


Abstract
We have recently developed a novel method for the affinity purification of the complete suite of translating mRNA from genetically labeled cell populations. This method permits comprehensive quantitative comparisons of the genes employed by each specific cell type. We provide a detailed description of tools for analysis of data generated with this and related methodologies. An essential question that arises from these data is how to identify those genes that are enriched in each cell type relative to all others. Genes relatively specifically employed by a cell type may contribute to the unique functions of that cell, and thus may become useful targets for development of pharmacological tools for cell-specific manipulations. We describe here a novel statistic, the specificity index, which can be used for comparative quantitative analysis to identify genes enriched in specific cell populations across a large number of profiles. This measure correctly predicts in situ hybridization patterns for many cell types. We apply this measure to a large survey of CNS cell-specific microarray data to identify those genes that are significantly enriched in each population Data and algorithms are available online (www.bactrap.org).
INTRODUCTION
The mammalian brain is the most complex organ of the body, containing hundreds of intermingled cell populations. These cells can be classified into types according to their morphology, projections, functions and gene expression profiles. Currently, in vivo analysis of gene expression and translation in particular cell types is often performed with methodologies that are non-parallel and difficult to quantify. Because of this, it remains a challenge to determine the complete set of proteins employed by a given cell type, determine which genes are expressed in or specific to a particular cell type relative to all others, or establish the degree to which a given cell population is unique in the nervous system.
Previously, we have described a method, translating ribosome affinity purification (TRAP, Supplementary Figure S1) for the isolation of translating mRNA from individual, genetically defined, cell types (1,2). In this method, transgenic mice are generated which express a fusion of eGFP and a ribosomal protein under the control of a bacterial artificial chromosome (BAC) (3) for a cell-specific ‘driver’ gene. A complete translational profile of all ribosome bound mRNAs is then generated from these labeled cells via brain homogenization and affinity purification with anti-eGFP antibodies. Relative quantities of the purified mRNAs are assessed via microarray or related technologies. Thus, for any cell type for which a driver gene can be identified, the methodology permits a comprehensive translational profile to be prepared for all genes. The TRAP protocol is rapid, simple, and requires no specialized equipment. This method permits the deconstruction of the complexity of the nervous system, allowing researchers to access individual cell types within the context of the whole brain, with sensitivity sufficient to study whole animal manipulations such as drug treatments, experimental injuries, or genetic manipulations (2).
The fundamental impetus for the development of the TRAP methodology was to allow the rapid and reproducible cell-specific assessment of RNA translation. Microarray analysis, as traditionally applied to the nervous system, results in data representing the aggregate RNA from all of the cell types present in the tissue (4), proportional to the percentage of those cells present and the relative amount of RNA they produce. This has several implications regarding the interpretation of these data (5). As the observed signal on the array represents an averaging of the levels of the transcript in each of these cell types, RNAs present in all cell types, even at moderate levels, will have fairly high observed values compared to RNAs present at high levels, but in rare cell types. In fact, such mRNAs may even be undetectable because they represent a small fraction of the total tissue RNA (1). Furthermore, as the RNAs from all the cell types are measured in aggregate, any changes in RNA levels measured in the whole tissue are not easily attributed to any particular cell type. Detected perturbations in RNA levels could be due to the death of one cell type, the arrival of another, and/or changes within some or all of the cell types present. Likewise, changes in one cell type could be masked by changes of opposite direction in another cell type. All of these factors clearly complicate the application of microarrays to assess changes in RNA due to experimental manipulations, especially those that may have their primary influence on rare cells. TRAP provides not only the ability to detect changes in rare cell types, but also enhanced ability to interpret the results, as it is known a priori which cells contain the tagged ribosomes. In addition, TRAP has the advantage over other approaches to cell-specific RNA profiling as it assesses translation, rather than expression, providing a better correlate of actual protein levels (6).
There are distinctions between microarray experiments from TRAP RNA compared to whole-tissue RNA, and these distinctions can have important impact on the assumptions regarding experimental design, normalization, analysis and interpretation. To aid researchers implementing cell-specific RNA-analysis technologies (1,7–10), we present here a preferred analytical method for TRAP translational profiling data. Importantly, the TRAP methodology provides in vivo quantitative comparative analysis of multiple cell types. Here, we have developed a robust analytical method for identifying and quantifying cell-specific and enriched mRNA’s across multiple cell populations, referred to as the specificity index (SI). We apply this to a large survey of CNS cell types and provide a simple perusable archive of plots of this measure across all cell types, for each gene.
MATERIALS AND METHODS
Dataset
TRAP data were generated as described (1,2), and are available for download from GEO: GSE13379. Etv1 data were not plotted because of known contamination with endothelial or lymphoblast cells (1). Other cell types and drivers are listed in Table 1. This dataset contains samples representing a variety of pure and mixed cell types from different structures of the mouse brain, as well as samples from the corresponding whole tissue. The purified samples are referred to as immunoprecipitates (IP). In parallel, RNA which did not bind to the antibody was also harvested to provide an assessment of the gene expression of the tissue as a whole. These samples are referred to as unbound RNA. Microarray analysis, as traditionally applied to the nervous system, results in samples that are most similar to unbound samples. As the immunoprecipitation does not lead to significant depletion of cell-specific RNAs, here we use the unbound samples as a measure for the total tissue homogenate RNA (referred to as Total).
Table 1.
