

Abstract
Synthetic channel-forming peptides that can restore chloride conductance across epithelial membranes could provide a novel treatment of channelopathies such as cystic fibrosis. Among series of 22-residue peptides derived from the second transmembrane segment of the glycine receptor α1-subunit (M2GlyR), p22-S22W (KKKKP ARVGL GITTV LTMTT QW) is particularly promising with robust membrane insertion and assembly. The concentration to reach one-half maximal short circuit current is reduced to 45±6 μM from that of 210±70 μM of peptide p22 (KKKKP ARVGL GITTV LTMTT QS). However, this is accompanied with nearly 50% reduction in conductance. Towards obtaining a molecular level understanding of the channel activities, we combine information from solution NMR, existing biophysical data, and molecular modeling to construct atomistic models of the putative pentameric channels of p22 and p22-S22W. Simulations in membrane bilayers demonstrate that these structural models, even though highly flexible, are stable and remain adequately open for ion conductance. The membrane-anchoring tryptophan residues not only rigidify the whole channel, suggesting increased stability, but also lead to global changes in the pore profile. Specifically, the p22-S22W pore has a smaller opening on average, consistent with lower measured conductance. Direct observation of several incidences of chloride transport suggests several qualitative features of how these channels might selectively conduct anions. The current study thus helps to rationalize the functional consequences of introducing a single C-terminal tryptophan. Availability of these structural models also paves the way for future work to rationally modify and improve M2GlyR-derived peptides toward potential peptide-based channel replacement therapy.
Keywords: anion selectivity, micelles, molecular dynamics, molecular modeling, ion channel, solution NMR, structure refinement, synthetic channel
Introduction
Epithelia act as barriers to the movement of small molecules, including ions and drugs, between body compartments. Ions cross the epithelial apical and basolateral membranes via tightly regulated ion-specific transporters and channels. Defective ion channels have been implicated in many human diseases including episodic ataxia, diabetes, epilepsy, cystic fibrosis and Alzheimer’s dementia1–3. Peptide-based channel replacement therapy has been proposed as a treatment modality for correcting genetically based diseases caused by defective channel proteins4–6. A particularly attractive strategy is to deliver an aqueous soluble monomeric surface-active channel-forming peptide to the apical surface of effected epithelia. Once bound to the cell membrane, the peptide spontaneously inserts and undergoes supramolecular assembly to form a homo-oligomeric anion-conducting pore. An optimal peptide sequence should also provide high anion conduction rates yet with high chloride selectivity and improved aqueous solubility with minimal solution aggregation. For this, series of channel-forming peptides derived from the second transmembrane (TM) domain of the α1–subunit of the glycine receptor (M2GlyR) have been designed and tested over recent years5–12. The p22 sequence (KKKKP ARVGL GITTV LTMTT QS) represents a leading candidate, in that it has high solubility in physiological buffers, controls the orientation of the inserted sequence, and, at the same time, has assembled channel properties similar to the full length M2GlyR segment10. In the open state, the channels are able to sustain anion transport rates across Madin-Darby canine kidney (MDCK) epithelial monolayers with a maximum short circuit current of Imax = 23.7 ± 5.6 μA cm−2. However, p22 forms soluble associations that greatly reduce the concentration of membrane active monomer, requiring a K1/2 = 210 ± 70 μM to reach one-half Imax. Whole cell patch clamp measurements of the current- voltage relation revealed inward rectification at positive voltages12, suggesting that the assembled pore has substantial degree of structural flexibility and can undergo conformational changes at different voltages. A later design that replaces Ser22 with a membrane anchoring tryptophan (S22W) eliminates non-productive solution assemblies and decreases the peptide concentration required for channel activity, such that pore formation can occur at a lower peptide concentration with a K1/2 = 46 ± 6 μM10,13. However, there is a substantial decrease of the ion current across the membrane (Imax =13.0 ± 1.0 μA cm−2) at saturating peptide concentrations, and the inward rectification observed for p22 is abolished. One might suspect that, in strengthening the membrane anchoring of the peptide C-terminus, certain conformational states (and flexibility) of the assembled channel are sacrificed. Importantly, M2GlyR-derived peptides do not appear to cause immune or inflammatory responses at clinically relevant doses14. However, a drawback shared by both p22 and p22-S22W is that the enhanced ion conductivity seems to be accomplished at the cost of much weaker anion permselectivity compared to channels formed by the full length M2GlyR segment12. This represents an important limitation for any potential therapeutic applications of such peptides; as such anion selectivity is necessary for maintaining the physiological asymmetrical cation distributions across epithelial membranes.
While much progress has been made through intuition-driven design of functional channel-forming peptides, further enhancement has been severely limited by a lack of molecular level understanding of how these synthetic channels function and how their functions might be modulated by various mutations as well as membrane environment. This is a key motivation of our recent efforts towards structural elucidation of selected M2GlyR-derived peptides13,15. Ultimately, one would desire experimental three-dimensional (3D) structures of the functional channels in membrane or membrane-like environments. However, this proves to be very challenging, particularly with the dynamical nature of the putative channels. Nonetheless, a range of experimental information can be obtained regarding the secondary structures and organization of monomers in the channel form13,16,17, which might allow one to start to construct reasonable 3D models of these channels. While such structural models will be largely conjectural in nature, they should allow one to explore modern molecular modeling techniques to derive insights on their structure, dynamics and function that have not been possible so far. In particular, recent years have witnessed dramatic improvement in all-atom molecular mechanics modeling of biomolecules18. Empirical all-atom classical force fields, combined with modern simulation methodologies, are now being routinely applied to understand a wide range of membrane systems including ion channels19–24.
The current work represents our initial efforts to construct, characterize and validate high-resolution structural models of the putative pentameric channels formed by peptides p22 and p22-S22W. The basic strategy is to first determine the monomeric structures of p22 and p22-S22W in SDS micelles using solution NMR spectroscopy. Combined with additional experimental data and theoretical considerations on the oligomerization state, pore-lining interface, and helix packing geometry, initial structural models of the putative channels can be then assembled. Molecular dynamics (MD) simulations are subsequently carried out to refine and validate these structural models, and to characterize the structural and dynamical properties of the channels in fully solvated model membrane bilayers. A key validation is to examine how a single S22W mutation impacts the channel characteristics and subsequently membrane insertion efficacy and channel activities. The ability to rationalize key experimentally observed physiological properties would provide an important (albeit inconclusive) validation of the structural models as constructed. It should be emphasized that similar strategies have been widely applied to model channel structures, by combining monomer structures (from solid state/solution NMR and/or homology modeling) and various theoretical and experimental considerations of peptide packing geometries20,25–29. An important challenge in such approaches is that de novo determination of the oligomerization state and helix-packing interface can be highly nontrivial30,31. As such, it is essential to incorporate sufficient experimental data to reduce the possible conformational space of pore assembly as much as possible.
