

Abstract
The paramyxoviruses define a diverse group of enveloped RNA viruses that includes a number of important human and animal pathogens. Examples include human respiratory syncytial virus and the human parainfluenza viruses, which cause respiratory illnesses in young children and the elderly; measles and mumps viruses, which have caused recent resurgences of disease in developed countries; the zoonotic Hendra and Nipah viruses, which have caused several outbreaks of fatal disease in Australia and Asia; and Newcastle disease virus, which infects chickens and other avian species. Like other enveloped viruses, paramyxoviruses form particles that assemble and bud from cellular membranes, allowing the transmission of infections to new cells and hosts. Here, we review recent advances that have improved our understanding of events involved in paramyxovirus particle formation. Contributions of viral matrix proteins, glycoproteins, nucleocapsid proteins, and accessory proteins to particle formation are discussed, as well as the importance of host factor recruitment for efficient virus budding. Trafficking of viral structural components within infected cells is described, together with mechanisms that allow for the selection of specific sites on cellular membranes for the coalescence of viral proteins in preparation of bud formation and virion release.
Keywords: virus budding, virus assembly, matrix protein, paramyxovirus, virus-like particle, polarized budding, lipid raft membranes, viral trafficking
1. Introduction
The paramyxoviruses define a large group of enveloped RNA viruses, some of which cause significant human and animal diseases. Examples include human respiratory syncytial virus (HRSV), human parainfluenza virus types 1-4 (hPIV 1-4), measles virus, mumps virus, Nipah virus, Hendra virus, and Newcastle disease virus (NDV). HRSV and hPIV types 1–3 are major contributors to respiratory infections in young children and the elderly (Counihan et al., 2001; Falsey, 1998; Hall et al., 2009; Heilman, 1990; Welliver, 2003). HRSV infection is the leading cause of severe pediatric respiratory tract disease, causing an estimated 64 million cases and 160,000 annual deaths globally (Hall et al., 2009; Welliver, 2003; WHO, 2009a; Wright et al., 2005; Zhang et al., 2002). There are currently no effective vaccines to prevent HRSV or hPIV infections. Although both measles virus and mumps virus infections are vaccine-preventable, these infections remain a health burden in developing countries, and several significant outbreaks attributed to low vaccination rates have occurred recently in the United Kingdom, Canada, and the United States (CDC, 2006a; CDC, 2006b; CDC, 2008; Dayan et al., 2008; Hviid et al., 2008; Peltola et al., 2007). Nipah virus and Hendra virus (Henipaviruses) cause deadly infections in humans, with severe and widespread vasculitis and encephalitis resulting in a mortality rate of about 40% (Bishop and Broder, 2008). The viruses are zoonotic, and the natural hosts are fruit bats, such as flying foxes. The viruses spread to humans mainly through intermediary hosts: horses in the case of Hendra virus, and pigs in the case of Nipah virus (Eaton et al., 2006). Twelve recognized outbreaks of Nipah virus infection in South Asia have occurred since its identification in 1999 (Epstein et al., 2006; Gurley et al., 2007; WHO, 2009) and a total of thirteen known outbreaks of Hendra virus in Australia have occurred, the first of which were recognized in 1994 (Bishop and Broder, 2008; ProMED-mail, 2009). There are currently no effective treatments or vaccines approved for Henipavirus infections, and outbreaks are likely to continue so long as humans and their domesticated animals encroach into geographic locations occupied by flying foxes. Such outbreaks have potential agricultural significance as well; the initial Malaysian epidemic caused by Nipah virus was contained only after 1.1 million pigs were culled (Mohd Nor et al., 2000). Other paramyxoviruses of agricultural importance include NDV, which causes a highly contagious respiratory and neurological disease in many avian species, including chickens (Alexander, 2009) and rinderpest virus, which causes disease in cattle (Roeder and Taylor, 2002). Additional viruses that have been widely used as laboratory models for the study of paramyxovirus entry and exit include parainfluenza virus 5 (PIV5, formerly SV5) and Sendai virus.
Virus particles are containers built within infected cells that are meant to transmit infection from cell-to-cell and from host-to-host. Enveloped virus particles form by budding from cellular membranes. Buds emerge from selected sites on the membranes where viral proteins and genomes have assembled together, then pinch off to achieve particle release (Fig. 1). The resulting virions have outer surfaces that consist of host-derived membrane, enriched with viral integral membrane glycoproteins (Fig. 2). The paramyxoviruses encode two glycoproteins: a fusion (F) protein, and an attachment [HN (hemagglutinin-neuraminidase), H (hemagglutinin), or G (glyco-)] protein. These proteins are packed very densely into the viral envelopes, forming “spike” layers that are visible by electron microscopy (Fig. 2B). Enclosed within paramyxovirus particles are the RNA genomes, bound with nucleocapsid (N or NP) proteins to form helical structures called ribonucleoproteins (RNPs). Directly underlying the viral membranes are the viral matrix (M) proteins, which bridge the viral glycoproteins and RNPs, thereby organizing virus assembly. Paramyxoviruses form particles that are mainly spherical, but sometimes filamentous, with considerable variation in size and shape. Particles typically range in size from 150 to 300 nm in diameter, but can reach diameters of greater than 1 μm in some cases (Goldsmith et al., 2003).
Figure 1. Budding of paramyxovirus particles.

(A) Schematic illustration of paramyxovirus structural components that have assembled together on a cellular membrane, in preparation for budding. (B) Thin section of a Sendai virus-infected MDCK cell, visualized by electron microscopy. Arrows indicate individual sites from which virus budding has initiated on the apical cell surface. The nucleocapsid is observed aligning below the areas of curving membrane. (Adapted from Rodriguez-Boulan and Sabatini, 1978, with the author’s permission).
Figure 2. Paramyxovirus virions.

(A) Schematic representation of a spherical paramyxovirus virion illustrating M protein lining the inner surface of the lipid envelope, the viral glycoproteins decorating the virion surface, and the helical RNP enclosed within. (B) A mumps virion purified by sucrose gradient centrifugation and imaged by transmission electron microscopy with negative staining. Glycoproteins are visible as a spike layer embedded in the envelope. (Adapted from Li et al., 2009, with the publisher’s permission).
Paramyxovirus infections (see Fig. 3) are initiated when virus particles bind to receptor molecules on the surfaces of target cells. For some paramyxoviruses, such as Sendai virus and mumps virus, attachment is mediated by HN proteins that bind to sialic acid receptors. These HN proteins also possess sialidase activities, which function later in the virus lifecycle to facilitate the separation of virions from infected cells and to prevent virion aggregation. Other paramyxovirus attachment proteins mediate binding to protein receptors. These include the H protein of measles virus and the G proteins of the Henipaviruses. Following virion attachment, viral F proteins are triggered, resulting in the fusion of virion membranes with target cell membranes via a process that is driven by the refolding of F proteins from initial metastable states into more stable hairpin structures (reviewed in Lamb and Parks, 2006; Russell and Luque, 2006). Completion of this process allows virion contents, including RNPs, to enter the cytoplasms of target cells. Viral transcription then occurs, with negative-sense viral genomic RNA within RNPs serving as templates for the production of mRNAs by viral RNA-dependent RNA polymerase complexes, composed of viral phospho- (P) protein and large (L) protein subunits. Transcription follows the “stop-start” model first described for vesicular stomatitis virus (VSV), the result of which is a gradient of transcription in which genes near the 3′ end of the genome are transcribed more abundantly than genes near the 5′ end (reviewed in Whelan et al., 2004). Later in the infectious cycle, the viral polymerase enters a replication mode in which viral genomes are not transcribed, but rather are replicated in a two-step process that involves first the production of positive-sense antigenomes from genomic templates, and subsequently the production of negative-sense genomes from antigenomic templates (reviewed in Lamb and Parks, 2006). Newly-synthesized viral proteins and RNPs assemble together at infected cell plasma membranes in preparation for particle budding, which completes the cycle. Additional functions, including the disabling of host innate antiviral responses, are carried out by viral V proteins and other accessory proteins (reviewed in Horvath, 2004).
Figure 3. Schematic illustration of a paramyxovirus life cycle.

For details, refer to the text. Viral transcription and genome replication occur in the cytoplasm. Following their synthesis, viral structural proteins and RNPs assemble together at the infected cell plasma membrane for budding. Additional processes mediated by the viral accessory proteins (shown surrounded by brackets) are not illustrated.
Here, recent progress in understanding paramyxovirus particle formation is reviewed. Contributions of various viral components, as well as host proteins, to virus assembly and release are discussed, as well as parameters that lead to successful trafficking of viral components to assembly sites from which virions bud.
2. Central role of M proteins in paramyxovirus particle formation
Infectious paramyxovirus particles can be formed only after all the structural components of the viruses, including viral glycoproteins and viral RNPs, have assembled together at selected sites on infected cell plasma membranes. Viral M proteins are the organizers of this assembly process. These highly abundant viral proteins bind directly to cellular membranes and occupy a central position that allows interaction both with viral RNP cores and also with viral glycoproteins via the cytoplasmic tails (Fig. 1A). Thus, M proteins are adapters that link together the structural components of virions, driving their assembly. Although structural studies of paramyxovirus M proteins have generally proved difficult, as these relatively hydrophobic proteins are prone to self-aggregation, these difficulties were recently overcome in the case of the HRSV M protein, allowing a determination of its atomic structure (Money et al., 2009). The HRSV M protein is composed of two domains, each consisting mainly of beta sheets. The domains are separated by a short, unstructured linker region. The overall structure is quite similar to that of Ebola virus matrix protein (Fig. 4A–B); (Dessen et al., 2000; Money et al., 2009). An extensive positively-charged surface extends across both domains as well as the linker region, and this likely directs electrostatic interactions between the M protein and the phospholipid head groups of membranes (Fig. 4C). This finding is generally consistent with previous biochemical studies, which implicated a combination of electrostatic and hydrophobic contributions to membrane binding by paramyxovirus M proteins (Caldwell and Lyles, 1986; Riedl et al., 2002; Stricker et al., 1994; Subhashri and Shaila, 2007), similar to membrane binding by Ebola virus VP40 (Jasenosky et al., 2001; Ruigrok et al., 2000) and by VSV M protein (Chong and Rose, 1993; Ye et al., 1994).
Figure 4. Atomic structure of HRSV M protein.
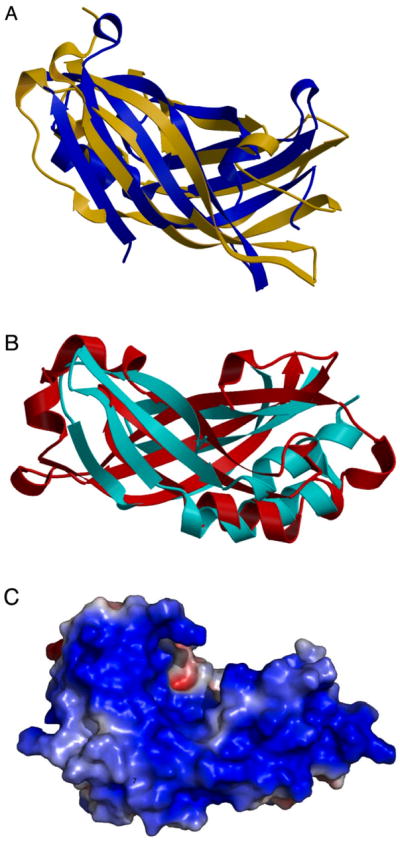
(A) Ribbon diagrams illustrating the similar beta-sheet arrangements of the HRSV M protein (blue) and Ebola virus VP40 (yellow) N-terminal domains. (B) Ribbon diagrams illustrating the similar beta-sheet arrangements of the HRSV M protein (red) and Ebola virus VP40 (cyan) C-terminal domains. (C) Space filling model of the HRSV M protein structure, with electrostatic surface potential depicted in colors ranging from red to blue. An extensive positively-charged (blue) surface is shown. (HRSV M protein PDB code 2VQP, Ebola virus VP40 PDB code 1ES6; figure adapted from Money et al., 2009, with the author’s permission).
Direct evidence for the critical role of M proteins in paramyxovirus assembly has been obtained through study of viruses harboring M protein mutations. Early studies relied on Sendai virus temperature-sensitive mutants in which M protein failed to accumulate to threshold levels at nonpermissive temperatures. This resulted in failure to produce virus particles (Kondo et al., 1993; Yoshida et al., 1979). Additional correlations between M protein defects and virus assembly impairments were made with altered measles virus isolates derived from patients with subacute sclerosing panencephalitis (SSPE). SSPE is a rare condition that results from persistent measles virus infection, causing fatal neurological disease years after initial acute infection with measles virus. SSPE strains of measles virus frequently encode hypermutated and unstable M proteins and are also found to exhibit severe defects in particle assembly (reviewed in Rima and Duprex, 2005). Unequivocal confirmation of the key roles played by paramyxovirus M proteins in particle assembly came with the advent of reverse genetics technology in the 1990s, allowing generation of negative-sense RNA viruses entirely from cloned cDNA (Conzelmann, 2004). Recombinant measles and recombinant Sendai viruses that completely lack M genes have been generated and both of these viruses exhibit near-complete defects in particle formation (Cathomen et al., 1998a; Inoue et al., 2003). Particle production defects have also been observed in recombinant measles virus in which wildtype M protein is replaced with hypermutated M protein derived from an SSPE strain (Patterson et al., 2001), recombinant measles virus in which a highly-conserved valine residue at position 101 has been changed to alanine, resulting in poor M protein stability (Runkler et al., 2007), and recombinant Sendai virus in which M protein accumulation can be prevented through the use of siRNA (Mottet-Osman et al., 2007). Taken together, these studies demonstrate convincingly that efficient paramyxovirus particle formation can occur only in the presence of a threshold level of functioning M protein.
