

Abstract
Variations in the balance between cell proliferation and apoptosis could contribute to the etiology of gingival overgrowth. The aim of this study was to test the hypothesis that, in fibrotic gingival lesions, fibroblast proliferation is stimulated and apoptosis is decreased. Apoptotic index, caspase 3 expression, the proliferative index, FOXO1 expression, and histological inflammation were measured in situ. Analysis of data showed that apoptosis decreased in all forms of gingival overgrowth examined (p < 0.05), and inflammation caused a small but significant increase compared with non-inflamed tissues (p < 0.05). The greatest decrease of apoptosis occurred in the most fibrotic tissues. Cell proliferation was elevated in all forms of gingival overgrowth tested, independent of inflammation (p < 0.05). To identify potential mechanisms of transcriptional regulation of apoptosis, we assessed FOXO1 and caspase 3 expression levels and found them to correlate well with diminished apoptosis. Analysis of data suggests that increased fibroblast proliferation and a simultaneous decrease in apoptosis contribute to gingival overgrowth.
Keywords: gingival overgrowth, fibrosis, fibroblast, apoptosis, FOXO1
INTRODUCTION
Cell division and death maintain the balance among cell populations in an organism (Chen and Zychlinsky, 1994). Apoptosis is a highly regulated form of programmed cell death, defined by distinct morphological and biochemical features (Vaux and Strasser, 1996; Peter et al., 1997b). Abnormal apoptosis has been implicated in various pathologies, e.g., cancer, AIDS, Alzheimer's disease, rheumatoid arthritis (Thompson, 1995; Peter et al., 1997a; Liu et al., 2006), periodontal diseases (Tonetti et al., 1998; Jarnbring et al., 2002; Bantel et al., 2005), and diabetes (Alikhani et al., 2005a; Graves et al., 2006). Apoptosis can be modulated by mediators of inflammation (Dalgleish and O'Byrne, 2006; Graves et al., 2006).
Intracellular signaling pathways regulate apoptosis, and caspase 3 plays a pivotal role regardless of the activation signal (Vaux and Strasser, 1996; Peter et al., 1997b; Alikhani et al., 2004; Bantel et al., 2005). The Akt/PI3 kinase pathway can regulate gene expression through control of the Forkhead (FOXO) transcription factors (Kaufmann, 1996; Brunet et al., 1998; Burgering and Kops, 2002; Schmidt et al., 2002; Stahl et al., 2002). Activation of FOXOs by dephosphorylation leads to cell cycle arrest (Medema et al., 2000) and alterations in energy metabolism (Barthel et al., 2002), and FOXOs play critical roles in the regulation of proliferation and oxidative stress (Birkenkamp and Coffer, 2003). FOXO activation can result in the induction of apoptosis (Ciechomska et al., 2003; Ghaffari et al., 2003; Hu et al., 2004; Seoane et al., 2004), while inactivation of FOXO proteins disrupts the critical balance between cell proliferation and cell death and contributes to tumorigenesis by promoting cell growth and cell survival (Arden, 2006). Inflammation can also increase FOXO expression, as demonstrated by tumor necrosis factor alpha (TNF-α), activating FOXO in vitro and in vivo, while TNF-α-stimulated fibroblast apoptosis was reduced 76% when FOXO was silenced (Alikhani et al., 2005b). FOXO is also associated with caspase-mediated apoptosis, as evidenced by inhibition of FOXO, which led to a reduced activation of caspases-3, −8, and −9 in response to TNF-α, indicating that FOXO-regulated genes played an important role in the signaling events leading to apoptosis, and identifying the activation of FOXO as a critical step in the capacity of TNF-α to stimulate the apoptosis of fibroblasts (Alikhani et al., 2005b).
