

Abstract
Point mutations in β-glucocerebrosidase (GCase) can result in a deficiency of both GCase activity and protein in lysosomes thereby causing Gaucher Disease (GD). Enzyme inhibitors such as isofagomine, acting as pharmacological chaperones (PCs), increase these levels by binding and stabilizing the native form of the enzyme in the endoplasmic reticulum (ER), and allow increased lysosomal transport of the enzyme. A high-throughput screen of the 50 000-compound Maybridge library identified two, non-carbohydrate-based inhibitory molecules, a 2,4-diamino-5-substituted quinazoline (IC50 5 μM) and a 5-substituted pyridinyl-2-furamide (IC50 8 μM). They raised the levels of functional GCase 1.5–2.5-fold in N370S or F213I GD fibroblasts. Immunofluorescence confirmed that treated GD fibroblasts had decreased levels of GCase in their ER and increased levels in lysosomes. Changes in protein dynamics, monitored by hydrogen/deuterium-exchange mass spectrometry, identified a domain III active-site loop (residues 243–249) as being significantly stabilized upon binding of isofagomine or either of these two new compounds; this suggests a common mechanism for PC enhancement of intracellular transport.
Keywords: chaperones, enzymes, glucocerebrosidase, high-throughput screening, hydrogen/deuterium exchange mass spectrometry
Introduction
Gaucher Disease (GD; MIM 230 800, 230 900, 2301 000) is the most common of the ~70 lysosomal storage diseases known.[1, 2] It is an autosomal recessive multisystem disorder with a high level of morbidity, and in severe cases is fatal at an early age. The biochemical hallmark of GD is the storage of glucosylceramide (GC), the precursor of 95 % of all cellular glycosphingolipids, primarily in the tissues of the reticuloendothelial system and the brain arising from deficiency of lysosomal β-glucocerebrosidase (GCase, EC 3.2.1.45) encoded by the GBA gene. Although the disorder represents a broad and continuous spectrum of clinical involvement, three main clinical phenotypes are generally recognized: type I, nonneuronopathic; II, acute neuronopathic: and III, subacute neuronopathic.[3] Type I GD (incidence 1/40 000–1/60 000) accounts for the bulk of the patients, who are generally mildly affected. The highest carrier frequency of type I GD occurs amongst Ashkenazi Jewish adults (1/11) with ~90% of these individuals carrying one of just four alleles—that is, N370S, F213I, L444P, or G202R.[4, 5] The N370S mutation alone accounts for 75% of these alleles.
Type I GD patients (N370S heterozygotes/homozygotes) have residual enzyme activity levels that are ~5–20 % of normal;[1, 6,7] this closely matches the critical threshold level of 11–15 % of normal activity required to prevent the storage of GC, which was determined using a murine macrophage cell line treated with conduritol-B-epoxide (CBE), an irreversible inhibitor of Gcase, as a model of type I GD.[6] Thus, like other lysosomal storage disorders, it appears that only a relatively small increase in GCase activity is necessary to prevent and/or reverse the clinical progression of the disease.
Type I[8] and to a lesser extent type II and III forms of GD[9,10] currently benefit from two existing therapeutic approaches. These include: 1) enzyme replacement therapy (ERT) and 2) substrate reduction therapy. ERT ameliorates many manifestations of GD and is both a safe and effective treatment. However, it is very costly at ~$ 200 000 per year for an average 70 kg adult.[11] SRT attempts to limit the storage of GC by using small molecules to inhibit its synthesis in vivo. Currently the only FDA-approved SRT-agent is N-butyl-deoxynojirimycin (NB-DNJ) (Miglustat or Zavesca®), which inhibits the first step in glycolipid synthesis and has shown some promise in treating GD type I. However, it is not as effective as ERT,[12] and the treatment is associated with unpleasant side effects, for example, severe diarrhea. Currently, a new therapeutic strategy, enzyme enhancement therapy (EET), is being evaluated in Phase I and II clinical trials. EET uses small molecule “pharmacological chaperones” (PCs) to stabilize the native conformation of a mutant enzyme as it folds in the endoplasmic reticulum (ER), allowing it to pass the ER quality control system (ER-QC) and avoiding the ER associated degradation system (ERAD), and be transported to the lysosome.[13,14] EET has shown promising preclinical results in at least four lysosomal enzyme deficiencies and could be applied to other lysosomal storage disorders.[15–18] To date successful PCs have also been competitive inhibitors of their target enzymes.[19] It is believed that once the PC–enzyme complex reaches the lysosome, the large amounts of stored substrate(s) will displace the PC and continue to stabilize the enzyme.[16] However, it is desirable to identify PCs that are most active at the neutral pH of the ER, in order to optimize binding strength and thus their ability to stabilize the folding process, and minimize their inhibitory properties once the complex enters the acidic environment of the lysosome, where stored substrate should continue to stabilize the enzyme.