List of the cell populations, relevant drivers and abbreviations
| Cell populations | Driver | Abbreviations used* |
|---|---|---|
| Drd1+ medium spiney neurons of neostriatum | Drd1 | CS.Drd1 |
| Drd2+ medium spiney neurons of neostriatum | Drd2 | CS.Drd2 |
| Cholinergic Interneurons of corpus striatum | Chat | CS.Chat |
| Motor neurons of brain stem | Chat | BS.Chat |
| Cholinergic neurons of basal forebrain | Chat | BF.Chat |
| Mature oligodendrocytes of cerebellum | Cmtm5 | Cb.Cmtm5 |
| Astroglia of cerebellum | Aldh1l1 | Cb.Aldh1L1 |
| Golgi neurons of cerebellum | Grm2 | Cb.Grm2 |
| Unipolar brush cells and Bergman glia of cerebellum | Grp | Cb.Grp |
| Stellate and basket cells of cerebellum | Lypd6 | Cb.Lypd6 |
| Granule cells of cerebellum | Neurod1 | Cb.Neurod1 |
| Oligodendroglia of cerebellum | Olig2 | Cb.Olig2 |
| Purkinje cells of cerebellum | Pcp2 | Cb.Pcp2 |
| Bergman glia and mature oligos. of cerebellum | Sept4 | Cb.Sept4 |
| Cck+ neurons of cortex | Cck | Ctx.Cck |
| Mature oligodendrocytes of cortex | Cmtm5 | Ctx.Cmtm5 |
| Cort+ interneurons of cortex | Cort | Ctx.Cort |
| Astrocytes of cortex | Aldh1l1 | Ctx.AldhL1 |
| Corticospinal, corticopontine neurons | Glt25d2 | Ctx.Glt25d2 |
| Corticothalamic neurons | Ntsr1 | Ctx.Ntsr1 |
| Oligodendroglia of cortex | Olig2 | Ctx.Olig2 |
| Pnoc+ neurons of cortex | Pnoc | Ctx.Pnoc |
| Motor neurons of the spinal cord | Chat | SC.Chat |
*Abbreviations used for Figures 4, 5, 7 and Supplementary Figures 7 and 8
Translating ribosome affinity purification
Additional TRAP experiments on wild-type mouse brains were conducted as described (2). RNA was quantified using the Ribogreen assay, according to manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA), and a Modulus single tube fluorometer from Turner Biosystems (Sunnyvale, CA, USA) with the blue optical kit.
R code
The scripts used for calculation of SI, are available from the bacTRAP website (www.bactrap.org).
SI for a given gene (n), in a given cell type (#1), compared to cell types, k = 2…m, is given by the formula:
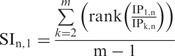 |
(1) |
where IP1,n is the expression value for gene n in cell type one, and rank(IP1,n / IP1,k) is the position, of gene n, in a descending-ordered list of ‘fold-change’ (IP1/IPk) values for all genes.
Note that SI is only calculated for those genes in cell type k with an absolute expression above 50 in IPk, and with log2(IPk/Totalk) values above a threshold
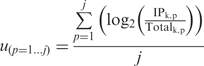 |
(2) |
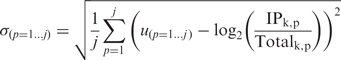 |
(3) |
| (4) |
where 1…j is a set of negative control genes known not to be expressed in this cell type, IPk,p is the expression value for gene p in the IP from cell type k. Totalk,p is the expression value for gene p in the total tissue RNA from the tissue cell type k was isolated from. Any gene for which log2(IPk/Totalk) < Thresholdk is excluded, with the caveat that Thresholdk was not allowed to exceed zero (Supplementary Data and Supplementary Figure S5).
Scoring allen brain atlas in situ hybridizations
For comparative analysis of TRAP data, we developed a blinded, unbiased scoring method (the SENU method). For the first application, for each of four cell types, 50 probesets were selected at random from the top 500 most enriched genes (IP/Total). For each cell-type, an additional 50 probesets were selected from the array at random, irrespective of IP/Total value. For each cell type, the 50 random and the 50 cell-enriched probesets were scrambled together and presented to three blinded judges, previously trained in the heuristics below until inter-rater reliability was above 60% on training sets.
Judges searched for each probeset in the Allen Brain Atlas (ABA) using the gene symbol and name. If no gene symbol or synonym could be found, the probeset was scored as absent. For probesets present in the ABA, judges first assessed overall quality of the in situ hybridization (ISH). If the ISH had no detectable signal or was of low quality for the given gene, the gene was scored as a ‘U’ (unscorable).
For probesets not scored U, judges evaluated potential expression in the four cell types. For each cell type the judges could assign one of three scores, ‘S’ (specific for cell-type within region), ‘E’ (expressed in cell type), and ‘N’ (clearly not expressed). Detailed heuristics for each are:
S: In situ must be of very good quality and show clear signal in cell type of interest that is at least three color levels with the Allen ‘expression viewer’ above any other cells in the same region.
E: In situ shows expression in cell type of interest but overall signal is weak or there is clear signal in surrounding cells as well. In situ may be moderate or good quality.
N: In situ must be of very good quality and clearly have (i) no signal in cell-type of interest and (ii) very good signal somewhere else in tissue.
As cell type is difficult to assign from colometric ISH alone, for each cell type, the pattern assayed was:
-
Purkinje cells: ISH pattern in cerebellum with evenly spaced large cells in the PCL.
Motor neurons: ISH pattern in brain stem in large cells at the approximate locations of the third, fifth and seventh motor nuclei.