In healing tissues, the transition from a granulation tissue to a remodeling tissue requires the apoptosis of fibroblasts. Wounds in which there is inadequate apoptosis may result in the formation of fibrotic tissues. Thus, modulation of apoptosis could contribute to the etiology of fibrosis in gingival tissues. The balance between cell proliferation and apoptosis may be characteristic of different forms of gingival overgrowth, and variations in this balance may provide insights into different etiologies in different forms of gingival overgrowth. For example, phenytoin-induced gingival overgrowth and inherited gingival fibromatosis appear to be more fibrotic than other forms of gingival overgrowth (Uzel et al., 2001; Casavecchia et al., 2004; Trackman and Kantarci, 2004; Kantarci et al., 2006). A working hypothesis is that, in hypercellular and fibrotic lesions, fibroblast proliferation may be stimulated, whereas in lesions containing high levels of extracellular matrix and fewer cells, fibroblast apoptosis may be stimulated. In this study, we investigated in situ the balance between apoptosis and proliferation in different forms of gingival overgrowth and the expression of caspase 3 and FOXO1, to gain potentially new insights into the etiology of different forms of gingival overgrowth.
MATERIALS & METHODS
Gingival Tissues
Gingival tissue samples were obtained from persons undergoing periodontal surgery in the Department of Periodontology and Oral Biology and the Clinical Research Center of Boston University, the Franciscan Children's Hospital and Rehabilitation Center, Boston, MA, and the Department of Periodontology of the University of Istanbul. In total, samples from 40 donors were included in the study. Classification of these samples was as follows: phenytoin-induced gingival overgrowth (n = 8), cyclosporin-A-induced gingival overgrowth (n = 7), nifedipine-induced gingival overgrowth (n = 7), hereditary gingival fibromatosis (n = 12), and control tissues (n = 6) from systemically healthy donors without gingival overgrowth. Consent from the donors was obtained prior to the study. The consent forms were approved by the Institutional Review Board of Boston University Medical Center, the Institutional Review Board of the Franciscan Children's Hospital and Rehabilitation Center, and the Department of Periodontology of the University of Istanbul. All participants in this study were 20 years of age or older. Age, gender, and clinical inflammation (gingival index and bleeding on probing) were recorded for each individual immediately prior to surgical procedures. Gingivectomy and other periodontal surgical procedures were performed after initial periodontal treatment, including professional elimination of supra- and subgingival plaque and maintenance of proper oral hygiene; overall plaque accumulation was brought to minimal (< 25% of all tooth surfaces). Upon excision, tissues were fixed in 4% paraformaldehyde in PBS at 4°C for 4 hrs and then incubated in 30% sucrose overnight. Tissues were then stored in 2-methylbutane at −80°C. At least 20 serial sections were made on a cryostat, and stored at −80°C.
Immunohistochemistry and Histomorphometric Analyses
Immunohistochemistry was carried out with a temperature-controlled staining system to standardize the staining conditions according to methods that we have previously described (Hong et al., 1999; Uzel et al., 2001; Kantarci et al., 2006). Primary antibodies were affinity-purified rabbit polyclonal antibodies against vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), active caspase-3 (Cell Signaling, Beverly, MA, USA), proliferative cell nuclear antigen (PCNA; Santa Cruz, CA, USA), active FKHR/FOXO1A (Abcam, Cambridge, MA, USA), and non-immune goat IgG controls. Quantitative histomorphometric analyses of fibrosis, inflammation, and of immunostained sections were performed as we have previously described (Uzel et al., 2001). Briefly, the orientation of each sample and identification of tissue sites were determined at 100X and 400X magnification; 5 sites with corresponding areas of 0.09 mm2 were defined and utilized for quantitative analyses of immunohistochemical staining. The sites were chosen to represent subepithelial connective tissue, subsulcular connective tissue, and deep connective tissue (Uzel et al., 2001). Quantitation was performed by computer-assisted image analysis (Image-Pro Plus 4.0, Media Cybernetics, Silver Spring, MD, USA), and data were collected from 3 to 5 serial sections per tissue specimen per assay. We evaluated the results by counting the number of stained cells per unit area, and used pre-immune stained slides from serial sections as controls, to determine background staining, which was low or negligible. We determined the number of inflammatory cells per 0.09 mm2 unit area from hematoxylin-stained sections to assess the relative degree of inflammation, and we classified tissues either as `highly inflamed' or as `not highly inflamed' (Uzel et al., 2001). Fibroblastic cells were identified and quantitated per unit area in hematoxylin-and-eosin-stained sections, based on morphological criteria in the same sections. The validity of the morphologic criteria for fibroblast identification was confirmed by quantitative analyses of serial sections stained with vimentin antibody.