Although ERT has been successfully used to treat type I GD patients, there are benefits to considering other therapeutic modalities such as SRT or EET. These could be used in lieu of or in combination with ERT. Small molecules are less expensive, can be given orally and usually cross the blood-brain barrier, which opens up the possibility of treating type II and III GD patients. As EET augments transit of the mutant GCase from the ER,[20–22] it also has the potential to attenuate the unfolded protein response and prevent ER stress that can lead to apoptosis and other inflammatory responses.[23] Recently, components of the ER-QC system have been implicated as factors involved in determining the clinical impact of GCase mutations.[24,25]
The degree to which the different GCase PCs enhance intra-cellular enzyme levels depends on the nature of the particular mutation.[26,27] For example, the GCase PC, N-octyl valienamine chaperones the F213I mutation better than the N370S mutation.[26] Overall the G202R substitution is most responsive to chaperoning, whereas the L444P mutation, associated with the neuronopathic form of GD in the homozygous form, thus far remains refractory to EET.[27,28] However, the intracellular activity of the L444P and G202R mutations can be increased by growing patients’ cells at a decreased temperature of 30°C;[27] this suggests that L444P might be “chaperoned” by other, as yet to be identified, compounds.
To date most Gaucher PCs consist of glucose-based azasu-gars either with an alkylated side chain, for example, NB-DNJ[29] or N-nonyl-deoxynojirimycin (NN-DNJ)[13] and derivatives thereof,[27] or without an alkylated side chain, for example, isofagomine (IFG).[20] IFG is currently undergoing Phase I and II clinical trials sponsored by Amicus Therapeutics (http://www.amicustherapeutics.com/clinicaltrials/at2101.asp). Although IFG is a nanomolar inhibitor, GCase activity is increased more than two-fold when GD type I patient fibroblasts are treated with 10–100 μM concentration of the compound. Other more potent and selective GCase inhibitors such as α-1-C-nonyl-1,5-dideoxy-1,5-imino-D-xylitol, with a Ki value of 2 nM, and 6-nonyl IFG, with an IC50 value of and 0.6 nM, have been described that also more than double GCase residual activity in Gaucher patients fibroblasts but act at nanomolar concentrations.[30, 31]
The mechanism by which NB-DNJ, NN-DNJ or IFG-binding stabilizes the wild-type enzyme has been explored by X-ray crystallography at acidic and/or neutral pH.[32,33] The general consensus is that residues from three loops (residues 311–319, 342–354 and 393–396), surrounding the substrate-binding pocket are stabilized upon binding of the glycone moiety of these PCs. The most striking finding of the crystallization studies was that PC-binding preferentially stabilizes a helical-turn conformation within a loop region located at the mouth of the active site (residues 311–319). It is proposed that the helical-like conformation is important for the chaperoning activity of IFG.[32] However, crystal, interchain or intermolecular contacts that occur solely as the result of protein crystallization, could have obscured the identification of additional regions of importance in chaperone-enhanced intracellular transport.
Hydrogen/deuterium exchange coupled with mass spectrometry (H/D-Ex) has been used to probe protein dynamics in solution in the presence and absence of ligand.[34] This procedure has been used to map ligand binding sites and to detect ligand-induced conformational and/or dynamic changes of a protein. Using this approach Kornhaber et al.[35] have detected changes in protein dynamics in several regions of GCase following IFG binding. Five of these regions, 119–127, 177–184, 230–240, 310–312 and 386–400, are consistent with the locations of residues involved in PC binding as determined by crystallography.[32,33] However, additional perturbations observed in regions 187–197, 243–249 and 414–417 were not previously seen. These results highlight the importance of examining the structural dynamic properties of GCase-PC complexes in solution.
We previously demonstrated that high-throughput screening (HTS) of large compound libraries of drug-like molecules for inhibitors of β-N-acetyl hexosaminidase (Hex) can identify non-carbohydrate (for example, non-iminosugar) based candidate PCs for Tay-Sachs disease.[36,37] Hits were subsequently verified in a cell-based assay for PC activity.[36] This approach has been applied to GD by Zheng et al. to identify three classes of GCase inhibitors.[38] To identify additional novel frameworks for GCase inhibitors we have screened a different library of small drug-like molecules, the 50000 compound Maybridge library, for inhibitors of purified GCase. The availability of additional frameworks for GCase inhibitors that also function as PCs could potentially increase the repertoire of GBA mutations responding to EET. Additionally, the examination of the effects that the binding of non-carbohydrate based PCs to GCase has on protein dynamics might be helpful in identifying the relevant region(s) of GCase that when stabilized, increase its intra-cellular transport efficiency.
Utilizing the above HTS strategy, two novel GCase inhibitors that functioned as PCs in cell-based assays were identified in the Maybridge library. Their effect on the conformational dynamics of wild-type GCase was determined by H/D-Ex, which revealed a single common region in GCase that was stabilized upon binding of IFG or either of these other two Maybridge compounds.
Results
Primary screen for identification of GCase inhibitors
Non-carbohydrate based PCs for GCase mutants were indirectly identified by first completing a primary high-throughput screen of the small molecule, drug-like Maybridge library. Inhibitors were identified through their ability to reduce hydrolysis of methylumbelliferyl-β-D-glucopyranoside (MUGlc), to the fluorogenic product methylumbelliferone (MU) by purified GCase. To evaluate the signal-to-noise ratio, the Z-statistic,[39] based on the activity of the enzyme in the presence the compound diluent, dimethyl sulfoxide (DMSO, high control), as compared to a known inhibitor, castanospermine (low control), was calculated. The resulting value of 0.75 indicated very good separation of the high and low controls. Following screening of 49586 compounds for activity against GCase, 680 hits were obtained based on a cut-off of three standard deviations from the mean of the activity. To facilitate screening of the hits in a secondary screen, the hit zone was empirically lowered to 30% of the mean, resulting in 108 hits.
