

Abstract
Background
Activation of both the type 1 (TNFR1) and type 2 (TNFR2) tumor necrosis factor (TNF) receptor confers cytoprotection in cardiac myocytes. Noting that the scaffolding protein tumor necrosis factor receptor associated factor 2 (TRAF2) is common to both TNF receptors, we hypothesized that the cytoprotective responses of TNF were mediated through TRAF2.
Methods and Results
Mice with cardiac-restricted overexpression of low levels of TNF (MHCsTNF3) and TRAF2 (MHC-TRAF2LC), and mice lacking TNFR1, TNFR2 and TNFR1/TNFR2 were subjected to ischemia (30 min) reperfusion (30 min) injury (I/R) ex vivo, using a Langendorff apparatus. MHC-sTNF3 mice were protected against I/R injury as shown by a significant ~ 30 % greater LV developed pressure, and ~ 80% lower creatine kinase (CK) release and Evans blue dye uptake compared to littermates (LM). The extent of I/R induced injury was similar in wild-type, TNFR1 and TNFR2 deficient mice; however, mice lacking TNFR1/TNFR2 had a significant ~ 40% lower LV developed pressure, a ~ 65 % greater CK release and ~ 40% greater Evans blue dye uptake compared to LM. Interestingly, MHC-TRAF2LC mice had a significant ~ 50 % lower LV developed pressure, a ~ 70% lower CK release and ~ 80% lower Evans blue dye uptake compared to LM controls following I/R injury. Biochemical analysis of the MHC-TRAF2LC hearts showed that there was activation of NF-κB but not JNK activation.
Conclusion
Taken together these results suggest that TNF confers cytoprotection in the heart through TRAF2 mediated activation of NF-κB.
Keywords: ischemia reperfusion injury, tumor necrosis factor, tumor necrosis factor receptor associated factor 2
INTRODUCTION
Previous studies have shown that the adult mammalian heart synthesizes an ensemble of proteins that modulate cardiac repair and remodeling following myocardial infarction and/or ischemia reperfusion (I/R) injury (reviewed in ref1). Recently, we and others have suggested that cytokines belonging to the innate immune system, a phylogenetically conserved host defense system, play a critical upstream role in terms of orchestrating the early myocardial response to tissue injury. Cytokines that comprise the innate stress response in the heart include: tumor necrosis factor (TNF), interleukin-1 (IL-1), IL-6, IL-18, cardiotrophin-1 (CT-1) and leukemia inhibitory factor (LIF). Although sustained overexpression of these molecules is sufficient to provoke LV dysfunction and LV remodeling, and can thus lead to overt cardiac decompensation, 1 there is increasing evidence which suggests that this same family of proteins plays an important role in limiting myocardial cell injury, at least in part, by conferring cytoprotective responses in cardiac myocytes (reviewed in ref 1). Prior studies have shown that TNF confers cytoprotective responses in the adult heart following coronary artery ligation and I/R injury. 2–7 However, the mechanisms that are responsible for the cytoprotective effects of TNF are not known. Germane to this discussion is the observation that mice lacking both the type 1 (TNFR1) and type 2 (TNFR2) TNF receptors have an increase in infarct size following acute ligation of the left tlineanterior descending artery, whereas infarct size was similar to wild-type in both TNFR1 and TNFR2 deficient mice.4 Moreover, gain of function studies have shown that activation of TNFR1 or TNFR2 confers cytoprotection in adult cardiac myocytes subjected to hypoxia/reoxygenation injury.2 Viewed together, these results suggest that the cytoprotective effects of TNF are conveyed through TNFR1 and/or TNFR2. Although it is conceivable that TNFR1 and TNFR2 activate disparate cytoprotective signal transduction pathways, the most parsimonious explanation for these experimental observations is that TNFR1 and TNFR2 transduce a common cytoprotective repertoire in the heart. Noting that the intracellular scaffolding protein tumor necrosis factor receptor associated factor 2 (TRAF2) was common to both TNFR1 and TNFR2, and recognizing that TRAF2 mediates cytoprotection in non-myocyte cell types,8 we hypothesized that the cytoprotective effects of TNF in the heart were mediated, at least in part, through TRAF2. To begin to test this hypothesis, we have generated lines of transgenic mice that overexpress TRAF2 in the cardiac compartment. Here we show through both gain and loss of function approaches, that endogenous TNF signaling confers cytoprotection in the heart following I/R injury ex vivo, and present the novel finding that TRAF2 is sufficient to phenocopy the cytoprotective effects of TNF in the heart following I/R injury.
MATERIALS AND METHODS
Generation and characterization of transgenic and knockout mice
MHCsTNF3 transgenic mice
The hemizygous line of transgenic mice with cardiac restricted overexpression of low levels of TNF (referred to as MHCsTNF3), have been described elsewhere in detail (C57BL/6 X ICR background).9 Briefly, these mice develop mild concentric hypertrophy by 8 weeks of age. However, in contrast to transgenic lines with high levels of expression of TNF,10 the MHCsTNF do not develop LV dilation nor LV dysfunction. The observation that lower levels of TNF expression lead to a milder cardiac phenotype is consistent with the observation by Higucji et al.,11 who showed that the adverse cardiac phenotype of high levels of TNF expression is attenuated in mice with a genetic deletion of the type 1 TNF receptor. Littermate controls lacking the TNF transgene were used as the appropriate controls.