Layer V cortical neurons: A laminar ISH pattern in cortex at approximately the position of layer 5, with, at most, labeling in one other layer.
Oligodendrocytes: Strong specific ISH pattern in the corpus callosum. Scattered labeling in cortex also permitted. Note: color criteria for ‘S’ had to be relaxed as oligodendrocytes are often too small to be recognized as cells by ABA expression viewer.
For the second round of SENU analysis, two additional cell types were added: granule cells and cortical interneurons. For each of the six cell types, 150 ISH were scored, 50 each from the top 250 of IP/Total, SI and random lists. If multiple ISH sets were available for the same gene, only the most recent sagital ISH set was used. Heuristics for an ISH pattern consistent with expression in granule cells or interneurons are:
Granule cells: clear expression exclusively in granule cell layer of cerebellum, in at least 50% of the cells.
Cortical interneurons: scattered, non-laminar expression in the cortex, with a cell number in the range between two reference ISH patterns, Cort and the GABA transporter Slc32a1.
For the third round of SENU analysis, all remaining cell types were evaluated (glial cells were only scored in cerebellum). For each cell type, all genes with SI P < 10e-5 were scrambled with an equal number of randomly selected genes and up to 40 genes per cell line were scored blindly as above, using the driver gene ISH as a reference pattern. It is worth noting, however, that several cell types had difficult to interpret ISH patterns (Cck), lacked appropriate signal even for the driver (Grp), represented small and scattered cells (Olig2, ALdh1L1), or were found in very cell dense regions (Neurod1). For many of these cell types, inter-rater reliability was correspondingly lower.
Immunofluorescence
Adult mice were perfused transcardially with PBS followed by 4% paraformaldehyde in PBS, cryoprotected in 30% sucrose PBS, frozen and sliced to 40 microns on a cryostat. Floating sections were blocked with 5% normal donkey serum in 0.25% Triton X-100 PBS and incubated overnight with chicken anti-GFP antibody (Abcam, Cambridge, MA, USA), and/or Grm1 (AB1551, Chemicon, Temecula, CA, USA) and Calb2 (6b3 Swant, Bellinzona, Switzerland) incubated 90 min with appropriately Alexa-conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA), and counterstained with DAPI. Images were acquired with a Zeiss LSM 510 inverted confocal microscope.
RESULTS
IPvTotal plots
The microarray data employed for these studies are from a published survey of CNS cell types generated with the TRAP methodology (1). The purified cell-specific RNA samples are referred to as IP. In parallel, RNA was also harvested to provide an assessment of the gene expression of the tissue as a whole (Total). As an initial assessment of TRAP data, described in detail below, we generated scatterplots with the log signal intensity for the IP on the x-axis and the Total on the y-axis (IPvTotal plot) for each cell population. Systematic examination of these plots revealed they could be used for quick visual assessment of the quality of the TRAP experiment, particularly for the level of non-specific background (Figure 1, Supplementary Figure S2). We first applied these plots for assessment of different metrics of normalization (Supplementary Figure S3, and ‘Supplementary Materials and Methods’ section). It also became apparent these plots may also indicate the rarity and/or uniqueness of the cell type within its tissue (Supplementary Figure S4, and ‘Supplementary Materials and Methods’ section). Finally, we assessed IP/Total as a measure to identify those RNAs that may be specific or enriched in a given cell type.
Figure 1.

Assessment of IPvTotal plots. (a) Scatterplot of immunoprecipitated (Purkinje cells, IP) versus unbound RNA (from whole cerebellum, Total) provides a basic measure of experiment quality. RNA for non-Purkinje cell genes (glial genes, red) are highly enriched in Total RNA, while RNAs determined to be in Purkinje cells (blue, Supplementary Table S1) are enriched in IP RNA. (b) Illustration of the interpretation of IPvTotal plots based on the locations of positive and negative control genes.
Figure 1a shows an example of this plot for Purkinje cells. A list of genes known from the literature to be glial specific (and thus not in Purkinje neurons) has been marked in red, and a variety of genes determined from ISH database (11,12) to be highly expressed in Purkinje cell layer, including the driver for this mouse line, Pcp2, have been marked in blue (Supplementary Table S1). From this plot, it is clear that RNAs known to be enriched in Purkinje cells have high ratios of IP/Total. Genes that are known not to be expressed in Purkinje cells, such as those that are specific to glia, are highly enriched in the Total RNA. They have low IP/Total ratios. Based on the locations of these positive and negative controls, we have developed a heuristic for the interpretation of IPvTotal plots, illustrated in Figure 1b. Essentially, from the top left corner of the plot to the bottom right, one has increasing confidence, first that the RNA derives from the targeted cell type, and then that it is highly enriched in that type. Note that probesets with low signal (bottom left corner) should be considered with caution, as they tend to have higher variability (13).