For many paramyxoviruses, M proteins expressed in the absence of other viral proteins are sufficient for the release of virus-like particles (VLPs) from transfected cells. M proteins of hPIV1 (Coronel et al., 1999), Sendai virus (Sugahara et al., 2004; Takimoto et al., 2001), NDV (Pantua et al., 2006), Nipah virus (Ciancanelli and Basler, 2006; Patch et al., 2007), and measles virus (Pohl et al., 2007; Runkler et al., 2007) are all able to elicit efficient particle budding from transfected cells when expressed alone. For NDV, membrane deformation and vesicle budding have even been reconstituted in vitro using purified M protein and unilamellar vesicles (Shnyrova et al., 2007), demonstrating that all of the activities necessary for inducing curvature and fission of a membrane are contained within M protein. In some cases, M protein-directed VLP production from transfected cells becomes even more efficient when the M proteins are co-expressed with other viral components, such as glycoproteins, nucleocapsid proteins, and C proteins (Table 1; and discussed below). Some paramyxovirus M proteins lack the ability to direct efficient VLP production when expressed alone in cells. PIV5 and rinderpest virus M proteins expressed by themselves in transfected cells do not elicit detectable VLP production (Schmitt et al., 2002; Subhashri and Shaila, 2007), and in this respect are similar to the influenza A virus matrix protein (Chen et al., 2007). Mumps virus M protein expression by itself leads to particle release that is barely detectable (Li et al., 2009). For PIV5 and mumps virus, VLP production becomes highly efficient when the M proteins are expressed together with other viral proteins (Table 1). Hence, the requirements for efficient VLP production differ among paramyxoviruses. However, M protein appears generally to be the major requirement for paramyxovirus VLP production, even in the cases where its expression is insufficient, as VLPs cannot be formed in its absence (Li et al., 2009; Schmitt et al., 2002; Teng and Collins, 1998), whereas for influenza A virus the major requirement for VLP budding is the hemagglutinin (HA) glycoprotein (Chen et al., 2007).
Table 1.
Differing requirements for paramyxovirus VLP production
| Virus | Viral proteins needed for efficient VLP production | Notes | References |
|---|---|---|---|
| hPIV1 | M | Nucleocapsid-like structures are incorporated into the VLPs when NP protein is expressed | Coronel et al., 1999 |
| Sendai virus | M | F protein enhances VLP production, but HN protein does not; C protein enhances VLP production, likely through recruitment of the host protein Aip1/Alix | Takimoto et al., 2001; Sugahara et al., 2004 |
| NDV | M | Glycoproteins and NP protein can be incorporated into VLPs, but do not enhance M-driven VLP production; incorporation of glycoproteins into VLPs becomes efficient when both glycoproteins are expressed | Pantua et al., 2006 |
| PIV5 | M, NP, and F; or M, NP, and HN | M protein expressed alone does not result in VLP production; F and HN proteins are necessary but redundant for VLP production | Schmitt et al., 2002 |
| Mumps virus | M, NP, and F | M protein expressed alone results in very little VLP production; F protein strongly enhances VLP production, but HN protein does not | Li et al., 2009 |
| Measles virus | M | F protein does not enhance M- driven VLP production | Pohl et al., 2007; Runkler et al., 2007 |
| Nipah virus | M | Glycoproteins and N protein can be incorporated into VLPs when expressed, but do not enhance M-driven VLP production | Ciancanelli and Basler, 2006; Patch et al., 2007 |
3. Cooperation among viral proteins during particle production
3.1 Viral glycoproteins
Paramyxovirus particles are covered with spike layers consisting of the viral attachment and fusion glycoproteins (Fig. 2). The viral glycoproteins assemble together with M proteins in infected cells, clustering on plasma membranes at locations from which virus particles will bud (Fig. 1). The cytoplasmic tails of paramyxovirus glycoproteins interact with the M proteins to organize virus assembly. Evidence for these interactions has been obtained through a variety of experimental approaches. Fluorescence microscopy experiments have documented co-localization of paramyxovirus M proteins and glycoproteins in virus-infected cells. In the case of PIV5, M proteins and glycoproteins co-localize in clusters on infected cell plasma membranes. However, this clustering is lost in cells infected with mutant viruses in which the cytoplasmic tail of HN protein, or the cytoplasmic tails of both HN and F proteins, have been truncated (Schmitt et al., 1999; Schmitt et al., 2005; Waning et al., 2002). In cells infected with the Edmonston vaccine strain of measles virus, M protein co-localizes with the viral F and H glycoproteins, but no co-localization could be observed between M protein and cytoplasmic tail-deleted viral glycoproteins (Moll et al., 2002). Co-localization studies have also been performed in cells transfected to produce the HRSV M and G proteins. Co-localization between the wildtype proteins was observed, but co-localization was lost upon removal of the first six amino acid residues of the G protein cytoplasmic tail (Ghildyal et al., 2005b). Co-immunoprecipitation studies using purified VLPs have been conducted to explore interactions involving the glycoproteins of NDV. Specific interactions between the NDV HN and M proteins, and also between the F and NP proteins, were detected. No evidence for direct interaction between the F and M proteins was obtained, however (Pantua et al., 2006). Further experimental support for specific interactions between viral glycoproteins and M proteins has been obtained by measuring the association of M proteins with detergent-resistant raft membranes (discussed further in section 5.2). Sendai virus M protein binds to cellular membranes intrinsically, but it does not associate strongly with detergent-resistant raft membranes when expressed by itself. The Sendai virus HN and F glycoproteins, however, are intrinsically sorted to raft membranes. When the viral glycoproteins are co-expressed together with M protein, M protein becomes raft membrane-associated (Ali and Nayak, 2000). Similar results were obtained with HRSV M protein, which also binds membranes intrinsically, but does not associate with detergent-resistant raft membranes unless the HRSV F protein is co-expressed, suggesting that the F protein pulls M protein into detergent-resistant membrane microdomains (Henderson, 2002). Together, these observations support a model in which viral glycoproteins and viral M proteins assemble together at specific locations on cellular membranes through interactions involving the glycoprotein cytoplasmic tails.
Proper assembly of paramyxovirus glycoproteins together with M proteins in infected cells is necessary for selective incorporation of the glycoproteins into budding particles, and in many instances is also necessary for the budding process itself to become efficient. In the case of PIV5, the F and HN glycoproteins have redundant functions important for efficient virus budding. Recombinant virus in which the HN protein cytoplasmic tail is truncated buds particles poorly, and this defect is more pronounced in double mutant virus in which both HN and F protein cytoplasmic tails have simultaneously been truncated (Schmitt et al., 1999; Waning et al., 2002). These findings parallel earlier results obtained with influenza A virus, in which simultaneous truncation of the HA and neuraminidase (NA) protein cytoplasmic tails resulted in severe particle production and particle morphology defects, while single cytoplasmic tail truncations led to only minor defects (Jin et al., 1997). For Sendai virus, the HN glycoprotein appears to be dispensable for the production of virus particles (Markwell et al., 1985; Portner et al., 1974; Stricker and Roux, 1991), although particle morphology may be altered in the absence of HN protein (Hirayama et al., 2006). Truncation of the Sendai virus F protein cytoplasmic tail led to inefficient virus release (Fouillot-Coriou and Roux, 2000). Some alterations to the HN protein led to deficient incorporation of the protein into virions, with no detrimental effects on virus budding, while severe truncation of the HN protein cytoplasmic tail caused poor virus release (Fouillot-Coriou and Roux, 2000). The importance of glycoproteins for measles virus particle formation has been inferred from observations with assembly-defective SSPE strains, which frequently harbor F proteins with mutated cytoplasmic tails (Cattaneo et al., 1988; Schmid et al., 1992). Alterations to the F and H protein cytoplasmic tails in recombinant viruses led to reduced incorporation of the viral glycoproteins into virions, along with increased nonspecific incorporation of cellular proteins (Cathomen et al., 1998b). These alterations also led to rapid and extensive syncytia formation within infected cell monolayers (Cathomen et al., 1998b). Interestingly, assembly-defective recombinant PIV5 with truncated HN protein cytoplasmic tail also caused rapid and extensive syncytia formation (Schmitt et al., 1999). Vaccine strains of measles virus, including the Edmonston strain, harbor adaptations in their M proteins that confer enhanced virus assembly and release compared with wildtype measles virus isolates (Tahara et al., 2007). These adaptations enhance interaction with the H protein cytoplasmic tail judged by co-localization studies, and reduce syncytia formation (Tahara et al., 2007). Together, these findings suggest that assembly of viral M proteins together with viral glycoproteins may benefit paramyxovirus particle formation and release, but at the same time may impair F protein-mediated cell-cell fusion. This may allow measles virus to optimize its mode of spread (cell-cell fusion versus budding) by acquiring mutations in the M protein, thereby modulating the interaction with the H protein cytoplasmic tail (Tahara et al., 2007).
The roles of viral glycoproteins in paramyxovirus assembly have also been examined in the context of VLPs produced from transfected cells. Although M proteins are sufficient for VLP production in many cases, expression of glycoproteins together with M proteins generally leads to production of VLPs that incorporate the viral glycoproteins and resemble paramyxovirus virions morphologically (Ciancanelli and Basler, 2006; Pantua et al., 2006; Patch et al., 2007). Some paramyxovirus glycoproteins have intrinsic exocytosis activities and are released as particles even when expressed alone. These include the Sendai virus F protein (Sugahara et al., 2004; Takimoto et al., 2001), the measles virus F protein (Pohl et al., 2007), and the Nipah virus F and G proteins (Ciancanelli and Basler, 2006; Patch et al., 2007). Other paramyxovirus glycoproteins lack inherent exocytosis activities, including the Sendai virus HN protein (Sugahara et al., 2004; Takimoto et al., 2001), and the glycoproteins of PIV5 (Schmitt et al., 2002), NDV (Pantua et al., 2006), and mumps virus (Li et al., 2009). In some cases, viral glycoproteins and M proteins cooperate with one another, resulting in VLP release that is more efficient than that obtained when either protein is expressed alone. For example, production of Sendai VLPs, driven by M protein, is made more efficient upon co-expression of the viral F protein (Sugahara et al., 2004; Takimoto et al., 2001). The Sendai virus HN protein, on the other hand, lacks the ability to enhance VLP production (Sugahara et al., 2004; Takimoto et al., 2001). Viral glycoprotein expression is required for efficient production of mumps VLPs and PIV5-like particles. Mumps VLP production is most efficient upon co-expression of the viral M, NP, and F proteins (Li et al., 2009). Co-expression of the mumps virus M, NP, and HN proteins leads to VLP production that is considerably less efficient. Hence, similar to the Sendai virus glycoproteins, the mumps virus glycoproteins are not equal contributors to virus assembly, with F protein playing the more important role. The PIV5 glycoproteins, in contrast, appear to have more equal, and seemingly redundant, functions during VLP production. Here, similar and highly efficient VLP production can be achieved either through co-expression of the M, NP, and F proteins, the M, NP, and HN proteins, or the M, NP, F, and HN proteins (Schmitt et al., 2002). Neither of the PIV5 glycoproteins can function for VLP production when their cytoplasmic tails have been truncated, in agreement with results obtained using recombinant viruses (Schmitt et al., 1999; Waning et al., 2002). These results suggest that some paramyxovirus glycoproteins, when present at virus assembly sites, can act to increase the efficiency of particle production. However, other viral glycoproteins, including those of NDV, Nipah virus, and measles virus, do not enhance the efficiency of M protein-driven VLP release from transfected cells (Ciancanelli and Basler, 2006; Pantua et al., 2006; Patch et al., 2007; Pohl et al., 2007). Hence, while assembly of paramyxovirus glycoproteins together with viral M proteins may have universal importance for efficient incorporation of the glycoproteins into budding particles, this assembly affects the efficiency of particle release for only a subset of paramyxoviruses (Table 1).