Materials and Methods
Peptide Synthesis and Purification
The peptides were chemically synthesized by solid-phase peptide synthesis methodology on an Applied Biosystems model 431A peptide synthesizer (Foster City, CA) using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry as previous described in ref. 5. CLEAR amide resin, (0.3 mmol/g) was purchased from Peptides International, Inc. (Louisville, KY) and Nα-Fmoc amino acids were purchased from Anaspec Inc. (San Jose, CA), Bachem (Torrance, CA), and Peptides International (Louisville, KY). All peptides were purified using a Phenomenex (Torrance, CA) reversed-phase C-18 column on a System Gold HPLC system (Beckman Instruments, Inc.; Fullerton, CA). Peptides were eluted from the column using a linear gradient of 3.0% min−1 of 10—90% acetonitrile containing 0.1% trifluoroacetic acid (TFA) at 1 mL min−1. HPLC-purified peptides were characterized by matrix assisted-laser desorption time-of-flight mass spectroscopy (MALDI-TOF/TOF) using a Bruker Ultraflex II mass spectrometer. After characterization, peptides were lyophilized and stored as dry powders until use.
Solution NMR Spectroscopy
Lyophilized peptide was dissolved in 1.0 mL of a (1:1) mixture of 1,1,1,3,3,3-Hexafluoro-2-propanol: 2,2,2-trifluoroethanol. The solution was then dried with a stream of nitrogen gas. The following day, the peptide sample was dissolved in 0.5 mL of 500 mM SDS-d25 in 90% H2O:10% D2O (Cambridge Isotope Labs, Andover MA). The final peptide concentration in the NMR sample was 2.5 mM. NMR spectra for p22 peptide were recorded at 45°C on a Bruker Avance 800 NMR spectrometer equipped with a cryo-probe (located in the Biomolecular NMR laboratory at the University of Kansas), and those of p22-S22W peptide were recorded at 40°C on a Bruker Avance 600 NMR spectrometer (located in the laboratory of Professor Yan Xu at the University of Pittsburgh). Elevated data acquisition temperatures were chosen to reduce the molecular tumbling time and improve spectral quality. CD measurements show minimal differences at these temperatures. We note that acquisition of the spectra for p22 was particularly challenging, as the spectra suffered from peak broadening and decreased in signal to noise ratio, making necessary the use a higher field NMR instrument. This also suggests that p22 is more flexible compared to p22-S22W in membrane-like environments. Data from both sets of experiments were processed using NMRPipe32 and analyzed using Sparky software33. Proton chemical shift assignments were made by standard techniques34 using 2D 1H-1H DQF-COSY and TOCSY spectra for intra residue spin systems and NOESY for inter residue connectivity. The NOE cross peaks were classified as string, medium, weak and very weak based on the observed relative intensity. Spin-lock time of 100 ms at B1 field strength of 7 KHz was used for TOCSY experiment, and mixing time of 300 ms and 400 ms were used for NOESY experiments. For both peptides, suppression of the solvent peak was achieved using the WATERGATE pulse scheme. The residual water peak (4.61 ppm for p22 and 4.56 ppm for p22-S22W) was used as reference for chemical shift values.
NMR Structure Calculation and Refinement
A previously established protocol was used for structure calculation13,35. Briefly, unambiguously identified NOE cross peaks were converted into inter-proton distance upper bounds using the following classifications: strong (2.7 Å), medium (3.5 Å), weak (4.0 Å), and very weak (5.0 Å)36. Upper distance limits for NOEs involving methyl protons and non stereo specifically assign methylene protons, were corrected appropriately for center averaging by adding 1 Å to the maximum distance constraint for center averaging36. Backbone dihedral angle restraints were derived from the distance restraints and 3JαHNH coupling constants using the program HABAS37. The obtained distance and dihedral restraints were used to calculate peptide structures using the Crystallography & NMR System (CNS) version 1.138, using a protocol that combines simulated annealing in torsional space and MD refinement in Cartesian space to produce structures with minimal restraint violations39. A total of 100 accepted structures were subjected to energy minimization in the AMBER force field40,41 using Sybyl 7.142. The minimized structures were then clustered based on mutual heavy-atom root mean-square deviation (RMSD). The 10 structures with no restraint violation and the lowest energies were selected from the largest cluster as the representative structures.
The average structures of the ensembles calculated by CNS were further refined using the GBSW implicit membrane model43 in CHARMM44,45. Distortions to covalent geometry due to averaging were corrected by short energy minimization while harmonic positional restrains on heavy atoms. Refinement in realistic solvent environments has been used to improve solution NMR structures and can lead to better hydrogen-bonding pattern and backbone dihedral distribution46–48. Such refinement might be particularly useful to remove potential structural distortions due to difference between micelle and bilayer membrane environments, which facilitates subsequent assembly of monomer structures into channels. The refinement protocol involves 100 ps restrained MD simulation at 300 K followed by energy minimization. Default GBSW implicit membrane parameters43 were used with a hydrophobic membrane thickness of 35 Å. All NMR restraints were imposed with force constants of 50 kcal/mol/Å2 for NOE distances and 100 kcal/mol/Å2 for dihedral angles during MD refinement. The force constants of NOE distances were increased to 100 kcal/mol/Å2 during the final energy minimization.
Channel Assembly and Molecular Simulation Protocols
Starting from refined monomer NMR structures, the peptides were assembled into pentameric forms using CHARMM based on a set of experimental data and theoretical considerations (see Results and Discussion for detail). Multiple initial models were generated for each sequence with different handiness of helix packing. The assembled channels were subject to energy minimization in presence of harmonic positional restraints on all heavy atoms and then solvated in a pre-equilibrated POPC bilayer membrane using the CHARMM-GUI automated Membrane Builder49,50. The final systems consist of 106 lipids and ~5350 water molecules besides the protein, with a box size of ~66 × 66 ×71 Å3. Forty Cl− ions and ten K+ ions were added to neutralize the whole system (with an effective ion concentration of 50 mM).
A 6-cycle equilibration protocol was applied to equilibrate the system at 303 K using CHARMM, during which various restraints were applied to selected atoms and with gradually decreasing restraint strength49. The full details on the restraint setups are provided in Table S1 (Supplementary Materials). All molecules were described by the CHARMM param22 force field with CMAP51–53. During equilibration, two cycles of NVT simulation (constant volume and temperature) were performed for 25 ps each, followed by three cycles of NPT simulation (constant pressure and temperature) of 25 to 100 ps. A final NPT equilibration cycle was performed for 100 ps where only the protein backbone atoms were held with weak harmonic positional restraints (k = 0.1 kcal/mol/Å2). SHAKE was used to fix the length of hydrogen-attaching bonds54 to allow for a 2 femtosecond (fs) dynamic time step during cycles 4–6. Periodic boundary conditions were enforced. Particle Mesh Ewald (PME)55 was used for treatment of long-range electrostatic interactions, and van der Waals interactions were smoothly switched off from 10 to 12 Å. The fluctuations in temperature, volume and energy were examined, and all were within reasonable ranges. The protein, water, and lipids were monitored during the equilibration to ensure that the water did not penetrate the lipid bilayer.