Tumor necrosis factor receptor 1 (TNFR1−/−), tumor necrosis factor receptor 2 (TNFR2−/−) and TNFR1/TNFR2 (TNFR1−/−/TNFR2−/−) null mice
Mice deficient in TNF receptor 1 (TNFR1−/−), TNF receptor 2 (TNFR2−/−), or both TNF receptors (TNFR1−/−/TNFR2−/−) were purchased from Jackson Laboratories (Bar Harbor, Maine). C57/BL6 wild-type mice were used as appropriate controls for the TNFR1−/− and TNFR2−/−, whereas B6129F2 wild-type mice were used as the appropriate controls for the TNFR1−/−/TNFR2−/− mice. The TNF receptor null mice exhibit a normal cardiac phenotype under baseline conditions.4
MHC-TRAF2 transgenic mice
Mice with cardiac restricted expression of tumor necrosis factor receptor associated factor 2 (TRAF-2) were generated for the present study using the alpha-myosin heavy chain (MHC) promoter (a gift from Jeff Robbins) to target murine TRAF2 to the cardiac myocyte (see data supplement for details).10 The MHC-TRAF2 mouse lines were characterized at 12 weeks of age using standard morphological and histological analyses, as described,10 as well as 2D-targeted M-mode echocardiography and hemodynamics using Millar catheters (see data supplement).
TRAF2 mediated signaling leads to activation of nuclear factor-kappaB (NF- κB) and c-Jun N-terminal kinase (JNK).12 Accordingly, we sought to determine whether these pathways were activated in the MHC-TRAF2LC mice using electromobility gel shift assays (EMSAs) to measure NF-κB activation, an in gel kinase assay to measure JNK activity (see data supplement for details). In preliminary control experiments (data supplement Figure S-1), we determined that the basal level of TGF-β-activated kinase (TAK1) activity, was minimal in the MHC-TRAF2LC and littermate control mice.
For all the studies reported herein, we used 10–12 week male mice. All mice were housed under standard environmental conditions and were fed commercial chow and tap water ad libitum. All studies conformed with the principles of the National Institutes of Health “Guide for the Care and Use of Laboratory Animals”, and were approved by the Baylor College of Medicine Animal Care and Use Committee.
Effects of Ischemia Reperfusion (I/R) Injury
Isolated heart perfusion studies
Hearts from MHC-sTNF3, TNFR1−/−, TNFR2−/−, TNFR1−/−/TNFR2−/−, MHC-TRAF2 and their respective littermate and/or wild-type controls, were isolated and perfused in the Langendorff mode as previously described.13 After a 30-minute stabilization period, hearts were subjected to no-flow ischemia (t = 0 min) for 30 minutes followed by reperfusion (t = 30 min) for up to 30 min (t = 60 min). All hearts were paced at 420 beats/min with pacing electrodes placed on the right atrium. Pacing was interrupted during ischemia and resumed 3 min after the start of reperfusion. Functional data were recorded at 1 kHz on a data acquisition system (PowerLab, ADInstruments). Left ventricular (LV) developed pressure (LVDP) was calculated as the difference between peak-systolic pressure and LV end-diastolic pressure.
Creatinine kinase assay
Coronary effluent was collected on ice during the first 30 minutes of reperfusion in the hearts subjected to I/R and during the last 30 min of perfusion in the control hearts. Creatine kinase (CK) activity was measured with a commercially available CK assay kit (Diagnostic Chemical, Charlottetown, PE, Canada) according to the manufacturer’s recommendations. CK activity was normalized for frozen-dry heart weight. Data are expressed as units per gram of cardiac tissue.
Evans blue staining
Because triphenyltetrazolium chloride (TTC) staining may underestimate the true extent of tissue injury within the first 3 hours of cardiac injury,14 we used Evans blue dye uptake to assess the degree of myocardial tissue injury following ischemia reperfusion injury (see data supplement). Evans blue is a cell impermeable diazo dye that has been used to study the integrity/permeability of blood vessels and cell membranes that become injured. In muscle cells with permeable membranes Evans blue dye crosses into the cell and accumulates in myofibrils, where it emits red auto-fluorescence when examined using fluorescence microscopy.15 Fluorescence microscopy (200x) was performed using a filter set with an excitation of 510 – 560 nm and an emission of 590 nm in order to assess the amount of Evans blue dye uptake in the myocardium at baseline and following I/R injury. Hearts were examined at the level of the papillary muscle, using a total of 30 microscopic fields per heart. Data are expressed as the percent area of the myocardium with red fluorescence.
Statistical Analysis
Data are expressed as a mean ± SEM. Two-way repeated measures analysis of variance (ANOVA) was used to test % LV developed pressure over time between groups. One way ANOVA was used to test for differences in CK release following reperfusion in the TNFR1−/−, TNFR2−/− and wild-type mice. Post-hoc ANOVA testing was performed, where appropriate, using the Tukey test. CK release, the area of the myocardium (%) with Evans blue uptake following I/R, and heart weight-to-body weight ratios were examined using a non-paired t-test. Significant differences were said to exist at p < 0.05.
RESULTS
Effect of cardiac restricted overexpression of TNF on I/R injury
To determine whether overexpression of low levels of TNF in the heart protects against I/R injury, we subjected hearts from MHC-sTNF3 mice to 30 minutes of no-flow ischemia followed by 30 minutes of reperfusion. Baseline LV developed pressure was not different in the MHC-sTNF3 and littermate control hearts (data not shown). Figure 1A shows that LV functional recovery was significantly greater after I/R injury in the MHC-sTNF3 mice when compared to the littermate control mice at 10, 20, and 30 minutes (p < 0.05/time) of reperfusion. To determine whether the increased functional recovery of MHC-sTNF3 hearts subjected to I/R injury was the result of decreased myocyte injury, we measured CK release and Evans blue dye uptake. There was a significant (p < 0.05) 6-fold decrease in CK release 30 minutes after I/R injury in the MHC-sTNF3 hearts when compared to littermate control hearts (Figure 1B). Similar findings were observed when Evans blue dye uptake was measured. As shown by the representative fluorescence photomicrographs (Figure 1C) and the results of group data (Figure 1D), there was a significant (p < 0.05) decrease in Evans blue dye uptake in the hearts of the MHC-sTNF3 mice 30 minutes after I/R injury. Viewed together, these results suggest that low levels of myocardial TNF protect cardiac myocytes against I/R injury ex vivo.