3.2 Viral nucleocapsid proteins
Paramyxovirus nucleocapsid proteins bind and encapsidate genomic RNA, forming helical RNP complexes in which the RNA is tightly packaged and resistant to exogenously added RNase. The structure of HRSV N protein bound to RNA was recently determined by x-ray diffraction and cryoelectron microscopy (Tawar et al., 2009). RNA is wrapped around a decameric ring of N protein, fitting along a basic surface groove. This arrangement is consistent with a model in which the viral RNA-dependent RNA polymerase is able to recognize its template even without disassembly of the RNP. From the analogy of tobacco mosaic virus, it is assumed that RNA encapsidation starts from a nucleocapsid protein-binding nucleation site. In virus-infected cells, nascent genomic RNA is efficiently encapsidated coupled with RNA synthesis, while viral mRNAs are not encapsidated. It is therefore assumed that the promoter sequences (the leader and trailer sequences) function as nucleation sites (Blumberg et al., 1983; Lamb and Parks, 2006). On the other hand, it is known that paramyxovirus nucleocapsid proteins expressed in mammalian cells bind intracellular host RNAs non-specifically and form nucleocapsid-like structures (Buchholz et al., 1993; Coronel et al., 1999; Errington and Emmerson, 1997; Juozapaitis et al., 2005; Schmitt et al., 2002; Spehner et al., 1991; Sugahara et al., 2004), indicating that RNA encapsidation may occur in some circumstances even without the aid of a nucleation site.
Once formed, RNPs must be incorporated into budding virus particles. Although paramyxovirus genomes are nonsegmented, it is not necessarily the case that all budding particles receive only a single copy of genomic RNA. Some virions are released that contain multiple genome copies (Loney et al., 2009; Rager et al., 2002). Selective incorporation of homologous genomes into virions has been demonstrated in experiments in which cells were transfected to produce the hPIV1 NP protein and also were infected with Sendai virus. Nucleocapsid structures containing hPIV1 NP protein were produced in the cells, but these were largely excluded from Sendai virions (Coronel et al., 2001). Selectivity of genome incorporation by genome length has also been reported; short defective-interfering genomes of Sendai virus were incorporated into budding particles much less efficiently than full-length genomes (Mottet and Roux, 1989). Selective genome incorporation based on the polarity of the viral RNA also occurs, with (−)-sense genomes incorporated more efficiently into budding particles than (+)-sense antigenomes. However, for paramyxoviruses such as Sendai virus, this packaging bias in favor of genomes is very weak, compared to the more stringent preferential packaging of (−)-sense genomes into rhabdovirus virions (Kolakofsky and Bruschi, 1975; Mottet and Roux, 1989). The Sendai virus C protein is thought to regulate viral RNA synthesis from the promoters, and excessive amounts of (+)-sense antigenomes are synthesized in partial C-knockout virus-infected cells. Virions released from the infected cells contain (−) and (+)-sense RNA genomes in a ratio similar to that present inside the infected cells, resulting in an overabundance of noninfectious virions containing (+)-sense antigenomic viral RNA (Irie et al., 2008a).
Incorporation of genomes into budding virions is likely driven by interactions between viral matrix proteins and nucleocapsids at virus assembly sites. Biochemical evidence supports a direct interaction between paramyxovirus matrix and nucleocapsid proteins (Iwasaki et al., 2009; Markwell and Fox, 1980; Yoshida et al., 1976). For measles virus, yeast two-hybrid binding assays have mapped this interaction to the C-terminal portion of the N protein (Iwasaki et al., 2009). In transfected cells, expression of measles virus M protein shifts the localization of wt N protein (but not N protein harboring a C-terminal mutation) so that a portion is plasma membrane associated and co-localized with M protein (Iwasaki et al., 2009). Similarly, RNPs are observed at the plasma membrane in cells infected with standard measles virus, but not in cells infected with recombinant measles virus harboring an unstable M protein (Runkler et al., 2007). Together, these studies support a model in which M protein is responsible for targeting RNPs to virus assembly sites on plasma membranes.
Incorporation of nucleocapsid-like structures into budding VLPs has been documented in transfected cells. For example, co-expression of the hPIV1 M and NP proteins in cells led to production of VLPs that enclose nucleocapsid-like structures (Coronel et al., 1999). Similar results were obtained with Sendai VLPs (Sugahara et al., 2004) and Nipah VLPs (Patch et al., 2007). Nucleocapsid-like structures can also be incorporated into NDV-like particles, however in this case nucleocapsid incorporation was poor unless viral glycoproteins were expressed in addition to the M and NP proteins (Pantua et al., 2006). For PIV5 and mumps virus, NP proteins not only incorporate into budding VLPs, they actually increase the efficiency of VLP production (Li et al., 2009; Schmitt et al., 2002). It is possible that for these viruses a requirement for NP protein during assembly could be beneficial through minimizing the release of noninfectious particles that lack viral genomes.
3.3 Viral C proteins
Some paramyxoviruses express accessory C proteins from their P genes via overlapping reading frames. For example, Sendai virus encodes a nested set of four C proteins from its P gene through use of alternative start codons that are recognized by translation machinery either by leaky scanning in the case of C and C′, or by a type of ribosomal shunting in the case of Y1 and Y2 (Lamb and Parks, 2006). C proteins appear to be multifunctional, with activities that regulate viral RNA synthesis (Cadd et al., 1996; Curran et al., 1992) and that counteract innate immune responses of host cells (Garcin et al., 1999; Gotoh et al., 1999). The longer C proteins (C and C′) contain N-terminal membrane-targeting sequences, while the shorter C proteins (Y1 and Y2) lack these sequences (Marq et al., 2007). A role for C proteins in Sendai virus budding was first suggested based on experiments with 4C(−) virus, which lacks the ability to produce any of the four C proteins (Kurotani et al., 1998). This virus replicates to titers that are about two logs less than wildtype Sendai virus and fails to assemble and release virus particles effectively, despite efficient synthesis of viral mRNAs, proteins, and genomic RNA (Hasan et al., 2000). Defective assembly of 4C(−) virus was observed in multiple cell types, including Vero cells that fail to produce IFN-β, suggesting a virus assembly function of C protein that is separate from its function in counteracting innate immune defenses. Separation of C protein assembly and immune-evasion functions is also supported by results obtained with altered Sendai virus Cm*, which harbors a C protein point mutation and fails to block the Jak/Stat pathway, yet exhibits no virus assembly defect (Kato et al., 2007). Although the above findings support a role for C protein in Sendai virus assembly, such a role could not be confirmed by Gosselin-Grenet et al., who used a somewhat different experimental system. Here, a 4C knockout virus was employed in which C protein fused to GFP was re-introduced as a separate transcriptional unit in the virus genome. GFP/C protein expression was ablated using siRNA, with no apparent effect on virus particle production (Gosselin-Grenet et al., 2007). This approach has an advantage in that it avoids passaging of a seriously debilitated virus, in which selective pressure could potentially cause unintended variants of the virus to emerge. The approach was also limited, however, in that siRNA depletion was incomplete (about 30% of normal GFP/C protein levels remained), raising the possibility that C protein could initially have been present in excess of what is needed for virus assembly. Further investigations on the importance of C proteins in Sendai virus assembly have been carried out in the context of VLPs produced from transfected cells. Expression of Sendai virus C protein together with M protein results in VLP production that is more efficient (by 2–3 fold) than that obtained when M protein is expressed alone (Sugahara et al., 2004). C and C′ proteins are each capable of enhancing Sendai VLP production, while the Y1 and Y2 proteins that lack membrane targeting sequences are unable to enhance VLP production (Irie et al., 2008b; Sakaguchi et al., 2005). A model has been proposed in which C proteins function for virus budding by interacting with the host Class E protein Aip1/Alix, recruiting it to Sendai virus assembly sites on plasma membranes (Irie et al., 2008b; Sakaguchi et al., 2005). Evidence in support of such a role for Aip1/Alix, and potential roles for other host proteins in paramyxovirus budding, are discussed below.
4. Role of host proteins in paramyxovirus budding
4.1 Class E proteins and ubiquitin
Enveloped viruses typically do not encode all of the machinery that is necessary to bud particles. Instead, host machinery is manipulated to allow efficient virus exit. Retroviruses in many cases employ protein-protein interaction sequences (late domains) within their Gag polypeptides to recruit host factors to virus assembly sites (reviewed in Bieniasz, 2006; Calistri et al., 2009). Absence of these sequences in some instances leads to defects in the very late stages of virus release, hence the term late domain. Late budding defects are characterized by accumulation of budding structures tethered to cellular membranes that have morphology consistent with virions, but cannot be released by membrane fission (reviewed in Demirov and Freed, 2004). Several distinct types of viral late domain sequences have been characterized from retroviral Gag proteins, including P(T/S)AP, PPxY, and YP(x)nL. Matrix proteins of some negative-strand RNA viruses such as VSV (Craven et al., 1999; Harty et al., 1999; Jayakar et al., 2000), rabies virus (Wirblich et al., 2008), Ebola virus (Harty et al., 2000; Licata et al., 2003; Martin-Serrano et al., 2001), LCMV (Perez et al., 2003), and Lassa fever virus (Perez et al., 2003) contain these same late domain sequences, suggesting that the overall strategy of recruiting host machinery via late domains is conserved even among distantly-related viruses. Paramyxoviruses, however, generally lack these well-characterized late domain sequences within their matrix proteins, indicating that the logistics of host factor recruitment must be different for these viruses.
Cellular factors recruited by late domains in many cases are Class E proteins that are part of the vacuolar protein sorting (Vps) pathway of the cell. These proteins form endosomal sorting complexes required for transport (ESCRTs) that allow for multivesicular body formation and the downregulation of cellular membrane proteins (reviewed in Stuffers et al., 2009). ESCRT complexes are dismantled and recycled with the aid of Vps4 AAA-ATPases, and disruption of the Vps sorting pathway through expression of dominant-negative (DN) Vps4 protein mutants blocks the budding of many retroviruses (reviewed in Bieniasz, 2006). Similar findings have been obtained with some paramyxoviruses. PIV5 budding (Schmitt et al., 2002) and mumps VLP budding (Li et al., 2009) were both inhibited upon expression of DN Vps4 proteins, suggesting that these viruses rely on ESCRT machinery during virus exit, even though they lack classical late domains that would mediate ESCRT recruitment. DN Vps4 protein expression also inhibits the budding of other negative-strand RNA viruses including Ebola virus (Licata et al., 2003), and Lassa fever virus (Urata et al., 2006), although in these cases classical late domain sequences are contained within the viral matrix proteins. Although it is clear that a wide range of enveloped viruses employ host ESCRT machinery during budding, it has recently become apparent that some enveloped viruses do not. Among paramyxoviruses, budding of HRSV (Utley et al., 2008) and budding of Nipah VLPs (Patch et al., 2008) occur efficiently even in the presence of DN Vps4 protein expression. These results parallel earlier observations of ESCRT-independent budding with VSV (Irie et al., 2004) and influenza virus (Chen et al., 2007), raising the possibility that some viruses employ host pathways during budding that are ESCRT-independent, or perhaps have the ability to bud even without the aid of host machinery (reviewed in Chen and Lamb, 2008).
Potential links between Sendai virus budding and host ESCRT machinery have been examined in some detail. The Sendai virus C protein, a nonstructural accessory protein of the virus, binds to Aip1/Alix, the same protein that is recruited by retroviral YP(x)nL late domains. However, Sendai virus C protein lacks YP(x)nL and instead binds to Aip1/Alix via a domain within its C-terminal region (Sakaguchi et al., 2005). Expression of C protein together with Aip1/Alix in transfected cells re-localized a portion of Aip1/Alix to the plasma membrane together with C protein (Irie et al., 2008b). Sendai VLP production, driven by M protein, was enhanced when M protein was expressed together with C protein (Sugahara et al., 2004). Mutant C proteins that fail to interact with Aip1/Alix (Sakaguchi et al., 2005) or that fail to bind membranes (Irie et al., 2008b) could not enhance VLP production. Independently of its interaction with C protein, Aip1/Alix has been shown to interact with the Sendai virus M protein (Irie et al., 2007). This interaction is mediated by a YLDL motif within M protein. Mutations in the YLDL motif or depletion of Aip1/Alix with siRNA inhibited the production of Sendai VLPs (Irie et al., 2007). These observations support a model in which Aip1/Alix can be recruited to sites of Sendai virus assembly on plasma membranes either through C protein interaction or through M protein interaction, resulting in enhanced VLP production. It is unclear at present why multiple mechanisms for Aip1/Alix recruitment seem to be necessary for optimal production of Sendai VLPs, though a similar potential redundancy has been reported for hPIV2, in which the V and NP proteins were found to bind Aip1/Alix independently (Nishio et al., 2007). Questions also remain concerning the importance of Aip1/Alix recruitment in the course of an actual Sendai virus infection, as conflicting results have been obtained when examining the effect of Aip1/Alix depletion on Sendai virus budding (Gosselin-Grenet et al., 2007; Sakaguchi et al., 2005). These reports also conflict regarding the effect of DN Vps4 protein expression on Sendai virus budding. Sakaguchi et al. (2005) found that transfection of cells with DN Vps4 protein reduced budding of virions from cells transfected with infectious Sendai virus nucleocapsids, while Gosselin-Grenet et al. (2007) observed no inhibition in the release of virions from cells transfected to express DN Vps4 protein and infected with Sendai virus, even though PIV5 virion release was substantially inhibited in parallel experiments. When examining VLP production, DN Vps4 protein expression was found to only affect the accelerated release of VLPs that occurs in the presence of the viral C protein, and not M-alone VLP production (Irie et al., 2008b), consistent with the possibility that C protein enhances VLP budding through recruitment of ESCRT machinery.
