

Abstract
Salt retention as a result of chronic, excessive dietary salt intake, is widely accepted as one of the most common causes of hypertension. In a small minority of cases, enhanced Na+ reabsorption by the kidney can be traced to specific genetic defects of salt transport, or pathological conditions of the kidney, adrenal cortex, or pituitary. Far more frequently, however, the salt retention may be the result of minor renal injury or small genetic variation in renal salt transport mechanisms. How the salt retention actually leads to the increase in peripheral vascular resistance (the hallmark of hypertension) and the elevation of blood pressure remain an enigma. Here we review the evidence that endogenous ouabain (an adrenocortical hormone), arterial smooth muscle α2 Na+ pumps, type-1 Na/Ca exchangers, and receptor- and store-operated Ca2+ channels play key roles in the pathway that links salt to hypertension. We discuss cardenolide structure-function relationships in an effort to understand why prolonged administration of ouabain, but not digoxin, induces hypertension, and why digoxin is actually anti-hypertensive. Finally, we summarize recent observations which indicate that ouabain upregulates arterial myocyte Ca2+ signaling mechanisms that promote vasoconstriction, while simultaneously downregulating endothelial vasodilator mechanisms. In sum, the reports reviewed here provide novel insight into the molecular mechanisms by which salt retention leads to hypertension.
Keywords: Salt-dependent hypertension, Calcium, Sodium Pump, Sodium/Calcium Exchanger, Receptor-operated Channel
1. Introduction
Hypertension, or chronic high blood pressure (BP) is a major contributor to ischemic heart disease, cerebrovascular disease, heart failure and renal failure, and is estimated to cause more than 7 million premature deaths per year worldwide [1]. Appropriate treatment, and even prevention, of hypertension depends upon better understanding of the underlying causes and mechanisms of the elevated BP. Despite extensive research during the past few decades, some critical questions about the pathogenesis of elevated BP remain unanswered. Here, we focus on recent findings that provide novel insight into the linkage between salt retention and hypertension.
2. Kidneys, salt retention and hypertension
The kidneys, which play the primary role in salt and water balance, have long been at the center of hypertension research. The kidney glomeruli of a 70 kg man filter ∼25,000 mEq of Na+ and 180 liters of water per day, and then reabsorb nearly 99.5% of this filtered load by a variety of Na+ transport mechanisms [2]. It is not surprising, therefore, that defects in any mechanisms that alter renal Na+ transport may contribute to the net gain or loss of salt (and water).
The association of hypertension with renal parenchymal diseases [3, 4], monogenic diseases of renal salt transport [5-7] and renal transplant studies [8-10], as well as Guyton's seminal work on the “over-riding dominance of the kidneys” in controlling BP [11-13], all point to the critical role of the kidneys in hypertension. Likewise, epidemiological studies as well as acute and chronic dietary studies [14, 15], volume expansion studies [13, 16-18], the efficacy of diuretic therapy [19], and monogenic diseases of renal salt transport [5, 6, 20], all indicate that NaCl retention and a tendency toward plasma volume expansion [21] play a fundamental role in the chronic elevation of BP. Conversely, genetic defects that reduce salt retention, such as those associated with Bartter's and Gittelman's syndromes, tend to lower BP and protect against the development of hypertension [22]. Nevertheless, the specific mechanism(s) responsible for salt retention in most forms of human essential hypertension (EH) is(are) unresolved. Perhaps subtle renal damage [23], which increases with age, including that which may result from obesity [24, 25], causes the salt retention. Importantly, any of several genetic variants (single nucleotide polymorphisms, SNPs), such as those in G-protein coupled receptor kinase type 4 [26-28], alpha-adducin [29, 30], or serine/threonine kinase (STK39) [31] genes may favor salt retention by the kidneys and, therefore, predispose the bearers of these genes to salt-dependent hypertension. It is apparent, however, that in virtually all of these situations, extracellular fluid (ECF) neither progressively increases nor decreases. Instead, homeostatic physiological (feedback) mechanisms come into play to protect against large ECF volume changes [21]. As we shall see, some of these mechanisms may alter BP to defend against changes in plasma (and ECF) volume.
3. Vascular tone
A related, unresolved issue in hypertension, and our main focus, is the specific mechanism(s) or “signaling pathway” by which salt retention actually elevates BP [32]. To explore this issue, we begin with some basic hemodynamic principles: Mean arterial BP is a function of cardiac output (CO), heart rate (HR), stroke volume (SV) and total peripheral vascular resistance (TPR) [33]. At constant CO, mean BP ≈ CO × TPR. CO, which is equal to HR × SV, is, in turn, directly related to the ECF volume and the volume of venous return to the heart. TPR is regulated dynamically by vasoconstriction/dilation in small “resistance” arteries by three groups of mechanisms: baroreflexes and other neuro-humoral mechanisms, endothelial mechanisms, and myogenic mechanisms [33]. The local (myocyte and endothelial) factors that maintain tonic arterial constriction, or ‘tone’, can be studied in isolated, cannulated small arteries. These arteries develop spontaneous ‘myogenic’ tone (MT) when the lumen is pressurized [34, 35]. Indeed, the level of tone in isolated arteries “is often comparable to that observed in the same vessels in vivo” [34, 35], and may even be used to predict BP changes [36].
Hypertension has often been associated with structural changes that decrease the lumen-to-wall thickness ratio and increase wall stiffness [37-39] due to vascular remodeling [40, 41]. It is not clear, however, whether this vascular remodeling is usually the cause or the effect of the hypertension. Recently, we reported that, in some hypertension models, most of the increase in TPR can be attributed to functional, and not structural, alterations in small resistance arteries [42]. Here, we will explore the basis of the dynamic, reversible arterial functional changes, the augmented tone and contractile responses that are observed in hypertension [43, 44]. To understand the generation of vascular tone, it is prudent to examine the fundamental mechanisms that influence arterial myocyte contraction. We will start with the mechanisms that regulate myocyte Ca2+ because contraction is activated by a rise in the cytosolic Ca2+ concentration, [Ca2+]CYT [45].
4. Ca2+ homeostasis and arterial constriction
Arterial constriction/dilation and, thus, BP are under neural control, and are also regulated by various endocrine and paracrine substances. Especially noteworthy is the role of the endothelium, which normally tends to restrict excessive vasoconstriction by secretion of nitric oxide (NO) and other vasodilatory factors [46]. In small “resistance arteries”, MT induced by intra-vascular pressure [34, 35], plays a key role in controlling BP.
At the cellular level, contraction depends directly on [Ca2+]CYT and the activation of myosin light chain kinase by Ca2+-calmodulin, as well as on modulation of the contractile apparatus' sensitivity to Ca2+ (e.g., by Rho/Rho kinase) [47, 48]. Myocyte [Ca2+]CYT is regulated by various Ca2+ entry, exit and storage systems [45]. Ca2+ enters myocytes from the ECF through voltage-gated, receptor-operated, store-operated and stretch-activated channels (VGCs, ROCs, SOCs and SACs, respectively; see Fig. 1). Most of the myocyte Ca2+ is sequestered in the sarcoplasmic reticulum (SR) by the sarco-/endoplasmic reticulum Ca2+ pump (SERCA). Myocytes can be activated by various hormones and neurotransmitters. For example, stimulation of the sympathetic nerves that innervate the arteries releases norepinephrine (NE), ATP and neuropeptide Y, all of which contribute to activation of the myocytes [49]. Myocytes can also be activated by increased intra-lumenal pressure and wall tension; this opens cation-selective SACs, which depolarize the myocytes, thereby opening Ca2+-selective VGCs. Neurotransmitter release, as well as NO release by the endothelium, are also activated by a rise in [Ca2+]CYT in the respective cell types, but the neurotransmitters promote myocyte contraction, while NO antagonizes contraction. Nevertheless, Ca2+ homeostasis in neurons and endothelial cells utilizes many of the same mechanisms that operate in arterial myocytes.
Fig. 1.
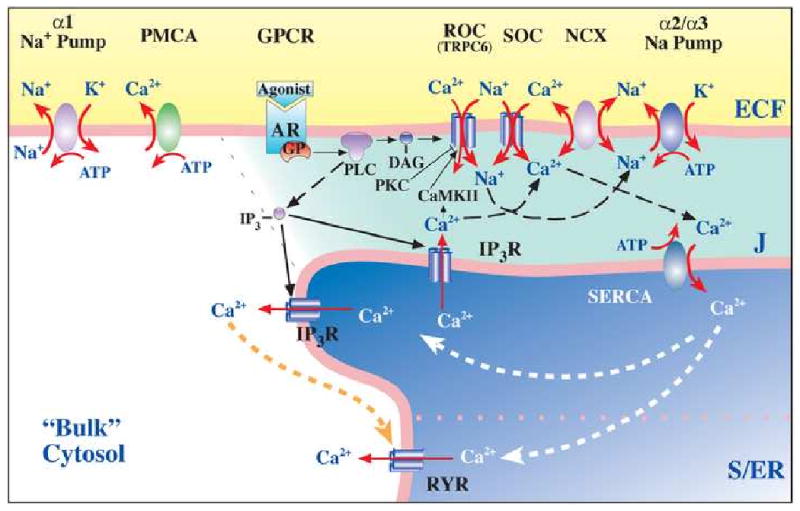
Model of the plasma membrane-junctional sarco-/endoplasmic reticulum (PM-jS/ER) region, the PLasmERosome, showing the location of key transport proteins involved in local control of jS/ER Ca2+ stores and Ca2+ signaling. The PLasmERosome consists of a PM microdomain, the adjacent jS/ER (with SERCA, IP3R and ryanodine receptors, RYR), and the intervening ‘diffusion-restricted’ junctional space (J). The PM microdomain contains agonist receptors, ARs (GPCRs), ROCs and SOCs (composed of various transient receptor potential channel proteins or TRPCs), α2 (in smooth muscle) or α3 Na+ pumps, and NCX. Activation of GPCRs and release of G-proteins (GPs) stimulates phospholipase C (PLC) to produce IP3 and diacylglycerol (DAG). DAG may activate ROCs directly. Na+ may enter locally, through ROCs, SOCs or, perhaps, SACs (not shown) to promote Ca2+ entry via NCX. Shading indicates relative Na+ and/or Ca2+ concentrations. Other regions of the PM contain α1 Na+ pumps and PMCA. Other abbreviations are defined in the text. Reprinted with permission [184].
Vasoconstrictors such as NE bind to agonist receptors (ARs), which are G-protein coupled receptors (GPCRs) located in the myocyte PM (Fig. 1). This induces the phospholipase C-mediated synthesis of inositol trisphosphate (IP3) and diacylglycerol (DAG). The IP3 interacts with its receptors/channels (IP3Rs) on the SR membrane, thereby releasing Ca2+ into the cytosol to activate contraction. Ca2+ (and Na+) [50, 51] can also enter the cytosol from the ECF through cation-selective ROCs (opened by DAG) and SOCs (opened by SR Ca2+ depletion). The Ca2+ can be re-sequestered in the SR by SERCA, or it can be extruded from the myocytes by ATP-driven PM Ca2+ pumps (PMCA). Importantly, Ca2+ can also either exit or enter the cells via the Na+/Ca2+ exchanger (NCX) which is driven by the Na+ electrochemical gradient across the PM under the control of the Na+ pumps [52]. NCX uniquely links Na+ metabolism to Ca2+ regulation and, thus, to arterial myocyte constriction. These mechanisms provide critical insight into question of how salt retention elevates BP.
6. Whole body autoregulation
A seminal advance in elucidating the pathophysiology of hypertension was the introduction of the concept of long-term1 “whole body autoregulation” of blood flow [53] and its experimental verification [16, 18, 54, 55]. These studies showed that salt retention and consequent plasma volume expansion initially elevates BP because of an increase in CO. With sustained volume expansion, even for just a few days, however, the CO declines and TPR increases to maintain the elevated BP. Thus, relatively normal CO and elevated TPR are routinely observed in established hypertension [11]. Nevertheless, in experimentally-induced hypertension, for example with mineralocorticoids [55] or renal artery clipping [56], a transient initial state of increased CO can often be detected. Failure to observe this high CO stage could be the result of compensatory mechanisms (“autoregulation”) that turn on very shortly after the volume starts to expand. In most humans with (essential) hypertension, the salt retention and (tendency toward) volume expansion likely occur gradually, over days to years. In that case, the mechanisms that tend to lower plasma volume and CO, including the rise in TPR and pressure natriuresis [12, 54, 57], likely operate simultaneously to prevent an overt increase in CO. This corresponds to a condition of “virtual hypervolemia,” however, because blood volume is still inappropriately high relative to the BP [21]. Importantly, the effects of volume expansion on TPR and BP are rapidly reversed if the stimulus (e.g., the volume load or the mineralocorticoid and salt) is withdrawn [54, 55, 57, 58]. This implies that the (initial) rise in TPR must be functional and not structural, and it must almost certainly be hormonal because this “autoregulation” involves all of the vasculature, veins as well as arteries, and pulmonary as well as systemic vessels [59].