Figure 1.
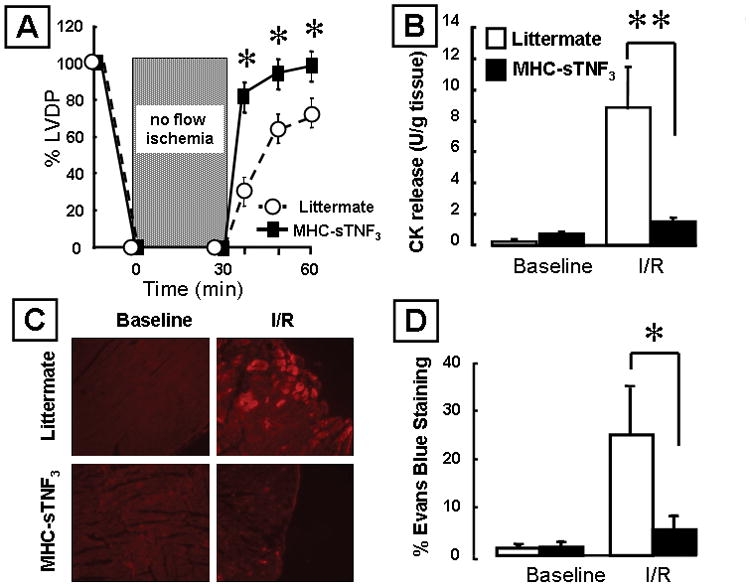
Effects of ischemia reperfusion (I/R) injury in mice over-expressing TNF in the heart (MHC-sTNF3) and littermate control hearts. (A) Percent developed left ventricular pressure (% LVDP) after I/R injury (n=5 hearts/group). (B) Creatine kinase (CK) release in the effluent at baseline and 30 minutes after I/R injury (n= 3–5 hearts/group). (C) Representative images of Evans blue dye uptake. The red coloration indicates uptake of Evans blue dye into necrotic/permeable cardiac myocytes. (D) Group data for Evans blue dye uptake at baseline and 30 minutes after I/R injury (n= 6 hearts/group). (Key *p< 0.05 and **p < 0.005 compared to littermate controls)
Effect of loss of endogenous TNF signaling on I/R injury
Acute coronary ligation results in increased cardiac myocyte cell death in TNF receptor null (TNFR1−/−/TNFR2−/−) mice that lack endogenous TNF signaling.4 To determine whether endogenous TNF receptor signaling was also important in the setting of I/R injury, we performed isolated heart I/R injury studies in TNF receptor null mice. Baseline LV developed pressure was not different in the TNF receptor null mice and littermate controls (data not shown). Figure 2A shows that LV functional recovery was significantly less at 10, 20 and 30 minutes after reperfusion injury (p < 0.05/time) in the TNF receptor null mice when compared to littermate control mice. To determine whether this decrease in functional recovery was secondary, at least in part, to increased cardiac myocyte cell injury we measured CK release and Evans blue dye uptake. There was a significant (p < 0.05) 2-fold increase in CK release in the effluent of the reperfused TNF receptor null mice (Figure 2B) that was accompanied by a significant (p < 0.05) increase in Evans blue dye uptake (Figures 2C and 2D) when compared to littermate controls.
Figure 2.
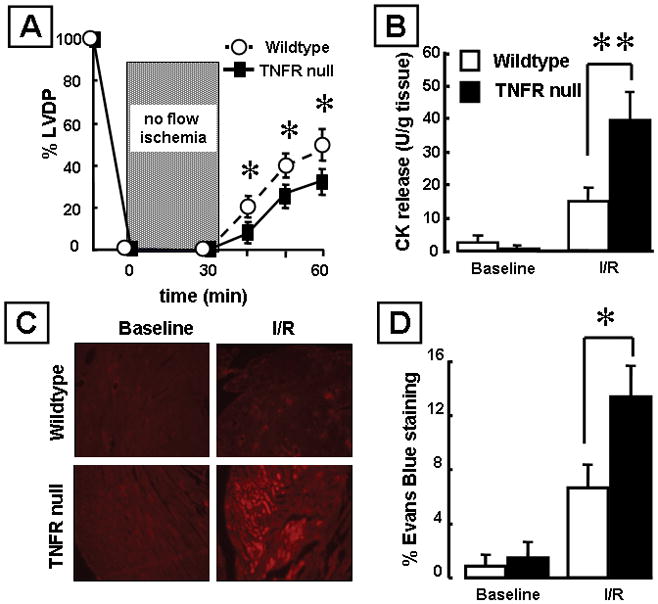
Effects of ischemia reperfusion (I/R) injury in mice null for both TNF receptors (TNFR1−/−/TNFR2−/−) and wild-type control hearts. (A) Percent developed left ventricular pressure (% LVDP) after I/R injury (n= 6 hearts/group). (B) Creatine kinase (CK) release in the effluent at baseline and 30 minutes after I/R injury (n= 5 hearts/group). (C) Representative images of Evans blue dye uptake. (D) Group data for Evans blue dye uptake at baseline and 30 minutes after I/R injury (n= 6 hearts/group). (Key *p< 0.05 and **p < 0.005 compared to littermate controls)
To determine which TNF receptors were responsible for the increased cell death in the TNFR null mice, we subjected hearts from TNFR1−/− or TNFR2−/− mice to 30 minutes no-flow ischemia followed by 30 minutes of reperfusion. Figure 3A shows that there was no significant difference (p = 0.60) in LV functional recovery in TNFR1−/− or TNFR2−/− hearts following I/R injury when compared to littermate control hearts at 10, 20 and 30 minutes of reperfusion. Similarly, the amount of myocardial CK release from TNFR1−/− or TNFR2−/− mice subjected to I/R injury was not significantly different (p= 0.90) when compared to I/R-injured littermate control hearts (Figure 3B). Taken together, these results suggest that complete loss of endogenous TNF receptor signaling in the heart leads to increased myocyte cell injury, and that endogenous signaling through either TNFR1 or TNFR2 is sufficient to protect against I/R injury ex vivo.