TUNEL
We used the in situ TUNEL assay to detect apoptotic fibroblasts, using the TACS 2 TdT (BTL) kit (Trevigen, Gaithersburg, MD, USA), following the manufacturer's instructions.
Apoptotic Index, Caspase 3 Expression, Proliferative Index
The apoptotic index was expressed as the percent of TUNEL-positive fibroblastic cells in 0.09 mm2 areas. Five areas per slide were measured in 3 to 5 serial sections per sample (Uzel et al., 2001). Caspase 3 data were expressed as the percent of positive fibroblasts, and data were collected in the same way as for the TUNEL assay. We determined the proliferative index by measuring the percentage of PCNA-positive fibroblasts per unit area in 5 areas per slide in 3 to 5 serial sections per sample.
Statistical Analysis
Results are expressed as mean ± standard error of the mean for each evaluated site. For statistical analysis, the Mann-Whitney test was used for the comparison of differences. Significance was defined as p < 0.05.
RESULTS
Apoptosis in Fibrotic Gingiva
Apoptosis in gingival tissues was assessed by TUNEL assays. We determined the number of TUNEL-positive fibroblasts and the total number of fibroblasts per unit area as a function of all forms of gingival overgrowth and inflammation, to assess for any differences. In addition to the total number of fibroblasts per unit area, TUNEL-positive fibroblasts were counted and are presented (Table, A). Data were also expressed as the apoptotic index, or the percent of TUNEL-positive fibroblasts (Fig. 1A). Results show that apoptosis was significantly reduced in phenytoin-induced gingival overgrowth and in non-drug-induced gingival fibromatosis tissues, compared with control and cyclosporin-A-induced gingival overgrowth samples. Cyclosporin A and nifedipine samples also showed significantly less apoptosis compared with controls. Inflammation led to increased apoptosis in non-fibrotic control samples and in cyclosporin A and nifedipine samples. In phenytoin and gingival fibromatosis samples, inflamed areas also showed significantly increased apoptosis compared with non-inflamed sites, but the effect of inflammation may be less than for control, cyclosporin A, and nifedipine samples (Fig. 1A).
Table.