An interaction between the PIV5 M protein and host protein angiomotin-like 1 (AmotL1), was characterized recently using yeast two-hybrid as well as co-immunoprecipitation assays (Pei et al., 2009). AmotL1 harbors two PPxY motifs, and recruitment of AmotL1 to virus assembly sites could in theory allow indirect recruitment of the same WW domain-containing host proteins that are directly recruited by viruses that employ PPxY late domains. PIV5 budding was inhibited by siRNA-mediated depletion of AmotL1 from cells, and overexpression of M-binding AmotL1-derived polypeptides blocked the production of PIV5-like particles (Pei et al., 2009). AmotL1 localizes to tight junctions and apical membranes of epithelial cells (Sugihara-Mizuno et al., 2007), and it has been suggested that this protein or other motin proteins could be involved in assembly of PIV5 on apical membranes (Pei et al., 2009).
Ubiquitin is thought to play a role in the budding of some enveloped viruses, likely stemming from its relationship with ESCRT machinery (reviewed in Martin-Serrano, 2007; Morita and Sundquist, 2004). Monoubiquitination or multiple monoubiquitination of cellular membrane proteins is a sorting signal for endocytosis and likely allows recognition by ESCRT proteins, several of which harbor well-characterized ubiquitin-binding domains (Hicke and Dunn, 2003; Shields et al., 2009). Several lines of evidence support a role for ubiquitin in the budding of certain retroviruses, including HIV-1. For example, ubiquitin has been detected in retrovirus particles (Ott et al., 2000; Putterman et al., 1990), multiple monoubiquitination of retroviral Gag proteins has been observed (Gottwein and Krausslich, 2005), and budding of some retroviruses is inhibited upon treatment of cells with proteasome inhibitors, which deplete cellular pools of free ubiquitin (Patnaik et al., 2000; Schubert et al., 2000). Ebola VP40 and VSV M protein can act as substrates for ubiquitin attachment in vitro, providing a link between ubiquitin and negative-strand RNA virus budding (Harty et al., 2000; Harty et al., 2001). In addition, VLP production driven by VSV M protein is proteasome inhibitor sensitive (Harty et al., 2001). Nedd4 mediated ubiquitination of Ebola VP40, detected in cells transfected to express VP40 and hemagglutinin-tagged ubiquitin (HA-Ub), is inhibited by expression of ISG15, a ubiquitin-like protein whose expression is induced by interferon (Malakhova and Zhang, 2008; Okumura et al., 2008). These findings support the ideas that enveloped virus budding may involve the ubiquitin-proteasome pathway, and that negative-strand RNA virus matrix proteins may be substrates for ubiquitin attachment.
Some evidence in support of ubiquitin involvement in paramyxovirus budding has been obtained. Release of PIV5 virions and VLPs from 293T cells is significantly impaired upon proteasome inhibitor treatments, suggesting that the ubiquitin-proteasome system may play an important role in some aspect of PIV5 assembly and/or budding (Schmitt et al., 2005). Potential ubiquitination of measles virus M protein has been observed in cells transfected to express measles virus M protein together with HA-Ub (Pohl et al., 2007). On the other hand, proteasome inhibitor treatments failed to inhibit the budding of HRSV from Hep-2 cells or the budding of Nipah VLPs from 293T cells, suggesting that differences may exist among the paramyxoviruses in the degree of dependence on ubiquitin for budding (Patch et al., 2008; Utley et al., 2008). For Sendai virus, sensitivity to proteasome inhibitor treatment was found to be cell type-dependent. Budding from LLC-MK2 cells was highly sensitive to proteasome inhibitor treatment, budding from CV-1 cells was less sensitive, and budding from A549 cells was proteasome inhibitor resistant (Watanabe et al., 2005). This cell type-dependent behavior underscores a more general complexity inherent in the study of host factors important for virus replication, in that the relative abundances of these factors are likely to differ among cell types, perhaps reinforcing the importance of studying these virus-host interactions, where possible, in cell types which most resemble those found in the natural hosts of infection. Overall, the observations described here are consistent with the involvement of ubiquitin in the assembly and release of a subset of paramyxoviruses, though an understanding of the specific role of ubiquitin will necessitate further examination.
4.2 M protein sequences that may function to allow host factor recruitment
Although paramyxovirus M proteins generally lack classical late domain sequences, a number of alternative sequences have been identified within M proteins that are important for budding activity and that may function to recruit host factors, similar to retroviral late domains. The sequence 20-FPIV-23 within PIV5 M protein was identified based on its ability to restore VLP budding function to PTAP-disrupted HIV-1 Gag protein (Schmitt et al., 2005). Mutation of either the phenylalanine or proline residues within this sequence drastically impaired the budding function of PIV5 M protein (Schmitt et al., 2005). Mumps virus M protein contains a similar sequence, 21-FPVI-24, and mumps VLP production was inhibited upon mutation of the proline residue within this sequence (Li et al., 2009). FPIV and FPIV-like sequences are presumed to function by mediating host factor recruitment to virus assembly sites, but the binding partner(s) for these sequences have not yet been identified.
Two sequences in NiV M protein that are important for VLP production have been identified: 62-YMYL-65 and 92-YPLGVG-97 (Ciancanelli and Basler, 2006; Patch et al., 2008). The YMYL sequence was able to functionally replace the PTAPPEY late domain of Ebola virus VP40, as appending this sequence to late domain-defective VP40 restored VLP budding function (Ciancanelli and Basler, 2006). Mutation of YMYL inhibited the VLP budding function of NiV M protein, and led to a re-localization of the protein to the cell nucleus (Ciancanelli and Basler, 2006). The sequence YPLGVG within NiV M protein contains residues that are well conserved among paramyxoviral M proteins (Patch et al., 2008). Mutation of this sequence disrupted M protein-driven VLP production, and deletion of the sequence led to nuclear localization of NiV M protein. YPLGVG was unable to rescue VLP budding function of late domain-defective Ebola virus VP40; however, the appearance of filamentous projections similar to those observed in cells expressing wt VP40 or wt NiV M protein was restored (Patch et al., 2008). Cellular binding partners for YMYL and YPLGVG sequences have not yet been identified. Interestingly, sequences resembling YPLGVG are found in the Ebola virus and Marburg virus VP40 proteins, and mutation of these sequences disrupted VP40 stability, intracellular localization, and VLP production (Liu et al., 2009). Thus, YPLGVG-like sequences appear to be important for the functions of divergent negative-strand RNA virus matrix proteins.
Sendai virus M protein possesses a sequence, 49-YLDL-52, which binds Alix/Aip1. Mutation of YLDL caused a reduction in VLP release that could not be rescued by the inclusion of other Sendai virus proteins (Irie et al., 2007). The YLDL sequence could not be replaced by P(T/S)AP, PPxY, or YP(x)nL late domains for the budding of Sendai VLPs. YLDL-Aip1/Alix interaction appears to be distinct from YP(x)nL-Aip1/Alix interaction, as the Sendai virus YLDL sequence interacts with Aip1/Alix residues 1–211 while YP(x)nL interacts with residues 409–715 (Irie et al., 2007).
5. Virus assembly sites
5.1 Paramyxovirus assembly on apical membranes of polarized cells
Polarized epithelial cells that line body surfaces possess apical and basolateral sides, with the apical sides facing outward and the basolateral sides facing inward towards the underlying tissue. Several paramyxoviruses target epithelial cells of the respiratory tract for infection and assemble and bud from the apical surfaces of these cells. HRSV, Sendai virus, PIV5, hPIV3, and measles virus all bud preferentially from the apical surfaces of polarized cells (Blau and Compans, 1995; Bose et al., 2001; Roberts et al., 1995; Rodriguez-Boulan and Sabatani, 1978). Other respiratory viruses, including influenza A virus, also bud apically (Rodriguez-Boulan and Sabatani, 1978), while VSV and Marburg virus bud basolaterally (Sänger et al., 2001). The direction of virus budding from polarized cells can influence whether a paramyxoviral infection remains localized to the respiratory epithelia, facilitating transmission between hosts, or disseminates systemically, which in some cases favors establishment of persistent infections (reviewed in Schmitt and Lamb, 2004; Takimoto and Portner, 2004).
In many cases, viral glycoproteins have intrinsic sorting signals that target them to apical or basolateral cell surfaces. However, it does not appear that viral glycoproteins generally control the polarity of paramyxovirus budding. Instead, budding polarity appears to be determined mainly by the viral M proteins. This is likely the case for HRSV, in which the F glycoprotein is intrinsically targeted to the apical surface (Batonick et al., 2008; Brock et al., 2005), yet recombinant HRSV that lacks the F gene still buds apically (Batonick et al., 2008). In fact, even HRSV lacking all three glycoproteins (F, G, and SH) buds apically, implying that internal viral proteins control the direction of budding (Batonick et al., 2008). An additional example is provided by measles virus, in which the viral glycoproteins intrinsically target to basolateral cell surfaces when expressed alone. In virus-infected cells, a substantial fraction of the measles virus F and H glycoproteins is re-routed to apical cell surfaces from which virus particles bud (Maisner et al., 1998). In cells infected with recombinant measles virus lacking an M gene, the measles virus glycoproteins fail to re-route to the apical side, indicating that the direction of measles virus budding, as well as the redirection of measles virus glycoproteins, is likely controlled by M protein (Naim et al., 2000). Intrinsic targeting of measles virus glycoproteins to basolateral membranes may be important for pathogenesis, as it favors cell-to-cell transfer of infection and may contribute to systemic spread in vivo (Moll et al., 2002). M protein is also thought to define the apical budding of Sendai virus. A pantropic mutant Sendai virus, F1-R, buds both apically and basolaterally and causes systemic infection in mice, whereas wt Sendai virus budding is restricted to apical surfaces, resulting in localized infection of the respiratory tract (Tashiro et al., 1996; Tashiro et al., 1992). The altered budding polarity is attributed to mutations in the M protein, as the mutant M protein disrupts the microtubule network of the cell and alters maintenance of cell polarity. Whereas wt Sendai virus M protein redirects the bipolar transport of mutant F protein from the F1-R virus to the apical cell surface, F1-R M protein does not (Tashiro et al., 1996). These findings all support a key role for paramyxovirus M proteins in the polarized assembly and release of virus particles.
Apical transport and recycling of host proteins in polarized cells is facilitated by the apical recycling endosome (ARE), which is enriched in Rab11a. Recent studies have demonstrated that efficient polarized budding of HRSV depends on proper ARE function (Brock et al., 2005; Utley et al., 2008). Expression of altered myosin Vb protein disrupted HRSV replication (Brock et al., 2005). The altered protein lacks its myosin motor domain, and expression of this protein in polarized cells disrupts basolateral-to-apical transcytosis. Reduction in HRSV yield was mainly caused by failure of virus assembly at the apical surface. HRSV replication could also be inhibited through expression of a DN Rab11-FIP2 protein that lacks its Rab-binding domain (Utley et al., 2008). Rab11 family interacting proteins (Rab11-FIPs) assist in the regulation of ARE-mediated protein sorting. Reduction in HRSV replication in this case was caused by a late domain-like defect, as judged by retention and significant elongation of assembled viral filaments on infected cells (Utley et al., 2008). Hence, FIP2 protein is required at a late stage of HRSV budding from polarized cells, most likely membrane fission. This may represent a novel, alternative pathway for the polarized budding of some viruses, such as HRSV, which exit from cells via a mechanism that seems to be completely independent of ESCRT machinery.