Figure 3.
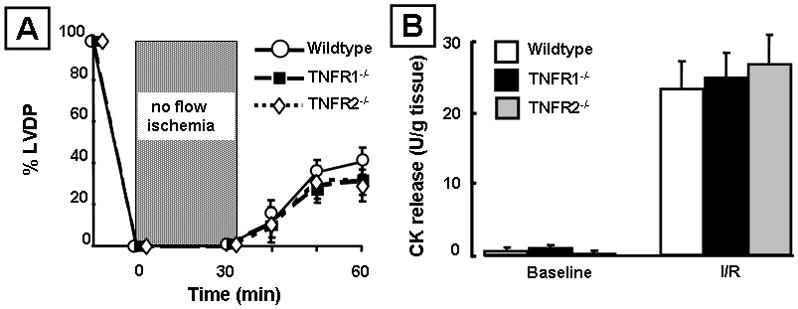
Effects of ischemia reperfusion (I/R) injury in mice lacking TNF receptor 1 (TNFR1−/−), TNF receptor 2 (TNFR2−/−) and wild-type control hearts. (A) Percent developed left ventricular pressure (% LVDP) after I/R injury (n= 6 hearts/group). (B) Creatine kinase (CK) release in the effluent at baseline and 30 minutes after I/R injury (n= 5 hearts/group).
Effects of cardiac restricted overexpression of TRAF2 on I/R injury
The results of the studies presented in Figures 1 and 2 suggest that TNF mediated signaling confers a cytoprotective signal(s) that is mediated either through the type 1 or the type 2 TNF receptor. Noting that TRAF2 was the only signaling molecule that was common to both TNF receptors, and recognizing that TRAF2 mediated signaling plays an important cytoprotective role in non-myocyte cell types,8 we sought to determine whether cardiac restricted overexpression of TRAF2 would confer cytoprotection in the setting of I/R injury ex vivo. To this end, we generated lines of transgenic mice with cardiac-restricted overexpression of TRAF2.
We obtained three founder lines: 329W (7 copies of TRAF2 transgene); 330W (15 copies of transgene); 335W (24 copies of transgene; Divakaran and Mann, manuscript in preparation). For the present study, we chose the founder line with the lowest number of copies (n= 7) of the transgene, which we refer to here as MHC-TRAF2LC. As shown in Figure 4A, TRAF2 proteins were readily detectable by Western blotting in the hearts of the MHC-TRAF2LC mice, whereas low levels of TRAF2 were present in the littermate control mice. As expected, the levels of FLAG-tagged TRAF2 proteins were readily detectable by Western blotting in the hearts of the MHC-TRAF2LC mice. Histological analysis showed that there was no obvious myofibrillar disarray nor evidence inflammatory cell infiltrates in the hearts of the hearts MHC-TRAF2LC mice when compared to littermate controls (Figure 4B). However, the heart-weight-to-body-weight ratio was significantly higher (p<0.005) in the MHC-TRAF2LC mouse hearts compared to littermate controls (Figure 4C). Further characterization of the mice using 2D-targeted M-mode echocardiography and Millar catheterization showed that there were no significant differences in LV structure between the MHC-TRAF2LC mice and control mice (data supplement Figure S-2). There were, however, small but significant differences in some indices of systolic and diastolic function in the MHC-TRAF2LC mice (data supplement Figures S-2 and S-3).
Figure 4.
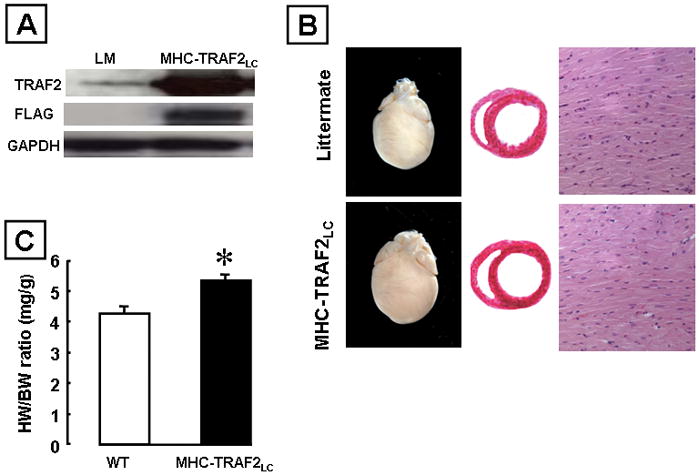
Characterization of transgenic mice expressing low levels of TRAF2 (MHC-TRAF2LC) in the cardiac compartment compared to littermate control mice. (A) Western blot analysis of hearts over-expressing flag tagged TRAF2. (B) Representative photographs of whole hearts and hematoxylin-and-eosin stained cross sections at the level of the papillary muscles and myocardial sections (x400). (C) Heart weight to body weight ratios (HW/BW) ratios. (Key *p < 0.005 compared to littermate controls)
To determine the effects of the cardiac-restricted overexpression of TRAF2 in the setting of I/R injury, we subjected MHC-TRAF2LC hearts to 30 minutes of no flow ischemia followed by 30 minutes of reperfusion. Figure 5A shows that hearts from the MHC-TRAF2LC mice had significantly (p < 0.05/time) improved LV function recovery at 10, 20 and 30 minutes of reperfusion when compared to littermate controls. Importantly, both the amount of myocardial CK release (Figure 5A) and the degree of uptake of Evans blue dye (Figures 5B and 5C) were significantly less (p < 0.005 and p < 0.05, respectively) in the I/R-injured MHC-TRAF2LC hearts when compared to littermate controls. These results suggest that TRAF2 mediated signaling is cytoprotective in the setting of I/R injury by preventing cardiac myocyte cell injury.
