

Abstract
Postmating reproductive isolation is often manifested as hybrid male sterility, for which X-linked genes are overrepresented (the so-called large X effect). In contrast, X-linked genes are significantly under-represented among testis-expressing genes. This seeming contradiction may be germane to the X:autosome imbalance hypothesis on hybrid sterility, in which the X-linked effect is mediated mainly through the misexpression of autosomal genes. In this study, we compared gene expression in fertile and sterile males in the hybrids between two Drosophila species. These hybrid males differ only in a small region of the X chromosome containing the Ods-site homeobox (OdsH) (also known as Odysseus) locus of hybrid sterility. Of genes expressed in the testis, autosomal genes were, indeed, more likely to be misexpressed than X-linked genes under the sterilizing action of OdsH. Since this mechanism of X:autosome interaction is only associated with spermatogenesis, a connection between X:autosome imbalance and the high rate of hybrid male sterility seems plausible.
Whole-genome expression studies have revealed that the X chromosome and autosomes do not play proportionately equal roles in gametogenesis (Lindsley and Lifschytz 1972; Strausberg et al. 1999; Reinke et al. 2000; Wang et al. 2001; Parisi et al. 2003; Sturgill et al. 2007). Most strikingly, the X chromosome genes tend to be expressed at lower rates in the testis (Reinke et al. 2000; Parisi et al. 2003; Sturgill et al. 2007). A plausible explanation is that the X chromosome spends only one-third of the time in males, as opposed to the autosomes, which spend equal time in both sexes. Since the X chromosome is associated with females more often, it may tilt toward female functions at the expense of male activities when genes experience sexual antagonism (Rice 1992). In this explanation, X-linked genes have a tendency to become “demasculinized” (Wu and Xu 2003).
The X-demasculinization hypothesis is one of several that have been invoked to explain differences between the X chromosome and the autosomes in gene expression and gene content (for other hypotheses, see Lercher et al. 2003; Khil et al. 2004). These expression and gene content differences include the precocious X inactivation in the germline of XY males (Wu and Xu 2003; Hense et al. 2007; Sturgill et al. 2007), the distribution of male-biased and testis-expressed genes between X and autosomes (Reinke et al. 2000; Meiklejohn et al. 2003; Parisi et al. 2003; Ranz et al. 2003; Sturgill et al. 2007), and the transposition of genes among chromosomes during evolution (Betran et al. 2002). Several studies have suggested that the expression of X-linked genes may be restricted to early spermatogenesis, when it shares features with early oogenesis (Xu 1996; Wang et al. 2001; Wu and Xu 2003; Reinke et al. 2004). In contrast, the expression of autosomal genes continues until meiosis.
The predominance of autosomal gene expression in spermatogenesis contrasts with the genetics of hybrid male sterility. Although X-linked genes make up a smaller portion of those involved in spermatogenesis, they nonetheless seem to play a larger role in male hybrid sterility (Hollocher and Wu 1996; True et al. 1996; Tao et al. 2003; Masly and Presgraves 2007). This phenomenon is sometimes referred to as “the large X-effect” of hybrid sterility. The large X effect is part of an ensemble of phenomena associated with interspecific hybridization (for details, see Wu and Davis 1993; Supplemental material).
To address the seeming incongruity in the genetics of hybrid male sterility and testicular gene expression, we investigate the influence of hybrid male sterility on gene expression. We did not use F1 hybrids in interspecific crosses in our study because F1 hybrid sterility is the manifestation of complex genic interactions between the entire genomes of two species. Instead, we ask the following question: Among those genes expressed in the testes, are autosomal genes more prone to be misregulated than the X-linked ones when males are sterile due to the effects of an X-linked gene?
For this purpose, we chose to study the transcriptional consequence of a single X-linked gene, the Ods-site homeobox (OdsH) (also known as Odysseus) gene, which causes male sterility in the Drosophila simulans background when cointrogressed with closely linked factors from Drosophila mauritiana (Ting et al. 1998). OdsH, a homeobox gene that has been suggested to encode a heterochromatin-binding protein (Bayes and Malik 2009), mainly functions in spermatogenesis; its absence reduces the fertility of young males (Sun et al. 2004). It is reasonable to suspect that the mechanism by which OdsH acts is through transcriptional regulation; indeed, misexpression of a number of genes in D. simulans–D. mauritiana sterile hybrid males does appear to be linked to the OdsH locus (Michalak and Noor 2004).