Secondary screen to validate PC activity of each hit
Three distinct characteristics of each hit were evaluated in the secondary screens using four assays and six different concentrations of the candidate compound (Figure 1). The characteristics evaluated for each hit were: 1) IC50, 2) ability to attenuate heat denaturation and 3) ability to function as specific, nontoxic PCs in GD cells. Each of the 108 hits from the primary screen showed a dose response curve with IC50 values ranging from single digit to more than 100 μM. (26 compounds had IC50 values ranging from 0.7–9.9 μM, 49 had IC50 values ranging between 10–50 μM, 16 hits had IC50 values between 50 and 100 μM and 17 compounds had IC50 values greater than 100 μM). We previously showed that inhibitory compounds identified by HTS that functioned as chaperones also increased the heat stability of the wild-type Hex.[36] Furthermore, N-substituted derivatives of deoxynojirimycin that function as PC have been shown to increase the thermostability of both wild-type GCase and N370S GCase.[39] Only 49 of the 108 hits attenuated the thermal denaturation of GCase to varying degrees. The remaining 59 hits were excluded as candidate PCs because 46 of the compounds had no effect and 13 resulted in increased thermal denaturation. Finally, to control for specificity and toxicity, changes in activity levels of both mutant intracellular GCase and wild-type Hex were monitored following treatment of GD cells with varying concentrations of each hit. After treating GD (N370S/N370S) cells for five days with each of the remaining hits, ~20 % produced a ≥1.4-fold increase in GCase activity relative to cells treated with DMSO. Of these 21 compounds, only two, MWP01127 (compound 1) and MAC1753 (compound 2) showed increased GCase activity over two concentration ranges and did not affect the activity of Hex (used as an indicator of toxicity) at the corresponding concentration (Figure 1). (Throughout this article compound 2 is referred to by the local library code, MAC1753, rather than the actual May-bridge library code, HTS 02324, so as to limit confusion with the acronym for high-throughput screening.)
Figure 1.

Screening strategy used to identify two GCase inhibitors in the Maybridge library of small, drug-like molecules. Firstly, each of 50 000 compounds was evaluated for its ability to reduce the activity of GCase to less than 30 % of that obtained from a DMSO control. Secondly, the 108 compounds (hits) were confirmed in a secondary inhibitory screen. Thirdly, each hit was evaluated for three characteristics by using four assays (y-axes are GCase activity relative to a DMSO control, that is, 1 =no change in activity; and x-axes represent the concentration [μM] of each compound used in the reaction). A) inhibition assay to confirm and determine IC50 values (0.8 mM MUGlc); B) heat denaturation attenuation assay: remaining GCase activity in the presence of the compound following heating (50 °C for 20 min); C) and D) changes in intracellular GCase and Hex activity levels in GD patient fibroblasts (N370S/N370S). Cells were treated for five days with the indicated concentration of test compound, and the activities were then measured. Curves for the two lead compounds, as well as for six other selected compounds, are shown. Finally, two lead compounds were selected for further study on the basis of their ability to increase GCase activity in treated patients’ cells without affecting Hex activity levels. The complete data set for all 108 hits is available as Table S1 (structures of hits) and Figure S1 (Inhibitory activity, attenuation thermal denaturation and intracellular GCase/Hex activity) in the Supporting Information.
Structure and selectivity of the lead GCase inhibitory compounds
The two lead compounds, 1 and 2, were found to be 5-((4-methylphenyl)thio)quinazoline-2,4-diamine and 5-(3,5-dichlorophenoxy)-N-(4-pyridinyl)-2-furamide, respectively (Figure 1). Although reminiscent of GCase inhibitory compounds consisting of nitrogen containing heterocycles,[38,40] 5-substituted 2,4-dia-minoquinazolines or 5-substituted pyridinyl-2-furamides have not been previously described as GCase inhibitors. Using the colourimetric substrate p-nitrophenyl-β-D-glucopyranoside (pNPGlc), 1 and 2 were found to be low micromolar inhibitors of GCase with IC50 values of 7.8 and 4.7 μM, respectively (Table 1). By comparison, IFG, a known carbohydrate-based GCase inhibitor, was found to have an IC50 of 30 nM using pNPGlc. Whereas 2 shows no inhibition towards other lysosomal enzymes such as human β-galactosidase (β-Gal), α-glucosidase (α-Glc) and Hex, 1 and IFG show detectable activity against these enzymes, albeit at concentrations more than a 100-fold higher. Both 1 and IFG also showed activity against human cytosolic β-glucosidase that also hydrolyses glucosylceramide and whose catalytic domain also consists of a (β/α)8 TIM barrel.[41] While both compounds 1 and IFG also enhanced GCase activity in N370S/N370S patient cells at 12.5 μM (Figure 1 and[32]), this value is below the 50 μM IC50 of 1 for neutral β-glucosidase but greater than the corresponding 1 μM IC50 value of IFG for this enzyme. Additionally both IFG and 1 inhibit almond β-glucosidase. However, while the IC50 of IFG towards the almond enzyme is nearly identical to that of the human enzyme, the IC50 of 1 for almond β-glucosidase is increased 24-fold relative to human GCase. In contrast, 2 is virtually non-inhibitory towards either the almond enzyme or the neutral β-glucosidase (Table 1). Thus, 1 and IFG have similar inhibitory profiles.
