation about an axis perpendicular to the heme plane. Coloring as in a. This figure was generated using PyMOL (http://www.pymol.org/)
Cld and photosystem II are the only enzymatic systems known to efficiently catalyze O–O bond formation. This is a reaction that is difficult to catalyze synthetically, even with chemically active oxygen atom donors. The crystal structure of Cld suggests that the speed and specificity of the Cld-catalyzed reaction lies in the ability of a tightly sequestered distal pocket to generate the electrophilic and nucleophilic oxygen atoms of compound I and hypochlorite, respectively, and to control their reaction to give products.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (R01 GM-66569 to C.M.W.; R03 ES-14390 and R01 GM-90260 to J.L.D.), and a Minnesota Partnership for Biotechnology and Medical Genomics grant SPAP-05-0013-P-FY06 to C.M.W. B.R.S. was supported by an Environmental Protection Agency STAR fellowship (FP-91690601-0). Computer resources were provided by the Basic Sciences Computing Laboratory of the University of Minnesota Supercomputing Institute, and we thank Can Ergenekan for his support. X-ray data were collected at the Kahlert Structural Biology Laboratory (KSBL) at The University of Minnesota and beamline 19-ID, Structural Biology Consortium–Collaborative Access Team, at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL, USA). Argonne National Laboratory is operated by University of Chicago Argonne LLC for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Ed Hoeffner for KSBL support and Steve Ginell and the staff at Sector 19, Advanced Photon Source, for their support.
Abbreviations
- ASU
Asymmetric unit
- Cld
Chlorite dismutase
- MES
2-(N-Morpholino)ethanesulfonate
- NCS
Noncrystallographic symmetry
- PDB
Protein Data Bank
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00775-010-0651-0) contains supplementary material, which is available to authorized users.
Contributor Information
Brandon R. Goblirsch, Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA
Bennett R. Streit, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA
Jennifer L. DuBois, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA
Carrie M. Wilmot, Email: wilmo004@umn.edu, Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA
References
- 1.Liu S, Suflita JM. Trends Biotechnol. 1993;11:344–352. doi: 10.1016/0167-7799(93)90157-5. [DOI] [PubMed] [Google Scholar]
- 2.Wackett LP. J Biol Chem. 2004;279:41259–41262. doi: 10.1074/jbc.R400014200. [DOI] [PubMed] [Google Scholar]
- 3.Wackett LP, Hershberger CD. Biocatalysis and biodegradation : microbial transformation of organic compounds. ASM Press; Washington: 2001. [Google Scholar]
- 4.Oremland RS, Kulp TR, Blum JS, Hoeft SE, Baesman S, Miller LG, Stolz JF. Science. 2005;308:1305–1308. doi: 10.1126/science.1110832. [DOI] [PubMed] [Google Scholar]
- 5.Oremland RS, Stolz JF. Science. 2003;300:939–944. doi: 10.1126/science.1081903. [DOI] [PubMed] [Google Scholar]
- 6.Stolz JF, Basu P, Santini JM, Oremland RS. Annu Rev Microbiol. 2006;60:107–130. doi: 10.1146/annurev.micro.60.080805.142053. [DOI] [PubMed] [Google Scholar]
- 7.Narasingarao P, Haggblom MM. Appl Environ Microbiol. 2007;73:3519–3527. doi: 10.1128/AEM.02737-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kashiwa M, Nishimoto S, Takahashi K, Ike M, Fujita M. J Biosci Bioeng. 2000;89:528–533. doi: 10.1016/s1389-1723(00)80051-1. [DOI] [PubMed] [Google Scholar]
- 9.Coates JD, Achenbach LA. Nat Rev Microbiol. 2004;2:569–580. doi: 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- 10.Coates JD, Chakraborty R, Lack JG, O'Connor SM, Cole KA, Bender KS, Achenbach LA. Nature. 2001;411:1039–1043. doi: 10.1038/35082545. [DOI] [PubMed] [Google Scholar]