In this study, we compared gene expression between D. simulans males that carried either a fertile or sterile introgression (referred to as F or S males, respectively) from D. mauritiana. The introgressions span approximately one-tenth of the X chromosome, but the difference between the two introgressions is only ∼3 kb, which contains exons 3–4 of OdsH. Thus, the sterile introgression harbors a full OdsH gene from D. mauritiana, whereas the fertile one has the critical homeodomain of OdsH from D. simulans (Ting et al. 1998; Sun et al. 2004). In short, F and S males have the same genetic background with respect to species origin, except the source of a portion of the OdsH gene. Therefore, any detected expression differences will not be confounded by differential hybridization efficiency due to sequence divergence between the lines.
Results
Testes were dissected from both F (for fertile introgression) and S (sterile introgression) males. RNA extracts from either sample were hybridized to Affymetrix expression arrays three times each, as described in Methods. In the testis samples, a total of 10,986 transcripts (60%), including five on the Y, 1683 on the X, and 9298 on the autosomes, were detected.
In Figure 1A, we plot the expression levels of genes from the S males against those of the F males. It is clear that the majority of genes are expressed at a comparable level (r2 = 0.9663) between the two samples, which differ in only ∼3 kb of their respective genomes. Of interest is the subset of genes that are differently expressed between the F and S males. A list of the genes that were found to differ significantly is shown in Supplemental Tables S1 and S2.
Figure 1.

The correlation in the expression level of F and S males in testis (A) and abdomen (B). B serves as the negative control (see text).
A salient observation of this study is the different representation of X-linked and autosomal genes that are misexpressed in S males. With Δ = 1.6 (corresponding to FDR < 1%), 14.5% (1348/9298) of the expressed genes on autosomes but only 7.66% (129/1683) of X-linked genes are misregulated in S males (Table 1). The difference is highly significant (P < 0.0005, Fisher's exact test). When each autosomal arm is examined individually, the percentages across the four arms are very close (ranging from 14.0% to 16.3% for Δ = 1.6), and each arm has more misexpressed genes than the X (all comparisons having P < 0.0005) (Supplemental Table S3).
Table 1.
Expression differences between males from the fertile (f) and sterile (s) introgression lines

aSee Methods.
bPercentage of genes that show an expression difference among those with detectable expression. Genes with log2(signal intensity) > 6.2 for the array data and with RKPM > 1 for the RNA-seq data are regarded as expressed genes (see Methods).
cDrosophila array version 1 was used for the abdomen samples, and Drosophila 2 array for the testis samples. Approximately 13,500 and 18,500 D. melanogaster transcripts are represented on the Drosophila 1 and 2 arrays, respectively (see Methods).
dP < 0.005 and P < 0.001 are based on the transformed RPKM data, as explained in Methods, for the purpose of comparing across the microarray and RNA-seq platforms. Both P < 0.005 and P < 0.001 in this table correspond to P < 10−8 in Supplemental Figure S3, which was based on the untransformed raw data.
We also use a less stringent set of criteria for misexpression in S males (Table 1). By this set, a gene is included in the S ≠ F category if P < 0.05 by t-test and |log2(f/s)| > 0.2, where f and s are the expression levels of F and S males, respectively. Indeed, the X-A contrast in the proportion of misregulated genes in S males is observed at all levels of expression (Fig. 2; see also Supplemental Fig. S1), thus ruling out the possibility of reduced sensitivity in detecting expression differences among X-linked genes as an explanation for the X-A contrast.
Figure 2.

Proportion of genes that are significantly misregulated in the testes of S males as a function of the cutoff in calling presence in expression. Misregulation is called by SAM at Δ = 1.6. See Supplemental Figure S2 for further analysis.