Fibroblast Counts, TUNEL-positive (A), PCNA-positive (B), and Caspase-3-positive (C) Fibroblasts in Gingival Samples in Inflamed and Non-inflamed Sites
| A | B | C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Fibroblast Counts | TUNEL(+) Fibroblasts | Total Fibroblast Counts | PCNA (+) Fibroblasts | Total Fibroblast Counts | Caspase 3(+) Fibroblasts | |||||||
| Inflamed | Non-inflamed | Inflamed | Non-inflamed | Inflamed | Non-inflamed | Inflamed | Non-inflamed | Inflamed | Non-inflamed | Inflamed | Non-inflamed | |
| Control | 231.3 ± 33.2a | 178.8 ± 24.6 | 8.9 ± 1.0§ | 1.4 ± 3.9 | 187.3 ± 51.2 | 177.4 ± 35.6 | 76.0 ± 16.7 | 62.4 ± 12.5 | 221.7 ± 31.2 | 165.8 ± 21.6 | 8.3 ± 2.0 | 7.3 ± 1.9 |
| Phenytoin | 162.2 ± 32.2*§ | 368.8 ± 62.2*# | 1.8 ± 0.8*†§ (21 ± 2%)λ | 0.2 ± 1.0* (15 ± 2%) | 154.7 ± 32.4*§ | 289.8 ± 32.8*# | 86.6 ± 11.8§ (114 ± 22%) | 167.4 ± 32.6*# (270 ±33%) | 142.2 ± 22.2*§ | 325.8 ± 27.2*# | 2.5 ± 1.7* (30 ± 2%) | 1.7 ± 1.4* (24 ± 7%) |
| Cyclosporin A | 211.5 ± 48.9 | 254.0 ± 47.8* | 3.6 ± 1.3*§ (41 ± 3%) | 0.5 ± 1.5* (35 ± 4%) | 171.4 ± 29.6 | 214.5 ± 34.8* | 69.8 ± 31.6 (92 ± 11%) | 81.8 ± 24.2 (132 ± 26%) | 181.5 ± 38.4 | 198.4 ± 48.8* | 6.3 ± 1.5 (76 ± 13%)(52 | 3.8 ± 1.6* (52 ± 9%) |
| Nifedipine | 186.8 ± 54.5§ | 322.0 ± 63.1* | 3.5 ± 1.4*§ (32 ± 4%) | 0.3 ± 1.4* (21 ± 2%) | 146.7 ± 36.3§ | 208.4 ± 35.7* | 78.3 ± 26.9§ (103 ± 16%) | 141.1 ± 21.7*# (227 ± 35%] | 176.4 ± 42.1§ | 212.0 ± 32.1* | 4.3 ± 1.7 (52 ± 11%) | 2.6 ± 1.9* (36 ± 6%) |
| Gingival Fibromatosis | 200.0 ± 34.5§ | 343.3 ± 47.1*# | 1.3 ± 1.8*†§ (15 ± 2%)(1 | 0.2 ± 0.8* (13 ± 5%) | 165.4 ± 21.7§ | 244.5 ± 51.2*# | 82.8 ± 24.7§ (108 ± 16%) | 164.0 ± 31.0*# (264 ± 41%) | 162.2 ± 24.5§ | 234.3 ± 43.1*# | 3.2 ± 1.6* (38 ± 9%) | 2.1 ± 1.3* (28 ± 7%) |
Data are expressed as mean ± SD per 0.09 mm2 area.
Independent samples per experimental group (n) are as follows: (for TUNEL) Control = 6, Phenytoin = 8, Cyclosporin A = 7, Nifedipine = 7, and Gingival Fibromatosis = 12; (for PCNA) Control = 6, Phenytoin = 6, Cyclosporin A = 6, Nifedipine = 6, Gingival Fibromatosis = 12; and (for Caspase 3 expression) Control = 6, Phenytoin = 7, Cyclosporin A = 7, Nifedipine = 6, and Gingival Fibromatosis = 11.
p < 0.05 compared with Control;
p < 0.05 compared with Cyclosporin A;
p < 0.05 compared with Cyclosporin A and Nifedipine;
p < 0.05 compared with non-inflamed. Inflamed areas are those having greater than 75 inflammatory cells/0.09 mm2; non-inflamed areas have fewer than 75 inflammatory cells/0.09 mm2.
Numbers in parentheses represent percent comparisons with control values.
Figure 1.