Despite the elapse of forty years since the demonstration of long-term autoregulation, efforts to elucidate the specific underlying mechanisms have been surprisingly scant. In the mid-1970's, we [60] and others [61] raised the possibility that an endogenous Na+ pump inhibitor, i.e., a ouabain-like compound with vasotonic action, might be secreted in response to salt retention and plasma volume expansion. In other words, this substance might be a missing hormonal link between salt retention, and the increased TPR and hypertension. Strict conservation of the high affinity ouabain-binding site amino acid sequence throughout mammalian evolution implies that there must be an endogenous ligand that interacts with this site. We suggested that partial Na+ pump inhibition by the endogenous inhibitor should promote the net gain of Ca2+ via the myocyte NCX, and thereby augment Ca2+ signaling and vasoconstriction [59, 60]. The central roles of these three molecular entities, the endogenous Na+ pump inhibitor, Na+ pumps and NCX, is described below. New evidence that certain TRPC proteins, components of Ca2+- and Na+- permeable ROCs and SOCs [50, 51], also make key contributions to the altered Ca2+ signaling [62], is discussed as well.
7. Endogenous ouabain and its receptor
The aforementioned ideas fueled the search for the postulated endogenous Na+ pump inhibitor, a ligand for the pump's cardiotonic steroid (CTS) binding site, that might mediate the vascular response. In 1991, we purified endogenous ouabain (EO) from human plasma; the endogenous substance was identified as ouabain by mass spectroscopy [63]. It is now possible to quantitate EO by liquid chromatography-tandem mass spectroscopy (LC-MS-MS) methods [64] starting from small (1 ml) samples of human or animal plasma [65, 66]. The idea that EO might be the 11β isomer of ouabain [67, 68] has been excluded because the 11-epimers of ouabain are chromatographically different [69].
Rat adrenal cortex is highly enriched with EO, and human and cow adrenals also contain very high levels [63]. Bilateral adrenalectomy greatly reduces EO in rat plasma; conversely, treatment of uni-nephrectomized rats with DOCA (deoxycorticosterone acetate) + salt greatly increases plasma EO and elevates BP [63]. These findings indicate that EO is an adrenocortical hormone. Other reports, however, suggest that EO may also be synthesized in, and secreted by, the hypothalamus [70, 71].
Numerous human and intact animal studies, as well as adrenocortical cell culture studies, indicate that EO is synthesized in the adrenal cortex, and that its synthesis and secretion is stimulated by adrenocorticotropic hormone (ACTH) [63, 72-83]. In humans [79] and animals [72, 75], ACTH-induced hypertension is associated with elevation of EO. Indeed, preliminary reports indicate that certain rare adrenocortical tumors, which are associated with severe hypertension, may produce prodigious amounts of EO [84, 85]. In ACTH-induced hypertension [75, 86], as well as in DOCA-salt hypertension [87] and reduced renal mass hypertension [88], BP is lowered by Digibind (digoxin-selective Fab fragments), which also binds ouabain with high affinity [89].
About 50% of humans with untreated essential hypertension and a majority of patients with adrenocortical adenomas and hypertension have significantly elevated plasma EO; moreover, BP correlates directly with plasma EO [90-93]. Even in normal human subjects, a high salt diet raises plasma EO [66], and a 10 min infusion of low dose ouabain increases vascular resistance and elevates BP for >60 min [94-96].
Critical support for the idea that EO might play a key role in the pathogenesis of hypertension was the demonstration that prolonged administration of ouabain to normal rats induces hypertension [97]. This observation has been replicated in many laboratories [98-100].
Plasma EO levels are elevated in several rodent salt-sensitive hypertension models [63, 88, 101-103], and chronic administration of low dose ouabain to normal rodents usually induces hypertension in 1-3 weeks (Fig. 2) [90, 97-99]. Also, sub-pressor doses of ouabain and DOCA act synergistically to induce hypertension [104]. Ouabain-induced BP elevation in rodents is counteracted by the ouabain antagonist, Rostafuroxin (PST-2238) [105, 106], and hypertension induced by ACTH or DOCA+salt is antagonized by Digibind [75, 86, 87].
Fig. 2.
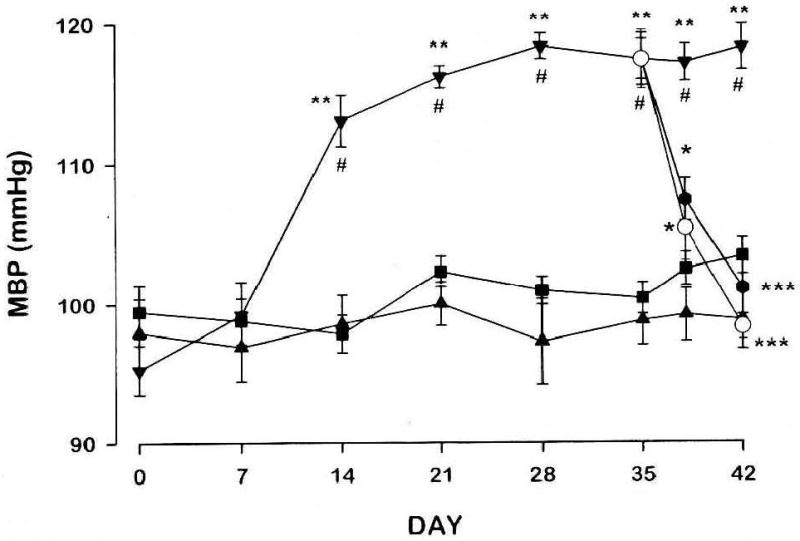
Oubain, but not digoxin, induces hypertension; digoxin and digitoxin reverse ouabain-induced hypertension. Rats were infused with vehicle (■), ouabain, 15 μg/kg/day (▼), or digoxin, 30 μg/kg/day (▲), for 42 days. From days 35 to 42, three groups of 8 ouabain-infused rats received a secondary infusion of digoxin, 30 μg/kg/day (●), digitoxin, 30 μg/kg/day (○), or vehicle (▼). Mean blood pressures (MBP) were obtained by tail cuff at weekly intervals or as indicated. *P<0.05 vs ouabain; ***P<0.001 vs ouabain; #P<0.005 vs vehicle; **P<0.001 vs digoxin. Reprinted with permission [99].
Effects of Ouabain, Digoxin and Digitoxin on BP
The aforementioned findings are strong evidence that circulating EO has a key role in the pathogenesis of salt-sensitive hypertension. Other studies suggest, however, that brain, not plasma, EO [70], or even marinobufagenin [70, 107], may be important.
Interestingly, low-dose ouabain increases TPR in dogs, but doesn't raise BP, presumably because heart rate and CO are markedly reduced [108]. Ouabain also doesn't induce hypertension in sheep [109] or in mineralocorticoid-resistant [110] Long-Evans rats [111]. Such exceptions not only show that the genetic background is important, but may provide novel information to help clarify the relationship between EO and hypertension.
Na+ pumps are widely accepted as the CTS receptor, but this greatly oversimplifies the situation. Na+ pumps are αβ heterodimers. The catalytic subunit, α, contains the Na+, K+, ATP and ouabain binding sites, and is phosphorylated during each pump cycle. β is essential for pump function; it stabilizes the α subunit conformation and chaperones the αβ complex to the PM [112-114]. The 4 mammalian α subunit isoforms (α1-α4) are products of different genes, but have nearly 90% sequence identity; they have different expression patterns and different kinetics, and are differently regulated [112, 115-121]. Many (most) cell types express Na+ pumps with an α1 subunit and Na+ pumps with a second α isoform [112, 119, 122]. Astroglia [123-125], endothelial cells [126], and all types of muscles [42, 112, 127-129] express Na+ pumps with an α2 subunit as well as pumps with an α1; most neurons express α1 and α3 [112, 123, 125, 129]. Renal epithelia express predominantly (>90-95%) Na+ pumps with α1, which mediate the final step in net transepithelial Na+ reabsorption [120, 130].
The Na+ pump α subunit CTS binding site has been highly conserved during the evolution of higher animals. Nevertheless, not all α subunit isoforms, nor the isoforms in all species, have the same high affinity for CTS. For example, rodent α1 Na+ pumps have unusually low affinity for CTS [112, 131]. Thus, it is important to understand better both the CTS and their interactions with their Na+ pump α2 subunit binding sites.
8. The myriad uses and roles of cardiotonic steroids
Recorded use of CTS dates back more than 1500 years. CTS have been employed not only as diuretics and cardiotonics, but as emetics, as abortion agents, and as poisons. For more than two centuries following William Withering's classic clinical study [132], Digitalis glycosides were the drugs of choice for the treatment of congestive heart failure and certain cardiac arrhythmias.
Recently, the novel roles of CTS and the Na+ pump in cancer therapeutics, and mood/behavioral [133] and neurological disorders [134] have been discussed. For example, one striking observation is that mortality from breast cancer was markedly reduced in patients on digitalis therapy [135]; this has prompted greatly renewed interest in CTS and their possible role in cancer therapy [136-140]. In addition, ideas about the action of EO as a natriuretic agent [60] have been revived [141, 142]. Furthermore, many observations now indicate that EO also is a growth hormone: EO may participate in a variety of kinase-mediated and other signaling pathways, independent of its effects on Na+ pump-mediated Na+ transport [143-150]. This might contribute, for example, to the target organ damage that often occurs in hypertension.
9. Cardiotonic steroid structure-activity relationships: hypertensinogenic and anti-hypertensinogenic cardenolides
Cardiotonic steroids have been widely used clinically to treat heart failure and cardiac arrhythmias. It has long been accepted that the cardiotonic effect of CTS results from their ability to inhibit Na pumps (Na,K-ATPase) [151] and thereby promote Ca2+ entry via NCX [152, 153]. The CTS include two structurally distinct groups – the cardenolides, in which the steroid is attached to a five member singly unsaturated lactone ring (Fig. 3, Table 1), and the bufadienolides, in which the lactone has six members and is doubly unsaturated. When one or more sugars are attached to the CTS at carbon 3, they are termed ‘cardiac glycosides’; common examples include ouabain and digoxin. With the exception of the bufanolide, proscillaridin, the steroid nucleus (aglycone) in the common bufadienolides is usually not glycosylated, but it may be conjugated with suberyl arginine or various other congeners.
Fig. 3.
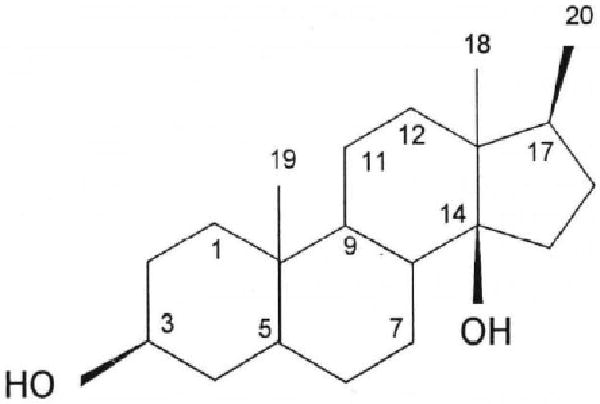
Prototypical cardenolide steroid skeleton. The primary feature is a steroid skeleton with the rings fused in a cis-trans-cis arrangement. The cardenolides discussed here have a 14βOH, an unsaturated lactone ring attached via C17 in the β configuration, and a methyl group at C18. When present, sugars are attached via the steroid 3βOH group. See Table 1 for the list of substituents in ouabain, ouabagenin, digoxin, digitoxin and Rostafuroxin. Reprinted with permission [99].
Table 1.