There may be two concerns about the technical validity of the observed X:A bias. The first one is the reliability of using microarrays for analyzing gene expression in D. simulans. We have therefore verified the microarray results by the RNA-seq procedure (Cloonan et al. 2008). A total of more than 50 million reads were collected from the RNAs of the fertile and sterile testes. Among them, 24,383,254 and 14,308,455 unique reads were mapped to the D. simulans genome in the fertile and sterile lines, respectively. In Methods and Supplemental Table S6, the quality and quantity of the data are evaluated in detail and the disparity in read numbers is explained. (The reason is technical, rather than biological.)
By Fisher's exact test, Supplemental Figure S3 shows that the X:A bias is evident at all P-values, ranging from 10−2 to 10−16. Given the large number of reads, the level of significance is necessarily much higher in the RNA-seq data than in the array data. Thus, in Table 1, we chose two levels of significance at which data from the two platforms can be compared. The X:A bias is, indeed, observable in the RNA-seq data, as in the array data (Table 1). The bias in the former is smaller, albeit only moderately so, than in the latter.
There is another notable aspect to the X:A bias. When the misexpressed genes in the sterile males are separated into over- and underexpressed categories, X-linked genes tend to be underexpressed, whereas autosomal genes are more evenly distributed between the two categories (see Supplemental Table S5). However, because the relative abundance of the two categories is strongly affected by the level of expression and by the expression differences, the robustness of this observation will need to be further explored.
A second concern with the results so far is the need for a negative control. As the OdsH gene is expressed primarily in testes (Sun et al. 2004) and affects male fertility but not viability (Perez et al. 1993), one would naturally expect little differences in gene expression between F and S males in nontesticular tissues. Due to the tedious and slow process of removing testes, we used abdominal samples for this purpose. In Figure 1B, the correlation of the abdominal expression in the F and S males is shown. The correlation in Figure 1B at r2 = 0.9866 is even higher than that in Figure 1A for testes because genes are not differentially expressed between F and S males in most tissues of the abdomen (except testes). In Table 1, the abdominal data indeed show much reduced numbers of misexpressed genes in the F/S comparison.
Interestingly, in the abdominal samples, of which a fraction is testis RNA, the X:A bias in misexpression is still weakly observable. By the t-test, 0.56% and 2.71% of genes on X and autosomes, respectively, are misexpressed in S males. The difference is significant (P < 0.001, Fisher's exact test). At Δ = 1.6, there are too few misexpressed genes to yield a significant test, but the trend is in the expected direction.
Discussion
The presence of the X-linked OdsH gene from D. mauritiana in the D. simulans background (which carries the introgression from D. mauritiana), indeed, causes the misexpression of several hundred genes. Perez et al. (1993) reported that the testes of sterile males are slightly smaller than those of fertile males. Their sperm is in characteristic disarray, and sperm bundles are defective. It should be noted that many of the changes in cell morphology become observable only after the completion of meiosis, when gene expression becomes quiescent (Perez et al. 1993). Hence, the changes in expression reported in Table 1 take place in the testes before the changes in cell morphology or composition. These expression changes eventually lead to spermatogenic failure (see also Sun et al. 2004).
Most interesting, these misexpressed genes are disproportionately more common on the autosomes than on the X. While OdsH is located on the X chromosome, the genes it regulates (directly or indirectly) tend not to be. In Supplemental Table S4, we list the proportion of such genes in each functional category according to the Gene Ontology (Ashburner et al. 2000) classification. The distribution of misregulated genes is rather broad across functional categories. Moreover, the proportion of genes located on the autosomes is comparably higher than on the X chromosome for almost every category.
The patterns discussed above suggest a general transcriptional basis for X:autosome imbalance whereby interaction between X-linked genes, which tend to be expressed early during spermatogenesis (Wang et al. 2001; Wu and Xu 2003), and autosomal genes, expressed later in spermatogenesis, leads to hybrid dysfunction. Specifically, factors such as OdsH that reside on the X chromosome, when expressed at early stages of sperm development, will fail to properly regulate genes on the autosomes, with cascading detrimental effects.