Figure 5.
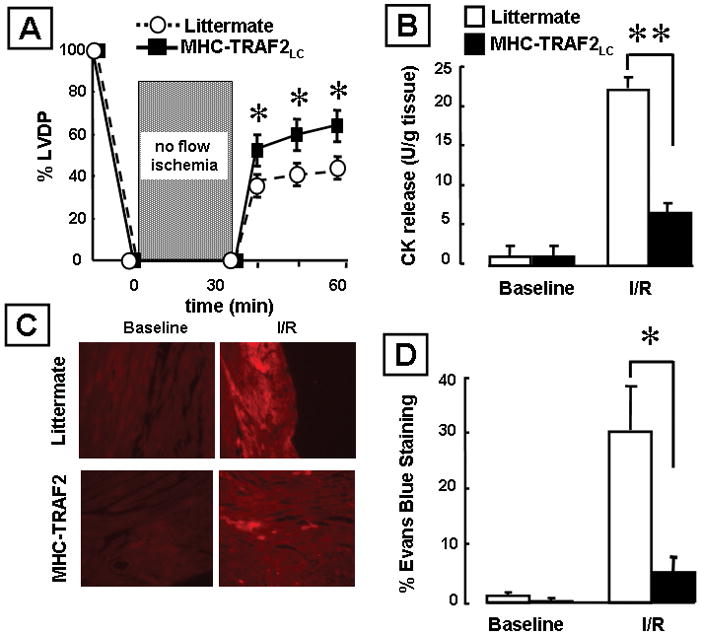
Effects of ischemia reperfusion (I/R) injury in transgenic mice expressing low levels of TRAF2 (MHC-TRAF2LC) and littermate controls. Effects of ischemia reperfusion (I/R) injury in mice over-expressing TNF in the cardiac compartment (MHC-sTNF3) and littermate control hearts. (A) Percent developed left ventricular pressure (% LVDP) after I/R injury (n= 6 hearts/group). (B) Creatine kinase (CK) release in the effluent at baseline and 30 minutes after I/R injury (n= 6 hearts/group). (C) Representative images of Evans blue dye uptake. (D) Group data for Evans blue dye uptake at 30 minutes at baseline and 30 minutes after I/R injury (n= 6 hearts/group). (Key *p< 0.05 and **p < 0.005 compared to littermate controls)
Previous studies suggest that the cytoprotective effects of TRAF2 in non-myocytes were mediated by activation of NF-κB and/or JNK.16 To determine whether these pathways were activated in the MHC-TRAF2LC hearts (12 weeks of age), we assessed NF-κB activation by EMSA and JNK activation using an in gel kinase assay. As shown, in Figure 6A the degree of NF-κB activation was greater in the MHC-TRAF2 than in littermate control hearts at baseline, as well as following I/R injury. The specificity of the DNA-protein interaction was determined by cold-chase experiments, which showed that the labeled NF-κB-DNA complexes were disrupted by a 50- fold excess of unlabeled oligonucleotide. The specificity of binding was further confirmed by supershift studies which showed that interesting finding that the nuclear extracts from the I/R hearts consisted predominately of p50- p50 homodimers. To confirm the fidelity of the p65 antibody used for these experiments, we repeated the EMSA and supershift assay in LPS treated hearts. Figure 6A shows that the p65 antibody supershifted the NF-κB complexes in the LPS hearts, suggesting that the antibody that was used was robust.
Figure 6.
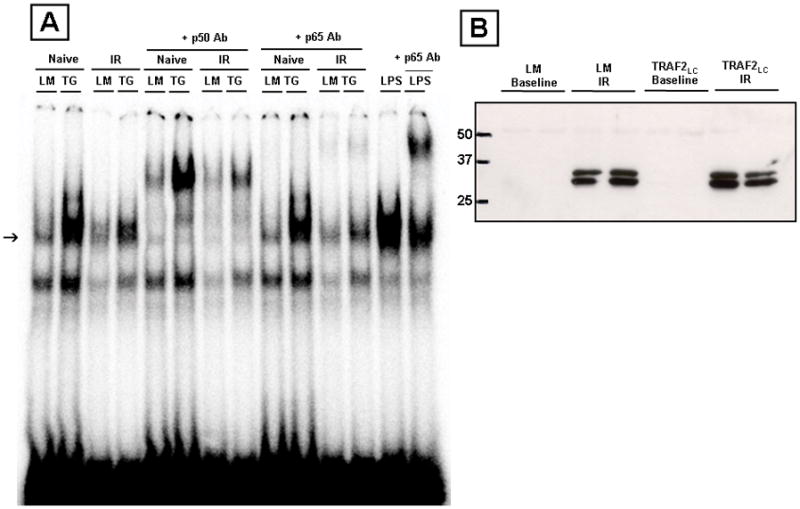
NF-κB and JNK activation in transgenic mice expressing low levels of TRAF2 (MHC-TRAF2LC) and littermate control mice. (A) Representative electromobility shift assay of NF-κB binding at baseline and after I/R injury (arrow). To determine the specificity of DNA-protein binding, nuclear extracts from TRAF2LC hearts were treated with a 20X excess of unlabelled oligonucleotides, as well as by supershift assays using polyclonal antibodies directed against the p50 and p65 components of NF-κB. The positive control for the p65 supershift was obtained from a nuclear extract from a heart of a wild-type mouse that was treated with LPS at 20mg/kg for 1 hour. (B) Representative JNK activity assay in littermate and TRAF2LC hearts (12 weeks) at baseline and after I/R injury.