Table 1.
Specificity of GCase inhibitory compounds.
Compounds 1 and 2 increase GCase protein levels in the lysosomes of GD cells
The effect of 1 and 2 on GCase and Hex levels in N370S/N370S patient fibroblasts was examined over a larger range of concentrations. Both compounds showed signs of toxicity at concentrations greater than 30 μM, as indicated by the parallel decrease in intracellular GCase and Hex activities (Figure 2A, B). Although maximum increase in GCase activity in patient cells was seen at a concentration of 12.5 μM for both compounds, 1 (Figure 2A, C) treatment resulted in a 2.5-fold increase in enzyme activity in comparison to the 1.5 fold rise seen with 2 (Figure 2B, C). In order to compare the efficacy of the compounds in patient fibroblasts with different GCase mutant alleles, the effect of IFG (25 μM), 1 (12.5 μM) or 2 (12.5 μM) on the F213I allele, more commonly found in GD patients of Asian descent, was examined. In these cell lines the efficacy of the two compounds was reversed relative to similarly treated GD cells expressing the N370S GCase allele. Treatment with 2 resulted in a 2.4-fold increase in GCase activity as compared to a 1.6-fold elevation observed in compound 1-treated cells (Figure 2C). The cellular localization of the enhanced GCase activity observed in 1- or 2-treated GD cells was probed by preparing lysosome-enriched fractions and examining their GCase and lysosomal-associated membrane protein-2 (Lamp-2) levels by Western blotting. A clear enrichment in GCase protein with little change in Lamp-2 levels was observed in the treated versus untreated cells (Figure 2D).
Figure 2.

Changes in GCase and Hex activity in GD patient fibroblasts after treatment with 1 or 2. A), B) GD patient cells carrying the N370S/N370S alleles were treated with 1 or 2 for five days. Activity levels are relative to cells treated with solvent only (DMSO), that is, a y-axis value of 1 indicates no change. Hex activity levels serve as a control for toxicity. Standard deviation (n =3) is shown for each point. C) Relative changes in GCase (black bars) and Hex (gray bars) activity following treatment of either N370S/N370S or F213I/L444P GD cells with IFG (25 μM), 1 (12.5 μM) or 2 (12.5 μM). D) 1 and 2 increase the levels of lysosomal GCase in N370S/N370S Gaucher patient fibroblasts. The iron–dextran colloid method was used to prepare a lysosome-enriched fraction from N370S/N370S GD cells treated with compounds 1 (12.5 μM) or 2 (12.5 μM) or vehicle (0.1 % DMSO). GCase or the lysosomal marker Lamp-2 were visualized by Western blotting.
1 and 2 are mixed-type inhibitors of GCase and are most efficient at neutral pH
The changes in apparent KM and Vmax values of GCase for MUGlc were determined at 5–7 different concentrations of 2 or 1 (Figure 3A, B). Unlike IFG that is a classic competitive inhibitor, the Vmax decreased along with an apparent increase in KM with increasing dose of either 1 or 2. These data are consistent with a mixed-type of inhibition, which has also been reported for other non-carbohydrate-based inhibitors of GCase.[38]
Figure 3.

Compounds 1 and 2 are mixed-type inhibitors that function optimally at a neutral pH. A) 1 (squares) and B) 2 (circles) were tested at five concentrations, two above and below their IC50 values, each in the presence of seven different concentrations of the substrate (MUGlc). The resultant apparent KM (mM; right y-axes, open symbols) and Vmax (relative fluorescence units (RFU) h−;1; left y-axes, filled symbols) values for each inhibitor concentration (x-axis, μM) are shown as linear graphics. C) The relative inhibitory activity of 1 (10 μM, squares) and 2 (12 μM, circles) were determined at different pH values.
Ideally inhibitory compounds acting as PC would be most active at the neutral pH found in the ER (where their binding increases the stability of the mutant enzyme, offsetting some of the destabilizing effects of the mutation) and least active in the lysosome (where they could continue to inhibit the activity of the cognate enzyme). Consequently, the inhibitory activity of each compound was evaluated over a pH range of 4.5 to 7. Both 1 and 2 are most active as inhibitors at neutral pH (Figure 3C).
Active derivatives of compounds 1 and 2
To evaluate the structure versus IC50, toxicity and PC-activity relationships of the compounds, a simple quinazoline derivative (1a; lacking any pendant hydrophobic groups) and a diethoxy quinozoline derivative (1b) of 1 were examined. Whereas the former compound exhibited a tenfold reduction, the latter derivative showed a two- to three-fold reduction in inhibitory activity (Table 2).
Table 2.