5.2 Budding of virus particles from lipid raft membranes
Microdomains within lipid bilayers, such as the cholesterol- and sphingolipid-rich raft microdomains, have the potential to act as platforms for virus assembly by providing nucleation points to allow concentration of viral proteins prior to budding (reviewed in Brown and London, 2000; Chazal and Gerlier, 2003; Schmitt and Lamb, 2005; Simons and Ikonen, 1997; Simons and Toomre, 2000; Takimoto and Portner, 2004). In paramyxovirus-infected cells, viral proteins in many cases sort to raft membranes selectively, judged by their association with detergent-resistant membrane (DRM) that resists solubilization with cold nonionic detergents such as Triton-X100. In Sendai virus-infected cells, for example, the viral structural proteins F, HN, M, N, and P are all found at least partially associated with DRM (Gosselin-Grenet et al., 2006). In transfected cells, the F and HN glycoproteins are associated with DRM when expressed alone or in combination (Sanderson et al., 1995), but the M protein is associated with DRM only when it is co-expressed with F or HN protein, suggesting that interaction between M protein and viral glycoprotein cytoplasmic tails is essential for proper targeting of M protein to raft membrane (Ali and Nayak, 2000). Measles virus structural proteins are also found associated with DRM in virus-infected cells (Manié et al., 2000; Vincent et al., 2000). Here, the F and M proteins are each capable of independently associating with DRM in transfected cells, although M protein DRM association was found to increase upon co-expression of F protein (Pohl et al., 2007). NDV HN, F, and NP proteins all associate with DRM during virus infection, and the DRM-associated viral proteins are ultimately found in released NDV virions (Laliberte et al., 2006). Lipid raft-associated proteins caveolin-1, flotillin-2, and actin are also found in NDV virions (Laliberte et al., 2006). NDV F protein is associated with DRM when expressed alone, and deletions of the F protein cytoplasmic tail inhibit DRM association (Dolganiuc et al., 2003). HRSV F, G, M, and N proteins all associate with DRM in virus-infected cells (Brown et al., 2004; Marty et al., 2004) and several lipid raft markers, including caveolin-1, GM-1, CD-55, and CD58, are found in released HRSV virions or in budding HRSV filaments (Brown et al., 2002; Brown et al., 2004; Jeffree et al., 2003). In transfected cells, HRSV M protein associates with raft membrane only when it is co-expressed with F protein (Henderson, 2002). In cells infected with canine distemper virus (CDV), the H and F glycoproteins are associated with DRM (Imhoff et al., 2007). In sum, the targeting of viral proteins to raft membranes for assembly appears to be a common strategy employed by a variety of different paramyxoviruses.
Evidence that lipid rafts are in fact important during paramyxovirus infections has been obtained mainly through experiments that disrupt raft microdomains by depleting cholesterol with the drug methyl-β-cyclodextrin (MβCD). In Sendai virus infection, MβCD treatment did not impair virion production, however the virions that were released were less infectious than virions released from untreated cells (Gosselin-Grenet et al., 2006). Treatment of CDV-infected cells with MβCD did not affect particle release or infectivity, though treatment of CDV virions with MβCD after they had been released resulted in a loss of infectivity that was restored upon supplementation with cholesterol (Imhoff et al., 2007). NDV particle release was found to be enhanced by MβCD treatment (Laliberte et al., 2006). However, the released particles were somewhat altered in polypeptide composition, displayed abnormal morphology, and were less infectious than particles released from untreated cells (Laliberte et al., 2006). Further characterization of these particles revealed a defect in virus-cell membrane fusion, and a loss of complexes formed between the HN and F glycoproteins in virion envelopes (Laliberte et al., 2007). Infectivity defects have also been observed for NDV virions released from Niemann-Pick syndrome type C cells, a cell type lacking lipid rafts (Laliberte et al., 2007). Overall, these results support a model in which assembly of paramyxovirus components at lipid raft platforms enhances the quality, but not necessarily the quantity, of virus particles that are released.
Recent reports illuminate changes in cholesterol biosynthesis that occur in paramyxovirus-infected cells. 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), an enzyme involved in the formation of endogenous cholesterol, and low-density lipoprotein receptor, involved in cholesterol homeostasis, are upregulated during HRSV infection, judged by microarray analysis (Martinez et al., 2007; Yeo et al., 2009). Levels of HMGCR and squalene monooxygenase, which functions in sterol biosynthesis, are altered during measles virus infection, and depletion of cholesterol from cells through simvastatin-mediated inhibition of HMGCR reduces that amount of infectious measles virus release in an acute infection (Robinzon et al., 2009). Intriguingly, several genes involved in cholesterol biosynthesis are downregulated in cells persistently infected with measles virus compared to those undergoing acute infection (Robinzon et al., 2009), and it has been suggested that the presence of a cellular mechanism to downregulate cholesterol during virus infection could serve as an innate antiviral mechanism aimed at limiting the budding of infectious particles (Robinzon et al., 2009).
5.3 Virus assembly and the cytoskeleton
Involvement of the actin cytoskeleton in paramyxovirus budding was first proposed based on the detection of large quantities of actin in purified preparations of Sendai virions and measles virions (Lamb et al., 1976; Tyrrell and Norrby, 1978). Other paramyxoviruses incorporate actin into virions as well, including NDV and HRSV (Laliberte et al., 2006; Marty et al., 2004). Sendai VLPs produced upon expression of either M protein or F protein contain actin, and both of these proteins contain sequences important for budding that resemble actin-binding domains (Takimoto et al., 2001). Both Sendai virus and NDV M proteins have been shown to physically associate with actin (Giuffre et al., 1982) and EM-based studies of measles virus and HRSV have revealed a tight association between actin filament formation and budding virions (Bohn et al., 1986; Jeffree et al., 2007).
Drug treatments that disrupt the cytoskeleton generally have deleterious effects on paramyxovirus replication. Multiple steps in the lifecycle of HRSV, for example, including viral entry and virus exit, are impaired upon disruption of the cytoskeleton, with severe disruptions in virus assembly and release following perturbation of either actin or microtubule networks (Burke et al., 1998; Kallewaard et al., 2005). Using molecular beacons to visually track RNPs, HRSV filament assembly and motion were recently examined in live cells, and myosin motor-driven transport on the actin cytoskeleton was postulated to drive the migration and/or rotational motion of the filaments (Santangelo and Bao, 2007). In the case of hPIV3, nocodazole-mediated disruption of microtubules significantly impairs the release of infectious virus from cells, although depolymerization of actin microfilaments with cytochalasin D had no effect on hPIV3 release (Bose et al., 2001). Microtubule disruption by nocodazole and colchicine treatments led to asymmetric budding of Sendai virus, similar to that observed with the pantropic F1-R mutant (Tashiro et al., 1993). Hence, efficient and directional budding of some paramyxoviruses depends on intact microtubules (Bose et al., 2001; Tashiro et al., 1993).
5.4 Trafficking of paramyxovirus M proteins through the cell nucleus
Although paramyxovirus replication takes place in the cytoplasm, various paramyxoviral proteins have been found to traffic to the nucleus during the course of infection (Coleman and Peeples, 1993; Ghildyal et al., 2009; Ghildyal et al., 2005a; Ghildyal et al., 2003; Peeples et al., 1992; Peeples, 1988; Rodriguez et al., 2004; Sato et al., 2006; Shaw et al., 2005; Watanabe et al., 1996; Yoshida et al., 1976). Early during infection, the M proteins of HRSV, NDV, and Sendai virus have all been observed in the nucleus, while later during infection they are localized mainly in the cytoplasm as well as associated with cell membranes (Ghildyal et al., 2003; Peeples et al., 1992; Peeples, 1988; Yoshida et al., 1976). As discussed earlier, altered Nipah virus M proteins with disruptions to the 62-YMYL-65 or 92-YPLGVG-97 sequences are retained in the nuclei of transfected cells, consistent with the possibility that Nipah virus M protein undergoes nuclear-cytoplasmic shuttling (Ciancanelli and Basler, 2006; Patch et al., 2008). By analogy with VSV, it has been suggested that transient nuclear localization of paramyxovirus M proteins in the early phase of infection could allow for M-mediated inhibition of cellular nuclear processes (Ghildyal et al., 2006). Transient nuclear localization of M proteins may also serve to delay the budding of particles until late times in infection, when sufficient quantities of viral glycoproteins and RNPs have accumulated. Functional nuclear localization signals (NLSs) of the NDV and HRSV M proteins have been characterized. The NLS of NDV M protein is a bipartite NLS composed of basic amino acid clusters (Coleman and Peeples, 1993). HRSV M protein is recruited into the nucleus through direct recognition by a nuclear import receptor, importin beta 1 (Ghildyal et al., 2005a).
Late in paramyxovirus infection, large quantities of M protein are needed outside the nucleus at locations of virus assembly. In the case of HRSV M protein, nuclear export is mediated by a leucine-rich nuclear export signal and is dependent on Crm-1 (exportin-1) (Ghildyal et al., 2009). Recombinant virus harboring a mutated nuclear export signal within its M protein could not be recovered, and treatment of HRSV-infected cells with a Crm-1 inhibitor, leptomycin B, prevented nuclear export of HRSV M protein and impaired virus production (Ghildyal et al., 2009). These findings support a model in which paramyxovirus M protein nuclear localization is meant to be transient, and failure to exit from the nucleus impairs accumulation of the protein at virus assembly sites at levels required for virus budding.
6. Conclusions
Substantial progress has been made in recent years towards understanding both unique and shared processes used by paramyxoviruses and other enveloped viruses during virus particle formation. Roles for paramyxovirus M proteins, glycoproteins, nucleocapsid proteins, and accessory proteins during virus assembly have been clarified. Mechanisms allowing for directional budding of paramyxoviruses from polarized cells have been defined, and the importance of lipid raft microdomains as virus assembly platforms has been demonstrated. However, questions regarding important aspects of the paramyxovirus assembly process remain unresolved. For example, although paramyxoviruses clearly differ from one another in the fundamental components required for particle formation, the biological implications of these differences remain unclear. Structural information is still lacking for most paramyxovirus M proteins, and consequently the molecular determinants that drive viral protein-protein interactions during virus assembly remain poorly understood. While host factors clearly play important roles in the budding of many paramyxoviruses, specific mechanisms of host factor recruitment remain largely undefined. Future efforts in these and related areas will provide a clearer understanding of the events which must occur for virions to form, and may contribute to the development of effective antiviral strategies aimed at blocking the late steps of paramyxovirus lifecycles.