The NAMD software package56 was used for the production simulation due to its better parallel scaling property. The final structure from the CHARMM equilibration simulation was used to initiate the NAMD runs. Another 100 ps equilibration with weak harmonic positional restraints on protein backbone atoms (k = 0.1 kcal/mol/Å2) in the NAMD simulations was used to suppress potential strains during subtle differences between CHARMM and NAMD. Setups equivalent or identical to those described above were used, unless otherwise noted below. Electrostatic interactions were evaluated based on a multiple-time-stepping algorithm56,57 implemented in NAMD, where bonded interactions were updated every time step, short-range nonbonded electrostatic and van der Waals interactions every two steps, and long-range interactions every four steps. After the system was equilibrated, all restraints were released. A NPAT (constant pressure and membrane area) production simulation was carried out for 20 nanoseconds (ns) for each structural model, with the coordinates saved every 1 ps.
The MD simulation protocol described above mainly serves to refine the initial channel structures constructed based on existing experimental data. While comprehensive sampling of the accessible conformational states of the channel assemblies (such as in a de novo channel structure prediction effort) would likely require simulations of several orders of magnitude longer, the current choice of 20-ns MD should be sufficient to further relax the constructed structure models in a realistic membrane environment and to determine the conformational flexibility (and dynamics) near the initial constructs. Similar simulation lengths (sometimes much shorter) are commonly chosen for the purpose of refining channel structures built based on experimental and/or theoretical considerations25,26,28,29. Indeed, all simulations carried out so far on various M2GlyR-derived channels including p22 and p22-S22W show that the initial channel structures relax quickly in membrane bilayers and reach (meta-) stable conformations within ~5 ns. Nonetheless, two independent simulations of the left-handed p22 and p22-S22W channels were carried in CHARMM, with the simulation of the less stable p22 channel extended to 50 ns. The purposes are to examine the longer timescale stability of the proposed structural models and to validate the suitability of the above NAMD simulation protocol with the more aggressive multiple-time-stepping algorithm. The results are included in the Supplementary Materials.
Data Analysis
All analysis was done using various modules in CHARMM. VMD58 was used for visualization and preparation of molecular images presented here. The pore profiles and estimated ion conductance were calculated using the program HOLE with default options59.
Results and Discussion
NMR Structures of Monomeric p22 and p22-S22W in Micelles
The chemical shift dispersion observed for the NH and CαH protons of both peptides supports the presence of a stable folded structure for in SDS micelles, as previously suggested by CD studies13. Proton chemical shift assignments for p22 and p22-S22W in SDS micelles are summarized in Table S2 of the Supplementary Materials. Figure 1 shows an example of the NH-CαH, NH-CβH, NH-CγH and NH-CδH cross-peak assignments in the finger print region of the TOCSY spectra used for residue identification. NOESY spectra allowed the identification of dαN(i,i+1) proton connectivity for sequential assignments and the disambiguation of repeated amino acid residues. Further analysis of the NOESY spectra revealed short and medium range connectivity including dNN(i,i+1), dαN(i,i+1), dβN(i,i+1), dαN(i,i+3) that provide structural information. An example of a NOESY spectra showing dNN(i,i+1) connectivity is provided in Figure S1 (Supplementary Materials. All sequential and medium range NOE connectivity for p22 and p22-S22W are summarized in Figure 2. For both peptides sets of dαN(i,i+2) and dαN(i,i+3) NOEs characteristic of helical conformation were observed from Ala6 to Thr20. These results suggest a continuous stretch of helical conformation for both p22 and p22-S22W in SDS micelles.
Figure 1.

Finger print region (HN-Hα/Side chain protons) of 1H-1H TOCSY spectrum of p22 in SDS micelles, showing individual amino acid spin systems connected with a vertical line and labeled with residue name and number.
Figure 2.

Summary of the NOE connectivity for peptides p22 and p22-S22W in SDS micelles. The line thickness indicates peak intensity, and dotted lines indicate ambiguity due to peak overlapping.
Summarized in Table I, a total of 117 distance and 18 dihedral angle restraints for p22 and 125 distance and 8 dihedral angle restraints for p22-S22W were eventually obtained and used to calculate structure ensembles in CNS. The clusters of 10 structures with the lowest total energies and no distance violation >0.5 A for peptides p22 and p22-S22W are shown in Figure 3a. The backbone ψ and ϕ angle distributions were evaluated using the PROCHECK program. The results show that >99% residues of both ensembles reside within the allowed regions (see Table I). The average structures after MD refinement in implicit membrane are shown in Figure 3b. The refinement led to minimal difference in the backbone conformation of the helical region (residues 6–20). The main structural differences are observed in the N-terminal tails, where refinement in the implicit membrane led to enhanced solvent exposure of lysine residues as expected. The overall heavy atom RMSD values from pre-refined structures are 2.0 A and 2.3 A for p22 and p22-S22W, respectively. Nonetheless, neither refined structure contains any severe NOE violation (> 0.5 Å). The total NOE distance violations are 0.5 A and 0.43 Å for p22 and p22-S22W, respectively. These refined monomer structures will served as the starting point to construct the putative pentameric channels.
Table I.
Structure Statistics of p22 and p22-S22W in SDS micelles.
| p22 | p22-S22W | |
|---|---|---|
| Experimental NMR Constraints | ||
| NOE Distance Constraints | ||
| Intraresidue | 38 | 80 |
| Sequential | 53 | 28 |
| Medium Range (1<|i–j|≤4) | 26 | 17 |
| Angle Constraints | 18 | 8 |
| Energy (kcal/mol−1) | ||
| Etotal | −52.9 ±16.08 | −32.36 ±10.52 |
| RMSD from the mean structure (Ǻ) | ||
| Backbone of the helical segment(residues 6–20) | 0.46±0.12 | 0.42±0.55 |
| Ramachandran statistics | ||
| Residues in allowed regions | > 99% | > 99% |
| Residues in generously allowed regions | < 1% | < 1% |
| Residues in disallowed regions | 0 | < 1% |
Figure 3.