Inhibitory activity of derivatives of 1 and 2.
| Compound | IUPAC name [compound symbol] | IC50 [μM][a] |
|---|---|---|
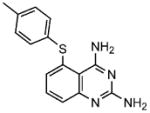 1 |
5-((4-methylphenyl)thio)- quinazoline 2,4-diamine) [1] |
7.8 |
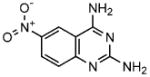 1a |
2,4,-diamino-6-nitro-quinazoline | 61 |
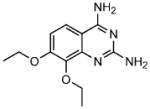 1b |
7,8-diethoxy-quinazoline 2,4-diamine | 22 |
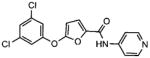 2 |
5-(3,5-dichlorophenoxy)-N-(4-pyridinyl)- 2-furamide [2] |
4.7 |
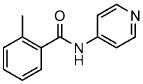 2a |
2-methyl-N-pyridin-4-yl-benzamide | 80 |
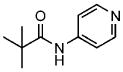 2b |
2,2-dimethyl-N-(4-pyridinyl)propanamide | >400[b] |
pNPGlc (1.6 mM).
Full dose–response could not be generated, estimated IC50 value.
These results suggest the importance of the size and identity of the hydrophobic group at the 4- and 5-positions. Although, in cultured cells 1 resulted in cell death at concentrations >17 μM, 1b showed no significant toxicity even at 800 μM. Whereas substitution of the phenoxyfuramide group in 2 with a phenyl ring (2a) reduced its inhibitory activity more than ten-fold, substitution with an alkyl group (2b) produced an essentially non-inhibitory compound. Derivative 1a was able to enhance GCase activity 1.5-fold in patient fibroblasts bearing either the N370S or F213I allele. On the other hand, compound 2a, did not significantly increase GCase activity in either of these cell lines. Thus, the parental compounds identified through HTS have lower IC50 values and greater PC activity in patient cells than the derivatives we have so far evaluated.
Treatment of GD patient cells with 1 or 2 changes the intra-cellular localization of GCase
The intracellular localization of mutant GCase before and after treatment with 1 or 2 was probed using indirect fluorescent immunostaining. Cells were co-stained with IgGs against GCase and either a marker for lysosomes, Lamp-1, or an ER marker, protein disulfide isomerase (PDI). In untreated cells (DMSO only), GCase staining was diffuse and distinct from the punctate staining pattern of Lamp-1 (Figure 4A, Top and bottom panels labelled N370S/N370S and F213I/L444P). Instead, GCase staining colocalized (indicated by yellow colour) with the ER marker PDI (Figure 4B, Top and bottom panels labelled N370S/N370S and F213I/L444P). However, when N370S or F213I patient cells were treated with either 1 (Figure 4A, 2nd row top/bottom panels) or 2 (Figure 4A, 3rd rows top/bottom panels), their GCase staining pattern increased in fluorescence intensity, became more punctate and exhibited a greater co-localization with Lamp-1, as indicated by the increased yellow colour in the Merge column (Figure 4A 3rd column in each panel). Furthermore there was a notable decrease in the overlap between GCase and PDI staining of N370S and F213I cells treated with 1. (That is, we observed a decreased amount of yellow in the merged GCase and PDI images). In the case of 1-treated N370S and F213I cells, there was also an observable decrease in the overall intensity level of PDI staining, suggesting a decrease in ER stress, for which PDI is also a marker, as compared to untreated cells (Figure 4B, second column, second row in all panels).
Figure 4.

Effects of compounds 1 and 2 on trafficking GCase from the ER to lysosomes in GD patient fibroblasts. N370S/N370S and L444P/F213I cells were treated with DMSO, 1 (10 μM) or 2 (12 μM). A) The primary IgGs against GCase or the lysosomal marker Lamp-1 are visualized as green or red, respectively. In the merged images, yellow denotes colocalization in lysosomes. B) The primary IgGs against GCase or the ER marker PDI are visualized as green or red, respectively. In the merged images, yellow denotes colocalization in the ER. Scale bars from left to right are 10 μm (DMSO), 13 μm (1) and 16 μm (2).
Profiling changes in protein dynamics within the GCase molecule upon PC-binding
Since PCs are proposed to stabilize mutant proteins by affecting their conformational dynamics, we used hydrogen/deuterium exchange mass spectrometry (H/D-Ex) to examine and map such changes within the GCase molecule. These experiments were performed in solution, in the absence or presence of a 59-fold molar excess of either ligands IFG, 1 or 2. The degrees and rates of deuteration of 26 distinct GCase peptides generated post D2O exposure were determined. In the presence of any of these compounds, there was a decrease in the deuteration of peptides surrounding the active site relative to the unliganded control. IFG-binding perturbed the largest area of GCase (16/26 of peptides), compared to 1 (6/26) and 2 that affected only one region, encompassed by peptide 243–248 (Figure 5A). Surprisingly, this is the only region that is most clearly and strongly perturbed by all three ligands. When examined over time, each of the regions that were affected by ligand binding were perturbed to different degrees by the three compounds (Figure 5B). The number of regions (16/26,6/26 and 1/26) perturbed by each of the ligands (IFG, 1, 2) parallels the maximal enhancement in GCase activity levels observed in N370S/N370S patient cells following treatment with the corresponding compounds (3.9-fold, 2.4-fold and 1.5-fold, respectively; Figure 2D).
Figure 5.