Acknowledgments
This work was supported in part by the Middle Atlantic Regional Center of Excellence (MARCE) for Biodefense and Emerging Infectious Disease Research NIH grant AI057168, and research grant AI070925 from the National Institute of Allergy and Infectious Diseases to A.P.S. This project is funded, in part, under a grant with the Pennsylvania Department of Health using Tobacco Settlement funds to A.P.S. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Abbreviations used
- AmotL1
angiomotin-like 1
- ARE
apical recycling endosome
- CDV
canine distemper virus
- Crm1
exportin1
- DN
dominant-negative
- DRM
detergent-resistant membrane
- ESCRT
endosomal sorting complex required for transport
- F
fusion
- G
glyco-
- H
hemagglutinin (paramyxovirus)
- HA
hemagglutinin (influenza virus)
- HA-Ub
hemagglutinin-tagged ubiquitin
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A
- HN
hemagglutinin-neuraminidase
- hPIV
human parainfluenza virus
- HRSV
human respiratory syncytial virus
- HSP
heat shock protein
- L
large
- M
matrix
- MVB
multivesicular body
- MβCD
methyl-β-cyclodextrin
- NA
neuraminidase
- NDV
Newcastle disease virus
- NLS
nuclear localization signal
- N
nucleocapsid
- NP
nucleocapsid
- P
phospho-
- PIV5
parainfluenza virus 5
- Rab11-FIP
Rab11 family interacting proteins
- RNP
ribonucleoprotein
- SSPE
subacute sclerosing panencephalitis
- VLP
virus-like particle
- Vps
vacuolar protein sorting
- VSV
vesicular stomatitis virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander DJ. OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Office of International Des Epizooties; Paris: 2009. Newcastle disease. [Google Scholar]
- Ali A, Nayak DP. Assembly of Sendai virus: M protein interacts with F and HN proteins and with the cytoplasmic tail and transmembrane domain of F protein. Virology. 2000;276:289–303. doi: 10.1006/viro.2000.0556. [DOI] [PubMed] [Google Scholar]
- Batonick M, Oomens AGP, Wertz GW. Human respiratory syncytial virus glycoproteins are not required for apical targeting and release from polarized epithelial cells. J Virol. 2008;82:8664–72. doi: 10.1128/JVI.00827-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology. 2006;344:55–63. doi: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- Bishop KA, Broder CC. Hendra and Nipah: Lethal Zoonotic Paramyxoviruses. In: Scheld WM, Hammer SM, Hughes JM, editors. Emerging Infections. Washington, D. C: American Society for Microbiology; 2008. pp. 155–87. [Google Scholar]
- Blau DM, Compans RW. Entry and release of measles virus are polarized in epithelial cells. Virology. 1995;210:91–9. doi: 10.1006/viro.1995.1320. [DOI] [PubMed] [Google Scholar]
- Blumberg BM, Giorgi C, Kolakofsky D. N protein of vesicular stomatitis virus selectively encapsidates leader RNA in vitro. Cell. 1983;32:559–67. doi: 10.1016/0092-8674(83)90475-0. [DOI] [PubMed] [Google Scholar]
- Bohn W, Rutter G, Hohenber H, Mannweiler K, Nobis P. Involvement of actin filaments in budding of measles virus: studies on cytoskeletons of infected cells. Virology. 1986;149:91–106. doi: 10.1016/0042-6822(86)90090-5. [DOI] [PubMed] [Google Scholar]
- Bose S, Malur A, Banerjee AK. Polarity of human parainfluenza virus type 3 infection in polarized human lung epithelial A549 cells: role of microfilament and microtubule. J Virol. 2001;75:1984–9. doi: 10.1128/JVI.75.4.1984-1989.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock SC, Heck JM, McGraw PA, Crowe JEJ. The transmembrane domain of the respiratory syncytial virus F protein is an orientation-independent apical plasma membrane sorting sequence. J Virol. 2005;79:12528–35. doi: 10.1128/JVI.79.19.12528-12535.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem. 2000;275:17221–4. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- Brown G, Aitken JD, Rixon HWM, Sugrue RJ. Caveolin-1 is incorporated into mature respiratory syncytial virus particles during virus assembly on the surface of virus-infected cells. J Gen Virol. 2002;83:611–21. doi: 10.1099/0022-1317-83-3-611. [DOI] [PubMed] [Google Scholar]
- Brown G, Jeffree CE, McDonald T, Rixon HWM, Aitken JD, Sugrue RJ. Analysis of the interaction between respiratory syncytial virus and lipid-rafts in Hep2 cells during infection. Virology. 2004;327:175–85. doi: 10.1016/j.virol.2004.06.038. [DOI] [PubMed] [Google Scholar]
- Buchholz CJ, Spehner D, Drillien R, Neubert WJ, Homann HE. The conserved N-terminal region of Sendai virus nucleocapsid protein NP is required for nucleocapsid assembly. J Virol. 1993;67:5803–12. doi: 10.1128/jvi.67.10.5803-5812.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke E, Dupuy L, Wall C, Barik S. Role of cellular actin in the gene expression and morphogenesis of human respiratory syncytial virus. Virology. 1998;252:137–48. doi: 10.1006/viro.1998.9471. [DOI] [PubMed] [Google Scholar]
- Cadd T, Garcin D, Tapparel C, Itoh M, Homma M, Roux L, et al. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J Virol. 1996;70:5067–74. doi: 10.1128/jvi.70.8.5067-5074.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell SE, Lyles DS. Dissociation of newly synthesized Sendai viral proteins from the cytoplasmic surface of isolated plasma membranes of infected cells. J Virol. 1986;57:678–83. doi: 10.1128/jvi.57.2.678-683.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calistri A, Salata C, Parolin C, Palù G. Role of multivesicular bodies and their components in the egress of enveloped RNA viruses. Rev Med Virol. 2009;19:21–45. doi: 10.1002/rmv.588. [DOI] [PubMed] [Google Scholar]
- Cathomen T, Mrkic B, Spehner D, Drillien R, Naef R, Pavlovic J, et al. A matrix-less measles virus is infectious and elicits extensive cell fusion: consequences for propagation in the brain. EMBO J. 1998a;17:3899–908. doi: 10.1093/emboj/17.14.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomen T, Naim HY, Cattaneo R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol. 1998b;72:1224–34. doi: 10.1128/jvi.72.2.1224-1234.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R, Schmid A, Eschle D, Baczko K, ter Meulen V, Billeter MA. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–65. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazal N, Gerlier D. Virus entry, assembly, budding, and membrane rafts. Microbiol Mol Biol Rev. 2003;67:226–37. doi: 10.1128/MMBR.67.2.226-237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology. 2008;372:221–32. doi: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BJ, Leser GP, Morita E, Lamb RA. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J Virol. 2007;81:7111–23. doi: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong LD, Rose JK. Membrane association of functional vesicular stomatitis virus matrix protein in vivo. J Virol. 1993;67:407–14. doi: 10.1128/jvi.67.1.407-414.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli MJ, Basler CF. Mutation of YMYL in the Nipah virus matrix protein abrogates budding and alters subcellular localization. J Virol. 2006;80:12070–8. doi: 10.1128/JVI.01743-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman NA, Peeples ME. The matrix protein of Newcastle disease virus localizes to the nucleus via a bipartite nuclear localization signal. Virology. 1993;195:596–607. doi: 10.1006/viro.1993.1411. [DOI] [PubMed] [Google Scholar]
- Conzelmann KK. Reverse genetics of mononegavirales. Curr Top Microbiol Immunol. 2004;283:1–41. doi: 10.1007/978-3-662-06099-5_1. [DOI] [PubMed] [Google Scholar]
- Coronel EC, Murti KG, Takimoto T, Portner A. Human parainfluenza virus type 1 matrix and nucleoprotein genes transiently expressed in mammalian cells induce the release of virus- like particles containing nucleocapsid-like structures. J Virol. 1999;73:7035–8. doi: 10.1128/jvi.73.8.7035-7038.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronel EC, Takimoto T, Murti KG, Varich N, Portner A. Nucleocapsid incorporation into parainfluenza virus is regulated by specific interaction with matrix protein. J Virol. 2001;75:1117–23. doi: 10.1128/JVI.75.3.1117-1123.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counihan ME, Shay DK, Holman RC, Lowther SA, Anderson LJ. Human parainfluenza virus-associated hospitalizations among children less than five years of age in the United States. Pediatr Infect Dis J. 2001;20:646–53. doi: 10.1097/00006454-200107000-00003. [DOI] [PubMed] [Google Scholar]
- Craven RC, Harty RN, Paragas J, Palese P, Wills JW. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J Virol. 1999;73:3359–65. doi: 10.1128/jvi.73.4.3359-3365.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J, Marq J-B, Kolakofsky D. The Sendai virus nonstructural C proteins specifically inhibit viral mRNA synthesis. Virology. 1992;189:647–56. doi: 10.1016/0042-6822(92)90588-g. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Dessen A, Volchkov V, Dolnik O, Klenk H-D, Weissenhorn W. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 2000;19:4228–36. doi: 10.1093/emboj/19.16.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolganiuc V, McGinnes LW, Luna EJ, Morrison TG. Role of the cytoplasmic domain of the Newcastle disease virus fusion protein in association with lipid rafts. J Virol. 2003;77:12968–79. doi: 10.1128/JVI.77.24.12968-12979.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BT, Broder CC, Middleton D, Wang LF. Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol. 2006;4:23–35. doi: 10.1038/nrmicro1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein JH, Field HE, Luby S, Pulliam JR, Daszak P. Nipah virus: impact, origins, and causes of emergence. Curr Infect Dis Rep. 2006;8:59–65. doi: 10.1007/s11908-006-0036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington W, Emmerson PT. Assembly of recombinant Newcastle disease virus nucleocapsid protein into nucleocapsid-like structures is inhibited by the phosphoprotein. J Gen Virol. 1997;78:2335–9. doi: 10.1099/0022-1317-78-9-2335. [DOI] [PubMed] [Google Scholar]
- Falsey AR. Respiratory syncytial virus infection in older persons. Vaccine. 1998;16:1775–8. doi: 10.1016/s0264-410x(98)00142-x. [DOI] [PubMed] [Google Scholar]
- Fouillot-Coriou N, Roux L. Structure-function analysis of the Sendai virus F and HN cytoplasmic domain: different role for the two proteins in the production of virus particle. Virology. 2000;270:464–75. doi: 10.1006/viro.2000.0291. [DOI] [PubMed] [Google Scholar]
- Garcin D, Latorre P, Kolakofsky D. Sendai virus C proteins counteract the interferon-mediated induction of an antiviral state. J Virol. 1999;73:6559–65. doi: 10.1128/jvi.73.8.6559-6565.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildyal R, Baulch-Brown C, Mills J, Meanger J. The matrix protein of Human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch Virol. 2003;148:1419–29. doi: 10.1007/s00705-003-0112-y. [DOI] [PubMed] [Google Scholar]
- Ghildyal R, Ho A, Dias M, Soegiyono L, Bardin PG, Tran KC, et al. The Respiratory Syncytial Virus Matrix Protein Possesses a Crm1-Mediated Nuclear Export Mechanism. J Virol. 2009;83:5353–62. doi: 10.1128/JVI.02374-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildyal R, Ho A, Jans DA. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol Rev. 2006;30:692–705. doi: 10.1111/j.1574-6976.2006.00025.x. [DOI] [PubMed] [Google Scholar]
- Ghildyal R, Ho A, Wagstaff KM, Dias M, Barton CL, Jans P, et al. Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry (Mosc) 2005a;44:12887–95. doi: 10.1021/bi050701e. [DOI] [PubMed] [Google Scholar]
- Ghildyal R, Li D, Peroulis I, Shields B, Bardin PG, Teng MN, et al. Interaction between the respiratory syncytial virus G glycoprotein cytoplasmic domain and the matrix protein. J Gen Virol. 2005b;86:1879–84. doi: 10.1099/vir.0.80829-0. [DOI] [PubMed] [Google Scholar]
- Giuffre RM, Toyell DR, Kay CM, Tyrrell DL. Evidence for an interaction between the membrane protein of a paramyxovirus and actin. J Virol. 1982;42:963–8. doi: 10.1128/jvi.42.3.963-968.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith CS, Whistler T, Rollin PE, Ksiazek TG, Rota PA, Bellini WJ, et al. Elucidation of Nipah virus morphogenesis and replication using ultrastructural and molecular approaches. Virus Res. 2003;92:89–98. doi: 10.1016/s0168-1702(02)00323-4. [DOI] [PubMed] [Google Scholar]
- Gosselin-Grenet AS, Marq J-B, Abrami L, Garcin D, Roux L. Sendai virus budding in the course of an infection does not require Alix and VPS4A host factors. Virology. 2007;365:101–12. doi: 10.1016/j.virol.2007.03.039. [DOI] [PubMed] [Google Scholar]
- Gosselin-Grenet AS, Mottet-Osman G, Roux L. From assembly to virus particle budding: pertinence of the detergent resistant membranes. Virology. 2006;344:296–303. doi: 10.1016/j.virol.2005.09.035. [DOI] [PubMed] [Google Scholar]
- Gotoh B, Takeuchi K, Komatsu T, Yokoo J, Kimura Y, Kurotani A, et al. Knockout of the Sendai virus C gene eliminates the viral ability to prevent the interferon-alpha/beta-mediated responses. FEBS Lett. 1999;459:205–10. doi: 10.1016/s0014-5793(99)01241-7. [DOI] [PubMed] [Google Scholar]
- Gottwein E, Krausslich HG. Analysis of human immunodeficiency virus type 1 Gag ubiquitination. J Virol. 2005;79:9134–44. doi: 10.1128/JVI.79.14.9134-9144.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley ES, Montgomery JM, Hossain MJ, Bell M, Azad AK, Islam MR, et al. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg Infect Dis. 2007;13:1031–7. doi: 10.3201/eid1307.061128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360:588–98. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Brown ME, McGettigan JP, Wang G, Jayakar HR, Huibregtse JM, et al. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J Virol. 2001;75:10623–9. doi: 10.1128/JVI.75.22.10623-10629.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci USA. 2000;97:13871–6. doi: 10.1073/pnas.250277297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harty RN, Paragas J, Sudol M, Palese P. A proline-rich motif within the matrix protein of vesicular stomatitis virus and rabies virus interacts with WW domains of cellular proteins: implications for viral budding. J Virol. 1999;73:2921–9. doi: 10.1128/jvi.73.4.2921-2929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan MK, Kato A, Muranaka M, Yamaguchi R, Sakai Y, Hatano I, et al. Versatility of the accessory C proteins of Sendai virus: contribution to virus assembly as an additional role. J Virol. 2000;74:5619–28. doi: 10.1128/jvi.74.12.5619-5628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilman CA. From the National Institute of Allergy and Infectious Diseases and the World Health Organization. Respiratory syncytial and parainfluenza viruses J Infect Dis. 1990;161:402–6. doi: 10.1093/infdis/161.3.402. [DOI] [PubMed] [Google Scholar]