- 11.Maixner F, Wagner M, Lucker S, Pelletier E, Schmitz-Esser S, Hace K, Spieck E, Konrat R, Le Paslier D, Daims H. Environ Microbiol. 2008;10:3043–3056. doi: 10.1111/j.1462-2920.2008.01646.x. [DOI] [PubMed] [Google Scholar]
- 12.Danielsson H, Stenklo TK, Karlsson J, Nilsson T. Appl Environ Microbiol. 2003;69:5585–5592. doi: 10.1128/AEM.69.9.5585-5592.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kengen SWM, Rikken GB, Hagen WR, Van Ginkel CG, Stams AJM. J Bacteriol. 1999;181:6706–6711. doi: 10.1128/jb.181.21.6706-6711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okeke BC, Frankenberger WT., Jr Microbiol Res. 2003;158:337–344. doi: 10.1078/0944-5013-00213. [DOI] [PubMed] [Google Scholar]
- 15.O'Connor SM, Coates JD. Appl Environ Microbiol. 2002;68:3108–3113. doi: 10.1128/AEM.68.6.3108-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewson WD, Hager LP. J Biol Chem. 1979;254:3175–3181. [PubMed] [Google Scholar]
- 17.Jakopitsch C, Spalteholz H, Furtmuller PG, Arnhold J, Obinger C. J Inorg Biochem. 2008;102:293–302. doi: 10.1016/j.jinorgbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Shahangian S, Hager LP. J Biol Chem. 1981;256:6034–6040. [PubMed] [Google Scholar]
- 19.Hollenberg PF, Rand-Meir T, Hager LP. J Biol Chem. 1974;249:5816–5825. [PubMed] [Google Scholar]
- 20.George P. J Biol Chem. 1953;201:413–426. [PubMed] [Google Scholar]
- 21.Streit BR, DuBois JL. Biochemistry. 2008;47:5271–5280. doi: 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AQ, Streit BR, Zdilla M, Abu-Omar MA, DuBois JL. Proc Natl Acad Sci USA. 2008 doi: 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Geus DC, Thomassen EA, Hagedoorn PL, Pannu NS, van Duijn E, Abrahams JP. J Mol Biol. 2009;387:192–206. doi: 10.1016/j.jmb.2009.01.036. [DOI] [PubMed] [Google Scholar]
- 24.Poulos TL. Curr Opin Biotechnol. 1993;4:484–489. doi: 10.1016/0958-1669(93)90016-p. [DOI] [PubMed] [Google Scholar]
- 25.Everse J, Everse KE, Grisham MB, editors. Peroxidases in chemistry and biology. Vol. 1. CRC Press; Boca Raton: 1990. [Google Scholar]
- 26.Jones P, Dunford HB. J Theor Biol. 1977;69:457–470. doi: 10.1016/0022-5193(77)90152-7. [DOI] [PubMed] [Google Scholar]
- 27.Palcic MM, Dunford HB. J Biol Chem. 1980;255:6128–6132. [PubMed] [Google Scholar]
- 28.Matsunaga I, Sumimoto T, Ayata M, Ogura H. FEBS Lett. 2002;528:90–94. doi: 10.1016/s0014-5793(02)03261-1. [DOI] [PubMed] [Google Scholar]
- 29.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 30.Savenkova MI, Kuo JM, Ortiz de Montellano PR. Biochemistry. 1998;37:10828–10836. doi: 10.1021/bi9725780. [DOI] [PubMed] [Google Scholar]
- 31.Newmyer SL, Ortiz de Montellano PR. J Biol Chem. 1995;270:19430–19438. doi: 10.1074/jbc.270.33.19430. [DOI] [PubMed] [Google Scholar]
- 32.Hager LP, Doubek DL, Silverstein RM, Hargis JH, Martin JC. J Am Chem Soc. 1972;94:4364–4366. doi: 10.1021/ja00767a068. [DOI] [PubMed] [Google Scholar]
- 33.Araiso T, Rutter R, Palcic MM, Hager LP, Dunford HB. Can J Biochem. 1981;59:233–236. doi: 10.1139/o81-031. [DOI] [PubMed] [Google Scholar]
- 34.Bakkenist AR, de Boer JE, Plat H, Wever R. Biochim Biophys Acta. 1980;613:337–348. doi: 10.1016/0005-2744(80)90088-1. [DOI] [PubMed] [Google Scholar]
- 35.Ullrich R, Hofrichter M. Cell Mol Life Sci. 2007;64:271–293. doi: 10.1007/s00018-007-6362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toy PH, Newcomb M, Hager LP. Chem Res Toxicol. 1998;11:816–823. doi: 10.1021/tx9800295. [DOI] [PubMed] [Google Scholar]