IP/Total for identification of enriched genes
As previously shown, if a RNA is specifically translated in the targeted cell type within a tissue, it should have a very high IP/Total ratio (1). As an independent, qualitative measure of the expression of specific mRNAs within a cell of interest, we compared our data to ISH data from the ABA (11). Since it is often difficult to establish cell identity by ISH data alone, we chose for this first comparative study four cell types that are relatively simple to identify by size and localization in colormetric ISH (brainstem motor neurons, cerebellar Purkinje cells, layer 5 cortical pyramidal cells, oligodendrocytes). For each cell type, a list of 50 ‘high IP/Total’ probesets was selected at random from the top 500 probesets, as ranked by IP/Total. Many of these mRNAs are only moderately enriched: minimum IP/Total ratios range from around two (motor neurons, layer V cortical neurons) to around four (Purkinje cells). For comparison, an additional 50 probesets were selected at random from the array, and scrambled with the list above. These lists were then presented to three blinded judges and the ISH for all genes were scored as specific (S), expressed (E), clearly not expressed (N) or unscorable (U) in the cell type of interest. Figure 2a shows examples of S, E, N and U scores for brain stem motor neurons. After excluding the ISH scored U, probesets for genes with high IP/Total were highly enriched by ISH in the cell type of interest (S), and less likely to appear not expressed (N) than the random list of 50 genes (chi-square, P < 0.0005 for each cell type). Typically, probesets with high IP/Total ratios were three to four times more likely to be scored S than random genes (Figure 2b). Although this analysis demonstrated that TRAP analysis results are concordant with the easily scored ISH data, the level of enrichment varied substantially between the cell types assessed. Given this fact, and the many factors that limit the utility of ISH data for detection of cell-specific changes in gene expression in complex tissues, we sought to develop an independent method for the quantitative measurement of the specificity of expression of any gene in a given cell type or condition relative to a large number of other cell types using comparative analysis of TRAP data from a variety of specific CNS cell types.
Figure 2.

High IP/Total can identify cell-specific genes. (a) Examples of in situ patterns from the ABA scored as specific, expressed, not expressed, or unscorable for brainstem motor neurons (Allen Mouse Brain Atlas [Internet]. Seattle (WA): Allen Institute for Brain Science. ©2008. Available from: http://mouse.brain-map.org). (b) For each of four cell types, 50 from the top 500 highest IP/Total ratio genes, and 50 random genes, were scrambled together and scored blindly by three judges trained in the rubric illustrated in a. Genes with high ratio for each cell type (grey bars) were more likely to be categorized as specific (center panel) and less likely to be categorized as Not Expressed (right panel) P < 0.0005, chi-squared test, all cell types. Genes with absent or unscorable ISH patterns (b, left panel) were not included in analysis.
The SI to identify cell-specific and enriched genes
As described above, the IP/Total metric can be used as a simple method to suggest cell-specific and enriched genes. However, there are three drawbacks to the method. First, there are cell types where logically it would be ineffective, such as granule cells of cerebellum or medium spiny neurons of striatum. Over 90% of the cells in the cerebellum are granule cells (14). As such, a comparison of a granule cell IP to total cerebellum will yield little enrichment of granule cell genes, as shown in Supplementary Figure S4b. In contrast, comparison of the granule cell IP data to the IP data obtained from Purkinje cells clearly reveals a high enrichment of the granule cell driver gene, Neurod1 (Supplementary Figure S4c). This demonstrates that the granule cell IP was robust, and illustrates the value of comparative analysis of TRAP derived from specific cell types. Likewise, comparison of the Drd1a+ or Drd2+ medium spiny neurons to total striatum, which is made primarily of medium spiny neurons, will identify very few striatally enriched genes (2). The second drawback is that a comparison of IP to Total will only yield information about enrichment relative to one particular dissected structure, and not the rest of the brain. To accurately determine the suite of cell-specific genes, one needs to make multiple comparisons across all available cell types and structures. Finally, IP/Total alone does not give a sense of how likely a particular ratio is to appear by chance, and at what threshold a gene should be considered enriched. Indeed, from the four cell types scored above, there were clear differences in fraction of specific genes found in the top 500 IP/Total (Figure 2b).
To overcome these problems, we developed a generic algorithm, the SI, to assess the specificity of a given RNA in one sample relative to all other samples analyzed. For each cell type, the SI is calculated in three steps, as illustrated in Figure 3a–e. First, following GCRMA normalization within replicates and global normalization across samples, the IP was compared to the total to filter out the non-specific background by setting a simple threshold based on negative controls (Supplementary Figure S5, and ‘Supplementary Materials and Methods’ section). For those cell types known to have significant background contamination, this threshold was left at one, so as to not filter too many probesets and create false negatives. Probesets with low signal were also removed, following standard practice with microarray data. Second, for the remaining probesets, this filtered IP was iteratively compared to each other (unfiltered) sample in the dataset and a ratio was calculated for each probeset. To prevent extreme outliers from skewing the subsequent analysis, and to make the analysis more robust for difficult to normalize datasets, the probesets were ranked from highest to lowest ratio within each comparison. Third, for each probeset, its ranks across all comparisons are averaged to give the SI. Thus, the SI is a measure of the specificity of expression for each probeset in a given cell type relative to all other cell types included in the analysis: how highly ranked on a gene list is this probeset, on average, in this cell type compared to all others. Note that the term ‘specificity’ has also been used in literature mining field for metrics assessing the precision of search algorithms in returning relevant literature (15). However, the use of term here, while related philosophically, is quite distinct from those metrics in both its mathematics and its applications.
Figure 3.

Illustration of algorithm for calculation of SI to identify cell-specific and enriched genes for a single cell type (Purkinje Cells, pink). (a) SI is a comparative analysis, thus multiple bacTRAP experiments are conducted for several classical cell types shown in this illustration from Cajal. (b) Data from each cell type are normalized and filtered to remove background, as illustrated in Supplementary Figure S5, prior to IP/IP calculation. (c) Normalized and filtered Purkinje cell data are compared to each other cell type (IP/IP). For each comparison (2…M), probesets are ranked from highest to lowest ‘fold change’. SI for each probeset is calculated as the average rank across all comparisons. (d) A P-value is assigned to a given SI value via a permutation testing, as illustrated in Supplementary Figure S6a. (e) A list of genes significantly enriched in Purkinje cells can be selected based on P-value.