The above mechanism of X:A imbalance suggests that the large X effect on hybrid male sterility and the predominant autosomal expression in spermatogenesis are not incongruous observations. Since X-linked genes tend to be expressed in early spermatogenesis (Wang et al. 2001; Parisi et al. 2003; Wu and Xu 2003), their effects on hybrid sterility may be correspondingly larger. On the other hand, the sterility effect tends to be exerted on autosomal genes, which continue to be expressed in later stages of spermatocytes after the X has been inactivated (Wu and Xu 2003; Reinke et al. 2004; Sturgill et al. 2007). The temporal difference in X versus autosomal expression is widespread, observable in mammals, Drosophila, and nematodes (Wu and Xu 2003).
The X:autosome imbalance hypothesis was originally proposed by Muller (1942) to explain Haldane's rule (Haldane 1922) whereby the hemizygous sex (XY or ZW) among interspecific hybrids is more severely affected than the homozygous sex (XX or ZZ). Muller's reasoning was that the hemizygous sex has the X chromosome from only one species, whereas the autosomes are from both species, potentially resulting in expression imbalance. (The homozygous sex has a more “balanced” genotype, with one full haploid genome from both species.)
Support for the X:A imbalance hypothesis on Haldane's rule declined, primarily in connection with experiments on hybrid sterility (for review, see Wu et al. 1996). There has been a long history of debate on the explanations for Haldane's rule and the several components of this rule. In the Supplemental material, we provide an account of this debate and the relevance of this current study to the various issues surrounding it. A main issue is that, in comparison with hybrid inviability or hybrid female sterility, hybrid male sterility has evolved 10–100 times faster in species with XX-XY sex determination (Wu 1991; Wu and Davis 1993; Wu et al. 1996). It has not been clear why X:autosome interactions should have a large effect on the fertility of XY males, but not on their viabilities. Our analysis of gene misexpression under the sterilizing effect of OdsH suggests that spermatogenesis may be particularly susceptible to the effect of X:autosome interactions, resulting in prevalent hybrid male sterility.
Methods
RNA preparation and oligonucleotide arrays
The fertile and sterile introgression lines were constructed and maintained as described in Ting et al. (1998). To obtain 5∼10 μg of RNA for one microarray hybridization, we collected on average 150 adult male flies aged 3–4 d from each line for testis dissection, and 50 flies for abdomen dissection. Flies were collected at different times in order to incorporate among-culture variation into the hybridizations. The testes were dissected on ice in RNALater and transferred into TRIzol reagent (Invitrogen). The abdomens were separated from whole body in dry ice and immediately homogenized in TRIzol. Total RNA was isolated according to the manufacturer's instructions, further purified with phenol–chloroform extraction, and precipitated with isopropanol. Three separate extractions and hybridizations were carried out for the RNA samples derived from testes and abdomens in each of two introgression lines.
GeneChip Drosophila Genome Arrays (Drosophila 1; Affymetrix) were used for the fly abdomen samples, and Drosophila Genome 2.0 Arrays (Drosophila 2; Affymetrix) for the testis samples. Target synthesis for GeneChips, hybridization, and scanning were performed according to the standard protocol outlined in the Affymetrix GeneChip expression Analysis Technical Manual, and the protocols are available at http://fgf.bsd.uchicago.edu/facility/microarray.htm.
Array data analysis
The probe sets represented on the Drosophila 1 and Drosophila 2 arrays are complementary to ∼13,500 and 18,500 Drosophila melanogaster transcripts (http://www.affymetrix.com), respectively. GeneChip images were quantified and gene expression values (signal intensity) were calculated by Affymetrix Microarray Suite Version 5.0 (MAS 5.0). Further details of the Affymetrix platform and their algorithms can be found at http://www.affymetrix.com. The GEO accession number for the array data is GSE21734.
The genetic background of the fertile and sterile lines is mostly that of D. simulans, which is significantly diverged from the D. melanogaster sequence on the array. For this reason, it may not be appropriate to use the assignments of presence and absence that depend on the comparison of perfect match and mismatch probes. Therefore, we calculated expression for each gene using robust multichip average (RMA) (Irizarry et al. 2003) as implemented in Bioconductor (Gentleman et al. 2004), which considers only the perfect match probes in its estimation of expression levels for each probe set. The gene expression values (log2-based) were estimated using convolution background correction, quantile normalization, and median-polish summarization. We applied a filtering procedure to expression array data from testes and abdomen. The transcripts with expression intensities over 6.2 (log2 value) were considered detected on the array. The choice of cutoff is arbitrary and does not affect the results of any analysis (Supplemental Fig. S2). The transcripts that do not have annotation information in FlyBase or are unmapped or regarded as heterochromatic sequences were excluded from further analysis.