Some relevant cardenolides and their substituents.
| Compound | Steroid substituent1 | Sugar at C3 | |||||
|---|---|---|---|---|---|---|---|
| 1β | 5β | 11α | 12β | 17α | 19 | ||
| Ouabain | OH | OH | OH | H | H | CH2OH | Rhamnose |
| Ouabagenin | OH | OH | OH | H | H | CH2OH | None |
| Digoxin | H | H | H | OH | H | CH3 | Digitoxose (3) |
| Digitoxin | H | H | H | H | H | CH3 | Digitoxose (3) |
| Digitoxigenin | H | H | H | H | H | CH3 | None |
| Rostafuroxin | H | H | H | H | OH | CH3 | None |
The common cardenolides have a 5-member singly-unsaturated lactone ring in the 17β position, but Rostafuroxan has a doubly-unsaturated furane substutent at this position.
Cardenolides and bufadienolides are synthesized in certain plants, some amphibians and insects, and possibly all higher animals. Crude as well as highly enriched extracts from plants and the parotid secretions of the toad have been used in homeopathic remedies to treat heart failure and some cancers, and as general tonics for metabolism and immune function, especially in China (e.g., Chan Su) and Japan (Senso). The advent of modern pharmacology, coupled with the desire to use purer preparations in therapy, led to extensive studies on the Digitalis and Strophanthus glycosides and their aglycones, and the more prominent entities in toad secretions, including bufalin and resibufagenin.
Most research on CTS, and on various natural and synthetic analogs, has focused on the positive inotropic response (enhanced contraction) of heart preparations, and on the inhibition of isolated kidney enzyme (Na,K-ATPase). Thus, the bulk of knowledge about the structure-activity relationships is relevant to the heart, or to (renal) Na+ pumps with an α1 catalytic subunit. Overall, the inotropic response appears to be correlated with the ability to bind and inhibit the Na+ pump. Introduction of various substitutions in the steroid nucleus and lactone ring indicate that the configuration of the steroid is crucial for these effects.
The classic adrenocortical, ovarian and testicular steroids lack the cis-trans-cis fusion of the AB, BC, and CD rings found in the CTS (Fig. 3 and Table 1), and do not bind to, or inhibit, the Na+ pump. Certain steroids with trans-trans-cis ring fusions are cardiotonic [154, 155], while those with cis-trans-trans ring fusions (e.g., common bile salts, 14α-digitoxigenin and 14α-artebufogenin) are either inactive or very weak [156, 157]. Addition of one or more sugars to the cardenolide steroid nucleus increases the potency, while inversion of the lactone configuration at C17 from β to α [158], or saturation (e.g., dihydroouabain and dihydrodigoxin) of the lactone ring, reduces the cardiotonic activity 10-30 fold. These fundamental relationships, obtained with cardiac preparations, have been widely assumed to be valid in other physiological systems.
Both ouabain and digoxin, when administered acutely in vivo, and often in high doses, induce vasoconstriction [95, 159-164]. Nanomolar ouabain, however, augments myogenic constriction in rodent isolated arteries [36, 42, 65, 165, 166]. The first experimental evidence of a previously-unrecognized cardenolide structure-activity relationship was the observation that the prolonged administration of digoxin, also an Na,K-ATPase inhibitor [167], does not raise BP in normal rats, whereas ouabain does (Fig. 2) [168]. This result has been confirmed by several investigators [99, 100, 169, 170]. Moreover, while digoxin itself does not raise BP [171], digoxin and a related CTS, digitoxin, are very effective in lowering the elevated BP in rats with ouabain-induced hypertension (Fig. 2) [99, 100]. Importantly, digoxin also is known to lower BP in hypertensive humans [172]. These remarkable observations can only be explained by structural differences between the Strophanthus (e.g., ouabain) and Digitalis steroids, even though they are ostensibly similar Na+ pump inhibitors. The sugar(s) is(are) not crucial for these effects: the aglycone of ouabain, ouabagenin (Table 1), is also pro-hypertensive [173], while Rostafuroxin, a derivative of digitoxigen [174], is anti-hypertensive in humans and rats [105, 106]. Thus, differences in the steroid moieties of digoxin/digitoxin and ouabain account for their disparate effects on long term BP. Excluding the common oxygen at C3, ouabain is hydroxylated in positions 1,5,11,14 and 19, while digoxin is hydroxylated in positions 12 and 14 (Table 1). The major structural difference between the two steroids therefore lies in the extensive hydroxylation of ouabain in the A and B rings (and well away from the lactone ring) and the 12 hydroxyl group in digoxin. Like digoxin and ouabain, Rostafuroxin has a steroid nucleus that is cis-trans-cis fused and has a 14β hydroxyl group. However, it lacks the ouabain hydroxyls at positions 1,5,11 and 19 and the lactone has been replaced with a doubly unsaturated furane [174].
In sum, the key structural components that underlie the long term pressor activity of the cardenolides appear to include a steroid nucleus whose rings are fused in a cis-trans-cis configuration with oxygenation of the AB ring at C5. The depressor activity of the cardenolides appears to be linked with the cis-trans-cis steroid configuration, deoxygenation of the AB ring at C5 and substituents at C17 that augment potency as Na+ pump inhibitors including unsaturated 5- and 6-member lactone rings. Many of the naturally occurring cardenolides are mixtures of structural features at opposite ends of the steroid nucleus that confer prolonged pressor and depressor activity in vivo. Synthetic analogs with either improved pressor or depressor activity, the latter exemplified by Rostafuroxin, may be of clinical relevance. Clearly, the physiology and pharmacology of these agents is still full of surprises.
9. The “PLasmERosome”: a structural basis for ouabain's action
The roles of the different α subunit isoforms were clarified by the discovery that, in the various cell types, Na+ pumps with the α2 or α3 subunit were confined to PM microdomains situated adjacent to “junctional” sarco-/endoplasmic reticulum (jS/ER) (Fig. 1) [117, 118]. Na/Ca exchangers are confined to the same PM microdomains (Fig. 1) [118], as are various TRPC proteins [175] that are components of ROCs and SOCs [176-178]. In contrast, Na+ pumps with an α1 subunit are more ubiquitously distributed in the PM, but are apparently excluded from these PM microdomains [122, 179, 180]. The functional as well as structural interrelationship of these transport proteins is supported by the remarkable observation that α2 (but not α1) Na+ pumps, NCX1, and TRPC6 and -1, are all upregulated by prolonged ouabain administration, both in vivo and in vitro [176].
The PM microdomains are separated by only 12-20 nm from the jS/ER [181], and these structures form a functional unit, the “PLasmERosome” [182, 183]. The volume of cytosol in the junctional space (J) between the PM and jS/ER of a single PLasmERosome (Fig. 1) is on the order of only 10-19 to 10-18 liters, and diffusion of Na+ and Ca2+ between this space and bulk cytosol is restricted. Thus, standing Na+ and Ca2+ concentration gradients between these compartments and bulk cytosol can be maintained [51, 127, 131, 179, 184, 185].
Differences in Na+ pump α subunit isoform kinetics are the key to PLasmERosome function. Rodent α1 has an unusually low affinity for ouabain (KOuabain > 100 μM, vs < 0.05 μM in humans) [112, 131]; thus, nanomolar ouabain inhibits only the α2 Na+ pumps in rodent arterial myocytes. Even in humans, however, where α1 Na+ pumps have high affinity for ouabain, partial inhibition of Na+ pumps by nanomolar ouabain will elevate [Na+] in the junctional space much more than in bulk cytosol. The reason is that the affinity of α2 Na+ pumps for Na+ is much lower (KNa ≈ 22 mM) than is the affinity of α1 Na+ pumps (KNa ≈ 12 mM) [121].
The broad distribution of α1 Na+ pumps implies that they control, primarily, [Na+] in bulk cytosol. In contrast, pumps with an α2 (in smooth muscle, for example) or α3 catalytic subunit regulate local [Na+] in the junctional space. Thus, these α2/α3 Na+ pumps control the local Na+ electrochemical gradient that influences Ca2+ transport by the adjacent NCX. The ROCs and SOCs located here (Fig. 1) are cation-selective channels that admit Na+ as well as Ca2+ [50]. This organizational arrangement (Fig. 1) links Na+ metabolism to cell Ca2+. Thus, the transporters in the PLasmERosome regulate not only [Ca2+] in the junctional space, but S/ER Ca2+ stores and global Ca2+ signaling in the cells as well [182, 183]. Therefore, modulation of α2 Na+ pumps in arterial myocyte PLasmERosomes by EO can influence arterial tone and BP. In the ensuing discussion we summarize data from recent experiments in which genetic engineering and pharmacological manipulation of mouse Na+ pumps and NCX have been used to examine the roles of these transporters in the long-term control of BP.
10. How does ouabain (EO) elevate blood pressure? The downstream effector mechanisms
α2 Na+ Pumps
The fact that chronic administration of exogenous ouabain induces hypertension in rodents has already been mentioned. The questions we now address are: How does ouabain (or EO) elevate BP? Is it due to inhibition of smooth muscle α2 Na+ pumps, as implied by the preceding discussion?
We have reported that acute application of nanomolar ouabain to isolated, pressurized rodent arteries with myogenic tone augments the tone. The EC50 is on the order of 1 nM ouabain in intact arteries, and even lower in arteries without endothelium [36, 42].
If circulating ouabain (or EO) elevates BP by inhibiting arterial smooth muscle (ASM) α2 Na+ pumps, reduced expression of α2 Na+ pumps should have a similar effect. Indeed, mice with a null mutation in one α2 Na+ pump allele (α2+/-) [128] express ∼50% of the normal complement of α2 in arteries [36, 127] and have elevated BP (Fig. 3) [36, 165]. Mice with a null mutant α1 allele (α1+/-) express half the normal complement of α1 Na+ pumps, but have normal BP (Fig. 4) [36, 165]. Moreover, mesenteric small arteries from the α2+/-, but not α1+/- mice, exhibit augmented myogenic reactivity and myogenic tone (MT).[36] The α2+/- mice are also “salt-sensitive”: a high salt diet increases BP much more in these mice than in their wild type littermates (Fig. 4).
Fig. 4.

Relative blood pressures of mice with genetically-engineered α2 Na+ pumps and NCX1. The data from several sources, are normalized to the BPs of the respective control wild type (WT) mice. Mice with a null mutation in one α2 Na+ pump allele (α2+/-) [36] or smooth muscle-specific α2 knockdown (α2SM/DN) (Song, Chen, Zhang, Lee, Kotlikoff and Blaustein, unpublished), or increased smooth muscle-specific NCX1 overexpression (NCX1SM/Tg) [166], had significantly elevated BP. A high salt diet augmented the elevated BP in α2+/- mice (4% NaCl × 2 weeks) and NCX1SM/Tg mice (8% NaCl + 1% NaCl in tap water × 4 weeks). Smooth muscle-specific overexpression of α2 Na+ pumps (α2SM/Tg)[187] or knockdown of NCX1 (NCX1SM-/-) [198] significantly reduced BP. * = P < 0.05, ** = P < 0.01 vs WT or the respective genotypes on a normal (0.5%) salt diet. Reprinted with permission [65].
The α2+/- mice are “global” single allele null mutants, but it is important to determine if the effects are the result of reduced α2 Na+ pump activity/expression in ASM. Recently, we found that expression of a short N-terminal segment of the α2 Na+ pump was dominant negative (DN) for expression of full-length α2 pumps [180]. Therefore, we generated mice (α2SM/DN) that expressed the N-terminal segment with a smooth muscle (SM)-specific myosin heavy chain promoter [186]. These mice, with greatly reduced α2 Na+ pump expression in artery smooth muscle, have elevated BP (Fig. 4). Conversely, mice that overexpress α2, but not those overexpress α1, Na+ pumps in smooth muscle, have, on average, significantly reduced BP compared to wild type (WT) mice (Fig. 4) [187].
The roles of ouabain/EO and α2 Na+ pumps in elevating BP was also examined in two other ways. We tested Rostafuroxin, which antagonizes the inhibitory action of ouabain on Na,K-ATPase [188]. In isolated arteries, Rostafuroxin counteracts the augmentation of MT by nanomolar ouabain, but not the (ouabain-independent) augmenting effect of reduced α2 expression on MT [36]. Rostafuroxin also lowers BP in ouabain-induced hypertension [105, 106] and in nearly 50% of humans with essential hypertension [105].
Alternatively, mice that expressed mutant, ouabain-resistant α2 pumps (α2R/R) [75, 115, 189] are resistant to ACTH-induced hypertension [75, 115] as well as to ouabain-induced hypertension [189]. These results demonstrate that ACTH-induced and ouabain-induced hypertension depend a high-affinity cardiotonic steroid binding site on the α2 Na+ pump. The hypertension also depends upon a water-soluble ligand that binds to this site because the plasma level of this ligand (presumably EO) is increased by ACTH and, like ouabain [89], bind to Digibind with high affinity [189].