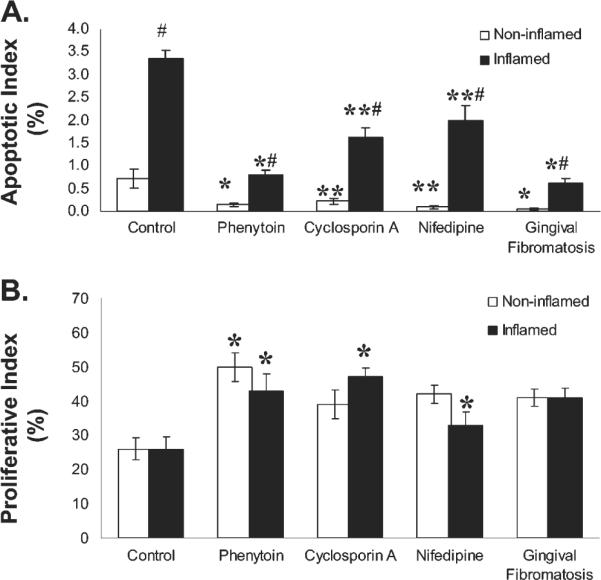
Apoptotic index (% TUNEL-positive fibroblasts) (A) and proliferative index (% PCNA-positive fibroblasts) (B) in gingival overgrowth and control tissue samples. Data are expressed as means ± SD. In (A), the numbers of independent samples (n) per group are: no overgrowth control, 6; phenytoin overgrowth, 8; cyclosporin A overgrowth, 7; nifedipine overgrowth, 7; and gingival fibromatosis, 12. *p < 0.05 compared with control, cyclosporin A, and nifedipine; **p < 0.05 compared with control; #p < 0.05 compared with non-inflamed. In (B), (n) per group is: control, 6; phenytoin, 6; cyclosporin A, 6; nifedipine, 6; gingival and fibromatosis, 12. *p < 0.05 compared with control.
Cell Proliferation in Connective Tissue
To investigate cell proliferation as a function of gingival overgrowth, we determined the levels of PCNA (Table, B). This factor is required for DNA synthesis and is a marker for actively dividing cells in vivo. We determined the proliferative index for all samples by normalizing the PCNA-positive fibroblasts to the total number of fibroblasts in each section (Fig. 1B). Phenytoin, cyclosporin A, nifedipine, and gingival fibromatosis samples showed significantly higher cell proliferation over control samples. No effect of inflammation was observed on the proliferative index, suggesting that all types of gingival overgrowth led to increased fibroblast proliferation.
To assess independently for apoptosis and to gain mechanistic insights, we next performed immunohisto chemistry to determine the in vivo expression of caspase 3. Measurement of caspase 3 was performed because it is a pivotal enzyme in apoptosis in the two best-characterized pathways (Graves et al., 2006). Data are presented as the total number of fibroblasts per unit area and the number of caspase-3-positive fibroblasts per unit area in inflamed and non-inflamed areas of tissues (Table, C). In addition, data are expressed as the percent caspase-3-positive fibroblasts, by analogy to the apoptotic index calculation (Fig. 2). Similar to TUNEL, phenytoin and gingival fibromatosis samples showed the lowest caspase 3 expressions among the groups, with significant differences compared with control and cyclosporin A samples. Nifedipine also contained significantly less caspase 3 than control samples (Fig. 2). Comparisons of inflamed and non-inflamed sites revealed a trend toward increased caspase 3 expression with inflammation, but the difference between caspase levels as a function of inflammation was not statistically significant. Thus, trends in the apoptotic index and the percent of caspase-3-positive cells were similar, though the absolute values of these measurements differed as expected, since the assays measured different stages of the process of apoptosis.
Figure 2.
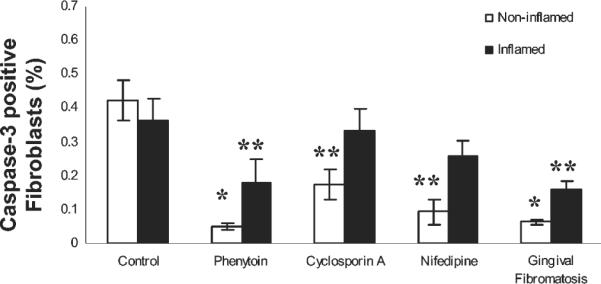
Caspase 3 expression in gingival tissues. Quantitative histomorphometric analyses of caspase 3 immunostaining in all forms of gingival overgrowth in inflamed and non-inflamed tissue areas (0.09 mm2). (n) per group is: control, 6; phenytoin, 8; cyclosporin A, 7; nifedipine, 7; and gingival fibromatosis, 12. *p < 0.05 compared with control and cyclosporin A; **p < 0.05 compared with control.