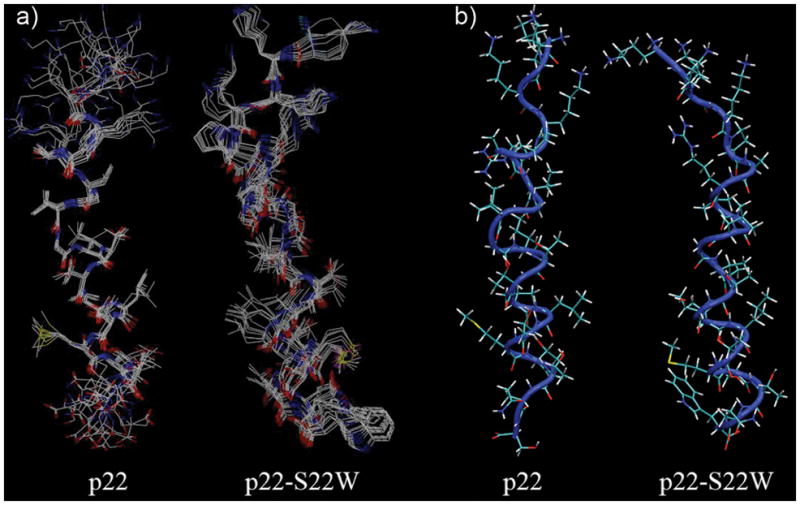
a) Clusters of 10 superimposed structures with the lowest energies of p22 and p22-S22W. b) Average structures of p22 and p22-S22W after MD refinement in an implicit membrane. Backbones are shown as a blue tube with licorice side chains.
These atomic structures clearly show that both p22 and p22-S22W are linear and mostly helical from residues 6–20 in SDS micelles. The N-terminal lysines are largely unstructured and apparently with substantial flexibility. These lysine residues are expected to be out of the micelle floating in the aqueous environment and/or interacting with the sulfate groups. The hydrophobic and hydrophilic side chains are segregated to different sides of the helix, as expected for channel forming TM segments. This is contrast to previous structures determined in a simple solvent mixture of 40% 2,2,2-trifluoroethanol (TFE) and 60% water, where p22-S22W adopts a bent conformation that shields the hydrophobic residues from solvent and presumably protects against aggregation13. Therefore, substantial conformation changes accompany the membrane insertion and assembly of p22-S22W, which might be a common mechanism that allows such peptides to co-exist stably in both aqueous and membrane environments. Another important insight derived from the solution NMR structural characterization is that p22 and p22-S22W most likely adopt similar conformations in the assembled pores, despite substantial structural differences in aqueous solution. This may allow substantial simplification in modeling of the pore structures of M2GlyR-derived peptides, as one should be able to derive reasonable models from a common scaffold through computational mutagenesis. Nonetheless, we note that some fine differences exist between the calculated structures of p22 and p22-S22W in SDS micelles. The backbone RMSD between the helical segments of p22 and p22-S22W (residues 6-20) is 1.6 A. The helical portion of p22-S22W structure appears to be better formed (and more linear): only a 41% of the backbone is in the canonical α-helix for p22, compared to that of 59% for p22-S22W. This might reflects an increased stability of p22-S22W in SDS micelles, due to the presence of membrane anchoring tryptophan residues. We further note that more stable p22-S22W helix is consistent with existing circular dichroism data12 and channel activity measurements12.
Assembly of Initial Pentameric Channel Structures
Based on the observations that p22 and p22-S22W adopt similar structures in micelles and that the p22 structure is less well defined due to greater flexibility, we first constructed the initial structures of p22-S22W channel and derived those of p22 channel by computational mutation of tryptophan to serine. This strategy might also minimize the impacts of potential artifacts of the micelle structures in the subsequent refinement simulations in fully solvated lipid bilayers. As described above, the key structural information obtained from solution NMR is really that both the peptides have α-helix structures within the C-terminal segments (residues 6–20), while the N-termini are largely unstructured. Additional experimental information and theoretical considerations, listed below, were then used to construct the initial structural models of p22-S22W channel. First, previous experimental work indicated that this family of peptides spontaneously assembled into pentameric structures when inserted into synthetic bilayers or cell membranes16. Second, cysteine-scanning experiments (Tomich, unpublished data) on a related peptide sequence (p22-T19R/S22W: KKKKP ARVGL GITTV LTMRT QW) have identified residues A6, L10, T13, and T17 as pore-lining residues. Third, solid state NMR and free energy calculations suggest that TM helix tilt angles most likely fall in the range 10 to 15 degrees60,61 for peptides of similar length, as a result of balance between helix procession entropy and peptide-membrane interactions. Subsequent simulations demonstrate that the channel structure can readily relax to adopt the optimal helix tilt in membrane bilayers, and the precise choice of initial tilt does not appear to be critical. Fourth, when the monomers are aligned to form the pentamer, the adjacent peptides should be in contact with each other while maintaining a pore cavity sufficiently large for ions to traverse. Finally, at this point, there is not reliable information on the handedness of helix assembly for either p22 or p22-S22W pores. For this, we constructed two models, shown in Figure 4, that satisfy all the above experimental and theoretical constraints, one with left-handed packing and the other with right-handed packing. While simulations presented in the current work were not sufficient to unambiguously identify apparent functionally relevant differences between these two models (see below), all three available crystal structures of members of the cys-loop ligand-gated ion channel superfamily (to which GlyR also belongs) contain left-handed packing of pore-forming TM helices62–64. It is plausible that the M2GlyR-derived synthetic channels might preserve the handiness of helix packing.
Figure 4.

Initial 3D structural models of the p22-S22W pentameric channel. Two of the key pore-lining residues, T13 and T17, are shown in van der Waals spheres. The sidechains of N-terminal lysines (K1-4) and the membrane anchoring tryptophan (W22) are also shown.
Relaxation of Channel Structures in POPC Bilayers
All channel structures (left-handed and right-handed packing for each sequence) are stable in the POPC membrane bilayers within the 20 ns NAMD production simulations. An additional 50-ns simulation of the left-handed structure of the presumably less stable p22 channel was also carried out using CHARMM. The results suggest that the structural models as constructed above are indeed stable after initial relaxation (see Supplementary Materials). For all the properties that we will examine in the rest of this paper, both p22 and p22-S22W channels appear to behavior similarly regardless the handedness of helix packing, even though there are indications that the right-handed channels might be less stable. As such, we will focus on presenting simulation results of the left-handed channel structures (see Supplementary Materials for additional information on characterization of the right-handed channels). Figure 5 shows the equilibrated structure of the left-handed p22-S22W channel in a solvated POPC bilayer and the final snapshot after the production simulation. Clearly, the whole channel drifts upward relative to the membrane, presumably to allow better solvent exposure of the charged N-terminal lysines. The positively charged lysine sidechains also interact extensively with lipid phosphate groups. The drift results in substantial distortions in the local membrane environment, particularly, thinning of the bilayer adjacent to the channel. As shown in Figure 6a–b), the channels relax rapidly during the production simulations, where the whole channel backbone RMSD from the initial structure increase quickly within a few nanoseconds before stabilizing at ~4.5 A for p22 and ~3.5 Å for p22-S22W. The backbone RMSD of each individual peptide from the initial (NMR) structure displays similar behavior. The increase in RMSD values is due mainly to unstructured N-terminal lysines and flexibility in the channel assembly. The helical segments of all five peptides in both channels are stable (at the secondary structure level). In fact, both channel structures become more helical compared to the monomeric NMR structures in SDS micelles, reaching approximately 75% for p22 and 65% for p22-S22W (see Figure 6c). The increase in helicity is due primarily to the formation of more regular helices within residues 6 to 20. The averaged helix tilt angles with respect to the membrane normal (computed using residues 6–20), shown in Figure 6d, increase to ~25° for p22 and ~20° for p22-S22W. These title angles are larger than what we initially expected for short helices like p22 and p22-S22W. Repulsion among positively charged side chains (K1–4 and R7), coupled with unstructured N-termini and local membrane thinning, is likely the main driving force for large helix tilt angles. This was also observed in previous simulations of a M2δ model channel20.