The studies on mice with genetically altered α2 Na+ pumps reveal that arterial myocyte α2 Na+ pumps mediate the effects of EO and play a role in the long-term regulation of BP. Genetically or pharmacologically reduced α2 activity elevates BP, whereas increased α2 activity lowers BP. It is not yet clear, however, how to reconcile these results with the evidence that isoouabain, with a saturated lactone ring tethered to C14 of steroid ring D, is hypertensinogenic, but a poor inhibitor of α1 Na+ pumps [173]. One possibility is that CTS structure-function relations may be different for α1 and α2 Na+ pumps.
NCX Type-1
The next question is: By what specific mechanism does the altered α2 Na+ pump activity influence BP? The answer appears to lie in Na/Ca exchange, which directly links Na+ to Ca2+ metabolism and is a distal regulator of cytosolic Ca2+. There are two classes of Na/Ca exchangers, those that co-transport K+ with Ca2+ (NCKX), and those that do not (NCX) [190]. Although NCKX has been found in ASM [191]. the dominant exchanger in arterial myocytes is NCX. There are three mammalian NCX isoforms (NCX1-NCX3), each the product of a different gene [192]. NCX1, which is expressed in ASM, has multiple splice variants; NCX1.3 is the dominant variant in ASM [193].
Studies on primary cultured rat arterial myocytes indicated that inhibition of Na+ pumps by nanomolar ouabain augments Ca2+ signaling without elevating bulk cytosolic Na+ [182]. Even knockout of α2 Na+ pumps in cultured cells (astrocytes) had only minimal effect of bulk cytosolic Na+, but a large effect on Ca2+ signaling [194]. These results are consistent with a functional linkage between α2 (but not α1) Na+ pumps and NCX1, and local reduction of the trans-PM Na+ gradient when α2 activity is reduced, as implied by the PLasmERosome model (Fig. 1). Also, recent pharmacological and genetic engineering studies reveal that NCX1 influences not only arterial myocyte Ca2+ metabolism, but long-term vascular tone and BP as well.
Mice in which NCX1 is overexpressed in smooth muscle (NCX1SM/Tg) have elevated BP that is markedly increased by a high salt diet (i.e., the mice are “salt-sensitive”) (Fig. 4) [166]. The elevated BP in the NCX1 overexpressors on high dietary salt is abolished by SEA0400, a selective NCX1 inhibitor [195], but not if the overexpressed NCX1 contains a G833C mutation [166], which specifically antagonizes the action of SEA0400 [196].
To perform the converse experiment, mice with floxed NCX1 (NCX1flx/flx) [197] were crossed with mice containing a Cre recombinase gene under the control of the smooth muscle myosin heavy chain promoter [186] to generate smooth muscle-specific NCX1 knockout (NCX1SM-/-) mice. These NCX1SM-/- mice have abnormally low blood pressure (Fig. 4), and isolated, pressurized small arteries from these mice have abnormally low MT[198]. Indeed, SEA0400 also lowers BP by about 5-10 mm Hg in WT mice [166] and reduces MT by about 10% in isolated arteries from these mice [36, 166]. Thus, NCX1 activity apparently makes a small, but direct, contribution to normal MT and BP. SEA0400 also attenuated the increased MT in arteries from α2+/- mice [36], indicating that NCX1 mediates effects that are distal to those of the α2 Na+ pumps. The BP and MT data from α2+/- and NCX1SM-/- mice support the view that MT in isolated arteries is an in vitro reflection of BP [34] and, most likely, TPR.
The mice with genetically engineered NCX1 demonstrate that this exchanger contributes to long-term BP regulation: increased NCX1 expression increases BP, while knockout of NCX1 reduces BP. This view is also supported by the effects of NCX blockers in several rodent models of salt-dependent or ACTH-induced hypertension. In DOCA+salt hypertensive rats, spontaneously hypertensive rats (SHR) on a high salt diet, and Dahl salt-sensitive rats on high salt, SEA0400 markedly reduced BP [166]. Also, KB-R7943, a less potent blocker, prevents ACTH from elevating BP in mice.[75] Moreover, although a null mutation in one NCX1 allele has negligible effect on BP (NCX+/- in Fig. 4) or MT[198], it does prevent the induction of hypertension by DOCA+salt [166]. Importantly, SEA0400 does not lower BP in several salt-independent rat hypertension models: SHR on a normal salt diet, stroke prone-SHR, and the renin-dependent two-kidney/one-clip rat [166]. The implication is that NCX1 contributes to the pathogenesis of salt-dependent hypertension, but not to salt-independent hypertension. As detailed elsewhere [65], these findings reveal that NCX1, along with SACs and L-type VGCs, contribute to the elevated [Ca2+]CYT that generates and maintains MT and, thus, influences TPR and BP.
11. The central role of Ca2+ signaling
At the outset, we noted that arterial myocyte contraction depends, ultimately, upon the availability of cytosolic Ca2+, and the sensitivity of the contractile apparatus to that Ca2+. NCX1, under the control of the Na+ gradient generated by the adjacent α2 Na+ pumps, helps regulate myocyte Ca2+ homeostasis (Fig. 1). For example, nanomolar ouabain-induced increases in MT are associated with increases in myocyte [Ca2+] [36]; conversely, reduction of MT by SEA0400 is associated with reduced myocyte [Ca2+] [166]. Thus, α2 Na+ pumps and NCX1 are relatively distal mechanisms in the final common path that links salt to vasoconstriction and hypertension. Indeed, all upstream vasoconstrictor and vasodilator mechanisms (neural and humoral) must, inevitably, be influenced by the activity of these two transporters [165].
As an alternative, it was recently suggested that activation of Rho/Rho kinase via the G12-G13-mediated G protein-coupled receptor pathway, which modulates the Ca2+ sensitivity of the contractile apparatus in ASM, is selective for salt-dependent hypertension [199]. Interference with the G12-G13 pathway, however, whether at the agonist receptor level [200], or at the level of Rho kinase [201], also lowers BP in salt-independent models such as the stroke-prone spontaneously hypertensive rat (SPSHR) [200] and the NO synthase-inhibited rat [201]. In our view, the findings of Wirth and colleagues [199] better fit the view that, once salt-sensitive NCX1-mediated Ca2+ entry has occurred [65], the G12-G13 pathway becomes a critical determinant of the increases in vascular tone and BP. The G12-G13 pathway is, therefore, downstream, and distinct from the key salt-sensitive steps in Na+-dependent hypertension.
12. Acute versus chronic effects of ouabain on the vasculature and blood pressure
Much of the preceding discussion concerns, primarily, the acute actions of ouabain on the vasculature. Nanomolar ouabain increases the myogenic reactivity of normal rodent arteries with a time course of seconds to minutes, and with an apparent EC50 (concentration for half-maximal effect) of 0.6-1.3 nM [36, 42]. A comparable effect is observed in arteries isolated from rats with ouabain-induced hypertension [42]. Nevertheless, in vivo ouabain administration (∼15-30 μg/kg/day), whether by injection, subcutaneous pellet, or osmotic minipump, elevates BP very slowly. BP usually rises with a delay, and takes about 14-21 days to plateau (Fig. 2) [97, 99]. A likely explanation for this slow rise, despite the increased myogenic reactivity, is that normal feedback mechanisms defend the BP and counteract the elevation. Important examples include the baroreceptor reflex and local endothelium-initiated vasodilator mechanisms [33]. With maintained administration, however, the BP slowly begins to rise (Fig. 2) [97, 99] as the chronic effects of ouabain become manifest and feedback controls are down-regulated or reset.
Ca2+ signaling is altered by prolonged ouabain treatment in both arterial smooth muscle and endothelium, but in different directions. Arterial smooth muscle from rats with ouabain-induced hypertension exhibits up-regulation of the protein components of the “Ca2+ signaling pathway” that includes the α2 Na+ pumps, NCX1, and TRPC6 and TRPC1 (the latter are components of ROCs and SOCs, respectively; see Fig. 2) [176]. The consequently enhanced Ca2+ signaling further augments myogenic reactivity and vasoconstrictor-evoked responses [42].
Acute administration of low dose ouabain also promotes Ca2+ signaling in the endothelium, and thereby augments vasodilator mechanisms such as the response to acetylcholine [126]. Importantly, however, these endothelial mechanisms are impaired in arteries from rats with ouabain-induced hypertension [126]. In other words, at the local (vascular) level, prolonged exposure to ouabain enhances the vasoconstrictor mechanisms in the arterial smooth muscle while, simultaneously, downregulating the endothelial feedback mechanisms that normally help prevent the BP from rising. The net effect, of course, is the development of hypertension.
These findings may have much broader relevance to the pathogenesis of hypertension. In many forms of human and animal hypertension, including the DOCA-salt model and the Dahl salt-sensitive model (both of which are associated with high EO levels), endothelial vasodilator mechanisms [202-205] and baroreflexes [206-208] are impaired, while vasoconstrictor responses are augmented [44, 203, 205].
13. Coda
In this review, we have explored some of the critical steps that link salt retention to the elevation of BP. Recent results, especially those from chemical analyses of human and rodent plasma samples, and from genetic engineering and pharmacological studies in rodents and rodent arteries, are summarized above. These studies give new insight into some of the molecular events that help regulate cytosolic Ca2+ and vascular tone. The data provide compelling evidence that EO, smooth muscle α2 Na+ pumps, NCX1, and TRPC channel proteins, are key molecular links in the pathway that leads from salt retention to hypertension.
These findings provide a framework, but the story is far from complete. A key area where knowledge is lacking is at the early steps between plasma volume expansion and the release of EO. The astonishing difference between the actions of ouabain and digoxin on BP demonstrate that cardenolide structure-activity relationships need to be better understood. Even the central role of the kidneys is still not completely resolved: For example, the renal and extra-renal arteries make apparently independent (and equal) contributions to the long-term regulation of BP [34, 209], but how the distal mechanisms, discussed above, affect the renal and extra-renal vasculature and renal function, and thereby contribute to BP control, is still unexplored. The progress outlined here should help identify new directions for hypertension research to resolve these issues.
Acknowledgments
We are indebted to our numerous collaborators whose key research contributions are cited and discussed in this review. We thank Mrs. K. Frankel for assistance with the manuscript. This work has been supported by National Institutes of Health grants HL-45215 (to MPB and JMH), HL-78870 (to MPB), and HL-75584 (to JMH).