FOXO1 Expression
FOXO1 is a regulator of apoptosis but, to our knowledge, has not been studied in fibrosis. Data expressed as FOXO1-positive fibroblasts per unit area (0.09 mm2) revealed that inflamed areas of control samples showed significantly higher immunostaining of FOXO1 (Fig. 3A). Phenytoin, cyclosporin A, nifedipine, and gingival fibromatosis samples also showed higher levels of FOXO1 in areas where the inflammation was high (Fig. 3B). All gingival fibrosis samples showed reduced FOXO1 expression in fibroblasts compared with controls.
Figure 3.

FOXO1 expression in gingival overgrowth and no overgrowth control tissues. (A) Representative sections for the immunohistochemical staining of FOXO1 expression in phenytoin and control tissues. Black arrows designate fibroblasts stained positive for FOXO1. Each bar represents 200 μm at a magnification of 200×. (B) Histomorphometric and quantitative analyses of FOXO1 immunostaining in all forms of gingival overgrowth in inflamed and non-inflamed tissue areas (0.09 mm2). (n) for control, 6; for phenytoin, 6; for cyclosporin A, 6; for nifedipine, 6; and for gingival fibromatosis, 8. *p < 0.05 compared with control; #p < 0.05 compared with non-inflamed.
DISCUSSION
This study demonstrated that fibroblast apoptosis is decreased in gingival overgrowth, and that this decrease may contribute to fibrosis, particularly in phenytoin-induced gingival overgrowth and in gingival fibromatosis tissues. Phenytoin-induced gingival overgrowth and gingival fibromatosis are the most fibrotic tissues, meaning that they contain the highest proportion of fibroblastic cells and connective tissue fibers (Uzel et al., 2001; Kantarci et al., 2006). Thus, increased fibroblast and extracellular matrix accumulation appears to be due, in part, to diminished fibroblast cell death in these tissues. Nifedipine and cyclosporin A tissues were more inflamed than phenytoin-induced gingival overgrowth and gingival fibromatosis tissues. The apoptotic indices of nifedipine and cyclosporin A tissues were higher than in phenytoin and gingival fibromatosis tissues, but lower than in normal control tissues. Analysis of the data showed that inflammation led to an increase in apoptosis in 'no overgrowth' control gingiva, and inflammation similarly appeared to stimulate apoptosis within the context of gingival overgrowth, but to a lesser degree. Inflammation was the lowest in phenytoin and gingival fibromatosis compared with other forms of gingival overgrowth (Uzel et al., 2001; Kantarci et al., 2006). Whether lower inflammation in phenytoin and gingival fibromatosis is a primary determining factor in the etiology of these forms of gingival overgrowth is not known at the present time. It is interesting that phenytoin-induced fibrosis and gingival fibromatosis contained high levels of CCN2/CTGF (connective tissue growth factor), a marker of increased TGF-β1 activity (Uzel et al., 2001; Kantarci et al., 2006). Because TGF-β1 suppresses the acquired immune response (Bottinger et al., 1997), it seems possible that stimulation of a TGF-β1/CTGF pathway in gingival fibromatosis and phenytoin-induced gingival overgrowth could, in turn, reduce gingival inflammation and fibroblast apoptosis, and thereby contribute to gingival overgrowth.
Fibroblast cell proliferation increased in all forms of gingival overgrowth tested, regardless of the level of inflammation. Thus, decreased apoptosis coupled with increased cell proliferation together appear to contribute to all forms of gingival overgrowth. Decreased fibroblast apoptosis appears to be particularly important in the development of the most fibrotic forms of gingival overgrowth. Analyses performed by both the TUNEL assay and determinations for caspase 3 levels support these findings.