Summary of the perturbations in H/D exchange from selected regions of GCase in the absence or presence of ligands. A) Regions that show significant (>10 %) change in H/D-Ex in the presence of IFG, 1 or 2 are colour-coded (see boxed legend). Deuterium buildups over time (30 to 3000 s) for different regions of GCase ±ligand are mapped onto the amino acid sequence of human GCase. β-Strands are shown as blue arrows, α-helices as pink tubes and selected loops as purple bars. Residues previously identified by crystallography as interacting with IFG are shown in red boxes. For clarity N- and C-terminal GCase sequences not showing any significant H/D-exchange perturbations have been omitted. B) Segments showing significant perturbations in the presence of IFG, 2 or 1 relative to the apo enzyme are colour coded according to position, and superimposed upon the cartoon ribbon diagram representation of the IFG-bound GCase X-ray crystal structure (2NSX). Surrounding the cartoon are representations of deuterium-buildup curves for GCase-segments 243–249 (green), 187–197(orange), 310–312 (red), 315–336 (pink), 130–134 (purple) and 386–396 (blue). Deuterium-buildup curves are shown for selected segments of GCase in the absence of ligand (+), or in the presence of excess IFG (△), 1 (□) or 2 (○). The illustration was generated with PyMOL (DeLano Scientific) and Kaleidagraph (Synergy Software). The deuterium-buildup curves for all segments are provided in Figure S2.
Discussion
By screening a library of drug-like compounds we identified 108 novel GCase inhibitors with IC50 values ranging from 6 μM to greater than 100 μM. Although each of these confirmed inhibitory compounds was tested for enhancing activity in GD patient cells, only two of the initial hits clearly increased GCase activity following treatment, a quinazoline-2,4-diamine (1) and a pyridinyl furamide (2; Figure 1 and 3). Only two of the 108 inhibitors functioned as PC in cells, which may be attributed to several factors. Whereas compounds 1 and 2, as well as 47 others, attenuated thermal denaturation of wild-type GCase, other inhibitors conferred no such benefit. In fact several of the other inhibitory compounds actually appeared to destabilize the enzyme, hence their apparent inhibitory effect. These compounds would not be expected to function as PCs. The other inhibitory compounds that also attenuated thermal denaturation may have failed as PCs due to toxicity, poor bio-availability and/or conversion to an inactive metabolite. The latter two properties may also explain why IFG, which is a 59 nM inhibitor of purified GCase in vitro, functions best as a PC at 10–30 μM in cultured cells. On the other hand both 1 and 2 function best as PCs at 12 μM, very close to their IC50 values. Currently all confirmed PCs for lysosomal storage diseases have been demonstrated to be inhibitory molecules that can stabilize the enzyme against thermal denaturation. However, the current results underscore the point that every inhibitory compound that does this, does not necessarily also function as a PC in patient cells.
We have shown that 1 and 2 function as PCs using three independent approaches. Firstly, 1 and 2 increase GCase activity 1.5–2.5-fold in GD patient cells bearing either the N370S or F213I alleles. Secondly, they specifically increase the levels of GCase in lysosomes of these cells by more than twofold. Lastly, using immunofluorescence it was shown that treatment of both sets of patient cells resulted in increased colocalization of GCase with Lamp-1, a lysosomal marker, and a corresponding decrease in GCase colocalization with the ER marker PDI. Additionally both compounds inhibit GCase best at the neutral pH of the ER. These data strongly support the hypothesis that these compounds enhance the folding and thus the intracellular transport of the GCase mutants from the ER to the lysosome.
Both 1 and 2 are heterocyclic compounds containing at least one nitrogen atom. 2 shares features with the phenyl and amino modified derivatives of pyridine that Li and Byers showed to function as single-digit mM inhibitors of almond β-glucosidase.[40] This and other imidazoles were evaluated due to their close resemblance to naturally occurring nitrogen containing heterocycles such as 1-deoxynojirimycin and castanospermine. Zheng et al. screened another library of 59815 small molecules for inhibitors that could function as PCs and described three classes of inhibitory nitrogen containing heterocycles, an N-substituted quinoline, an N-substituted-1,3,5-triazin-2-ylamino ethanol and a 1,3,4-thiadazol-2-yl-4-(phenylsulfonamido) benzamide derivative.[38] Although these compounds differ from 1 and 2, the pyridinyl moiety in the quinoline derivative and the triazine group are reminiscent of the pyrimidine ring found in 1. Whereas 1 and 2 have IC50 values of 8 and 5 μM for GCase, the three classes of inhibitors identified by Zheng et al. functioned as inhibitors at 30, 103 and 430 nM. However, concentrations of 13–40 μM of these compounds were required to increase the activity of N370S GCase in GD patient cells, similar to the optimal 12 μM PC concentration we report for 1 and 2.[38]
Although cytosolic β-glucosidase shares some of the residues found in the GCase active site, it is only inhibited by 1 at a tenfold higher concentration relative to GCase.[42] Consequently, the activity levels of the neutral cytosolic β-glucosidase would not be expected to be affected by the concentration of 1 (12 μM) that was shown to enhance GCase activity. It is interesting that IFG like 1 also inhibits neutral β-glucosidase, suggesting that the two compounds may interact with the active site in a similar manner. The quinazoline framework in 1 is found in drugs such as trimetrexate, antifungal and antineoplastic agents that function as dihydrofolate reductase (DHFR) inhibitors, as well as doxazocin, a selective α-1-adrenergic blocker. Both these compounds do not inhibit GCase activity (data not shown), possibly due to substitutions on the amino groups or the 6-position of quinazoline. Previously 2,4-diamino-5-substituted quinazolines have been shown to act as inhibitors of human and bacterial DHFR.[43,44] This may explain the observed toxicity at concentrations greater than 30 μM. The fact that substituents on the 6- and 7-positions of 2,4-diaminoquinazoline result in decreased GCase inhibition and an increase in DHFR inhibition suggests that selectivity of 1 congeners for GCase over DHFR could be increased by modifying the substituents on the 5-position of quinazoline. Although 1 did not inhibit human lysosomal Hex, it is interesting that pyrimethamine, a known Plasmodium falciparum DHFR inhibitor also functions as a PC for mutant forms of Hex found in late onset GM2-gangliosidosis,[45] and like 1 contains a 2,4-diamino-pyrimidine moiety.