Footnotes
This long-term, or day-to-day, whole body autoregulation, can be distinguished temporally and, therefore, almost certainly mechanistically, from the minute-to-minute autoregulation that maintains constant blood flow in, for example, the brain or kidney vasculature.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Whitworth JA. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21:1983–1992. doi: 10.1097/00004872-200311000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Koeppen BM, Stanton BA. Renal Physiology. Mosby; 2001. p. 50. [Google Scholar]
- 3.Campese VM, Mitra N, Sandee D. Hypertension in renal parenchymal disease: why is it so resistant to treatment? Kidney Int. 2006;69:967–973. doi: 10.1038/sj.ki.5000177. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan NM. Kaplan's Clinical Hypertension. Lippincott Williams & Wilkins; Philadelphia: 2002. pp. 1–24.pp. 36–135. [Google Scholar]
- 5.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 6.Lifton RP. Genetic dissection of human blood pressure variation: common pathways from rare phenotypes. Harvey Lect 2004-2005. 2004-2005;100:71–101. [PubMed] [Google Scholar]
- 7.Newhouse SJ, Wallace C, Dobson R, Mein C, Pembroke J, Farrall M, Clayton D, Brown M, Samani N, Dominiczak A, Connell JM, Webster J, Lathrop GM, Caulfield M, Munroe PB. Haplotypes of the WNK1 gene associate with blood pressure variation in a severely hypertensive population from the British Genetics of Hypertension study. Hum Mol Genet. 2005;14:1805–1814. doi: 10.1093/hmg/ddi187. [DOI] [PubMed] [Google Scholar]
- 8.Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, Diethelm AG. Remission of essential hypertension after renal transplantation. N Engl J Med. 1983;309:1009–1015. doi: 10.1056/NEJM198310273091702. [DOI] [PubMed] [Google Scholar]
- 9.Guidi E, Menghetti D, Milani S, Montagnino G, Palazzi, Bianchi G. Hypertension may be transplanted with the kidney in humans: a long-term historical prospective follow-up of recipients grafted with kidneys coming from donors with or without hypertension in their families. J Am Soc Nephrol. 1996;7:1131–1138. doi: 10.1681/ASN.V781131. [DOI] [PubMed] [Google Scholar]
- 10.Rettig R, Grisk O. The kidney as a determinant of genetic hypertension: evidence from renal transplantation studies. Hypertension. 2005;46:463–468. doi: 10.1161/01.HYP.0000178189.68229.8a. [DOI] [PubMed] [Google Scholar]
- 11.Cowley AWJ. Long-term regulation of arterial blood pressure. Physiol Rev. 1992;72:231–300. doi: 10.1152/physrev.1992.72.1.231. [DOI] [PubMed] [Google Scholar]
- 12.Guyton AC. Blood pressure control--special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 13.Guyton AC, Jones CE, Coleman TG. Circulatory Physiology Cardiac Output and its Regulation. Saunders; Philadelphia, PA: 1973. [Google Scholar]
- 14.Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 15.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–490. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 16.Guyton AC, Coleman TG. Quantitative analysis of the pathophysiology of hypertension. Circ Res. 1969;24(5 Suppl):1–19. [PubMed] [Google Scholar]
- 17.Guyton AC, Coleman TG, Cowley AVJ, Scheel KW, Manning RDJ, Norman RAJ. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1872;52:584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 18.Norman RA, Jr, Coleman TG, Wiley TL, Jr, Manning RD, Jr, Guyton AC. Separate roles of sodium ion concentration and fluid volumes in salt-loading hypertension in sheep. Am J Physiol. 1975;229:1068–1072. doi: 10.1152/ajplegacy.1975.229.4.1068. [DOI] [PubMed] [Google Scholar]
- 19.Freis ED. Mechanism of the antihypertensive effects of diuretics. Possible role of salt in hypertension. Clin Pharmacol Ther. 1960;1:337–344. doi: 10.1002/cpt196013337. [DOI] [PubMed] [Google Scholar]
- 20.Rinehart J, Kahle KT, de Los Heros P, Vazquez N, Meade P, Wilson FH, Hebert SC, Gimenez I, Gamba G, Lifton RP. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl-cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci U S A. 2005;102:16777–16782. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamlyn JM, Blaustein MP. Sodium chloride, extracellular fluid volume, and blood pressure regulation. Am J Physiol. 1986;251:F563–575. doi: 10.1152/ajprenal.1986.251.4.F563. [DOI] [PubMed] [Google Scholar]
- 22.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson RJ, Rodriguez-Iturbe B, Nakagawa T, Kang DH, Feig DI, Herrera-Acosta J. Subtle renal injury is likely a common mechanism for salt-sensitive essential hypertension. Hypertension. 2005;45:326–330. doi: 10.1161/01.HYP.0000154784.14018.5f. [DOI] [PubMed] [Google Scholar]
- 24.Davy KP, Hall JE. Obesity and hypertension: two epidemics or one. Am J Physiol Regul Integr Comp Physiol. 2004;286:R803–R813. doi: 10.1152/ajpregu.00707.2003. [DOI] [PubMed] [Google Scholar]
- 25.Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41:625–633. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]
- 26.Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA. Functional genomics of the dopaminergic system in hypertension. Physiol Genomics. 2004;19:233–246. doi: 10.1152/physiolgenomics.00127.2004. [DOI] [PubMed] [Google Scholar]
- 27.Felder RA, Sanada H, Xu J, Yu PY, Wang Z, Watanabe H, Asico LD, Wang W, Zheng S, Yamaguchi I, Williams SM, Gainer J, Brown NJ, Hazen-Martin D, Wong LJ, Robillard JE, Carey RM, Eisner GM, Jose PA. G protein-coupled receptor kinase 4 gene variants in human essential hypertension. Proc Natl Acad Sci U S A. 2002;99:3872–3877. doi: 10.1073/pnas.062694599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng C, Eisner GM, Felder RA, Jose PA. Dopamine receptor and hypertension. Curr Med Chem Cardiovasc Hematol Agents. 2005;3:69–77. doi: 10.2174/1568016052773289. [DOI] [PubMed] [Google Scholar]
- 29.Bianchi G, Ferrari P, Staessen JA. Adducin polymorphism: detection and impact on hypertension and related disorders. Hypertension. 2005;45:331–340. doi: 10.1161/01.HYP.0000156497.39375.37. [DOI] [PubMed] [Google Scholar]
- 30.Lanzani C, Citterio L, Jankaricova M, Sciarrone MT, Barlassina C, Fattori S, Messaggio E, Serio CD, Zagato L, Cusi D, Hamlyn JM, Stella A, Bianchi G, Manunta P. Role of the adducin family genes in human essential hypertension. J Hypertens. 2005;23:543–549. doi: 10.1097/01.hjh.0000160210.48479.78. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, O'Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, Shi X, Pan L, Rampersaud E, Shen H, Kim JD, Subramanya AR, Steinle NI, Parsa A, Ober CC, Welling PA, Chakravarti A, Weder AB, Cooper RS, Mitchell BD, Shuldiner AR, Chang YP. From the Cover: Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A. 2009;106:226–231. doi: 10.1073/pnas.0808358106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidlin O, Sebastian AF, Morris RC., Jr What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49:1032–1039. doi: 10.1161/HYPERTENSIONAHA.106.084640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy MN, Pappano AJ. Cardiovascular Physiology. 9th. Mosby Elsevier; Philadelphia, PA: 2007. [Google Scholar]
- 34.Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- 35.Hill MA, Zou J, Potocnik SJ, Meininger GA, Davis MJ. Arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol. 2005;569:243–256. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Payne RA, Webb DJ. Arterial blood pressure and stiffness in hypertension: is arterial structure important? Hypertension. 2006;48:366–367. doi: 10.1161/01.HYP.0000237668.31786.1f. [DOI] [PubMed] [Google Scholar]
- 38.Schiffrin EL. Remodeling of resistance arteries in essential hypertension and effects of antihypertensive treatment. Am J Hypertens. 2004;17:1192–1200. doi: 10.1016/j.amjhyper.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 39.Touyz RM. Molecular and cellular mechanisms in vascular injury in hypertension: role of angiotensin II. Curr Opin Nephrol Hypertens. 2005;14:125–131. doi: 10.1097/00041552-200503000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Feihl F, Liaudet L, Waeber B, Levy BI. Hypertension: a disease of the microcirculation? Hypertension. 2006;48:1012–1017. doi: 10.1161/01.HYP.0000249510.20326.72. [DOI] [PubMed] [Google Scholar]
- 41.Levy BI, Ambrosio G, Pries AR, Struijker-Boudier HA. Microcirculation in hypertension: a new target for treatment? Circulation. 2001;104:735–740. doi: 10.1161/hc3101.091158. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, Hamlyn JM, Karashima E, Raina H, Mauban JR, Izuka M, Berra-Romani R, Zulian A, Wier WG, Blaustein MP. Low-dose ouabain constricts small arteries from ouabain-hypertensive rats: implications for sustained elevation of vascular resistance. Am J Physiol Heart Circ Physiol. 2009;297:H1140–1150. doi: 10.1152/ajpheart.00436.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidlin O, Sebastian AF, Morris RC., Jr What initiates the pressor effect of salt in salt-sensitive humans? Observations in normotensive blacks. Hypertension. 2007;49:1032–1039. doi: 10.1161/HYPERTENSIONAHA.106.084640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bohr DF, Dominiczak AF, Webb RC. Pathophysiology of the vasculature in hypertension. Hypertension. 1991;18:III69–75. doi: 10.1161/01.hyp.18.5_suppl.iii69. [DOI] [PubMed] [Google Scholar]
- 45.Rüegg JC. Calcium in Muscle Contraction: Cellular and Molecular Physiology. second. Springer-Verlag; Berlin, Heidelberg, New York, London, Paris, Tokyo, Hong Kong, Barcelona, Budapest: 1992. pp. 206–212. [Google Scholar]
- 46.Vanhoutte PM, Mombouli JV. Vascular endothelium: vasoactive mediators. Prog Cardiovasc Dis. 1996;39:229–238. doi: 10.1016/s0033-0620(96)80003-x. [DOI] [PubMed] [Google Scholar]
- 47.Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 48.Maguire JJ, Davenport AP. Regulation of vascular reactivity by established and emerging GPCRs. Trends Pharmacol Sci. 2005;26:448–454. doi: 10.1016/j.tips.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 49.Wier WG, Zang WJ, Lamont C, Raina H. Sympathetic neurogenic Ca2+ signalling in rat arteries: ATP, noradrenaline and neuropeptide Y. Exp Physiol. 2009;94:31–37. doi: 10.1113/expphysiol.2008.043638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnon A, Hamlyn JM, Blaustein MP. Na+ entry via store-operated channels modulates Ca2+ signaling in arterial myocytes. Am J Physiol Cell Physiol. 2000;278:C163–C173. doi: 10.1152/ajpcell.2000.278.1.C163. [DOI] [PubMed] [Google Scholar]
- 51.Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient Receptor Potential Channel 6 Mediated, Localized Cytosolic [Na+] Transients Drive Na+/Ca2+ Exchanger Mediated Ca2+ Entry in Purinergically Stimulated Aorta Smooth Muscle Cells. Circ Res. 2007;101:1030–1038. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 52.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 53.Borst JG, Borst-De Geus A. Hypertension explained by Starling's theory of circulatory homoeostasis. Lancet. 1963;1:677–682. doi: 10.1016/s0140-6736(63)91443-0. [DOI] [PubMed] [Google Scholar]
- 54.Coleman TG, Guyton AC. Hypertension caused by salt loading in the dog. 3. Onset transients of cardiac output and other circulatory variables. Circ Res. 1969;25:153–160. doi: 10.1161/01.res.25.2.153. [DOI] [PubMed] [Google Scholar]
- 55.Schalekamp MA, Man in't Veld AJ, Wenting GJ. The second Sir George Pickering memorial lecture. What regulates whole body autoregulation? Clinical observations. J Hypertens. 1985;3:97–108. doi: 10.1097/00004872-198504000-00001. [DOI] [PubMed] [Google Scholar]