Expression differences were calculated using significance analysis of microarrays (SAM) (Tusher et al. 2001). For each gene, SAM calculates the relative difference, d(i), between the two sample groups:
where 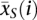 and
and 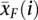 are the mean expression levels of a given gene in the sterile and fertile lines, respectively, s(i) is the standard deviation of repeated measurements for each gene, and s0 is a factor chosen to minimize the effect of expression level on variance in d(i). The d(i) values are ordered so that d(1) ≥ d(2) ≥ … ≤ d(p). The sample groupings are then permuted, d(i) values are calculated for each gene, which are again ordered, and the mean of the ordered d(i)s from these permutations is used as the expected value. Genes are called significantly differentially expressed if the difference between d(i)expected and d(i)observed exceeds a chosen threshold Δ. While choice of Δ is somewhat arbitrary, false discovery rates can be calculated for each value of Δ, allowing for the determination of an appropriate cutoff. In this study, we use Δ = 1.6 and Δ = 4.1 as the false discovery rate is nearly constant beyond these points in the testis and abdomen, respectively (see Supplemental Fig. S2). Our results of the expression difference between fertile and sterile lines are not sensitive to the choice of Δ.
are the mean expression levels of a given gene in the sterile and fertile lines, respectively, s(i) is the standard deviation of repeated measurements for each gene, and s0 is a factor chosen to minimize the effect of expression level on variance in d(i). The d(i) values are ordered so that d(1) ≥ d(2) ≥ … ≤ d(p). The sample groupings are then permuted, d(i) values are calculated for each gene, which are again ordered, and the mean of the ordered d(i)s from these permutations is used as the expected value. Genes are called significantly differentially expressed if the difference between d(i)expected and d(i)observed exceeds a chosen threshold Δ. While choice of Δ is somewhat arbitrary, false discovery rates can be calculated for each value of Δ, allowing for the determination of an appropriate cutoff. In this study, we use Δ = 1.6 and Δ = 4.1 as the false discovery rate is nearly constant beyond these points in the testis and abdomen, respectively (see Supplemental Fig. S2). Our results of the expression difference between fertile and sterile lines are not sensitive to the choice of Δ.
We calculated the difference (|log2(f/s)|) of the gene expression level in fertile and sterile lines. Since there are few genes expressed differently in abdomen, in order to include an adequate number of genes for comparison, we used a cutoff of 0.2 (log2 value subtraction, equal to f/s of 1.15).
RNA-seq library construction and sequencing
The testes with accessory glands from about 500 males (2–4 d after eclosion) of each line were dissected on ice and were homogenized in TRIzol reagent (Invitrogen). Approximately 100 μg of total RNA of each line was extracted according to the manufacturer's instructions. Poly(A)+ mRNA was isolated from total RNA using Oligotex (QIAGEN). The trancriptome libraries construction for fertile and sterile lines commences with RNA fragmentation using RNase III. The 100–200-bp RNA fragments were isolated after gel electrophoresis. Single-strand cDNA was made from RNA fragments using reverse transcriptase followed by second-strand synthesis using the Applied Biosystems SOLiD Whole Transcriptome Analysis Kit as described in the Whole Transcriptome Library Protocol (updated 4/29/09; http://solid.appliedbiosystems.com). Emulsion PCR and SOLiD sequencing were performed according to the Applied Biosystems SOLiD System manufacturer's instructions. The 50-base sequences were obtained on a 1/4 SOLiD v3 slide for each line.