The regions in the wild-type GCase structure that are stabilized by our two new PCs were identified by H/D-Ex experiments and compared to those regions affected by IFG. At a 59-fold molar excess of 1, 2 or IFG, specific regions of the enzyme were rigidified (reduced the extent of H/D exchange). Although the stabilizing effects of the compounds were examined using wild-type enzyme, these results likely extend to the mutant enzyme.
Kornhaber et al.[35] have recently used H/D-Ex to identify the regions that undergo stabilization following IFG binding. Consistent with the crystal structures of GCase:ligand complexes, loops encompassing residues 311–319 (labeled loop1311–319 in ref. [32] and loop3312–319 in ref. [33]), 342–350 (labeled loop2342–354 in ref. [32] and loop1341–350 in ref. [32]) and 393–396 (labeled loop3393–396 in ref. [33]) showed decreased levels of deuteration in the presence of IFG and hence increased rigidification of the regions. A similar albeit more limited, perturbation pattern was seen for the same regions in the presence of either 1 or 2. Each of the three loops contains residues that form hydrogen bonds with IFG.[32,33]
Although IFG binding induced the greatest degree of perturbation in the three loops, 1 had a greater overall impact on the rate of hydrogen/deuterium exchange than 2. The only region demonstrating a significant reduction in the rate of hydrogen/deuterium exchange by all three PCs was the segment encompassing residues 243–249. This is a rather surprising observation given that none of the crystal structures of GCase: ligand complexes have shown any residues in this region making a direct contact with the bound glycone moiety. However, this region does contain Leu241 which forms a hydrophobic contact with the alkyl chain present in GCase:NN-DNJ complex.[33] Furthermore, computational docking studies of a truncated GC ligand derivative into the active site of the enzyme (2NSX) suggested that the alkyl chain would lie in a shallow hydrophobic groove between residues 311–317 and 235–252.[33] Although one could envision a hydrophobic group such as 4-methylphenylthiol in 1 or 3,5-dichlorophenoxy in 2 lying in this groove, IFG lacks a hydrophobic group that could have such an effect on residues 235–252.
The rigidification observed in residues 235–252 may arise indirectly as a result of the concerted movement brought about by the direct interaction of the ligands with one of these loops. Alternatively, differences between the two experimentally derived binding profiles may be due to the constraints placed on the breathing motions of the protein monomers in the crystal lattice versus the protein in solution.[35] The conformational differences in loop341–350 observed in the first two crystallographically derived structures of GCase are attributed to crystal contact differences.[46] It is interesting to note that in all structures to date with the exception of 2NSX, residues Trp348 (2NT1, 2J25, 2V3E, 2V3D) or Asp353 (2NT0) in loop342–354, make crystal contacts through a hydrogen-bond with Ser242 that is part of loop235–242 in the adjacent monomer in the crystal lattice (PISA-webserver).[47] Thus one could speculate that the lack of observed differences in these regions upon ligand binding in the crystal structures may be due to the constraints imposed by the crystal contacts between the two loops in adjacent monomers.
The degree to which each of the three PCs (IFG, 1 or 2) are able to enhance GCase activity in GD patient cells with the N370S mutation appears to most closely correlate with perturbation effects on H/D-Ex in loop243–248. The importance of this loop in the formation of a functional GCase is also indicated by the fact that its residues are conserved to greater degree across a wider phylogenetic distance (tetropods, fugu, honeybee and Caenorhabditis elegans) than residues found in loop311–319.[48] Our H/D-Ex data on this limited set of PCs are consistent with the crystallographic data correlating an enhancement in global stability with the rigidification of loop311–319 and/or loop342–354. However, it is difficult to reconcile whether the conformational changes observed in these loops are more relevant to allosteric control of GCase activity[33] if conformational fluctuations lead to recognition by the ER-QC machinery and ER retention. The two conformers of the enzyme may relate to conformational changes that GCase is proposed to undergo following activation via its interaction with SapC.[49] The existence of two conformational states in GCase is analogous to the conformational states of the delta opioid receptor. One of these conformations is stabilized by agonists and the other by antagonists.[50] Interestingly, both agonists and antagonists also enhance surface expression of the delta opioid receptors.[51] It would therefore be interesting to see whether or not a compound that stabilizes an alternative conformer of GCase at neutral pH would also function as a PC.
The H/D-Ex experiments on GCase in the presence and absence of IFG, 1 or 2 underscore the importance of other regions of GCase, such as loop 235–252, in mediating the transport enhancing effects of PCs on N370S and F213I mutants. As a technique H/D-Ex serves to highlight structural regions of GCase that undergo important conformational changes. These regions may be the focus of other higher resolution techniques such as NMR to generate in atomic detail, the dynamic and conformational changes that the protein undergoes in solution in the presence and absence of ligands.