- 56.Ledingham JM, Cohen RD. Changes in the Extracellular Fluid Volume and Cardiac Output during the Development of Experimental Renal Hypertension. Can Med Assoc J. 1964;90:292–294. [PMC free article] [PubMed] [Google Scholar]
- 57.Guyton AC, Granger HJ, Coleman TG. Autoregulation of the total systemic circulation and its relation to control of cardiac output and arterial pressure. Circ Res. 1971;28 1:93–97. [PubMed] [Google Scholar]
- 58.Distler A, Just HJ, Philipp T. Studies on the mechanism of aldosterone-induced hypertension in man. Clin Sci Mol Med. 1973;45:743–750. doi: 10.1042/cs0450743. [DOI] [PubMed] [Google Scholar]
- 59.Blaustein MP. Physiological effects of endogenous ouabain: control of intracellular Ca2+ stores and responsiveness. Am J Physiol Cell Physiol. 1993;264:C1367–C1387. doi: 10.1152/ajpcell.1993.264.6.C1367. [DOI] [PubMed] [Google Scholar]
- 60.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol. 1977;232:C165–C173. doi: 10.1152/ajpcell.1977.232.5.C165. [DOI] [PubMed] [Google Scholar]
- 61.Haddy FJ, Overbeck HW. The role of humoral agents in volume expanded hypertension. Life Sci. 1976;19:935–947. doi: 10.1016/0024-3205(76)90284-8. [DOI] [PubMed] [Google Scholar]
- 62.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol. 298:H263–274. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci U S A. 1991;88:6259–6263. doi: 10.1073/pnas.88.14.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail. 2006;8:179–186. doi: 10.1016/j.ejheart.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 65.Blaustein MP, Zhang J, Chen L, Song H, Raina H, Kinsey SP, Izuka M, Iwamoto T, Kotlikoff MI, Lingrel JB, Philipson KD, Wier WG, Hamlyn JM. The pump, the exchanger, and endogenous ouabain: signaling mechanisms that link salt retention to hypertension. Hypertension. 2009;53:291–298. doi: 10.1161/HYPERTENSIONAHA.108.119974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol. 2006;290:R553–559. doi: 10.1152/ajpregu.00648.2005. [DOI] [PubMed] [Google Scholar]
- 67.Huang M, Hester RL, Coleman TG, Smith MJ, Guyton AC. Development of hypertension in animals with reduced total peripheral resistance. Hypertension. 1992;20:828–833. doi: 10.1161/01.hyp.20.6.828. [DOI] [PubMed] [Google Scholar]
- 68.O'Connell DP, Ragsdale NV, Boyd DG, Felder RA, Carey RM. Differential human renal tubular responses to dopamine type 1 receptor stimulation are determined by blood pressure status. Hypertension. 1997;29:115–122. doi: 10.1161/01.hyp.29.1.115. [DOI] [PubMed] [Google Scholar]
- 69.Hong BC, Kim S, Kim TS, Corey EJ. Synthesis and properties of several isomers of the cardioactive steroid ouabain. Tetrahedron Letters. 2006;47:2711–2715. [Google Scholar]
- 70.Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol. 2006;21:295–304. doi: 10.1097/01.hco.0000231398.64362.94. [DOI] [PubMed] [Google Scholar]
- 71.Murrell JR, Randall JD, Rosoff J, Zhao JL, Jensen RV, Gullans SR, Haupert GT., Jr Endogenous ouabain: upregulation of steroidogenic genes in hypertensive hypothalamus but not adrenal. Circulation. 2005;112:1301–1308. doi: 10.1161/CIRCULATIONAHA.105.554071. [DOI] [PubMed] [Google Scholar]
- 72.Yamada K, Goto A, Omata M. Adrenocorticotropin-induced hypertension in rats: role of ouabain-like compound. Am J Hypertens. 1997;10:403–408. [PubMed] [Google Scholar]
- 73.Laredo J, Hamilton BP, Hamlyn JM. Secretion of endogenous ouabain from bovine adrenocortical cells: role of the zona glomerulosa and zona fasciculata. Biochem Biophys Res Commun. 1995;212:487–493. doi: 10.1006/bbrc.1995.1996. [DOI] [PubMed] [Google Scholar]
- 74.Boulanger BR, Lilly MP, Hamlyn JM, Laredo J, Shurtleff D, Gann DS. Ouabain is secreted by the adrenal gland in awake dogs. American Journal of Physiology. 1993;264:E413–E419. doi: 10.1152/ajpendo.1993.264.3.E413. [DOI] [PubMed] [Google Scholar]
- 75.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA. 2005;102:15845–15850. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hamlyn JM, Laredo J, Shah JR, Lu ZR, Hamilton BP. 11-hydroxylation in the biosynthesis of endogenous ouabain: multiple implications. Ann N Y Acad Sci. 2003;986:685–693. doi: 10.1111/j.1749-6632.2003.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 77.Hamlyn JM, Lu ZR, Manunta P, Ludens JH, Kimura K, Shah JR, Laredo J, Hamilton JP, Hamilton MJ, Hamilton BP. Observations on the nature, biosynthesis, secretion and significance of endogenous ouabain. Clin Exp Hypertens. 1998;20:523–533. doi: 10.3109/10641969809053230. [DOI] [PubMed] [Google Scholar]
- 78.Laredo J, Hamilton BP, Hamlyn JM. Ouabain is secreted by bovine adrenocortical cells. Endocrinology. 1994;135:794–797. doi: 10.1210/endo.135.2.8033829. [DOI] [PubMed] [Google Scholar]
- 79.Goto A, Yamada K, Hazama H, Uehara Y, Atarashi K, Hirata Y, Kimura K, Omata M. Ouabainlike compound in hypertension associated with ectopic corticotropin syndrome. Hypertension. 1996;28:421–425. doi: 10.1161/01.hyp.28.3.421. [DOI] [PubMed] [Google Scholar]
- 80.Lichtstein D, Steinitz M, Gati I, Samuelov S, Deutsch J, Orly J. Biosynthesis of digitalis-like compounds in rat adrenal cells: hydroxycholesterol as possible precursor. Life Sci. 1998;62:2109–2126. doi: 10.1016/s0024-3205(98)00186-6. [DOI] [PubMed] [Google Scholar]
- 81.el-Masri MA, Clark BJ, Qazzaz HM, Valdes R., Jr Human adrenal cells in culture produce both ouabain-like and dihydroouabain-like factors. Clin Chem. 2002;48:1720–1730. [PubMed] [Google Scholar]
- 82.Sophocleous A, Elmatzoglou I, Souvatzoglou A. Circulating endogenous digitalis-like factor(s) (EDLF) in man is derived from the adrenals and its secretion is ACTH-dependent. J Endocrinol Invest. 2003;26:668–674. doi: 10.1007/BF03347027. [DOI] [PubMed] [Google Scholar]
- 83.Gooz M, Toth M, Vakkuri O, Gooz P, Smolka AJ, de Chatel R, Szalay KS. Endogenous ouabain-like factor (OLF) secretion is modulated by nicotinic mechanisms in rat adrenocortical cells. Life Sci. 2004;74:2111–2128. doi: 10.1016/j.lfs.2003.07.058. [DOI] [PubMed] [Google Scholar]
- 84.Manunta P, Evans G, Hamilton BP, Gans D, Reseau J, Hamlyn JM. A new syndrome with elevated plasma ouabain and hypertension secondary to an adrenocortical tumor. (Abstract) J Hypertens. 1992;10 4:827. [Google Scholar]
- 85.Evans G, Manunta P, Hamlyn JM, Hamilton BP, Gann DS. Ouabainoma: A new syndrome of endocrine hypertension caused by a ouabain-secreting cortical adenoma. Endocrine Society 75th Annual Meeting; Las Vegas. June 9-12, 1993; 1993. p. 291. Abstract. [Google Scholar]
- 86.Li M, Wen C, Whitworth JA. Hemodynamic effects of the Fab fragment of digoxin antibody (digibind) in corticotropin (ACTH)-induced hypertension. Am J Hypertens. 1997;10:332–336. doi: 10.1016/s0895-7061(96)00318-4. [DOI] [PubMed] [Google Scholar]
- 87.Krep H, Price DA, Soszynski P, Tao QF, Graves SW, Hollenberg NK. Volume sensitive hypertension and the digoxin-like factor. Reversal by a Fab directed against digoxin in DOCA-salt hypertensive rats. Am J Hypertens. 1995;8:921–927. doi: 10.1016/0895-7061(95)00181-N. [DOI] [PubMed] [Google Scholar]
- 88.Kaide J, Ura N, Torii T, Nakagawa M, Takada T, Shimamoto K. Effects of digoxin-specific antibody Fab fragment (Digibind) on blood pressure and renal water-sodium metabolism in 5/6 reduced renal mass hypertensive rats. Am J Hypertens. 1999;12:611–619. doi: 10.1016/s0895-7061(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 89.Pullen MA, Brooks DP, Edwards RM. Characterization of the neutralizing activity of digoxin-specific Fab toward ouabain-like steroids. J Pharmacol Exp Ther. 2004;310:319–325. doi: 10.1124/jpet.104.065250. [DOI] [PubMed] [Google Scholar]
- 90.Manunta P, Rogowski AC, Hamilton BP, Hamlyn JM. Ouabain-induced hypertension in the rat: relationships among plasma and tissue ouabain and blood pressure. J Hypertens. 1994;12:549–560. [PubMed] [Google Scholar]
- 91.Manunta P, Stella P, Rivera R, Ciurlino D, Cusi D, Ferrandi M, Hamlyn JM, Bianchi G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension. 1999;34:450–456. doi: 10.1161/01.hyp.34.3.450. [DOI] [PubMed] [Google Scholar]
- 92.Pierdomenico SD, Bucci A, Manunta P, Rivera R, Ferrandi M, Hamlyn JM, Lapenna D, Cuccurullo F, Mezzetti A. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am J Hypertens. 2001;14:44–50. doi: 10.1016/s0895-7061(00)01225-5. [DOI] [PubMed] [Google Scholar]
- 93.Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, Pessina AC. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertens. 1995;13:1181–1191. doi: 10.1097/00004872-199510000-00013. [DOI] [PubMed] [Google Scholar]
- 94.Mason DT, Braunwald E. Studies on Digitalis. X. Effects of Ouabain on Forearm Vascular Resistance and Venous Tone in Normal Subjects and in Patients in Heart Failure. J Clin Invest. 1964;43:532–543. doi: 10.1172/JCI104939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schulte KL, van Gemmeren D, Thiede HM, Meyer-Sabellek W, Gotzen R, Distler A. Ouabain-induced elevation in forearm vascular resistance, calcium entry and alpha-adrenoceptor blockade, and release and removal of noradrenaline. J Hypertens Suppl. 1987;5:S215–218. [PubMed] [Google Scholar]
- 96.Robinson BF, Phillips RJ, Wilson PN, Chiodini PL. Effect of local infusion of ouabain on human forearm vascular resistance and on response to potassium, verapamil and sodium nitroprusside. J Hypertens. 1983;1:165–169. doi: 10.1097/00004872-198308000-00009. [DOI] [PubMed] [Google Scholar]
- 97.Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ, Pamnani MB. Long-term ouabain administration produces hypertension in rats. Hypertension. 1993;22:178–187. doi: 10.1161/01.hyp.22.2.178. [DOI] [PubMed] [Google Scholar]
- 98.Kurashina T, Kirchner KA, Granger JP, Patel AR. Chronic sodium-potassium-ATPase inhibition with ouabain impairs renal haemodynamics and pressure natriuresis in the rat. Clin Sci (Lond) 1996;91:497–502. doi: 10.1042/cs0910497. [DOI] [PubMed] [Google Scholar]
- 99.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res. 2000;23(Suppl):S77–85. doi: 10.1291/hypres.23.supplement_s77. [DOI] [PubMed] [Google Scholar]
- 100.Huang BS, Kudlac M, Kumarathasan R, Leenen FH. Digoxin prevents ouabain and high salt intake-induced hypertension in rats with sinoaortic denervation. Hypertension. 1999;34:733–738. doi: 10.1161/01.hyp.34.4.733. [DOI] [PubMed] [Google Scholar]
- 101.Ferrandi M, Manunta P, Balzan S, Hamlyn JM, Bianchi G, Ferrari P. Ouabain-like factor quantification in mammalian tissues and plasma: comparison of two independent assays. Hypertension. 1997;30:886–896. doi: 10.1161/01.hyp.30.4.886. [DOI] [PubMed] [Google Scholar]
- 102.Ferrandi M, Manunta P, Rivera R, Bianchi G, Ferrari P. Role of the ouabain-like factor and Na-K pump in rat and human genetic hypertension. Clin Exp Hypertens. 1998;20:629–639. doi: 10.3109/10641969809053241. [DOI] [PubMed] [Google Scholar]
- 103.Takada T, Nakagawa M, Ura N, Kaide J, Yoshida H, Shimamoto K. Endogenous immunoreactive ouabain-like and digoxin-like factors in reduced renal mass hypertensive rats. Hypertens Res. 1998;21:193–199. doi: 10.1291/hypres.21.193. [DOI] [PubMed] [Google Scholar]
- 104.Sekihara H, Yazaki Y, Kojima T. Ouabain as an amplifier of mineralocorticoid-induced hypertension. Endocrinology. 1992;131:3077–3082. doi: 10.1210/endo.131.6.1446641. [DOI] [PubMed] [Google Scholar]
- 105.Ferrari P, Ferrandi M, Valentini G, Bianchi G. Rostafuroxin: an ouabain antagonist that corrects renal and vascular Na+-K+- ATPase alterations in ouabain and adducin-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:R529–535. doi: 10.1152/ajpregu.00518.2005. [DOI] [PubMed] [Google Scholar]