RNA-seq data analysis
The 50-bp reads from the sterile or fertile lines were generated on the 1/4 slide of SOLiD and were mapped to the D. simulans genome (droSiml, Apr.2005) by the software provided by Applied Biosystem (Corona_lite_v4.0r2.0; http://solidsoftwaretools.com/gf/project/corona/). The 50-bp sequencing reads were first mapped to the genome, allowing for five color base mismatches. Reads that cannot be mapped to the genome were mapped again after trimming to 45 bp with four mismatches, then 40 bp with four mismatches, and finally 35 bp with three mismatches. The Short Read Archive accession number for the RNA-seq data is SRA012675.
After filtering multiple mapped reads, the unique mapped reads were annotated to RefSeq genes from UCSC (http://hgdownload.cse.ucsc.edu/goldenPath/droSim1/database/) by mapping location. In total, 24,383,254 and 14,308,455 unique reads were mapped to the genomes of the fertile and sterile lines, respectively (Supplemental Table S6). The difference was mainly due to the lower proportion of beads suitable for emulsion PCR when preparing the sterile sample for sequencing. Other than bead numbers, RNAs from the fertile and sterile lines are of comparable quality by three criteria. First, the measurements of RNA quality are indistinguishable between the fertile and sterile samples (e.g., the OD ratio at wavelength 260 vs. 280 is 2.10 and 2.06 for S and F lines, respectively). Second, as shown in Supplemental Figure S4a, the correlation of expression level in the two lines is high, at r = 0.894. Third, Supplemental Table S6 further shows that the genomic distributions of reads in exons, introns, exon–intron junctions, and non-coding regions are also nearly identical between the two lines.
Using the mapped reads of nearly 40 million between the fertile and sterile lines, 20%–40% of genes are differently expressed by Fisher's exact test at different P-values of Supplemental Figure S3. The X:A bias is observed at all P-values. With the large number of reads, the sensitivity in detecting expression differences by the RNA-seq method is extremely high (see Supplemental Fig. S3). In order to compare these results with the array data, we also used a conservative method as described below. Reads located in exons or exon–intron junction regions (Supplemental Table S6) were used to calculate RPKM (reads per kilobase of exon model per million mapped reads) for each gene using the standard formula:
where C is the number of unique mapped reads in the merged exonic region, N is the total number of unique mapped reads in the experiment, and L is the length of the exons (Mortazavi et al. 2008). Levels of significance of expression differences were then calculated using Fisher's exact test on the rounded RPKM values. The numbers of differently expressed genes under this conservative method are given in Table 1 for P < 0.005 and P < 0.001. The DEGseq package (Wang et al. 2010) was used in the statistical analysis. Scatterplot and the MA plot that compare expression profiles of sterile and fertile lines in Supplemental Figure S4 were also done by this package.
Acknowledgments
We thank Xinmin Li and Jamie Zhou of the University of Chicago Functional Genomics Facility for work with the microarrays, and members of the Wu laboratory, Kai Zeng, and Justin Borevitz for helpful discussions. This work was supported by National Institutes of Health grant to C.I.W., by National Natural Science Foundation of China (30600064) to X.M.L., by National Science Council (Taiwan, ROC) to C.T.T., and by a predoctoral fellowship from the Howard Hughes Medical Institute to J.A.S.