Validation of SI and comparison to IP/Total
To determine if the SI succeeds in selecting cell-specific genes in those cases where IP/Total comparisons fail, we first examined the expression of genes predicted by each method to be translated in granule cells. Figure 4a shows a comparison of eGFP immunohistochemistry for GENSAT BAC transgenics (16) for two genes selected by IP/Total and two selected for a high SI. The genes selected by SI clearly have an expression pattern that is more consistent with highly enriched expression in cerebellar granule cells: labeling of many cell bodies in the cerebellar granule cell layer, with fibers filing the molecular layer, where granule cell axons project.
Figure 4.

The SI provides a robust method for identifying cell-specific and enriched mRNAs. (a) Specificity index performs better at selecting granule cell-specific mRNAs. Right panels: examples of GENSAT eGFP expression patterns for two mRNAs with SI P < 10−5, but low IP/Total (<2-fold) show robust expression in cerebellar granule cells. Left panels: examples of two mRNAs high IP/Total (>3-fold), but non-significant specificity indices show little expression in cerebellum. (b) Blind scoring by three judges of ABA ISH for 50 random, 50 high IP/Total and 50 high SI genes, across six cell types, reveals that SI generally performs better than IP/Total in predicting specific ISH patterns, among those ISH that are scorable. (c) Plot for combined SI P-values (blue bars, −log10 scale) and IP/Total values (red bars, log2) across all cell populations for a probeset of Neurod1. SI clearly identifies Neurod1 as a marker for granule cells (P < 10−5) across all cell types. Axis on left shows log scale. Axes on right show corresponding P-values in blue, and IP/Total ratio in red. Cell types are in same order, and with same abbreviations as Table 1. (d) Post hoc analysis across all judges and cell types reveals that more than 75% of those genes with SI P < 10e-5 are scored as specific, compared to 15% of those with P > 0.1.
To determine how effectively the SI index performs in general compared to IP/Total in selecting cell-specific and enriched genes, we repeated our SENU analysis of ABA ISH patterns for 150 probesets for each of six cell types (Figure 4b). Fifty probesets were chosen randomly each from the top 250 probesets of SI and IP/Total, as well as 50 random probesets from the array. These were scrambled and scored by three blinded judges, as above. As before, chi-squared tests revealed that TRAP data performed significantly better than chance at predicting specific gene expression (P < 0.01 to 10−99, across either metric in each cell type). As expected, SI outperforms IP/Total for those cases where the TRAPed cell type makes up a significant fraction of the total, such as Neurod1 positive granule cells. Quite surprisingly SI also out-performed IP/Total with Purkinje cells (Cb.Pcp2), cortical oligodendrocytes (Ctx.Cmtm5) and cortical interneurons (Ctx.cort). In the worst case, that of layer V cortical projection neurons (Ctx.Glt25d2), IP/Total or SI both yielded ∼50% more than the amount of specific patterns expected by chance. There were no cell types where IP/Total clearly performed significantly better than SI. Thus, we determined that the SI is a useful and robust metric for identifying cell-specific and enriched genes.
The SI as a statistical measure
The SI is influenced by both the variations in the number of transcripts that are enriched in each cell type being analyzed, and the purity and recovery of TRAP mRNA collected for each cell type. The range of the rankings is dependent on the number of probesets in the comparison, and that number depends on the number of genes expressed and the level of filtering in each particular cell type. Consequently, raw SI values are not directly comparable across cell types. In addition, the SI alone does not provide a sense of how likely a given rank is to occur by chance. Therefore, for each SI we calculate a P-value via permutation testing as illustrated in Supplementary Figure S6a: for each IP, the filtered expression values are randomly shuffled many times and SIs are calculated for all probesets, to determine the frequency of a particular SI value appearing. This creates a simulated probability distribution. The probability of any given SI from the true distribution can be assigned from the simulated distribution. Thus one can derive a list of genes that are significantly specific to, or enriched in, any particular cell type, with a known probability (Supplementary Figure S6b). We note that for each cell type, the number of genes that reach a given statistical threshold is different. However, since these probabilities are comparable across cell types, they can be plotted to permit assessment of the specificity for a given probeset across all cell types analyzed, as illustrated for the granule cell driver Neurod1 in Figure 4c.
To determine whether the SI is an accurate relative measure of the specificity of expression of each gene relative to all others for the cell types analyzed, we next performed a post hoc analysis of our judges’ ratings in the SENU analysis pooled across all six cell types from Figure 4b. For P < 0.00001, over 75% of scorable ISH were scored ‘specific,’ compared to ∼15% of those P > 0.1 (Figure 4d). Even with extensive training in detailed heuristics and blind scoring there is substantial subjectivity in the interpretation of ISH, and only 55% of the 900 ISH had identical scores from all three judges (for 95% however, at least two judges agreed on the score). Of these ISH on which all three judges agreed, 100% of the genes with P < 0.00001 were scored as specific (not shown). This analysis provides a potential heuristic for the interpretation of various SI P-values for a gene across cells types: while any P < 0.1 suggests some enrichment, as P-values continue to decrease, enrichment increases until the majority, if not all genes at extremely low P-values are highly specific (Figure 4d).