- Henderson G. Sorting of the Respiratory Syncytial Virus Matrix Protein into Detergent-Resistant Structures Is Dependent on Cell-Surface Expression of the Glycoproteins. Virology. 2002;300:244–54. doi: 10.1006/viro.2002.1540. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–72. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hirayama E, Hattori M, Kim J. Specific binding of heat shock protein 70 with HN-protein inhibits the HN-protein assembly in Sendai virus-infected Vero cells. Virus Res. 2006;120:199–207. doi: 10.1016/j.virusres.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Horvath CM. Weapons of STAT destruction. Interferon evasion by paramyxovirus V protein Eur J Biochem. 2004;271:4621–8. doi: 10.1111/j.1432-1033.2004.04425.x. [DOI] [PubMed] [Google Scholar]
- Imhoff H, von Messling V, Herrier G, Haas L. Canine distemper virus infection requires cholesterol in the viral envelope. J Virol. 2007;81:4158–65. doi: 10.1128/JVI.02647-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Tokusumi Y, Ban H, Kanaya T, Shirakura M, Tokusumi T, et al. A new Sendai virus vector deficient in the matrix gene does not form virus particles and shows extensive cell-to-cell spreading. J Virol. 2003;77:6419–29. doi: 10.1128/JVI.77.11.6419-6429.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie T, Licata JM, McGettigan JP, Schnell MJ, Harty RN. Budding of PPxY-containing rhabdoviruses is not dependent on host proteins TGS101 and VPS4A. J Virol. 2004;78:2657–65. doi: 10.1128/JVI.78.6.2657-2665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie T, Nagata N, Yoshida T, Sakaguchi T. Paramyxovirus Sendai virus C proteins are essential for maintenance of negative-sense RNA genome in virus particles. Virology. 2008a;374:495–505. doi: 10.1016/j.virol.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Irie T, Nagata T, Yoshida T, Sakaguchi T. Recruitment of Alix/AIP1 to the plasma membrane by Sendai virus C protein facilitates budding of virus-like particles. Virology. 2008b;371:108–20. doi: 10.1016/j.virol.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Irie T, Shimazu Y, Yoshida T, Sakaguchi T. The YLDL sequence within Sendai virus M protein is critical for budding of virus-like particles and interacts with Alix/AIP1 independently of C protein. J Virol. 2007;81:2263–73. doi: 10.1128/JVI.02218-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki M, Takeda M, Shirogane Y, Nakatsu T, Nakamura T, Yanagi Y. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J Virol. 2009;83:10374–83. doi: 10.1128/JVI.01056-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasenosky LD, Neumann G, Lukashevich I, Kawaoka Y. Ebola virus VP40-induced particle formation and association with the lipid bilayer. J Virol. 2001;75:5205–14. doi: 10.1128/JVI.75.11.5205-5214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakar HR, Murti KG, Whitt MA. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. J Virol. 2000;74:9818–27. doi: 10.1128/jvi.74.21.9818-9827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffree CE, Brown G, Aitken JD, Su-Yin DY, Tan B-H, Sugrue RJ. Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology. 2007;369:309–23. doi: 10.1016/j.virol.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Jeffree CE, Rixon HWM, Brown G, Aitken JD, Sugrue RJ. Distribution of the attachment (G) glycoprotein and GM1 within the envelope of mature respiratory syncytial virus filaments revealed using field emission scanning electron microscopy. Virology. 2003;306:254–67. doi: 10.1016/s0042-6822(02)00016-8. [DOI] [PubMed] [Google Scholar]
- Jin H, Leser GP, Zhang J, Lamb RA. Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 1997;16:1236–47. doi: 10.1093/emboj/16.6.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juozapaitis M, Slibinskas R, Staniulis J, Sakaguchi T, Sasnauskas K. Generation of Sendai virus nucleocapsid-like particles in yeast. Virus Res. 2005;108:221–4. doi: 10.1016/j.virusres.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Kallewaard NL, Bowen AL, Crowe JEJ. Cooperativity of actin and microtubule elements during replication of respiratory syncytial virus. Virology. 2005;331:73–81. doi: 10.1016/j.virol.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Kato A, Kiyotani K, Kubota T, Yoshida T, Tashiro M, Nagai Y. Importance of the anti-interferon capacity of Sendai virus C protein for pathogenicity in mice. J Virol. 2007;81:3264–71. doi: 10.1128/JVI.02590-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakofsky D, Bruschi A. Antigenomes in Sendai virions and Sendai virus-infected cells. Virology. 1975;66:185–91. doi: 10.1016/0042-6822(75)90189-0. [DOI] [PubMed] [Google Scholar]
- Kondo T, Yoshida T, Miura N, Nakanishi M. Temperature-sensitive phenotype of a mutant Sendai virus strain is caused by its insufficient accumulation of the M protein. J Biol Chem. 1993;268:21924–30. [PubMed] [Google Scholar]
- Kurotani A, Kiyotani K, Kato A, Shioda T, Sakai Y, Mizomoto K, et al. Sendai virus C proteins are categorically nonessential gene products but silencing their expression severely impairs viral replication and pathogenesis. Genes Cells. 1998;3:111–24. doi: 10.1046/j.1365-2443.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- Laliberte JP, McGinnes LW, Morrison TG. Incorporation of functional HN-F glycoprotein-containing complexes into newcastle disease virus is dependent on cholesterol and membrane lipid raft integrity. J Virol. 2007;81:10636–48. doi: 10.1128/JVI.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laliberte JP, McGinnes LW, Peeples ME, Morrison TG. Integrity of membrane lipid rafts is necessary for the ordered assembly and release of infectious Newcastle disease virus particles. J Virol. 2006;80:10652–62. doi: 10.1128/JVI.01183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb RA, Mahy BW, Choppin PW. The synthesis of sendai virus polypeptides in infected cells. Virology. 1976;69:116–31. doi: 10.1016/0042-6822(76)90199-9. [DOI] [PubMed] [Google Scholar]
- Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia: Lippincott, Williams and Wilkins; 2006. pp. 1449–96. [Google Scholar]
- Li M, Schmitt PT, Li Z, McCrory TS, He B, Schmitt AP. Mumps virus matrix, fusion, and nucleocapsid proteins cooperate for efficient production of virus-like particles. J Virol. 2009;83:7261–72. doi: 10.1128/JVI.00421-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata JM, Simpson-Holley M, Wright NT, Han Z, Paragas J, Harty RN. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4. J Virol. 2003;77:1812–9. doi: 10.1128/JVI.77.3.1812-1819.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cocka L, Okumura A, Zhang YA, Sunyer JO, Harty RN. Conserved Motifs within Ebola and Marburg Virus VP40 Proteins are Important for Stability, Localization, and Subsequent Budding of VLPs. J Virol. 2009:2294–303. doi: 10.1128/JVI.02034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loney C, Mottet-Osman G, Roux L, Bhella D. Paramyxovirus ultrastructure and genome packaging: cryo-electron tomography of sendai virus. J Virol. 2009;83:8191–7. doi: 10.1128/JVI.00693-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisner A, Klenk H-D, Herrler G. Polarized Budding of Measles Virus Is Not Determined by Viral Surface Glycoproteins. J Virol. 1998;72:5276–8. doi: 10.1128/jvi.72.6.5276-5278.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhova OA, Zhang D-E. ISG15 Inhibits Nedd4 Ubiquitin E3 Activity and Enhances the Innate Antiviral Response. J Biol Chem. 2008;283:8783–7. doi: 10.1074/jbc.C800030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manié SN, de Breyne S, Vincent S, Gerlier D. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol. 2000;74:305–11. doi: 10.1128/jvi.74.1.305-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwell MA, Fox CF. Protein-protein interactions within paramyxoviruses identified by native disulfide bonding or reversible chemical cross-linking. J Virol. 1980;33:152–66. doi: 10.1128/jvi.33.1.152-166.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwell MA, Portner A, Schwartz AL. An alternative route of infection for viruses: entry by means of the asialoglycoprotein receptor of a Sendai virus mutant lacking its attachment protein. Proc Natl Acad Sci USA. 1985;82:978–82. doi: 10.1073/pnas.82.4.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marq J-B, Brini A, Kolakofsky D, Garcin D. Targeting of the Sendai virus C protein to the plasma membrane via a peptide-only membrane anchor. J Virol. 2007;81:3187–97. doi: 10.1128/JVI.02465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J. The role of ubiquitin in retroviral egress. Traffic. 2007;8:1297–303. doi: 10.1111/j.1600-0854.2007.00609.x. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Martinez I, Lombardia L, Garcia-Berreno B, Dominguez O, Melero JA. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J Gen Virol. 2007;8:570–81. doi: 10.1099/vir.0.82187-0. [DOI] [PubMed] [Google Scholar]
- Marty A, Meanger J, Mills J, Shields B, Ghildyal R. Association of matrix protein of respiratory syncytial virus with the host cell membrane of infected cells. Arch Virol. 2004;149:199–210. doi: 10.1007/s00705-003-0183-9. [DOI] [PubMed] [Google Scholar]
- Mohd Nor MN, Gan CH, Ong BL. Nipah virus infection of pigs in peninsular Malaysia. Rev Sci Tech. 2000;19:160–5. doi: 10.20506/rst.19.1.1202. [DOI] [PubMed] [Google Scholar]
- Moll M, Klenk H-D, Maisner A. Importance of the cytoplasmic tails of the measles virus glycoproteins for fusogenic activity and the generation of recombinant measles viruses. J Virol. 2002;76:7174–86. doi: 10.1128/JVI.76.14.7174-7186.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Money VA, McPhee HK, Mosley JA, Sanderson JM, Yeo RP. Surface features of a Mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci USA. 2009;106:4441–6. doi: 10.1073/pnas.0805740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Mottet G, Roux L. Budding efficiency of Sendai virus nucleocapsids: influence of size and ends of the RNA. Virus Res. 1989;14:175–87. doi: 10.1016/0168-1702(89)90037-3. [DOI] [PubMed] [Google Scholar]
- Mottet-Osman G, Iseni F, Pelet T, Wiznerowics M, Garcin D, Roux L. Suppression of the Sendai virus M protein through a novel short interfering RNA approach inhibits viral particle production but does not affect viral RNA synthesis. J Virol. 2007;81:2861–8. doi: 10.1128/JVI.02291-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naim HY, Ehler E, Billeter MA. Measles virus matrix protein specifies apical virus release and glycoprotein sorting in epithelial cells. EMBO J. 2000;19:3576–85. doi: 10.1093/emboj/19.14.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio M, Tsurudome M, Ishihara H, Ito M, Ito Y. The conserved carboxyl terminus of human parainfluenza virus type 2 V protein plays an important role in virus growth. J Virol. 2007;362:85–98. doi: 10.1016/j.virol.2006.12.017. [DOI] [PubMed] [Google Scholar]
- Okumura A, Pitha PM, Harty RN. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc Natl Acad Sci USA. 2008;105:3974–9. doi: 10.1073/pnas.0710629105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott DE, Coren LV, Chertova EN, Gagliardi TD, Schubert U. Ubiquitination of HIV-1 and MuLV Gag. Virology. 2000;278:111–21. doi: 10.1006/viro.2000.0648. [DOI] [PubMed] [Google Scholar]
- Pantua HD, McGinnes LW, Peeples ME, Morrison TG. Requirements for the assembly and release of Newcastle disease virus-like particles. J Virol. 2006;80:11062–73. doi: 10.1128/JVI.00726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch JR, Crameri G, Wang LF, Eaton BT. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol J. 2007;4:1. doi: 10.1186/1743-422X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch JR, Han Z, McCarthy SE, Yan L, Wang LF, Harty RN, et al. The YPLGVG sequence of the Nipah virus matrix protein is required for budding. Virol J. 2008;5:1. doi: 10.1186/1743-422X-5-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik A, Chau V, Wills JW. Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci USA. 2000;97:13069–74. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, Cornu TI, Redwine J, Dales S, Lewicki H, Holz A, et al. Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology. 2001;291:215–25. doi: 10.1006/viro.2001.1182. [DOI] [PubMed] [Google Scholar]
- Peeples ME. Differential detergent treatment allows immunofluorescent localization of the Newcastle disease virus matrix protein within the nucleus of infected cells. Virology. 1988;162:255–9. doi: 10.1016/0042-6822(88)90418-7. [DOI] [PubMed] [Google Scholar]
- Peeples ME, Wang C, Gupta KC, Coleman N. Nuclear entry and nucleolar localization of the Newcastle disease virus (NDV) matrix protein occur early in infection and do not require other NDV proteins. J Virol. 1992;66:3263–9. doi: 10.1128/jvi.66.5.3263-3269.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Z, Bai Y, Schmitt AP. PIV5 M protein interaction with host protein angiomotin-like 1. Virology. 2009 doi: 10.1016/j.virol.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Craven RC, de la Torre JC. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci USA. 2003;100:12978–83. doi: 10.1073/pnas.2133782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl C, Duprex WP, Krohne G, Rima BK, Schneider-Schaulies S. Measles virus M and F proteins associate with detergent-resistant membrane fractions and promote formation of virus-like particles. J Gen Virol. 2007;88:1243–50. doi: 10.1099/vir.0.82578-0. [DOI] [PubMed] [Google Scholar]