Analysis of the data suggests that FOXO1 plays a role in the regulation of apoptosis in fibrotic gingiva. FOXO1 is an important regulator of apoptosis. As noted earlier, FOXO1 has other biological roles as well (Barthel et al., 2002; Birkenkamp and Coffer, 2003). To our knowledge, however, FOXO1 expression has not been studied in the context of fibrotic diseases. Since FOXO activation leads to pro-apoptotic gene expression and, ultimately, enhanced caspase 3 activation (Alikhani et al., 2005b), the finding that tissues with low FOXO1 activation also have low caspase-3 activation is intriguing. To our knowledge, the present study provides the first evidence of FOXO1 regulation in gingival tissues.
In conclusion, the data presented here provide evidence that decreased apoptosis possibly mediated through diminished FOXO and caspase-3 expression, together with increased proliferative activity in fibroblasts, could contribute to fibrotic overgrowth of gingival tissues. The similarity between phenytoin-induced gingival overgrowth and non-medication-induced gingival fibromatosis suggests that decreased gingival fibroblast apoptosis may have different initiating causes, but is a particular characteristic of the most fibrotic tissues. Moreover, inflammation increased apoptosis to a certain degree in all gingival tissues. Further in vitro work is required to identify the pathways of inhibition of apoptosis in gingival fibroblasts and tissues, and an increased understanding of these pathways could ultimately provide opportunities for therapeutic interventions.
ACKNOWLEDGMENTS
This research was supported by NIH DE11004, DE07559, and M01 RR00533. We thank Dr. Selva Sume for assistance with quantitative analyses of stained histology slides, and Dr. Weicheng Wu for technical assistance.
REFERENCES
- Alikhani M, Alikhani Z, Raptis M, Graves DT. TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol. 2004;201:341–348. doi: 10.1002/jcp.20067. [DOI] [PubMed] [Google Scholar]
- Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT. Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem. 2005a;280:12087–12095. doi: 10.1074/jbc.M406313200. [DOI] [PubMed] [Google Scholar]
- Alikhani M, Alikhani Z, Graves DT. FOXO1 functions as a master switch that regulates gene expression necessary for tumor necrosis factor-induced fibroblast apoptosis. J Biol Chem. 2005b;280:12096–12102. doi: 10.1074/jbc.M412171200. [DOI] [PubMed] [Google Scholar]
- Arden KC. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Exp Gerontol. 2006;41:709–717. doi: 10.1016/j.exger.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Bantel H, Beikler T, Flemmig TF, Schulze-Osthoff K. Caspase activation is involved in chronic periodontitis. FEBS Lett. 2005;579:5559–5564. doi: 10.1016/j.febslet.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Kruger KD, Roth RA, Joost HG. Regulation of the forkhead transcription factor FKHR (FOXO1a) by glucose starvation and AICAR, an activator of AMP-activated protein kinase. Endocrinology. 2002;143:3183–3186. doi: 10.1210/endo.143.8.8792. [DOI] [PubMed] [Google Scholar]
- Birkenkamp KU, Coffer PJ. FOXO transcription factors as regulators of immune homeostasis: molecules to die for? J Immunol. 2003;171:1623–1629. doi: 10.4049/jimmunol.171.4.1623. [DOI] [PubMed] [Google Scholar]
- Bottinger EP, Letterio JJ, Roberts AB. Biology of TGF-beta in knockout and transgenic mouse models. Kidney Int. 1997;51:1355–1360. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- Brunet CL, Gunby RH, Benson RS, Hickman JA, Watson AJ, Brady G. Commitment to cell death measured by loss of clonogenicity is separable from the appearance of apoptotic markers. Cell Death Differ. 1998;5:107–115. doi: 10.1038/sj.cdd.4400334. [DOI] [PubMed] [Google Scholar]