- 106.Ferrari P, Torielli L, Ferrandi M, Padoani G, Duzzi L, Florio M, Conti F, Melloni P, Vesci L, Corsico N, Bianchi G. PST2238: A new antihypertensive compound that antagonizes the long-term pressor effect of ouabain. J Pharmacol Exp Therap. 1998;285:83–94. [PubMed] [Google Scholar]
- 107.Anderson DE, Fedorova OV, Morrell CH, Longo DL, Kashkin VA, Metzler JD, Bagrov AY, Lakatta EG. Endogenous sodium pump inhibitors and age-associated increases in salt sensitivity of blood pressure in normotensives. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1248–1254. doi: 10.1152/ajpregu.00782.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hildebrandt DA, Montani JP, Heath BJ, Granger JP. Chronic ouabain (QUA) infusion alters systemic hemodynamics in normal dogs. FASEB J. 1999;9:A297. [Google Scholar]
- 109.Pidgeon GB, Lewis LK, Yandle TG, Richards AM, Nicholls MG. Endogenous ouabain, sodium balance and blood pressure. J Hypertens. 1996;14:169–171. doi: 10.1097/00004872-199602000-00003. [DOI] [PubMed] [Google Scholar]
- 110.Rowland NE. NaCl appetite in two strains of rat reported to be resistant to mineralocorticoid-induced hypertension. Physiol Behav. 1998;64:49–56. doi: 10.1016/s0031-9384(98)00018-3. [DOI] [PubMed] [Google Scholar]
- 111.Wang J, Tempini A, Schnyder B, Montani JP. Regulation of blood pressure during long-term ouabain infusion in Long-Evans rats. Am J Hypertens. 1999;12:423–426. doi: 10.1016/s0895-7061(98)00250-7. [DOI] [PubMed] [Google Scholar]
- 112.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol. 1998;275:F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 113.Geering K. Subunit assembly and functional maturation of Na,K-ATPase. J Membr Biol. 1990;115:109–121. doi: 10.1007/BF01869450. [DOI] [PubMed] [Google Scholar]
- 114.Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens. 2008;17:526–532. doi: 10.1097/MNH.0b013e3283036cbf. [DOI] [PubMed] [Google Scholar]
- 115.Dostanic-Larson I, Lorenz JN, Van Huysse JW, Neumann JC, Moseley AE, Lingrel JB. The physiological role of the á1 and á2 isoforms of the Na,K-ATPase and biological significance of their cardiac glycoside binding site. Am J Physiol Regul Integr Comp Physiol. 2006;290(3):R524–R528. doi: 10.1152/ajpregu.00838.2005. [DOI] [PubMed] [Google Scholar]
- 116.Feraille E, Barlet-Bas C, Cheval L, Rousselot M, Carranza ML, Dreher D, Arystarkhova E, Doucet A, Favre H. Presence of two isoforms of Na, K-ATPase with different pharmacological and immunological properties in the rat kidney. Pflugers Arch. 1995;430:205–212. doi: 10.1007/BF00374651. [DOI] [PubMed] [Google Scholar]
- 117.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann N Y Acad Sci. 1997;834:524–536. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 118.Juhaszova M, Blaustein MP. Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proc Natl Acad Sci U S A. 1997;94:1800–1805. doi: 10.1073/pnas.94.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lingrel JB, Orlowski J, Shull MM, Price EM. Molecular genetics of Na,K-ATPase. Prog Nucleic Acid Res Mol Biol. 1990;38:37–89. doi: 10.1016/s0079-6603(08)60708-4. [DOI] [PubMed] [Google Scholar]
- 120.Summa V, Camargo SM, Bauch C, Zecevic M, Verrey F. Isoform specificity of human Na+, K+-ATPase localization and aldosterone regulation in mouse kidney cells. J Physiol. 2004;555:355–364. doi: 10.1113/jphysiol.2003.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zahler R, Zhang ZT, Manor M, Boron WF. Sodium kinetics of Na,K-ATPase alpha isoforms in intact transfected HeLa cells. J Gen Physiol. 1997;110:201–213. doi: 10.1085/jgp.110.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lencesova L, O'Neill A, Resneck WG, Bloch RJ, Blaustein MP. Plasma membrane-cytoskeleton-endoplasmic reticulum complexes in neurons and astrocytes. J Biol Chem. 2004;279:2885–2893. doi: 10.1074/jbc.M310365200. [DOI] [PubMed] [Google Scholar]
- 123.McGrail KM, Phillips JM, Sweadner KJ. Immunofluorescent localization of three Na,K-ATPase isozymes in the rat central nervous system: both neurons and glia can express more than one Na,K-ATPase. J Neurosci. 1991;11:381–391. doi: 10.1523/JNEUROSCI.11-02-00381.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moseley AE, Lieske SP, Wetzel RK, James PF, He S, Shelly DA, Paul RJ, Boivin GP, Witte DP, Ramirez JM, Sweadner KJ, Lingrel JB. The Na,K-ATPase alpha 2 isoform is expressed in neurons, and its absence disrupts neuronal activity in newborn mice. J Biol Chem. 2003;278:5317–5324. doi: 10.1074/jbc.M211315200. [DOI] [PubMed] [Google Scholar]
- 125.Blaustein MP, Juhaszova M, Golovina VA, Church PJ, Stanley EF. Na/Ca exchanger and PMCA localization in neurons and astrocytes: functional implications. Ann N Y Acad Sci. 2002;976:356–366. doi: 10.1111/j.1749-6632.2002.tb04762.x. [DOI] [PubMed] [Google Scholar]
- 126.Cao C, Payne K, Lee-Kwon W, Zhang Z, Lim SW, Hamlyn J, Blaustein MP, Kwon HM, Pallone TL. Chronic ouabain treatment induces vasa recta endothelial dysfunction in the rat. Am J Physiol Renal Physiol. 2009;296:F98–F106. doi: 10.1152/ajprenal.90429.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shelly DA, He S, Moseley A, Weber C, Stegemeyer M, Lynch RM, Lingrel J, Paul RJ. Na+ pump alpha 2-isoform specifically couples to contractility in vascular smooth muscle: evidence from gene-targeted neonatal mice. Am J Physiol Cell Physiol. 2004;286:C813–C820. doi: 10.1152/ajpcell.00389.2003. [DOI] [PubMed] [Google Scholar]
- 128.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,K-ATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell. 1999;3:555–563. doi: 10.1016/s1097-2765(00)80349-4. [DOI] [PubMed] [Google Scholar]
- 129.Lingrel JB. Na,K-ATPase: isoform structure, function, and expression. J Bioenerg Biomembr. 1992;24:263–270. doi: 10.1007/BF00768847. [DOI] [PubMed] [Google Scholar]
- 130.Lucking K, Nielsen JM, Pedersen PA, Jorgensen PL. Na-K-ATPase isoform (alpha 3, alpha 2, alpha 1) abundance in rat kidney estimated by competitive RT-PCR and ouabain binding. Am J Physiol. 1996;271:F253–260. doi: 10.1152/ajprenal.1996.271.2.F253. [DOI] [PubMed] [Google Scholar]
- 131.O'Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys. 1994;310:32–39. doi: 10.1006/abbi.1994.1136. [DOI] [PubMed] [Google Scholar]
- 132.Withering W. An account of the foxglove and some of its medical uses: with practical remarks on dropsy and other diseases. J and J Robinson, London. 1785 [Google Scholar]
- 133.Riegel RE, Valvassori SS, Elias G, Reus GZ, Steckert AV, de Souza B, Petronilho F, Gavioli EC, Dal-Pizzol F, Quevedo J. Animal model of mania induced by ouabain: Evidence of oxidative stress in submitochondrial particles of the rat brain. Neurochem Int. 2009;55:491–495. doi: 10.1016/j.neuint.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 134.Ashmore LJ, Hrizo SL, Paul SM, Van Voorhies WA, Beitel GJ, Palladino MJ. Novel mutations affecting the Na, K ATPase alpha model complex neurological diseases and implicate the sodium pump in increased longevity. Hum Genet. 2009;126:431–447. doi: 10.1007/s00439-009-0673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stenkvist B. Is digitalis a therapy for breast carcinoma? Oncol Rep. 1999;6:493–496. [PubMed] [Google Scholar]
- 136.Al-Ghoul M, Valdes R., Jr Mammalian cardenolides in cancer prevention and therapeutics. Ther Drug Monit. 2008;30:234–238. doi: 10.1097/FTD.0b013e31816b90ff. [DOI] [PubMed] [Google Scholar]
- 137.Newman RA, Yang P, Pawlus AD, Block KI. Cardiac glycosides as novel cancer therapeutic agents. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]
- 138.Winnicka K, Bielawski K, Bielawska A, Surazynski A. Antiproliferative activity of derivatives of ouabain, digoxin and proscillaridin A in human MCF-7 and MDA-MB-231 breast cancer cells. Biol Pharm Bull. 2008;31:1131–1140. doi: 10.1248/bpb.31.1131. [DOI] [PubMed] [Google Scholar]
- 139.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Simpson CD, Mawji IA, Anyiwe K, Williams MA, Wang X, Venugopal AL, Gronda M, Hurren R, Cheng S, Serra S, Zavareh RB, Datti A, Wrana JL, Ezzat S, Schimmer AD. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Cancer Res. 2009;69:2739–2747. doi: 10.1158/0008-5472.CAN-08-2530. [DOI] [PubMed] [Google Scholar]
- 141.Loreaux EL, Kaul B, Lorenz JN, Lingrel JB. Ouabain-Sensitive alpha1 Na,K-ATPase enhances natriuretic response to saline load. J Am Soc Nephrol. 2008;19:1947–1954. doi: 10.1681/ASN.2008020174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nesher M, Dvela M, Igbokwe VU, Rosen H, Lichtstein D. Physiological roles of endogenous ouabain in normal rats. Am J Physiol Heart Circ Physiol. 2009;297:H2026–2034. doi: 10.1152/ajpheart.00734.2009. [DOI] [PubMed] [Google Scholar]
- 143.Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol. 2007;293:C509–536. doi: 10.1152/ajpcell.00098.2007. [DOI] [PubMed] [Google Scholar]
- 144.Sweadner KJ. A third mode of ouabain signaling. Focus on “Regulation of ERK1/2 by ouabain and Na-K-ATPase-dependent energy utilization and AMPK activation in parotid acinar cells”. Am J Physiol Cell Physiol. 2008;295:C588–589. doi: 10.1152/ajpcell.00388.2008. [DOI] [PubMed] [Google Scholar]
- 145.Soltoff SP, Hedden L. Regulation of ERK1/2 by ouabain and Na-K-ATPase-dependent energy utilization and AMPK activation in parotid acinar cells. Am J Physiol Cell Physiol. 2008;295:C590–599. doi: 10.1152/ajpcell.00140.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci U S A. 2008;105:11218–11223. doi: 10.1073/pnas.0801661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008;23:205–211. doi: 10.1152/physiol.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang S, Malmersjo S, Li J, Ando H, Aizman O, Uhlen P, Mikoshiba K, Aperia A. Distinct role of the N-terminal tail of the Na,K-ATPase catalytic subunit as a signal transducer. J Biol Chem. 2006;281:21954–21962. doi: 10.1074/jbc.M601578200. [DOI] [PubMed] [Google Scholar]
- 149.Schoner W, Scheiner-Bobis G. Endogenous cardiac glycosides: hormones using the sodium pump as signal transducer. Semin Nephrol. 2005;25:343–351. doi: 10.1016/j.semnephrol.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 150.Li Z, Xie Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflugers Arch. 2009;457:635–644. doi: 10.1007/s00424-008-0470-0. [DOI] [PubMed] [Google Scholar]
- 151.Schatzmann HJ. Cardiac glycosides as inhibitors of active potassium and sodium transport by erythrocyte membrane. Helv Physiol Pharmacol Acta. 1953;11:346–354. [PubMed] [Google Scholar]
- 152.Baker PF, Blaustein MP, Hodgkin AL, Steinhardt RA. The influence of calcium on sodium efflux in squid axons. J Physiol. 1969;200:431–458. doi: 10.1113/jphysiol.1969.sp008702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Langer GA. Mechanism of action of the cardiac glycosides on the heart. Biochem Pharmacol. 1981;30:3261–3264. doi: 10.1016/0006-2952(81)90597-9. [DOI] [PubMed] [Google Scholar]
- 154.Patnaik GK, Dhawan BN. Pharmacological investigations on asclepin--a new cardenolide from Asclepius curassavica. Part I: Cardiotonic activity and acute toxicity. Arzneimittelforschung. 1978;28:1095–1099. [PubMed] [Google Scholar]
- 155.Patnaik GK, Kohler E. Pharmacological investigation on asclepin--a new cardenolide from Asclepias curassavica. Part II. Comparative studies on the inotropic and toxic effects of asclepin, g-strophantin, digoxin and digitoxin) Arzneimittelforschung. 1978;28:1368–1372. [PubMed] [Google Scholar]