Finally, to generalize this finding to all remaining cell types, we examined the ISH pattern for all cell types for those genes with P < 0.00001. This represented a challenge as most of these cell types cannot be unambiguously identified by position information alone. For each cell type, the P < 0.00001 genes were scrambled with an equal number of randomly selected genes and up to 40 genes per cell type were scored blindly by three judges. For this analysis, genes were scored as specific if their ISH pattern matched that of the driver for the TRAP line. Across nearly all cell types most of these P < 0.00001 genes had patterns consistent with specific expression in the correct cell types (Figure 5a), representing in all cases a highly significant enrichment relative to randomly selected genes (chi-squared P < 0.0005 to 10−24).
Figure 5.

SI concurs well with ISH pattern for most cell types. (a) A higher percentage of scorable ISH patterns scored are specific for those genes with SI P < 10e-5 for these cell types, relative to randomly selected genes (chi-squares from P < 0.0004 to <10e-21). ‘n’ is the number of P < 10e-5 genes for a given cell type. (b) For three cell types, ISH analysis did not show significant enrichment in specific genes by SI. Left panel: Etv1 data are known to be contaminated with blood or endothelial cells, reflected here in the large fraction of not expressed scores. Middle panel: Cck data are from a mix of many different cortical neuronal cell types, limiting interpretability of both TRAP and ISH data. Right panel: unipolar brush cells are difficult to identify by ISH. (c) Confocal immunofluoresence on GENSAT Nmb eGFP line reveals clear GFP expression in both Calb2+ and Grm1+ unipolar brush cells (18). (Grm1 labels only the brush of these cells. Calb2 labels cytoplasm. Z stacks, not shown, confirm all GFP+ cells are positive for either Grm1 or Calb2). Nmb was scored as ‘not expressed’ by two of three judges based on ISH alone, suggesting ISH may have limited sensitivity for some cell types.
However, for three cell types, SI did not perform well at predicting ISH patterns, and we will discuss these briefly because they are each illustrative of an important point regarding this analysis (Figure 5b). First, for the ISH patterns for genes from the line Etv1, which expresses the eGFP-L10a transgene in layer 5b projection neurons, over 70% of the P < 0.00001 were specific to blood vessels. This strongly suggests that the bacTRAP construct is also expressed in endothelial cells or some component of the blood in this line. This illustrates the point that careful anatomical characterization of TRAP lines is essential. Minor contamination by rare cell types will be very apparent following SI analysis, and confirmation with ISH databases.
Second, for the Cck TRAP line, which is expressed broadly in multiple layers of cortex and in both pyramidal cells and interneurons, we observed no significant enrichment for specific ISH patterns (chi-square, P = 0.3). We believe this reflects the fact that this line includes so many neuron types that nearly any gene expressed in neurons will be present in the IP. This illustrates the difficulty in assessing ISH results for a TRAP driver that is broadly expressed.
Third, in the Cb.Grp data, representing a mix of unipolar brush cells (UBC) and Bergman glia, SI identified genes did not show enrichment for specific ISH patterns (chi-squared P = 0.14). As Bergman glia are represented in both the Cb.Aldh1L1 and Cb.Sept4 datasets, the most specific genes for this data should come from the UBCs, a small excitatory interneuron found primarily in the granule cell layer of the posterior lobules of the cerebellum (17). However, since even the driver, Grp, did not have a specific ISH pattern, we were suspicious that ISH may have reduced sensitivity for detecting messages in this scattered population of small neurons in the cell dense cerebellar granule layer. Some SI identified genes, such as Nmb, did show a scattered precipitate in lobule X of the cerebellum, but the particles were too small to be clearly identifiable as cells by our judges. To provide an independent dataset, we examined the GENSAT database (16) for the SI identified genes in the five lines for which adult data were available online (Grp, Nmb, Ntf3, Otx2 and Eomes). Three of these five lines clearly expressed GFP in cells with the distinct morphology and position of UBCs in the online database. We further confirmed, in the NMB line, that these were indeed UBC’s by confocal triple immunofluorescence for GFP and the UBC markers Calb2 and Grm1 (Figure 5c) (18).
In general, we note that, despite the concordance between the TRAP data and ISH results for genes whose expression is easily detected by ISH, a significant fraction of the genes determined to be enriched in a specific cell type by TRAP analysis could not be scored from the ISH data (Figure 2b). RT–PCR on genes without detectable signal (U) by ISH reveals that in these cases the RNA is indeed present in the brain, and enriched in the TRAP samples from the cell types of interest (1). This is not surprising, since successful ISH is dependant on many factors, including expression level of the gene, hybridization kinetics of the probe and availability of unique sequence for probe design. We conclude that negative results on ISH should be interpreted with caution. Of course, for any specific case, differences in ISH patterns and TRAP measurements could also indicate there is a difference between transcription and translation of a given gene.
Finally, the mixed oligodendrocyte data (Olig2 line) only showed 43% specific ISH patterns. While this is still significantly better (chi-square, P < 10−5) than the 12% identified by chance, we were curious if this reflected the fact that of the 26 cell types included in the SI calculation, at least four contained information primarily from oligodendrocytes (ctx.olig2, cb.olig2, ctx.cmtm5, cb.cmtm5). Thus, we repeated the SI analysis on our data set, but excluded three of these four samples collected from oligodendrocytes so only one, unique oligodendrocyte sample remained. As shown in Figure 6, this resulted in two major effects: first, as the oligodendrocyte data became more unique in the analysis, there were now twice as many genes with P < 0.00001 by SI; second, when the ISH for these genes were scored blindly as above, 70% of them showed specific ISH patterns. This demonstrates the fact SI is a relative measure that is influenced by the composition of the entire dataset, and that one should carefully consider which datasets to include for the specific experimental question being addressed.