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27:352–360. doi: 10.1016/s0968-0004(02)02113-8. [DOI] [PubMed] [Google Scholar]
- Casavecchia P, Uzel MI, Kantarci A, Hasturk H, Dibart S, Hart TC, et al. Hereditary gingival fibromatosis associated with generalized aggressive periodontitis: a case report. J Periodontol. 2004;75:770–778. doi: 10.1902/jop.2004.75.5.770. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zychlinsky A. Apoptosis induced by bacterial pathogens. Microb Pathog. 1994;17:203–212. doi: 10.1006/mpat.1994.1066. [DOI] [PubMed] [Google Scholar]
- Ciechomska I, Pyrzynska B, Kazmierczak P, Kaminska B. Inhibition of Akt kinase signalling and activation of Forkhead are indispensable for upregulation of FasL expression in apoptosis of glioma cells. Oncogene. 2003;22:7617–7627. doi: 10.1038/sj.onc.1207137. [DOI] [PubMed] [Google Scholar]
- Dalgleish AG, O'Byrne K. Inflammation and cancer: the role of the immune response and angiogenesis. Cancer Treat Res. 2006;130:1–38. [PubMed] [Google Scholar]
- Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci USA. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Liu R, Alikhani M, Al-Mashat H, Trackman PC. Diabetes-enhanced inflammation and apoptosis—impact on periodontal pathology. J Dent Res. 2006;85:15–21. doi: 10.1177/154405910608500103. [DOI] [PubMed] [Google Scholar]
- Hong HH, Uzel MI, Duan C, Sheff MC, Trackman PC. Regulation of lysyl oxidase, collagen, and connective tissue growth factor by TGF-beta1 and detection in human gingiva. Lab Invest. 1999;79:1655–1667. [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Jarnbring F, Somogyi E, Dalton J, Gustafsson A, Klinge B. Quantitative assessment of apoptotic and proliferative gingival keratinocytes in oral and sulcular epithelium in patients with gingivitis and periodontitis. J Clin Periodontol. 2002;29:1065–1071. doi: 10.1034/j.1600-051x.2002.291203.x. [DOI] [PubMed] [Google Scholar]
- Kantarci A, Black SA, Xydas CE, Murawel P, Uchida Y, Yucekal-Tuncer B, et al. Epithelial and connective tissue cell CTGF/CCN2 expression in gingival fibrosis. J Pathol. 2006;210:59–66. doi: 10.1002/path.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH. Proteolytic cleavage during chemotherapy-induced apoptosis. Mol Med Today. 1996;2:298–303. doi: 10.1016/1357-4310(96)10023-x. [DOI] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, et al. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res. 2006;85:510–514. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Peter ME, Ehret A, Berndt C, Krammer PH. AIDS and the death receptors. Br Med Bull. 1997a;53:604–616. doi: 10.1093/oxfordjournals.bmb.a011633. [DOI] [PubMed] [Google Scholar]
- Peter ME, Heufelder AE, Hengartner MO. Advances in apoptosis research. Proc Natl Acad Sci USA. 1997b;94:12736–12737. doi: 10.1073/pnas.94.24.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, et al. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Tonetti MS, Cortellini D, Lang NP. In situ detection of apoptosis at sites of chronic bacterially induced inflammation in human gingiva. Infect Immun. 1998;66:5190–5195. doi: 10.1128/iai.66.11.5190-5195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trackman PC, Kantarci A. Connective tissue metabolism and gingival overgrowth. Crit Rev Oral Biol Med. 2004;15:165–175. doi: 10.1177/154411130401500305. [DOI] [PubMed] [Google Scholar]
- Uzel MI, Kantarci A, Hong HH, Uygur C, Sheff MC, Firatli E, et al. Connective tissue growth factor in drug-induced gingival overgrowth. J Periodontol. 2001;72:921–931. doi: 10.1902/jop.2001.72.7.921. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Strasser A. The molecular biology of apoptosis. Proc Natl Acad Sci USA. 1996;93:2239–2244. doi: 10.1073/pnas.93.6.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]