Figure 5.

Snapshots of p22-S22W left-handed channel in a fully solvated POPC bilayer before and after the 20 ns production simulation. The protein is shown in blue cartoon and lipid molecules shown in the green licorice with phosphorus atoms shown as green spheres.
Figure 6.

Backbone RMSD as a function of time during 20 ns production simulation of the left-handed channel structures of a) p22 and b) p22-S22W. The thick traces correspond to whole channel RMSD, and thin traces plot those for individual helices. c) Relative helicities and d) average helix tilt angles with respect to membrane normal of both channels during the production simulation. Helical residues were identified using the secondary structure analysis module in CHARMM. The tilt angle shown is the average value of the tilts of all five individual helical segments (residues 6–20).
Flexibility, Pore Profiles and Implications in Channel Activities
To directly test that the membrane-anchoring tryptophan residue at the C-terminus increases the channel stability and rigidity, we first calculated the whole channel Cα root mean square fluctuation (RMSF) using the last 5 ns of the production simulations. Shown in Figure 7a–b), the results support that the p22 pore is more flexible than that of p22-S22W, either with left-handed or right-handed helix packing. Interestingly, the presence of the C-terminal tryptophan not only reduces the channel flexibility near the C-terminus, but also leads to a global reduction of flexibility including the unstructured N-termini. As shown in Figure 7c–d), the C-terminal tryptophan mainly restricts the translational motion along the membrane normal and might thus enhance the overall integrity of the channel assembly. Tryptophan residues might also contribute further to the channel stability by restricting rotation around the helix principle axis65. While reduced flexibility does not necessarily reflect increased stability, these two properties are often positively correlated. An increased stability of the p22-S22W channel as suggested by reduced flexibility is consistent with the difference in concentration dependence of short circuit current induced by p22 and p22-S22W measured in MDCK cell monolayers12,13. Considering a simple scenario where channel assembly is described by the equilibrium between the soluble monomeric state and the functional pentameric channel in membrane, 5 P1 ⇔ P5, the concentration of functional channels is given by [P5] = Keq [P1]5 for a given monomer concentration. Therefore, the steeper slope of the concentration dependence of short circuit current induced by p22-S22W (see Figure 1 in Cook et al., Biophys J, 2004 13) reflects a larger Keq and thus increased stability of the p22-S22W channel. We note that the actual mechanism of channel assembly is not known and will expectedly be more complex than the simple two-state system discussed above. An alternative explanation of the steeper short circuit current-concentration slope, which does not involve increased stability of the p22-S22W channel, is that W22 reduces the stability of putative non-functional assemblies.
Figure 7.

Cα RMSF of a) left-handed and b) right-handed channels of p22 and p22-S22W, computed from the last 5 ns of the production simulations, and the centers of mass and N-/C-terminal positions of individual monomers of the left-handed channels along the membrane normal with respect to the lipid center of mass as functions of time for c) p22 and d) p22-S22W. The N- and C-termini are represented by Cα atoms of K1 and S22, respectively. The monomer center of mass was computed using all heavy atoms.
To further assess the structure-activity relationship of the channels, pore profiles along the channel principal axis were computed using snapshots sampled every 200 ps during the last 5 ns of production. The snapshot profiles were then averaged to reduce the sensitivity to instantaneous side chain configuration of pore-lining residues. The average profiles are shown in Figure 8, along with representative snapshot and initial pore profiles. Consistent with reduced flexibility of the p22-S22W channel as demonstrated above, the snapshot pore profiles of the p22-S22W channels show smaller fluctuation (see Figure 8a and b). On average, the p22 channel appears to maintain a larger opening, not only at the narrowest region (near Thr17), but also throughout the rest of the pore. The time-averaged configurations of these two channels, shown in Figure 8d–e), show substantial differences: the p22 channel expands in the plane of the membrane towards the N-terminus relative to the initial configuration, while the p22-S22W structure contracts. The observed differences in the pore openings could explain the experimentally observed difference in Imax induced by p22 and p22-S22W in MDCK epithelial cell monolayers. Assuming that the maximum numbers of channels that can be incorporated in the epithelial monolayer are the same (or similar) for both peptides, Imax then reflects the average conductance of individual channels. The larger p22 pore would give rise to larger conductance and thus higher maximum short circuit current at saturating peptide concentrations. Reliable quantitative calculation of the single-channel conductance are challenging18. Nonetheless, it has been shown previously that simple geometric consideration of the pore dimension can be used to predict the true conductance with a 0.90 correlation66. It can be estimated that the observed narrowing of p22-S22W pore would lead to slightly over 30% reduction in conductance, from the ratio of geometric factors ~ΣR−2 along the channel computed from the average pore radius profiles shown in Figure 8. This estimate is in reasonable agreement with ~50% reduction of measured Imax.
Figure 8.

Pore profiles along the channel principal axis computed from snapshots taken every 0.5 ns of the last 5 ns production simulation of left-handed channel of a) p22 and b) p22-S22W. c) Averaged pore profiles of p22 and p22-S22W, show in red and blue traces, respectively, in comparison the initial profile (shown in black, same for both channels). Average channel structures obtained from the last 5 ns of the production simulations for d) p22 and e) p22-S22W, together with the initial (equilibrated) structures shown in grey cartoon.
Chloride conductance and anion selectivity mechanism
During the course of the simulations, several incidences of spontaneous Cl− diffusion through the channels were observed. This is probably due to substantial local Cl− concentration enrichment at the N-terminal opening of the channels due to the presence of large number of lysine residues. It allows us to qualitatively understand the nature of chloride conduction and possible mechanism of anion selectivity of these synthetic channels. Figure 9 shows the time evolution of the position of a single Cl− along the membrane normal during an incidence of its spontaneous diffusion through the left-handed p22 channel, together with a snapshot where this particular Cl− locates inside the channel around the narrowest point (near Thr17). Other incidences show qualitatively similar diffusion process and are thus not shown. Clearly, Cl− ions remain highly solvated throughout the process. In fact, both p22 and p22-S22W pores remain solvated throughout the simulations. Similar observations have also been made in a recent simulation of model channels derived from the cationic human R7 nicotinic acetylcholine receptor and the anionic R1 glycine receptor21. The notion that Cl− ions do not need to be significantly de-solvated for transport is also consistent with the high conductivity of these synthetic channels, as desolvation might lead to higher free energy barriers. The resident time of Cl− ions in the channel is estimated to be on the ns timescale, during which the ions seem to pause in the neighborhood of residues L10, T13 and T17. In particular, Cl− appears to interact with the T17 and T13 hydroxyl side chains with distances less than 3 Å, as expected for a typical hydrogen bond. As shown previously, serine and threonine sidechains preferably stabilizes Cl− with respect to Na+ and K+21. It is plausible that the threonine residues at the narrowest part of these pores (Thr13 and Thr17) contribute to the weak anion selectivity (2– to 10-fold) observed experimentally12. While the hydrogen bonding of chlorides to NH and OH has been recognized among inorganic chemists67,68, it has received little attention as a possible contributing factor in anion selective protein channels. The size of the p22 and p22-S22W pore allows for the permeation of a hydrated chloride. A rapid reversible binding of a chloride to one or more threonine hydroxyls makes for a plausible selectivity filter. The positively charged residues at the C-terminus may also contribute substantially to anion selectivity. At this point, it is not clear how much each structural element contributes to the overall charge selectivity of these channels. Ongoing free energy calculations will provide a more quantitative base for resolving this question.