Figure 6.

Outcome of SI analysis depends on composition of dataset. When SI is calculated with only one oligodendrocyte sample in the dataset (b), more genes arrive at P < 10e-5 (n of 80 instead of 40), and a higher fraction of those genes show a clear specific ISH pattern, compared to SI calculated with four oligodendrocytes samples in the dataset (a).
An archive of SI for all genes
To provide a resource to permit researchers to examine the specificity of the translation of any gene across all cell types included in this analysis, we have created SI plots for all genes on the array using updated chip definition files (19) that provide one measure per ENTREZ gene ID (20) (Supplementary Figure S7). Figure 7 illustrates SI P-values, as well as IP/Total values for six representative genes, across the 24 cell populations from the original studies. This includes examples for genes known to be enriched in a few cell types (Slc18a3, the vesicular acetylcholine transporter, in cholinergic cells, Dlx1 in interneurons); metabolic genes expressed ubiquitously, though not equally, across the brain (Actb, Rpl8); and two genes implicated in autism, Nrxn2 and Nrxn1, which show broad but variable expression, or enrichment in a limited set of cortical neurons and granule cells, respectively. SI plots for all genes are available as a downloadable archive from www.bactrap.org. Simply browsing through images can highlight remarkable biology. For example, the GalNAc transferase family is a group of golgi apparatus enzymes that catalyze the addition of oligosaccharides to protein receptors destined for the cell surface. Supplementary Figure S8 shows plots for six members of this family, Galnt2,3,4,6,14,L2, five of which show remarkable cellular specificity to either oligodendrocyte progenitors, astroglia, mature oligodendrocytes, layer 5 cortical projection neurons or granule cells. As many of these enzymes have affinities for distinct donors and acceptors (21), cell-specific expression of these proteins may result in distinct cell surface moieties.
Figure 7.

SI P-values and IP/Totals for a selection of representative mRNAs. (a–f) Combined SI P-values (blue bars, −log10 scale) and IP/Total values (red bars, log2) across all cell populations. (a) The acetylcholine transporter, Slc18a3 is significantly specific to all four cholinergic cell populations assessed. (b) The interneuronal marker Dlx1 is translated specifically in the Cort and Pnoc bacTRAP lines. (c and d) Ubiquitously expressed genes B-actin(Actb) and ribosomal protein L8(Rpl8) are not specific to any cell type, though translation does vary across cell types. (e and f) The Neurexin autism candidate genes Nrxn1, and Nrxn2, have differential patterns of translation. Nrxn2 is more broadly translated, while Nrxn1 has low to moderate enrichment in cerebellar granule cells and some cortical neuron types.
DISCUSSION
We present here a set of analytical procedures that have been developed for analysis of TRAP translational profiling data. These approaches are specifically designed to accommodate features of TRAP translational profiling data that arise from the cell-specific nature of the TRAP data, and to provide a robust framework for comparative analysis of data obtained from large numbers of cell types. In particular, we report the development of a SI to provide a relative and quantitative measure for the specificity of expression of all genes across the cell types being studied. In general, results of this analysis are concordant with easily evaluated ISH data from the ABA (11). However, our data also indicate that TRAP translational profiling can reveal cell enriched expression for a large number of genes and cell types that are not easily assessed by ISH.
The SI
The impetus for the development of the SI was to accommodate the facts that there are dramatic differences in mRNA profiles between different cell types, and that it was evident that a new method for comparative and quantitative analysis of TRAP data was needed. Although analysis with standard tools for identifying statistically significant differences between samples also can apply to TRAP data (22–26) comparisons of widely divergent cell types, such as astrocytes and Purkinje neurons, results in >60% of probesets reaching statistical significance (P < 0.05) using the empirical Limma (25) module of Bioconductor (26) with FDR multiple testing correction. This number of statistically significant changes demonstrates the limited utility of such methods for selecting small numbers of targets for biological follow-up studies from such dramatically different cell types. The SI we have described here is robust, and uses a permutation-based statistical approach to compensate for any irregularities in the distributions of the data, allowing direct comparison of P-values across samples with quite varied distributions. As shown above, this measure provides results consistent with published data, and independent assays of gene expression provided in the ABA. However, the SI is clearly dependent on the number and nature of the samples included in the analysis (Figure 6). Consequently, the design of the SI analysis should be tailored to the biological question at hand. However, as we anticipate that many researchers will be interested primarily in the output of this analysis, rather than its implementation, we provide an archive of SI histograms for all genes across all cell types included in this study to permit in silico interrogation of cell-specific and enriched mRNA translation.
TRAP compared to ISH, immunohistochemistry and BAC transgenesis for assessment of gene expression
The TRAP methodology is complementary to other methods of examining gene expression and protein translation in the CNS, though it has several distinct advantages. ISH and immunohistochemistry both require the laborious development and optimization of gene specific reagents, and depending on the size, location, expression level and subcellular localization of the target, may not provide sufficient information to unambiguously identify the cell type labeled. BAC transgenesis with an eGFP transgene provides comprehensive information about morphology and projections of the labeled cells, as well as a living reagent for further study (3), but requires a substantial time investment. Of the four methods, only TRAP can provide, in a single experiment, interrogation of the entire translated genome. TRAP data is quantitative, with the highest potential sensitivity and dynamic range. However, for the examination of a single gene across the entire CNS, ISH, immunohistochemistry, or BAC transgenesis will remain the methods of choice.