Although we observed a slight increase in JNK activation in MHC-TRAF2 hearts at 4 and 8 weeks of age (data not shown), we did not observe JNK activation using in the MHC-TRAF2 hearts at 12 weeks of age (Figure 6B), consistent with the known effects of NF-κB on dampening sustained JNK mediated signaling.17 Further, JNK activity was increased to a similar degree in the littermate control and TRAF2LC mice following IR injury. Taken together, the results of these studies suggest (but do not prove) that the cytoprotective effects of TRAF2 are mediated by NF- κB rather than JNK.
DISCUSSION
The results of this study, in which we employed both gain and loss of function approaches, suggest that endogenous TNF signaling confers cytoprotection in the heart following ischemia reperfusion (I/R) injury. This study further suggests that TRAF2, a phylogenetically conserved scaffolding protein that is common to both TNFR1 and TNFR2, is sufficient to confer cytoprotective responses in the heart following I/R injury. Three distinct lines of evidence support these statements. First, LV functional recovery was significantly better and the extent of LV tissue injury was significantly less following I/R injury in lines of transgenic mice (MHC-sTNF3) with low levels of expression of TNF when compared to littermate control mice (Figure 1). Second, loss of endogenous TNF signaling in mice that were null for TNF receptor signaling resulted in significantly worse LV functional recovery and significantly greater LV tissue injury following I/R injury when compared to littermate control mice with endogenous TNF receptor signaling intact (Figure 2), consistent with our prior in vivo observations in TNF receptor null mice subjected to acute coronary artery occlusion.4 To determine which TNF receptors were responsible for conveying cytoprotective responses in the heart, we subjected hearts from TNFR1−/− or TNFR2−/− mice to I/R injury. Interestingly, both LV recovery and myocardial CK release were similar in wild-type, TNFR1−/− and TNFR2−/− mice subjected to I/R injury (Figures 3A, 3B), suggesting that signaling through either TNFR1 or TNFR2 alone is sufficient to protect adult murine cardiac myocytes following I/R injury. Third, a transgenic mouse line (MHC-TRAF2LC) with low levels of cardiac restricted expression of TRAF2, resulted in significantly improved LV functional recovery and significantly less LV tissue injury following I/R injury when compared to littermate control mice with wild-type levels of endogenous TRAF2 expression (Figure 5A). Although the scope of the present study was not intended to define the cytoprotective signaling pathways that were downstream from TRAF2-mediated signaling, our provisional results suggest an important role for p50-p50 NF-κB homodimers. Insofar as p50 does not contain a transactivation domain, and is therefore considered to be transcriptionally inactive, it bears emphasis that p50/p50 homodimers can inhibit transcription by displacing transcriptional activators (e.g. Sp1 or p50/p65 heterodimers) from NF-κB binding sites, or can act as a transcriptional activator by displacing repressor transcriptional factors from from NF-κB binding sites.18,19 Additional studies will be required to determine the ensemble of cytoprotective genes that are modulated by TRAF2 activation.
Cytoprotective Effects of Tumor Necrosis Factor
The results of the present study both confirm and expand upon prior in vitro,2 ex vivo3,7,20,21 and in vivo4–6 studies which suggest that TNF confers cytoprotective signaling in the adult mammalian heart. Thus far, three mechanisms have been proposed in order to explain the cytoprotective effects of TNF in the heart. In models of ischemic preconditioning it has been suggested that TNF-induced activation of the neutral sphingomyelinase pathway confers cytoprotection through generation of ceramide and/or sphingosine 1-phosphate.5 A second mechanism that has been proposed with respect to TNF mediated pre-conditioning suggests that the cytoprotective effects of TNF are mediated through activation of the JAK-STAT (Janus kinase/Signal Transducers and Activators of Transcription) pathway, insofar as TNF-induced pre-conditioning was sensitive to pharmacologic inhibition of the JAK/STAT pathway.21 Similar results were reported recently by another group of investigators who observed decreased LV myocardial recovery post-ischemia, as well as decreased mRNA and protein levels of STAT3 and SOCS3 (supressor of cytokine signaling) in TNFR2−/− deficient mice. 22 The results of the current study suggest that TNF-TRAF2 mediated activation of NF-κB is responsible for provoking cytoprotective responses in the heart following I/R injury (Figure 6). While we cannot formally exclude that TRAF2-induced activation of JNK signaling contributed to the observed cytoprotection, our results suggest that this signaling pathway is down-regulated in chronic TRAF2 signaling (Figure 6), consistent with the observation that NF-κB suppresses JNK signaling, 23 and that the increase in JNK activation following I/R injury is not different in littermate control and MHC-TRAF2LC mice. Although our findings with respect to TRAF2 mediated activation of NF-κB appear to conflict with the aforementioned studies that suggest a cytoprotective role for free radicals and/or JAK/STAT signaling, it is important to recognize that free radicals activate NF-κB, and that there is cross talk between JAK signaling and NF-κB activation. 24,25 Thus, it is possible the NF-κB may represent a final common pathway that allows for the convergence of multiple upstream TNF induced cytoprotective signaling pathways. Further studies will be necessary to determine whether these pathways converge on the canonical NF-κB pathway that is activated via TRAF2, as well as whether these effects are mediated via p50-p50 homodimers.