Figure 9.

a) A snapshot that shows the interactions between Cl− (shown as the green sphere) and water molecules and Thr17 hydroxyl side chains inside the channel. Only those water molecules located within 5 Å of chloride are shown. b) Position of a Cl−along the membrane normal as a function of simulation time during an incidence of spontaneous diffusion through the left-handed p22 channel. The green lines mark the approximate locations of membrane boundaries, and the black lines mark the boundaries of the periodic simulation box. Key residues that interacted with this particular Cl− during its spontaneous diffusion through the channel are also marked.
Conclusion
M2GlyR-derived peptides have been proposed as promising candidates for channel replacement therapy for treating channelopathies such as cystic fibrosis. Previous intuition-based designs have led to much progress in improving the solubility and channel activities compared to the original pore-forming TM sequence of GlyR channel, which suffered from strong aggregation in aqueous solution and poor membrane insertion efficacy. One of the key remaining limitations is insufficient chloride selectivity, and further improvement has been severely hindered by a lack of molecular level understanding of how these synthetic channels function. Faced with formidable challenges in direct determination of the structures of the putative channels, we first determined the structures of monomeric p22 and p22-S22W peptides in SDS micelles using solution NMR spectroscopy. The calculated structures show that both p22 and p22-S22W are linear and mostly helical from residues 6–20 in SDS micelles, even though p22-S22W appears to be less flexible and contains a more regular α-helical segment. This is in contrast to previous solution NMR structures in 40% TFE solution, where p22-S22W adopts a bent conformation that shields the hydrophobic residues from solvent and prevents aggregation13. Similarity of p22 and p22-S22W structures in SDS micelles also point to structural similarity of the assembled pores in membrane. This allows substantial simplification in modeling of the pore structure formed by any M2GlyR-derived peptide: one does not need to re-solve the solution structure of the monomer and might be able to derive reasonable models from a common scaffold through computational mutagenesis.
A range of additional experimental data and theoretical considerations were then utilized to assemble the monomer structure into channels, including oligomerization state, pore-lining interface, helix packing distance and tile angle. In particular, experimental identification of the pore-lining residues greatly reduces uncertainty of the channel assembly and allows the construction of reliable initial structural models. All-atom MD simulations in fully solvated POPC bilayers were subsequently carried out to refine these structural models and to characterize and validate the structural and dynamical properties of the channels. The results demonstrate that the channel structures as constructed are adequately stable within the simulation time frame (up to 50 ns) and remain sufficiently open for ion conductance. All initial structures quick relax to (meta-) stable structures within a few nanoseconds. Analysis of the relaxed structures of p22 and p22-S22W reveal important differences in their structural and dynamical properties, providing a structural basis for understanding the differences in channel activities. Specifically, the structural characterization supports the initial postulation that the S22W mutation helps to anchor the pore in membrane and reduces the flexibility of the whole assembly. The implied increased stability of the p22-S22W channel explains the steeper short circuit current-concentration slope measured experimentally. Furthermore, introduction of the C-terminal tryptophan appears to lead to global changes of the channel structure. The p22-S22W channel maintains a smaller opening at the narrowest region and throughout the rest of the pore compared to the p22 channel. The smaller size of p22-S22W could effectively reduce its ion conductance by a magnitude that is consistent with a nearly 50% reduction in measured Imax. The ability to recapitulate these differences in key physiological properties observed experimentally is an important validation of the proposed structural models. Nonetheless, it is should be emphasized that these structures are largely conjecture models in nature. Further testing, such as examining the ability to predict the structural and functional consequences of other sequence alteration and/or different membrane environments, is necessary and will provide important feedback on the reliability and usefulness of such conjecture models. A joint computational and experimental effort is ongoing along this line and results will be reported in forthcoming publications.
The structures of monomeric p22 and p22-S22W peptides in SDS micelles as well as the structures of putative channels formed by these peptides are the main outcomes of the current work. In simulations of the channels in POPC bilayers, we also serendipitously observed that Cl− could spontaneously diffuse through the channels. This allows one to draw a few direct, albeit qualitative, observations on how these channels might function. Two main features emerge: first, the chloride ions remain largely solvated during transport; and second, the ions mainly interact with one of the hydroxyl side chains of Thr13 and Thr17 at the narrowest regions of the channels. These observations provide useful hints to the possible mechanisms of anion selectivity. In particular, it supports the hypothesis that a rapid reversible binding of a chloride to one or more threonine hydroxyls makes for a plausible, albeit weak, selectivity filter. We expect that the positively charged C-termini might also contribute to the charge selectivity. However, it is not clear how much each structural element contributes to the overall charge selectivity of these channels, and further understanding requires rigorous free energy calculations of both anion and cation transport through these channels. The current efforts in construction and validation of reliable channel structures provide a necessary foundation for such future work. Available of reasonable structure models also opens a door to computational exploration of other residue replacements and additions to study their structural, dynamical and functional consequences on the resulting pores. Such studies will be useful for continual efforts in developing better functional channels that satisfy all the necessary requirements for potential clinical applications.
Supplementary Material
Acknowledgments
This study is supported in part by the National Institutes of Health (Grants GM 074096 and P20 RR017686). The authors thank Dr. Wonpil Im for useful discussions as well as assistance with the CHARMM-GUI Membrane Builder (http://www.charmmgui.org). Ms. Asma Al-Rawi is acknowledged for help with the preparation of several figures. This article is contribution 10-269-J from the Kansas Agriculture Research Station.
Footnotes
Supplementary data associated with this work can be found in the online version of this article.