Footnotes
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE21734, and the RNA-seq data have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/Traces/sra) under accession no. SRA012675.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.076620.108.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes JJ, Malik HS 2009. Altered heterochromatin binding by a hybrid sterility protein in Drosophila sibling species. Science 326: 1538–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betran E, Thornton K, Long M 2002. Retroposed new genes out of the X in Drosophila. Genome Res 12: 1854–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. 2008. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5: 613–619 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R10 doi: 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldane JBS 1922. Sex ratio and unisexual sterility in animal hybrids. J Genet 12: 101–109 [Google Scholar]
- Hense W, Baines JF, Parsch J 2007. X chromosome inactivation during Drosophila spermatogenesis. PLoS Biol 5: 2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollocher H, Wu CI 1996. The genetics of reproductive isolation in the Drosophila simulans clade: X vs. autosomal effects and male vs. female effects. Genetics 143: 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobb B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- Khil PP, Smirnova NA, Romanienko PJ, Camerini-Otero RD 2004. The mouse X chromosome is enriched for sex-biased genes not subject to selection by meiotic sex chromosome inactivation. Nat Genet 36: 642–646 [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Urrutia AO, Hurst LD 2003. Evidence that the human X chromosome is enriched for male-specific but not female-specific genes. Mol Biol Evol 20: 1113–1116 [DOI] [PubMed] [Google Scholar]
- Lindsley DL, Lifschytz E 1972. The genetic control of spermatogenesis in Drosophila. In Proceedings of the International Symposium on the Genetics of the Spermatazoon (ed. Beatty RA and Waelsch SG), pp. 203–222 Edinburgh [Google Scholar]
- Masly JP, Presgraves DC 2007. High-resolution genome-wide dissection of the two rules of speciation in Drosophila. PLoS Biol 5: e243 doi: 10.1371/journal.pbio.0050243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci 100: 9894–9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak P, Noor MA 2004. Association of misexpression with sterility in hybrids of Drosophila simulans and D. mauritiana. J Mol Evol 59: 277–282 [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Muller HJ 1942. Isolating mechanisms, evolution, and temperature. Biol Symp 6: 71–125 [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299: 697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DE, Wu C-I, Johnson NA, Wu M-L 1993. Genetics of reproductive isolation in the Drosophila simulans clade: DNA-marker assisted mapping and characterization of a hybrid male sterility gene, Odysseus (Ods). Genetics 134: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745 [DOI] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Doren CV, Begley R, Jones SJM, Scherer S, Ward S, Kim SK 2000. A global profile of germline gene expression in C. elegans. Mol Cell 6: 605–616 [DOI] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K 2004. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131: 311–323 [DOI] [PubMed] [Google Scholar]
- Rice WR 1992. Sexually antagonistic genes: Experimental evidence. Science 256: 1436–1439 [DOI] [PubMed] [Google Scholar]
- Strausberg RL, Feingold EA, Klausner RC, Collins FS 1999. The mammalian gene collection. Science 286: 455–457 [DOI] [PubMed] [Google Scholar]
- Sturgill C, Zhang Y, Parisi M, Oliver B 2007. Demasculinization of X chromosomes in the Drosophila genus. Nature 450: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Ting CT, Wu CI 2004. The normal function of a speciation gene, Odysseus, and its hybrid sterility effect. Science 305: 81–83 [DOI] [PubMed] [Google Scholar]
- Tao Y, Chen S, Hartl DL, Laurie CC 2003. Genetic dissection of hybrid incompatibilities between Drosophila simulans and D. mauritiana. I. Differential accumulation of hybrid male sterility effects on the X and autosomes. Genetics 164: 1383–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting C-T, Tsaur S-C, Wu M-L, Wu C-I 1998. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282: 1501–1504 [DOI] [PubMed] [Google Scholar]
- True JR, Weir BS, Laurie CC 1996. A genome-wide survey of hybrid incompatibility factors by the introgression of marked segments of Drosophila mauritiana chromosomes into Drosophila simulans. Genetics 142: 819–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PJ, McCarrey JR, Yang F, Page DC 2001. An abundance of X-linked genes expressed in spermatogonia. Nat Genet 27: 422–426 [DOI] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X 2010. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26: 136–138 [DOI] [PubMed] [Google Scholar]
- Wu C-I 1991. Inferences of species phylogeny in relation to segregation of ancient polymorphisms. Genetics 127: 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-I, Davis AW 1993. Evolution of postmating reproductive isolation: The composite nature of Haldane's rule and its genetic bases. Am Nat 142: 187–212 [DOI] [PubMed] [Google Scholar]
- Wu CI, Xu EY 2003. Sexual antagonism and X inactivation—the SAXI hypothesis. Trends Genet 19: 243–247 [DOI] [PubMed] [Google Scholar]
- Wu C-I, Johnson NA, Palopoli MF 1996. Haldane's rule and its legacy: Why are there so many sterile males? Trends Ecol Evol 11: 281–284 [DOI] [PubMed] [Google Scholar]
- Xu EY 1996. The X chromosome and spermatogenesis in Drosophila melanogaster: A molecular and genetic analysis University of Chicago, Chicago, IL [Google Scholar]