- 156.Zucher W, Weiss-Berg E, Tamm C. 14-epi-digitoxigenin and 3-desoxydigitoxigenin. 19. Reactions with microorganisms. Helv Chim Acta. 1969;52:2449–2458. doi: 10.1002/hlca.19690520826. [DOI] [PubMed] [Google Scholar]
- 157.Ragab MS, Linde HHA, Meyer K. 14 β-Artebufogenin Ueber Kroetengifte, 29. Mitteilung Helvetica Chimica Acta. 1962;45:1794–1799. [Google Scholar]
- 158.Guntert TW, Linde HHA. Chemistry and Structure-Activity Relationships of Cardioactive Steroids. In: Greef K, editor. Cardiac Glycosides, Part 1 Experimental Pharmacology. 1. Vol. 56. Springer Verlag; New York: 1981. pp. 13–24. [Google Scholar]
- 159.Levinsky RA, Lewis RM, Bynum TE, Hanley HG. Digoxin induced intestinal vasoconstriction. The effects of proximal arterial stenosis and glucagon administration. Circulation. 1975;52:130–136. doi: 10.1161/01.cir.52.1.130. [DOI] [PubMed] [Google Scholar]
- 160.Hess T, Scholtysik G, Salzmann R, Riesen W. Digoxin-specific antibody fragments and a calcium antagonist for reversal of digoxin-induced mesenteric vasoconstriction. J Pharm Pharmacol. 1983;35:647–651. doi: 10.1111/j.2042-7158.1983.tb02858.x. [DOI] [PubMed] [Google Scholar]
- 161.Hulthen UL, Bolli P, Kiowski W, Buhler FR. Forearm vasoconstrictor response to ouabain: studies in patients with mild and moderate essential hypertension. J Cardiovasc Pharmacol. 1984;6 1:S75–81. [PubMed] [Google Scholar]
- 162.Mecca TE, Elam JT, Caldwell RW. Mechanism of the pulmonary vasoconstrictor action of digoxin in the dog. J Cardiovasc Pharmacol. 1985;7:833–840. doi: 10.1097/00005344-198509000-00004. [DOI] [PubMed] [Google Scholar]
- 163.Cooke JP, Shepherd JT, Vanhoutte PM. Vasoconstriction induced by ouabain in the canine coronary artery: contribution of adrenergic and nonadrenergic responses. Cardiovasc Drugs Ther. 1988;2:255–263. doi: 10.1007/BF00051242. [DOI] [PubMed] [Google Scholar]
- 164.Indolfi C, Piscione F, Russolillo E, Villari B, Golino P, Ambrosini V, Condorelli M, Chiariello M. Digoxin-induced vasoconstriction of normal and atherosclerotic epicardial coronary arteries. Am J Cardiol. 1991;68:1274–1278. doi: 10.1016/0002-9149(91)90230-i. [DOI] [PubMed] [Google Scholar]
- 165.Blaustein MP, Zhang J, Chen L, Hamilton BP. How does salt retention raise blood pressure? Am J Physiol Regul Integr Comp Physiol. 2006;290:R514–R523. doi: 10.1152/ajpregu.00819.2005. [DOI] [PubMed] [Google Scholar]
- 166.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nature Med. 2004;10:1193–1199. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 167.Bodemann HH. The current concept for the cardiac glycoside receptor. Clin Cardiol. 1981;4:223–228. doi: 10.1002/clc.4960040502. [DOI] [PubMed] [Google Scholar]
- 168.Manunta P, Tyzack J, Hamilton BP, Hamlyn JM. Augmentation and antagonism of ouabain-induced hypertension. Hypertension. 1993;22:432. [Google Scholar]
- 169.Kimura K, Manunta P, Hamilton BP, Hamlyn JM. Different effects of in vivo ouabain and digoxin on renal artery function and blood pressure in the rat. Hypertens Res. 2000;23(Suppl):S67–76. doi: 10.1291/hypres.23.supplement_s67. [DOI] [PubMed] [Google Scholar]
- 170.Wang H, Lu Z, Yuan W. Comparative study of the effects of ouabain and digoxin on blood pressure of rats. Chin Med J (Engl) 1997;110:911–914. [PubMed] [Google Scholar]
- 171.Gheorghiade M, van Veldhuisen DJ, Colucci WS. Contemporary use of digoxin in the management of cardiovascular disorders. Circulation. 2006;113:2556–2564. doi: 10.1161/CIRCULATIONAHA.105.560110. [DOI] [PubMed] [Google Scholar]
- 172.Abarquez RF., Jr Digitalis in the treatment of hypertension. A prelminary report. Acta Med Philipp. 1967;3:161–170. [PubMed] [Google Scholar]
- 173.Manunta P, Hamilton BP, Hamlyn JM. Structure-activity relationships for the hypertensinogenic activity of ouabain: role of the sugar and lactone ring. Hypertension. 2001;37:472–477. doi: 10.1161/01.hyp.37.2.472. [DOI] [PubMed] [Google Scholar]
- 174.Quadri L, Bianchi G, Cerri A, Fedrizzi G, Ferrari P, Gobbini M, Melloni P, Sputore S, Torri M. 17 beta-(3-furyl)-5 beta-androstane-3 beta, 14 beta, 17 alpha-triol (PST 2238). A very potent antihypertensive agent with a novel mechanism of action. J Med Chem. 1997;40:1561–1564. doi: 10.1021/jm970162e. [DOI] [PubMed] [Google Scholar]
- 175.Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol. 2005;564:737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Up-regulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain hypertensive rats. Am J Physiol Heart Circ Physiol. 2009;298:H263–274. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 178.Harteneck C, Plant TD, Schultz G. From worm to man: three subfamilies of TRP channels. Trends Neurosci. 2000;23:159–166. doi: 10.1016/s0166-2236(99)01532-5. [DOI] [PubMed] [Google Scholar]
- 179.Lee MY, Song H, Nakai J, Ohkura M, Kotlikoff MI, Kinsey SP, Golovina VA, Blaustein MP. Local subplasma membrane Ca2+ signals detected by a tethered Ca2+ sensor. Proc Natl Acad Sci U S A. 2006;103:13232–13237. doi: 10.1073/pnas.0605757103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Song H, Lee MY, Kinsey SP, Weber DJ, Blaustein MP. An N-terminal sequence targets and tethers Na+ pump alpha2 subunits to specialized plasma membrane microdomains. J Biol Chem. 2006;281:12929–12940. doi: 10.1074/jbc.M507450200. [DOI] [PubMed] [Google Scholar]
- 181.Somlyo AV, Franzini-Armstrong C. New views of smooth muscle structure using freezing, deep-etching and rotary shadowing. Experientia. 1985;41:841–856. doi: 10.1007/BF01970000. [DOI] [PubMed] [Google Scholar]
- 182.Arnon A, Hamlyn JM, Blaustein MP. Ouabain augments Ca2+ transients in arterial smooth muscle without raising cytosolic Na+ Am J Physiol Heart Circ Physiol. 2000;279:H679–H691. doi: 10.1152/ajpheart.2000.279.2.H679. [DOI] [PubMed] [Google Scholar]
- 183.Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin and Exp Hypertens. 1998;20:691–703. doi: 10.3109/10641969809053247. [DOI] [PubMed] [Google Scholar]
- 184.Blaustein MP, Wier WG. Local sodium, global reach: filling the gap between salt and hypertension. Circ Res. 2007;101:959–961. doi: 10.1161/CIRCRESAHA.107.164459. [DOI] [PubMed] [Google Scholar]
- 185.Rembold CM, Chen XL. The buffer barrier hypothesis, [Ca2+]i homogeneity, and sarcoplasmic reticulum function in swine carotid artery. J Physiol. 1998;513(Pt 2):477–492. doi: 10.1111/j.1469-7793.1998.477bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xin HB, Deng KY, Rishniw M, Ji G, Kotlikoff MI. Smooth muscle expression of Cre recombinase and eGFP in transgenic mice. Physiol Genomics. 2002;10:211–215. doi: 10.1152/physiolgenomics.00054.2002. [DOI] [PubMed] [Google Scholar]
- 187.Pritchard TJ, Bullard DP, Lynch RM, Lorenz JN, Paul RJ. Transgenic Mice Expressing Na+-K+ ATPase in Smooth Muscle Decreases Blood Pressure. Am J Physiol Heart Circ Physiol. 2007;293:H1172–1182. doi: 10.1152/ajpheart.00279.2007. [DOI] [PubMed] [Google Scholar]
- 188.Ferrari P, Ferrandi M, Tripodi G, Torielli L, Padoani G, Minotti E, Melloni P, Bianchi G. PST 2238: A new antihypertensive compound that modulates Na,K-ATPase in genetic hypertension. J Pharmacol Exp Ther. 1999;288:1074–1083. [PubMed] [Google Scholar]
- 189.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The alpha2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol. 2005;288:H477–H485. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 190.Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- 191.Dong H, Jiang Y, Triggle CR, Li X, Lytton J. Novel role for K+-dependent Na+/Ca2+ exchangers in regulation of cytoplasmic free Ca2+ and contractility in arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2006;291:H1226–H1235. doi: 10.1152/ajpheart.00196.2006. [DOI] [PubMed] [Google Scholar]
- 192.Quednau BD, Nicoll DA, Philipson KD. The sodium/calcium exchanger family-SLC8. Pflugers Arch. 2004;447:543–548. doi: 10.1007/s00424-003-1065-4. [DOI] [PubMed] [Google Scholar]
- 193.Nakasaki U, Iwamotot T, Hanada H, Imagawa T, Shigekawa M. Cloning of the rat aortic smooth muscle Na+/Ca2+ exchanger and tissue-specific expression of isoforms. J Biochem (Tokyo) 1993;114:528–534. doi: 10.1093/oxfordjournals.jbchem.a124211. [DOI] [PubMed] [Google Scholar]
- 194.Golovina VA, Song H, James PF, Lingrel JB, Blaustein MP. Na+ pump alpha 2-subunit expression modulates Ca2+ signaling. Am J Physiol Cell Physiol. 2003;284:C475–C486. doi: 10.1152/ajpcell.00383.2002. [DOI] [PubMed] [Google Scholar]
- 195.Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, Takahashi K, Takahashi T, Suzuki T, Ota T, Hamano-Takahashi A, Onishi M, Tanaka Y, Kameo K, Baba A. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–256. [PubMed] [Google Scholar]
- 196.Iwamoto T, Kita S, Uehara A, Imanaga I, M T, Baba A, Katsuragi T. Molecular determinants of Na+/Ca2+ exchange (NCX1) inhibition by SEA0400. J Biol Chem. 2004;279:7544–7553. doi: 10.1074/jbc.M310491200. [DOI] [PubMed] [Google Scholar]
- 197.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+ - Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002;90:305–308. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- 198.Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR, Blaustein MP. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current, and lowers blood pressure. Am J Physiol Heart Circ Physiol. 2009 doi: 10.1152/ajpheart.00964.2009. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- 200.Wagner J, Drab M, Bohlender J, Amann K, Wienen W, Ganten D. Effects of AT1 receptor blockade on blood pressure and the renin-angiotensin system in spontaneously hypertensive rats of the stroke prone strain. Clin Exp Hypertens. 1998;20:205–221. doi: 10.3109/10641969809053215. [DOI] [PubMed] [Google Scholar]
- 201.Seko T, Ito M, Kureishi Y, Okamoto R, Moriki N, Onishi K, Isaka N, Hartshorne DJ, Nakano T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ Res. 2003;92:411–418. doi: 10.1161/01.RES.0000059987.90200.44. [DOI] [PubMed] [Google Scholar]
- 202.Bragulat E, de la Sierra A. Salt intake, endothelial dysfunction, and salt-sensitive hypertension. J Clin Hypertens (Greenwich) 2002;4:41–46. doi: 10.1111/j.1524-6175.2002.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kunes J, Hojna S, Kadlecova M, Dobesova Z, Rauchova H, Vokurkova M, Loukotova J, Pechanova O, Zicha J. Altered balance of vasoactive systems in experimental hypertension: the role of relative NO deficiency. Physiol Res. 2004;53 1:S23–34. [PubMed] [Google Scholar]
- 204.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- 205.Ponnuchamy B, Khalil RA. Cellular mediators of renal vascular dysfunction in hypertension. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1001–1018. doi: 10.1152/ajpregu.90960.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gordon FJ, Mark AL. Impaired baroreflex control of vascular resistance in prehypertensive Dahl S rats. Am J Physiol. 1983;245:H210–217. doi: 10.1152/ajpheart.1983.245.2.H210. [DOI] [PubMed] [Google Scholar]
- 207.Ferrari AU, Gordon FJ, Mark AL. Primary impairment of cardiopulmonary baroreflexes in Dahl salt-sensitive rats. J Hypertens Suppl. 1984;2:S401–403. [PubMed] [Google Scholar]
- 208.Miyajima E, Bunag RD. Impaired sympathetic baroreflexes in prehypertensive Dahl hypertension-sensitive rats. Clin Exp Hypertens A. 1986;8:1049–1061. doi: 10.3109/10641968609044085. [DOI] [PubMed] [Google Scholar]
- 209.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]