References
- 1.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic-fibrosis. Cell. 1993;73(7):125–254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82(2):503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 3.Cannon SC. Physiologic principles underlying ion channelopathies. Neurotherapeutics. 2007;4(2):174–183. doi: 10.1016/j.nurt.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DP, Tomich JM, Iwamoto T, Henderson K, Grantham JJ, Sullivan LP. A synthetic peptide derived from glycine-gated Cl− channel induces transepithelial Cl− and fluid secretion. Am J Physio-Cell Physio. 1997;41(5):C1672–C1679. doi: 10.1152/ajpcell.1997.272.5.C1672. [DOI] [PubMed] [Google Scholar]
- 5.Tomich JM, Wallace D, Henderson K, Mitchell KE, Radke G, Brandt R, Ambler CA, Scott AJ, Grantham J, Sullivan L, Iwamoto T. Aqueous solubilization of transmembrane peptide sequences with retention of membrane insertion and function. Biophys J. 1998;74(1):256–267. doi: 10.1016/S0006-3495(98)77784-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallace DP, Tomich JM, Eppler JW, Iwamoto T, Grantham JJ, Sullivan LP. A synthetic channel-forming peptide induces Cl− secretion: modulation by Ca2+-dependent K+ channels. Biochimica Et Biophysica Acta-Biomembranes. 2000;1464(1):69–82. doi: 10.1016/s0005-2736(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 7.Broughman JR, Mitchell KE, Sedlacek RL, Iwamoto T, Tomich JM, Schultz BD. NH2-terminal modification of a channel-forming peptide increases capacity for epithelial anion secretion. Am J Physio-Cell Physio. 2001;280(3):C451–C458. doi: 10.1152/ajpcell.2001.280.3.C451. [DOI] [PubMed] [Google Scholar]
- 8.Gao L, Broughman JR, Iwamoto T, Tomich JM, Venglarik CJ, Forman HJ. Synthetic chloride channel restores glutathione secretion in cystic fibrosis airway epithelia. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2001;281(1):L24–L30. doi: 10.1152/ajplung.2001.281.1.L24. [DOI] [PubMed] [Google Scholar]
- 9.Broughman JR, Shank LP, Prakash O, Schultz BD, Iwamoto T, Tomich JM, Mitchell K. Structural implications of placing cationic residues at either the NH2- or COOH-terminus in a pore-forming synthetic peptide. J Membrane Biol. 2002;190(2):93–103. doi: 10.1007/s00232-002-1027-3. [DOI] [PubMed] [Google Scholar]
- 10.Broughman JR, Shank LP, Takeguchi W, Schultz BD, Iwamoto T, Mitchell KE, Tomich JM. Distinct structural elements that direct solution aggregation and membrane assembly in the channel-forming peptide M2GlyR. Biochemistry. 2002;41(23):7350–7358. doi: 10.1021/bi016053q. [DOI] [PubMed] [Google Scholar]
- 11.Broughman JR, Brandt RM, Hastings C, Iwamoto T, Tomich JM, Schultz BD. Channel-forming peptide modulates transepithelial electrical conductance and solute permeability. Am J Physio-Cell Physio. 2004;286(6):C1312–C1323. doi: 10.1152/ajpcell.00426.2002. [DOI] [PubMed] [Google Scholar]
- 12.Shank LP, Broughman JR, Takeguchi W, Cook G, Robbins AS, Hahn L, Radke G, Iwamoto T, Schultz BD, Tomich JM. Redesigning channel-forming peptides: Amino acid substitutions that enhance rates of supramolecular self-assembly and raise ion transport activity. Biophys J. 2006;90(6):2138–2150. doi: 10.1529/biophysj.105.070078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook GA, Prakash O, Zhang K, Shank LP, Takeguchi WA, Robbins A, Gong YX, Iwamoto T, Schultz BD, Tomich JM. Activity and structural comparisons of solution associating and monomeric channel-forming peptides derived from the glycine receptor M2 segment. Biophys J. 2004;86(3):1424–1435. doi: 10.1016/S0006-3495(04)74212-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Ginkel FW, Iwamoto T, Schultz BD, Tomich JM. Immunity to a self-derived, channel-forming peptide in the respiratory tract. Clinical and Vaccine Immunology. 2008;15(2):260–266. doi: 10.1128/CVI.00319-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnston JM, Cook GA, Tomich JM, Sansom MSP. Conformation and environment of channel-forming peptides: A simulation study. Biophys J. 2006;90(6):1855–1864. doi: 10.1529/biophysj.105.069625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy GL, Iwamoto T, Tomich JM, Montal M. Synthetic Peptides and 4-Helix Bundle Proteins as Model Systems for the Pore-Forming Structure of Channel Proteins .2. Transmembrane Segment M2 of the Brain Glycine Receptor Is a Plausible Candidate for the Pore-Lining Structure. J Biol Chem. 1993;268(20):14608–14615. [PubMed] [Google Scholar]
- 17.Montal MO, Iwamoto T, Tomich JM, Montal M. Design, Synthesis and Functional-Characterization of a Pentameric Channel Protein That Mimics the Presumed Pore Structure of the Nicotinic Cholinergic Receptor. FEBS Lett. 1993;320(3):261–266. doi: 10.1016/0014-5793(93)80599-p. [DOI] [PubMed] [Google Scholar]
- 18.Roux B, Allen T, Berneche S, Im W. Theoretical and computational models of biological ion channels. Q Rev Biophys. 2004;37(1):15–103. doi: 10.1017/s0033583504003968. [DOI] [PubMed] [Google Scholar]
- 19.Bond PJ, Sansom MSP. Bilayer deformation by the Kv channel voltage sensor domain revealed by self-assembly simulations. Proc Natl Acad Sci U S A. 2007;104(8):2631–2636. doi: 10.1073/pnas.0606822104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law RJ, Tieleman DP, Sansom MSP. Pores formed by the nicotinic receptor M2 delta peptide: A molecular dynamics simulation study. Biophys J. 2003;84(1):14–27. doi: 10.1016/S0006-3495(03)74829-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov I, Cheng XL, Sine SM, McCammon JA. Barriers to ion translocation in cationic and anionic receptors from the Cys-loop family. J Am Chem Soc. 2007;129(26):8217–8224. doi: 10.1021/ja070778l. [DOI] [PubMed] [Google Scholar]
- 22.Roux B. Ion conduction and selectivity in K+ channels. Annu Rev Bioph Biom. 2005;34:153–171. doi: 10.1146/annurev.biophys.34.040204.144655. [DOI] [PubMed] [Google Scholar]
- 23.Saiz L, Klein ML. The transmembrane domain of the acetylcholine receptor: Insights from simulations on synthetic peptide models. Biophys J. 2005;88(2):959–970. doi: 10.1529/biophysj.104.049726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pastor RW, Venable RM, Feller SE. Lipid bilayers, NMR relaxation, and computer simulations. Acc Chem Res. 2002;35(6):438–446. doi: 10.1021/ar0100529. [DOI] [PubMed] [Google Scholar]
- 25.Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R. Misfolded amyloid ion channels present mobile beta-sheet subunits in contrast to conventional ion channels. Biophys J. 2009;97(11):3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng MHY, Cascio M, Coalson RD. Theoretical studies of the M2 transmembrane segment of the glycine receptor: Models of the open pore structure and current-voltage characteristics. Biophys J. 2005;89(3):1669–1680. doi: 10.1529/biophysj.105.060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang P, Mandal PK, Xu Y. NMR structures of the second transmembrane domain of the human glycine receptor alpha(1) subunit: Model of pore architecture and channel gating. Biophys J. 2002;83(1):252–262. doi: 10.1016/S0006-3495(02)75166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng MHY, Cascio M, Coalson RD. Homology modeling and molecular dynamics simulations of the alpha 1 glycine receptor reveals different states of the channel. Proteins. 2007;68(2):581–593. doi: 10.1002/prot.21435. [DOI] [PubMed] [Google Scholar]