- 37.Jankowski JJ, Kieber DJ, Mopper K. Photochem Photobiol. 1999;70:319–328. doi: 10.1562/0031-8655(2000)071<0431:daious>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 38.Goblirsch BR, Streit BR, DuBois JL, Wilmot CM. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2009;65:818–821. doi: 10.1107/S1744309109026785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collaborative Computational Project N. Acta Crystallogr Sect D Biol Crystallogr. 1994;50:760–763. [Google Scholar]
- 40.Emsley P, Cowtan K. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 42.Winn MD, Isupov MN, Murshudov GN. Acta Crystallogr Sect D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 43.Ebihara A, Okamoto A, Kousumi Y, Yamamoto H, Masui R, Ueyama N, Yokoyama S, Kuramitsu S. J Struct Funct Genomics. 2005;6:21–32. doi: 10.1007/s10969-005-1103-x. [DOI] [PubMed] [Google Scholar]
- 44.Streit BR, Blanc B, Lukart-Rodgers GS, Rodgers KL, DuBois JL. J Am Chem Soc. 2010 doi: 10.1021/ja9082182. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yi J, Heinecke J, Tan H, Ford PC, Richter-Addo GB. J Am Chem Soc. 2009;131(50):18119–18128. doi: 10.1021/ja904726q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams PA, Fulop V, Garman EF, Saunders NF, Ferguson SJ, Hajdu J. Nature. 1997;389:406–412. doi: 10.1038/38775. [DOI] [PubMed] [Google Scholar]
- 47.Yi J, Safo MK, Richter-Addo GB. Biochemistry. 2008;47:8247–8249. doi: 10.1021/bi801015c. [DOI] [PubMed] [Google Scholar]
- 48.Valentine JS, Sheridan RP, Allen LC, Kahn PC. Proc Natl Acad Sci USA. 1979;76:1009–1013. doi: 10.1073/pnas.76.3.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poulos TL, Fenna RE. Met Ions Biol Syst. 1994;30:25–75. [Google Scholar]
- 50.Candeias LP, Folkes LK, Wardman P. Biochemistry. 1997;36:7081–7085. doi: 10.1021/bi970384e. [DOI] [PubMed] [Google Scholar]
- 51.Poulos TL, Finzel BC. Pept Protein Rev. 1984;4:115–171. [Google Scholar]
- 52.Rodriguez-Lopez JN, Smith AT, Thorneley RN. J Biol Chem. 1996;271:4023–4030. doi: 10.1074/jbc.271.8.4023. [DOI] [PubMed] [Google Scholar]
- 53.Rodriguez-Lopez JN, Smith AT, Thorneley RN. J Biol Chem. 1997;272:389–395. doi: 10.1074/jbc.272.1.389. [DOI] [PubMed] [Google Scholar]
- 54.Henriksen A, Schuller DJ, Meno K, Welinder KG, Smith AT, Gajhede M. Biochemistry. 1998;37:8054–8060. doi: 10.1021/bi980234j. [DOI] [PubMed] [Google Scholar]
- 55.Midda S, Das AK. Theochem. 2005;713:101–106. [Google Scholar]
- 56.Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- 57.Fita I, Rossmann MG. J Mol Biol. 1985;185:21–37. doi: 10.1016/0022-2836(85)90180-9. [DOI] [PubMed] [Google Scholar]
- 58.Smith AT, Veitch NC. Curr Opin Chem Biol. 1998;2:269–278. doi: 10.1016/s1367-5931(98)80069-0. [DOI] [PubMed] [Google Scholar]
- 59.Ortiz de Montellano PR, Choe YS, DePillis G, Catalano CE. J Biol Chem. 1987;262:11641–11646. [PubMed] [Google Scholar]
- 60.Zdilla MJ, Lee AQ, Abu-Omar MM. Inorg Chem. 2009;48:2260–2268. doi: 10.1021/ic801681n. [DOI] [PubMed] [Google Scholar]
- 61.Pelletier H, Kraut J. Science. 1992;258:1748–1755. doi: 10.1126/science.1334573. [DOI] [PubMed] [Google Scholar]
- 62.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Science. 1989;245:738–740. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 63.Bonagura CA, Bhaskar B, Shimizu H, Li H, Sundaramoorthy M, McRee DE, Goodin DB, Poulos TL. Biochemistry. 2003;42:5600–5608. doi: 10.1021/bi034058c. [DOI] [PubMed] [Google Scholar]
- 64.Goodin DB, McRee DE. Biochemistry. 1993;32:3313–3324. [PubMed] [Google Scholar]
- 65.Johnson BJ, Cohen J, Welford RW, Pearson AR, Schulten K, Klinman JP, Wilmot CM. J Biol Chem. 2007;282:17767–17776. doi: 10.1074/jbc.M701308200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:282. [Google Scholar]
- 67.Cruikshank DW. In: International tables for crystallography. Rossmann MG, Arnold E, editors. Springer; New York: 2006. pp. 403–418. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.