The thesis that NF-κB mediates the cytoprotective effects of TNF is consistent with a prior study from this laboratory, wherein we showed an increase in infarct size following acute coronary occlusion in mice harboring a dominant negative IkBα construct in which the critical phosphorylation (serine-32 and serine-36) and ubiquitination sites (lysine-21 and lysine-22) were removed, thereby rendering the mutant protein resistant to proteolytic degradation.26 This point of view is also consistent with previous studies that have suggested a cytoprotective role for NF-κB in hypoxic cardiac myocytes in vitro16 and in late pre-conditioning in vivo. 27–29 Our findings are also consistent with a recent study which showed that there was adverse post-infarct cardiac remodeling in mice with targeted disruption of p50.30 Nonetheless, our findings would appear to conflict with a series of well done studies in models of cardiac I/R and/or coronary ligation, which have shown salutary effects by inhibiting NF-κB, either through an intracoronary infusion of a synthetic (double-stranded DNA) NF-κB decoy, 31 through pharmacologic inhibition of IKKβ,32 through genomic deletion of p50,33 or using a genetically engineered mouse that expressed a cardiac restricted mutated dominant negative IkBα (Ser32Ala,Ser 36Ala,Tyr42Phe) “3M” construct.34 While the immediate explanation for these disparate views with respect to the role of NF-κB in cardiac injury is not obvious, it may relate, at least in part, to differences in spatial NF-κB activation in the cardiac injury models that were studied. For example, the use of oligonucleotide decoys, pharmacologic inhibition of IKKβ, or genomic deletion of p50 would be expected to inhibit NF-κB activation in cardiac myocytes, fibroblasts, smooth muscle cells, as well as endothelial cells, whereas the cardiac restricted expression of IκBα dominant negative would be expected to inhibit NF-κB activation in cardiac myocytes alone. Thus, activation of NF-κB in the vascular compartment during I/R injury might result in increased influx of inflammatory cells and/or increased activation of serum proteases (e.g., complement), and hence increased tissue damage that would offset the cytoprotective effects mediated by NF-κB in cardiac myocytes. Further, we cannot exclude the formal possibility that differences in the genetic strain of the mice used may have influenced the results.
Conclusion
In summary, the results of this study suggest that TNF mediated signaling confers cytoprotection in the heart through TRAF2 mediated activation of NF-κB. Apart from the novel mechanistic insights provided by this report, the results of this study have also identified a potentially important role for TRAFs in the heart. Given that TRAF1, 2 and 6 are expressed in the heart, 35 and that TRAF proteins are involved in mediating inflammation, cell signaling, antirviral responses and apoptosis, it is likely that TRAFs will have a variety of pleiotropic effects in the heart. Accordingly, further studies will be necessary to delineate the potentially important role that these proteins play in the heart in health and disease.
Supplementary Material
Acknowledgments
Sources of Funding: This research was supported by research funds from the N.I.H. (RO1 HL58081, RO1 HL61543, RO1 HL-42250, HL 076661, and HL 089792)
Footnotes
Disclosures
None.
References
- 1.Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol. 3 A.D;65:81–101. doi: 10.1146/annurev.physiol.65.092101.142249. [DOI] [PubMed] [Google Scholar]
- 2.Nakano M, Knowlton AA, Dibbs Z, Mann DL. Tumor necrosis factor-α confers resistance to injury induced by hypoxic injury in the adult mammalian cardiac myocyte. Circulation. 1998;97:1392–1400. doi: 10.1161/01.cir.97.14.1392. [DOI] [PubMed] [Google Scholar]
- 3.Eddy LJ, Goeddel DV, Wong GHW. Tumor necrosis factor-α pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem Biophys Res Commun. 1992;184:1056–1059. doi: 10.1016/0006-291x(92)90698-k. [DOI] [PubMed] [Google Scholar]
- 4.Kurrelmeyer K, Michael L, Baumgarten G, Taffet G, Peschon J, Sivasubramanian N, Entman ML, Mann DL. Endogenous myocardial tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci U S A. 2000;290:5456–5461. doi: 10.1073/pnas.070036297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lecour S, Smith RM, Woodward B, Opie LH, Rochette L, Sack MN. Identification of a novel role for sphingolipid signaling in TNF alpha and ischemic preconditioning mediated cardioprotection. J Mol Cell Cardiol. 2002;34:509–518. doi: 10.1006/jmcc.2002.1533. [DOI] [PubMed] [Google Scholar]
- 6.Deuchar GA, Opie LH, Lecour S. TNFalpha is required to confer protection in an in vivo model of classical ischaemic preconditioning. Life Sci. 2007;80:1686–1691. doi: 10.1016/j.lfs.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Lecour S, Rochette L, Opie L. Free radicals trigger TNF alpha-induced cardioprotection. Cardiovasc Res. 2005;65:239–243. doi: 10.1016/j.cardiores.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–725. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 9.Sekiguchi K, Tian Q, Ishiyama M, Burchfield J, Gao F, Mann DL, Barger PM. Inhibition of PPARα activity in mice with cardiac-restricted expression of Tumor Necrosis Factor: potential role of TGFβ/Smad3. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.01056.2006. [DOI] [PubMed] [Google Scholar]