- 29.Yi MG, Cross TA, Zhou HX. Conformational heterogeneity of the M2 proton channel and a structural model for channel activation. Proc Natl Acad Sci U S A. 2009;106(32):13311–13316. doi: 10.1073/pnas.0906553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bu L, Im W, Brooks CL., 3rd Membrane assembly of simple helix homo-oligomers studied via molecular dynamics simulations. Biophys J. 2007;92(3):854–863. doi: 10.1529/biophysj.106.095216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Chen JH, Brooks CL, Im WP. Application of solid-state NMR restraint potentials in membrane protein modeling. J Magn Reson. 2008;193(1):68–76. doi: 10.1016/j.jmr.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 33.Goddard TD, Kneller DG. SPARKY 3. San Francisco: University of California; 2004. [Google Scholar]
- 34.Wüthrich K. NMR of proteins and nucleic acids. xv. New York: Wiley; 1986. p. 292. [Google Scholar]
- 35.Kirnarsky L, Prakash O, Vogen SM, Nomoto M, Hollingsworth MA, Sherman S. Structural effects of O-glycosylation on a 15-residue peptide from the mucin (MUC1) core protein. Biochemistry. 2000;39(39):12076–12082. doi: 10.1021/bi0010120. [DOI] [PubMed] [Google Scholar]
- 36.Wüthrich K, Billeter M, Braun W. Pseudo-structures for the 20 common amino acids for use in studies of protein conformations by measurements of intramolecular proton-proton distance constraints with nuclear magnetic resonance. J Mol Biol. 1983;169(4):949–961. doi: 10.1016/s0022-2836(83)80144-2. [DOI] [PubMed] [Google Scholar]
- 37.Guntert P, Braun W, Billeter M, Wuthrich K. Automated sterospecif H-1 NMR assignments and their impact on the precision of protein structure determinations in solution. J Am Chem Soc. 1989;111(11):3997–4004. [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54 ( Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Stein EG, Rice LM, Br\”unger AT. Torsion-Angle Molecular Dynamics as a New Efficient Tool for NMR Structure Calculation. J Magn Reson. 1997;124:154–164. doi: 10.1006/jmre.1996.1027. [DOI] [PubMed] [Google Scholar]
- 40.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz JKM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 41.D.A. Case DAP. Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER. 2002;7 [Google Scholar]
- 42.Tripos Inc. SYBYL7.1. 1699 South Harley Rd, St. Louis, Missouri, 63144, USA: Tripos Inc; 2005. [Google Scholar]
- 43.Im W, Feig M, Brooks CL., 3rd An implicit membrane generalized born theory for the study of structure, stability, and interactions of membrane proteins. Biophys J. 2003;85(5):2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry. 1983;4(2):187–217. [Google Scholar]
- 45.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia B, Tsui V, Case DA, Dyson HJ, Wright PE. Comparison of protein solution structures refined by molecular dynamics simulation in vacuum, with a generalized Born model, and with explicit water. J Biomol NMR. 2002;22(4):317–331. doi: 10.1023/a:1014929925008. [DOI] [PubMed] [Google Scholar]
- 47.Chen J, Im W, Brooks CL., 3rd Refinement of NMR structures using implicit solvent and advanced sampling techniques. J Am Chem Soc. 2004;126(49):16038–16047. doi: 10.1021/ja047624f. [DOI] [PubMed] [Google Scholar]
- 48.Chen JH, Won HS, Im WP, Dyson HJ, Brooks CL. Generation of native-like protein structures from limited NMR data, modern force fields and advanced conformational sampling. J Biomol NMR. 2005;31(1):59–64. doi: 10.1007/s10858-004-6056-z. [DOI] [PubMed] [Google Scholar]
- 49.Jo S. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE. 2007;2(9):e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jo S, Kim T, Iyer VG, Im W. Software news and updates - CHARNIM-GUI: A web-based grraphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 51.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 52.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25(11):1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 53.MacKerell AD, Feig M, Brooks CL. Improved treatment of the protein backbone in empirical force fields. J Am Chem Soc. 2004;126(3):698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 54.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- 55.Darden T, York D, Pedersen L. Particle Mesh Ewald - an N. Log(N) Method for Ewald Sums in Large Systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 56.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kal L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han G, Deng Y, Glimm J, Martyna G. Error and timing analysis of multiple time-step integration methods for molecular dynamics. Comput Phys Commun. 2007;176(4):271–291. [Google Scholar]
- 58.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14(1):33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 59.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MSP. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J Mol Graph Model. 1996;14(6):354. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 60.Lee J, Im W. Understanding of Transmembrane Helix Tilting by Potential of Mean Force Calculation. Phys Rev Lett. 2008 doi: 10.1103/PhysRevLett.100.018103. (in press) [DOI] [PubMed] [Google Scholar]
- 61.Montal M, Opella SJ. The structure of the M2 channel-lining segment from the nicotinic acetylcholine receptor. Biochimica Et Biophysica Acta-Biomembranes. 2002;1565(2):287–293. doi: 10.1016/s0005-2736(02)00575-8. [DOI] [PubMed] [Google Scholar]
- 62.Hilf RJC, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457(7225):115–U122. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 63.Hilf RJC, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452(7185):375–U312. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 64.Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457(7225):111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 65.White SH, von Heijne G. Transmembrane helices before, during, and after insertion. Curr Opin Struc Biol. 2005;15(4):378–386. doi: 10.1016/j.sbi.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 66.Smart OS, Breed J, Smith GR, Sansom MSP. A novel method for structure-based prediction of ion channel conductance properties. Biophys J. 1997;72(3):1109–1126. doi: 10.1016/S0006-3495(97)78760-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emmeluth C, Poad BLJ, Thompson CD, Bieske EJ. Interactions between the chloride anion and aromatic molecules: Infrared spectra of the Cl--C6H5CH3, Cl--C6H5NH2 and Cl--C6H5OH complexes. Journal of Physical Chemistry A. 2007;111(31):7322–7328. doi: 10.1021/jp070453r. [DOI] [PubMed] [Google Scholar]
- 68.Kang SO, Begum RA, Bowman-James K. Amide-based ligands for anion coordination. Angewandte Chemie-International Edition. 2006;45(47):7882–7894. doi: 10.1002/anie.200602006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.