- Portner A, Marx PA, Kingsbury DW. Isolation and characterization of Sendai virus temperature-sensitive mutants. J Virol. 1974;13:298–304. doi: 10.1128/jvi.13.2.298-304.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ProMED-mail. Hendra Virus, Human, Equine - Australia (05): (Queensland) ProMED-mail; 2009. Sept 10, [Accessed March 17, 2009]. 20090910.3189. http://www.promedmail.org. [Google Scholar]
- Putterman D, Pepinsky RB, Vogt VM. Ubiquitin in avian leukosis virus particles. Virology. 1990;176:633–7. doi: 10.1016/0042-6822(90)90035-p. [DOI] [PubMed] [Google Scholar]
- Rager M, Vongpunsawad S, Duprex WP, Cattaneo R. Polyploid measles virus with hexameric genome length. EMBO J. 2002;21:2364–72. doi: 10.1093/emboj/21.10.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl P, Moll M, Klenk H-D, Maisner A. Measles virus matrix protein is not cotransported with the viral glycoproteins but requires virus infection for efficient surface targeting. Virus Res. 2002;83:1–12. doi: 10.1016/s0168-1702(01)00379-3. [DOI] [PubMed] [Google Scholar]
- Rima BK, Duprex WP. Molecular mechanisms of measles virus persistence. Virus Res. 2005;111:132–47. doi: 10.1016/j.virusres.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Roberts SR, Compans RW, Wertz GW. Respiratory syncytial virus matures at the apical surfaces of polarized epithelial cells. J Virol. 1995;69:2667–73. doi: 10.1128/jvi.69.4.2667-2673.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinzon S, Dafa-Berger A, Dyer MD, Paeper B, Proll SC, Teal TH, et al. Impaired cholesterol biosynthesis in a neuronal cell line persistently infected with measles virus. J Virol. 2009;83:5495–504. doi: 10.1128/JVI.01880-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JJ, Cruz CD, Horvath CM. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J Virol. 2004;78:5358–67. doi: 10.1128/JVI.78.10.5358-5367.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Sabatani DD. Asymmetric budding of viruses in epithelial monolayers: A model system for study of epithelial polarity. Proc Natl Acad Sci USA. 1978;75:5071–5. doi: 10.1073/pnas.75.10.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeder PL, Taylor WP. Rinderpest. Vet Clin North Am Food Anim Pract. 2002;18:515–47. doi: 10.1016/s0749-0720(02)00035-x. [DOI] [PubMed] [Google Scholar]
- Ruigrok RW, Schoehn G, Dessen A, Forest E, Volchkov V, Dolnik O, et al. Structural characterization and membrane binding properties of the matrix protein VP40 of Ebola virus. J Mol Biol. 2000;300:103–12. doi: 10.1006/jmbi.2000.3822. [DOI] [PubMed] [Google Scholar]
- Runkler N, Pohl C, Schneider-Schaulies S, Klenk H-D, Maisner A. Measles virus nucleocapsid transport to the plasma membrane requires stable expression and surface accumulation of the viral matrix protein. Cell Microbiol. 2007;9:1203–14. doi: 10.1111/j.1462-5822.2006.00860.x. [DOI] [PubMed] [Google Scholar]
- Russell CJ, Luque LE. The structural basis of paramyxovirus invasion. Trends Microbiol. 2006;14:243–6. doi: 10.1016/j.tim.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi T, Kato A, Sugahara F, Shimazu Y, Inoue M, Kiyotani K, et al. AIP1/Alix is a binding partner of Sendai virus C protein and facilitates virus budding. J Virol. 2005;81:8933–41. doi: 10.1128/JVI.79.14.8933-8941.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson CM, Avalos R, Kundi A, Nayak DP. Interaction of Sendai viral F, HN, and M proteins with host cytoskeletal and lipid components in Sendai virus-infected BHK cells. Virology. 1995;209:701–7. doi: 10.1006/viro.1995.1308. [DOI] [PubMed] [Google Scholar]
- Sänger C, Mülberger E, Ryabchikova E, Kolesnikova L, Klenk H-D, Becker S. Sorting of Marburg virus surface protein and virus release take place at opposite surfaces of infected polarized epithelial cells. J Virol. 2001;75:1274–83. doi: 10.1128/JVI.75.3.1274-1283.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo PJ, Bao G. Dynamics of filamentous viral RNPs prior to egress. Nucleic Acids Res. 2007;35:3602–11. doi: 10.1093/nar/gkm246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Masuda M, Miuri R, Yoneda M, Kai C. Morbillivirus nucleoprotein possesses a novel nuclear localization signal and a CRM1-independent nuclear export signal. Virology. 2006;352:121–30. doi: 10.1016/j.virol.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Schmid A, Spielhofer P, Cattaneo R, Baczko K, ter Meulen V, Billeter MA. Subacute sclerosing panencephalitis is typically characterized by alterations in the fusion protein cytoplasmic domain of the persisting measles virus. Virology. 1992;188:910–5. doi: 10.1016/0042-6822(92)90552-z. [DOI] [PubMed] [Google Scholar]
- Schmitt AP, He B, Lamb RA. Involvement of the cytoplasmic domain of the hemagglutinin-neuraminidase protein in assembly of the paramyxovirus simian virus 5. J Virol. 1999;73:8703–12. doi: 10.1128/jvi.73.10.8703-8712.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, Lamb RA. Escaping from the Cell: Assembly and Budding of Negative-Strand RNA Viruses. Curr Top Microbiol Immunol. 2004;283:145–96. doi: 10.1007/978-3-662-06099-5_5. [DOI] [PubMed] [Google Scholar]
- Schmitt AP, Lamb RA. Influenza virus assembly and budding at the viral budozone. Adv Virus Res. 2005;64:383–416. doi: 10.1016/S0065-3527(05)64012-2. [DOI] [PubMed] [Google Scholar]
- Schmitt AP, Leser GP, Morita E, Sundquist WI, Lamb RA. Evidence for a new viral late domain core sequence, FPIV, necessary for budding of a paramyxovirus. J Virol. 2005;79:2988–97. doi: 10.1128/JVI.79.5.2988-2997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, Leser GP, Waning DL, Lamb RA. Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol. 2002;76:3952–64. doi: 10.1128/JVI.76.8.3952-3964.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Ott DE, Chertova EN, Welker R, Tessmer U, Princiotta MF, et al. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci USA. 2000;97:13057–62. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ML, Cardena WB, Zamarin D, Palese P, Basler CF. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J Virol. 2005;79:6078–88. doi: 10.1128/JVI.79.10.6078-6088.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SB, Oestreich AJ, Winistorfer S, Nguyen D, Payne JA, Katzmann DJ, et al. ESCRT ubiquitin-binding domains function cooperatively during MVB cargo sorting. J Cell Biol. 2009;185:213–24. doi: 10.1083/jcb.200811130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnyrova AV, Ayllon J, Mikhalyov II, Villar E, Zimmerberg J, Frolov VA. Vesicle formation by self-assembly of membrane-bound matrix proteins into a fluidlike budding domain. Cell Biol. 2007;179:627–33. doi: 10.1083/jcb.200705062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–72. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–9. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Spehner D, Kirn A, Drillien R. Assembly of nucleocapsidlike structures in animal cells infected with a vaccinia virus recombinant encoding the measles virus nucleoprotein. J Virol. 1991;65:6296–300. doi: 10.1128/jvi.65.11.6296-6300.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker R, Genevieve M, Roux L. The Sendai virus matrix protein appears to be recruited in the cytoplasm by the viral nucleocapsid to function in viral assembly and budding. J Gen Virol. 1994;75:1031–42. doi: 10.1099/0022-1317-75-5-1031. [DOI] [PubMed] [Google Scholar]
- Stricker R, Roux L. The major glycoprotein of Sendai virus is dispensable for efficient virus particle budding. J Gen Virol. 1991;72:1703–7. doi: 10.1099/0022-1317-72-7-1703. [DOI] [PubMed] [Google Scholar]
- Stuffers S, Brech A, Stenmark H. ESCRT proteins in physiology and disease. Exp Cell Res. 2009;315:1619–26. doi: 10.1016/j.yexcr.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Subhashri R, Shaila MS. Characterization of membrane association of Rinderpest virus matrix protein. Biochem Biophys Res Commun. 2007;355:1096–101. doi: 10.1016/j.bbrc.2007.02.088. [DOI] [PubMed] [Google Scholar]
- Sugahara F, Uchiyama T, Watanabe H, Shimazu Y, Kuwayama M, Fujii Y, et al. Paramyxovirus Sendai virus-like particle formation by expression of multiple viral proteins and acceleration of its release by C protein. Virology. 2004;325:1–10. doi: 10.1016/j.virol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- Sugihara-Mizuno Y, Adachi M, Kobayashi Y, Hamazaki Y, Nishimura M, Imai T, et al. Molecular characterization of angiomotin/JEAP family proteins: interaction with MUPP1/Patj and their endogenous properties. Genes Cells. 2007;12:473–86. doi: 10.1111/j.1365-2443.2007.01066.x. [DOI] [PubMed] [Google Scholar]
- Tahara M, Takeda M, Yanagi Y. Altered interaction of the matrix protein with the cytoplasmic tail of hemagglutinin modulates measles virus growth by affecting virus assembly and cell-cell fusion. J Virol. 2007;81:6827–36. doi: 10.1128/JVI.00248-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto T, Murti KG, Bousse T, Scroggs RA, Portner A. Role of matrix and fusion proteins in budding of sendai virus. J Virol. 2001;75:11384–91. doi: 10.1128/JVI.75.23.11384-11391.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takimoto T, Portner A. Molecular mechanism of paramyxovirus budding. Virus Res. 2004;106:133–45. doi: 10.1016/j.virusres.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Tashiro M, McQueen NL, Seto JT, Klenk H-D, Rott R. Involvement of the mutated M protein in altered budding polarity of a pantropic mutant, F1-R, of Sendai virus. J Virol. 1996;70:5990–7. doi: 10.1128/jvi.70.9.5990-5997.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro M, Seto JT, Choosakul S, Yamakawa M, Klenk H-D, Rott R. Budding site of Sendai virus in polarized epithelial cells is one of the determinants for tropism and pathogenicity in mice. Virology. 1992;187:413–22. doi: 10.1016/0042-6822(92)90443-s. [DOI] [PubMed] [Google Scholar]
- Tashiro M, Seto JT, Klenk H-D, Rott R. Possible involvement of microtubule disruption in bipolar budding of a Sendai virus mutant, F1-R, in epithelial MDCK cells. J Virol. 1993;67:5902–10. doi: 10.1128/jvi.67.10.5902-5910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagné N, et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science. 2009;326:1279–83. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- Teng MN, Collins PL. Identification of the respiratory syncytial virus proteins required for formation and passage of helper-dependent infectious particles. J Virol. 1998;72:5707–16. doi: 10.1128/jvi.72.7.5707-5716.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell DL, Norrby EJ. Structural plypeptides of measles virus. J Gen Virol. 1978;39:219–29. doi: 10.1099/0022-1317-39-2-219. [DOI] [PubMed] [Google Scholar]
- Urata S, Noda T, Kawaoka Y, Yokosawa H, Yasuda J. Cellular factors required for Lassa virus budding. J Virol. 2006;80:4191–5. doi: 10.1128/JVI.80.8.4191-4195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley TJ, Ducharme NA, Varthakavi V, Shepherd BE, Santangelo PJ, Lindquist ME, et al. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci USA. 2008;105:10209–14. doi: 10.1073/pnas.0712144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent S, Gerlier D, Manié SN. Measles virus assembly within membrane rafts. J Virol. 2000;74:9911–5. doi: 10.1128/jvi.74.21.9911-9915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waning DL, Schmitt AP, Leser GP, Lamb RA. Roles for the cytoplasmic tails of the fusion and hemagglutinin-neuraminidase proteins in budding of the paramyxovirus simian virus 5. J Virol. 2002;76:9284–97. doi: 10.1128/JVI.76.18.9284-9297.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Tanaka Y, Shimazu Y, Sugahara F, Kuwayama M, Hiramatsu A, et al. Cell-specific inhibition of paramyxovirus maturation by proteasome inhibitors. Microbiol Immunol. 2005;49:835–44. doi: 10.1111/j.1348-0421.2005.tb03672.x. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kawano M, Tsurudome M, Kusagawa S, Nishio M, Komada H, et al. Identification of the sequences responsible for nuclear targeting of the V protein of human parainfluenza virus type 2. J Gen Virol. 1996;77:327–38. doi: 10.1099/0022-1317-77-2-327. [DOI] [PubMed] [Google Scholar]
- Welliver DJ. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J Pediatr. 2003;143:S112–7. doi: 10.1067/s0022-3476(03)00508-0. [DOI] [PubMed] [Google Scholar]
- Whelan SP, Barr JN, Wertz GW. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immunol. 2004;283:61–119. doi: 10.1007/978-3-662-06099-5_3. [DOI] [PubMed] [Google Scholar]
- WHO. Nipah virus. Fact Sheet 262. 2009. [Google Scholar]
- Wirblich C, Tan GS, Papaneri A, Godlewski PJ, Orenstein JM, Harty RN, et al. PPEY motif within the rabies virus (RV) matrix protein is essential for efficient virion release and RV pathogenicity. J Virol. 2008;82:9730–8. doi: 10.1128/JVI.00889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Sun W, Suryanarayana K, Justice P, Robinson D, Wagner RR. Membrane-binding domains and cytopathogenesis of the matrix protein of vesicular stomatitis virus. J Virol. 1994;68:7386–96. doi: 10.1128/jvi.68.11.7386-7396.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo DS-Y, Chan R, Brown G, Ying L, Sutejo R, Aitken JD, et al. Evidence that selective changes in the lipid composition of raft-membranes occur during respiratory syncytial virus infection. Virology. 2009:386. doi: 10.1016/j.virol.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Nagai Y, Maeno K, Linuma M, Hamaguchi M, Matsumoto T, et al. Studies on the role of M protein in virus assembly using a ts mutant of HVJ (Sendai virus) Virology. 1979;92:139–54. doi: 10.1016/0042-6822(79)90220-4. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Nagai Y, Yoshii S, Maeno K, Matsumoto T. Membrane (M) protein of HVJ (Sendai virus): its role in virus assembly. Virology. 1976;71:143–61. doi: 10.1016/0042-6822(76)90101-x. [DOI] [PubMed] [Google Scholar]