- 10.Sivasubramanian N, Coker ML, Kurrelmeyer K, DeMayo F, Spinale FG, Mann DL. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;2001:826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 11.Higuchi Y, McTiernan CF, Frye CB, McGowan BS, Chan TO, Feldman AM. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation. 2004;109:1892–1897. doi: 10.1161/01.CIR.0000124227.00670.AB. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 13.Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, Tacheuchi O, Akira S, Mann DL. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292:H503–H509. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 14.Birnbaum Y, Hale SL, Kloner RA. Differences in reperfusion length following 30 minutes of ischemia in the rabbit influence infarct size, as measured by triphenyltetrazolium chloride staining. J Mol Cell Cardiol. 1997;29:657–666. doi: 10.1006/jmcc.1996.0308. [DOI] [PubMed] [Google Scholar]
- 15.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regula KM, Baetz D, Kirshenbaum LA. Nuclear factor-kappaB represses hypoxia-induced mitochondrial defects and cell death of ventricular myocytes. Circulation. 2004;110:3795–3802. doi: 10.1161/01.CIR.0000150537.59754.55. [DOI] [PubMed] [Google Scholar]
- 17.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 18.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I, Scheidereit C. Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Mol Cell Biol. 1998;18:1266–1274. doi: 10.1128/mcb.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson SK, Wong GHW, McCord JM. Leukemia inhibitory factor and tumor necrosis factor induce manganese superoxide dismutase and protect rabbit hearts from reperfusion injury. J Mol Cell Cardiol. 1995;27:223–229. doi: 10.1016/s0022-2828(08)80021-1. [DOI] [PubMed] [Google Scholar]
- 21.Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, Huisamen B, Opie LH. Pharmacological preconditioning with tumor necrosis factor-alpha activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal-regulated kinase) Circulation. 2005;112:3911–3918. doi: 10.1161/CIRCULATIONAHA.105.581058. [DOI] [PubMed] [Google Scholar]
- 22.Wang M, Crisostomo PR, Markel TA, Wang Y, Meldrum DR. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation. 2008;118:S38–S45. doi: 10.1161/CIRCULATIONAHA.107.756890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D’Adamio L, Franzoso G. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6:146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 24.Jones WK, Brown M, Ren X, He S, McGuinness M. NF-kappaB as an integrator of diverse signaling pathways: the heart of myocardial signaling? Cardiovasc Toxicol. 2003;3:229–254. doi: 10.1385/ct:3:3:229. [DOI] [PubMed] [Google Scholar]
- 25.Digicaylioglu M, Lipton SA. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature. 2001;412:641–647. doi: 10.1038/35088074. [DOI] [PubMed] [Google Scholar]
- 26.Misra A, Haudek SB, Knuefermann P, Vallejo JG, Chen ZJ, Michael LH, Sivasubramanian N, Olson EN, Entman ML, Mann DL. Nuclear factor-kappaB protects the adult cardiac myocyte against ischemia-induced apoptosis in a murine model of acute myocardial infarction. Circulation. 2003;108:3075–3078. doi: 10.1161/01.CIR.0000108929.93074.0B. [DOI] [PubMed] [Google Scholar]
- 27.Maulik N, Sato M, Price BD, Das DK. An essential role of NFkappaB in tyrosine kinase signaling of p38 MAP kinase regulation of myocardial adaptation to ischemia. FEBS Lett. 1998;429:365–369. doi: 10.1016/s0014-5793(98)00632-2. [DOI] [PubMed] [Google Scholar]
- 28.Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ, Bolli R. Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res. 1999;84:1095–1109. doi: 10.1161/01.res.84.9.1095. [DOI] [PubMed] [Google Scholar]
- 29.Zhao TC, Kukreja RC. Late preconditioning elicited by activation of adenosine A(3) receptor in heart: role of NF- kappa B, iNOS and mitochondrial K(ATP) channel. J Mol Cell Cardiol. 2002;34:263–277. doi: 10.1006/jmcc.2001.1510. [DOI] [PubMed] [Google Scholar]
- 30.Timmers L, Henriques JP, de Kleijn DP, Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek JJ, Doevendans PA, Pasterkamp G, Hoefer IE. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol. 2009;53:501–510. doi: 10.1016/j.jacc.2008.10.033. [DOI] [PubMed] [Google Scholar]
- 31.Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- 32.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, Muto S, Itai A, Isobe M. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;63:51–59. doi: 10.1016/j.cardiores.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Frantz S, Hu K, Bayer B, Gerondakis S, Strotmann J, Adamek A, Ertl G, Bauersachs J. Absence of NF-kappaB subunit p50 improves heart failure after myocardial infarction. FASEB J. 2006;20:1918–1920. doi: 10.1096/fj.05-5133fje. [DOI] [PubMed] [Google Scholar]
- 34.Brown M, McGuinness M, Wright T, Ren X, Wang Y, Boivin GP, Hahn H, Feldman AM, Jones WK. Cardiac-specific blockade of NF-kappaB in cardiac pathophysiology: differences between acute and chronic stimuli in vivo. Am J Physiol Heart Circ Physiol. 2005;289:H466–H476. doi: 10.1152/ajpheart.00170.2004. [DOI] [PubMed] [Google Scholar]
- 35.Brink R, Lodish HF. Tumor necrosis factor receptor (TNFR)-associated factor 2A (TRAF2A), a TRAF2 splice variant with an extended RING finger domain that inhibits TNFR2-mediated NF-kappaB activation. J Biol Chem. 1998;273:4129–4134. doi: 10.1074/jbc.273.7.4129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.