CONCLUSIONS
A variety of different tools have been generated for the analysis of microarray data (13,22–24). For those interested in developing or applying other array analysis methods to TRAP data, it is advisable to first test those methods on our most robust datasets, such as the Purkinje cell data, where a variety of positive and negative control genes can be used as standards (Supplementary Table S1), and the cells can be more easily identified by ISH. Furthermore, for most experiments, there are two important considerations for data analysis: first, quantile normalization should only be applied to that which would be expected to have similar mRNA distributions (from the same cell type or region, see Supplementary Figure S3, and ‘Supplementary Materials and Methods’ section); second, comparisons of IPvTotal data can be used to remove non-specific background prior to IPvIP comparisons, regardless of the source of this background (Supplementary Figures S2, 5, 9 and ‘Supplementary Materials and Methods’ section). Ongoing improvements in the molecular methodology are likely to remove most of the non-specific background deriving from interaction of purification reagents with untagged ribosomes seen in this first survey, making in many cases the filtering steps for calculating SI unnecessary in the future, though this filtering approach may still be applicable for dealing with low level expression of the transgene in cell types of secondary interest to the study. To aide in this normalization, lists of recommended negative control probesets are included in Supplementary Table S2 (although any genes known not to be expressed in the cell type of interest may be used). Standard statistical methods (22–26) remain essential for detecting more subtle differences, such as the changes within a single cell type following exposure of the animal to a drug (2).
Given the improved sensitivity and anatomic specificity obtained using TRAP and related methodologies, we anticipate wide application of these methodologies for gene expression studies in the mouse nervous system. The methods outlined here provide analytical tools for those researchers employing these methodologies, as a well as those interested in mining published TRAP datasets for cell-specific and enriched mRNAs. Continued experimental and analytical developments will enhance the value of the methods and data provided here. Nonetheless this methodology provides a systematic approach to the expressed genes that determine the unique properties of specific neural cell types, and to identify candidate genes to serve as markers and pharmacological targets.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute; Adelson Program in Neural Rehabilitation and Repair; the Simons Foundation; Conte Center (NIH/NIMH 5P50MH074866 P2); Croll Charitable Trust. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank P. Greengard, J. P. Doyle, J. C. Earnhart, S. Gayawali, M. Heiman, S. Kriaucionis and members of the Geschwind and Heintz Laboratories for discussions and advice, and R. Shah for blinded scoring of ISH data. We would also like to thank The Rockefeller University Bioimaging Facility, and Genomics Resource Center.
REFERENCES
- 1.Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat. Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- 4.Sandberg R, Yasuda R, Pankratz DG, Carter TA, Del Rio JA, Wodicka L, Mayford M, Lockhart DJ, Barlow C. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc. Natl Acad. Sci. USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geschwind DH. Mice, microarrays, and the genetic diversity of the brain. Proc. Natl Acad. Sci. USA. 2000;97:10676–10678. doi: 10.1073/pnas.97.20.10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 8.Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006;9:99–107. doi: 10.1038/nn1618. [DOI] [PubMed] [Google Scholar]
- 10.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 11.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 12.Magdaleno S, Jensen P, Brumwell CL, Seal A, Lehman K, Asbury A, Cheung T, Cornelius T, Batten DM, Eden C, et al. BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006;4:e86. doi: 10.1371/journal.pbio.0040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 14.Eccles JC, Itåo M, Szentâagothai J. The cerebellum as a neuronal machine. Berlin, New York etc: Springer; 1967. [Google Scholar]
- 15.Jensen LJ, Saric J, Bork P. Literature mining for the biologist: from information retrieval to biological discovery. Nat. Rev. Genet. 2006;7:119–129. doi: 10.1038/nrg1768. [DOI] [PubMed] [Google Scholar]
- 16.Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 17.Mugnaini E, Floris A. The unipolar brush cell: a neglected neuron of the mammalian cerebellar cortex. J. Comp. Neurol. 1994;339:174–180. doi: 10.1002/cne.903390203. [DOI] [PubMed] [Google Scholar]
- 18.Nunzi MG, Shigemoto R, Mugnaini E. Differential expression of calretinin and metabotropic glutamate receptor mGluR1alpha defines subsets of unipolar brush cells in mouse cerebellum. J. Comp. Neurol. 2002;451:189–199. doi: 10.1002/cne.10344. [DOI] [PubMed] [Google Scholar]
- 19.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2007;35:D26–D31. doi: 10.1093/nar/gkl993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wandall HH, Hassan H, Mirgorodskaya E, Kristensen AK, Roepstorff P, Bennett EP, Nielsen PA, Hollingsworth MA, Burchell J, Taylor-Papadimitriou J, et al. Substrate specificities of three members of the human UDP-N-acetyl-alpha-D-galactosamine:Polypeptide N-acetylgalactosaminyltransferase family, GalNAc-T1, -T2, and -T3. J. Biol. Chem. 1997;272:23503–23514. doi: 10.1074/jbc.272.38.23503. [DOI] [PubMed] [Google Scholar]
- 22.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2:RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatti C, Karsten SL, Geschwind DH. Thresholding rules for recovering a sparse signal from microarray experiments. Math. Biosci. 2002;176:17–34. doi: 10.1016/s0025-5564(01)00102-x. [DOI] [PubMed] [Google Scholar]
- 25.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 26.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.