Conclusions
These experiments have validated HTS for inhibitors as a general approach to identify additional frameworks for generating PCs for glycosidases deficient in lysosomal storage disorders. The fact that only two compounds out of the initial 108 hits for GCase inhibitors were ultimately identified as PCs underscored the importance of testing each candidate compound in patient cells. Although H/D-Ex does not produce the high resolution data obtainable by crystallography, its use allows for rapid identification and comparison of regions in a protein that are stabilized in solution upon binding of various ligands. In the case of GCase, the previously unobserved correlation between the stabilization of amino acid residues 242–253 and PC activity was elucidated.
Experimental Section
Chemicals and reagents
A total of 49586 drug-like compounds from the Maybridge collection (Maybridge PLC, Cornwall, UK) were used in the initial screen. Compounds were evaluated in the secondary screen and their derivatives were re-ordered from Maybridge PLC or Chembridge (San Diego, CA, USA) and solubilized using DMSO or water. Human GCase (cerezyme) was purchased from Genzyme (Cambridge, MA, USA). The concanavalin A-binding fraction of human placental lysate was used as a source for lysosomal enzymes β-Gal and α-Glc. Human Hex was purified from placenta as described.[52] Almond β-glucosidase was purchased from Sigma (USA). Human neutral β-glucosidase kindly provided by N. Juge (Biosciences FRE-3005-CNRS Universite Paul Cezanne Aix Marseille III, France) was expressed and purified from Pichia pastoris as described.[42] Fluorogenic substrates purchased from SIGMA (USA) included; 4-methylumbelliferyl-β-D-glucopyranoside (MUGlc), GCase; 4-methylumbelliferyl-β-D-galactopyranoside (MUGal), β-Gal; 4-methylumbelliferyl-α-D-glucopyranoside (MU-α-Glc), α-Glc; 4-methylumbelliferyl-β-N-acetylglucosamine (MUG), Hex. The colourimetric substrate p-nitrophenyl-β-D-glucopyranoside (pNPGlc) (SIGMA, USA) was also used to monitor GCase, human cytosolic β-glucosidase and almond β-glucosidase activity.
Cell lines
The following cell lines were used: “N370S” fibroblast cell line from a patient diagnosed with the Type I Gaucher disease homozygous for the N370S mutation (Molecular Diagnostics Laboratory, SickKids, Toronto, Ont., Canada); “F213I” fibroblast cell line from a patient diagnosed with type I Gaucher bearing the F213I/L444P alleles (kindly provided by F. Choy, University of Victoria). All cell lines were grown in α-minimal essential media (α-MEM; Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS; Sigma, USA), and antibiotics Penicillin/Streptomycin (Invitrogen, USA) at 37°C in a humidified CO2 incubator.
Primary screening
Human GCase was screened against the 49586 compound library of drug-like small molecules (Maybridge PLC, Cornwall, UK) in duplicate in 384-well plate format. The screen was fully automated on a SAIGAIN core system (Beckman-Coulter Inc., Fullerton, CA) with an ORCA arm for labware transportation, a Biomek FX (Beckman-Coulter) for liquid handling, and an Analyst HT (Molecular Devices Corp., Sunnyvale, CA) for fluorescence detection (λex = 330 nm; λem = 460 nm). All liquid handling and activity detection was done at room temperature. Each 384-well assay plate was read nine times, with 105 s between each read. Reaction rates (RFUs−1) were calculated as the slope of the data of the second to ninth data point, inclusive. Each reaction consisted of GCase (72 μg mL−1), taurodeoxycholate (TdC, 0.24 %), human serum albumin (0.1 %), MUGlc substrate (625 μM) and compounds in 20 mM citrate-phosphate (CP) buffer. Library compounds dissolved in DMSO were added to a final concentration of 20 μM. Each 80 compound set from the library was analyzed in duplicate using two quadrants of the 384 well plate. Eight replicate high (2 % DMSO) and low controls (2 % DMSO, castanospermine (45 μM)) were included in each quadrant of the 384 well plate. The residual activity (RA) of the enzyme in the presence of each of the compounds was determined as previously described.[36] To obtain an estimate of the variability of the assay, eight replicate high and low controls were used to generate a Z-factor,[53] which measures the variability of the rate values for GCase. A Z-factor of 0.75 was obtained for the primary GCase screen. (That is, a very good separation of the high and low controls was observed).
Secondary screening
The dose-response curves of the 108 hits selected from the primary screen were determined by the endpoint GCase assay, in the presence of seven concentrations (0.1–100 μM) of the putative inhibitor diluted in DMSO. IC50 values were determined as described previously.[36] Compounds exhibiting sigmoidal dose response curves were selected as bona fide inhibitors.
GCase and other glycosidase activity assays
GCase activity was measured by release of 4-methylumbelliferyl fluorophore from MUGlc. Assays (50 μL) contained CP (20 mM, pH 5.5), TdC (0.2 %) and MUGlc (0.8 mM). For the endpoint assay, the reaction at 37°C was terminated by raising the pH to 10.5, above the pKa of 4-MU by adding 2-amino-2-methyl-1-propanol (0.1M, 200 μL). The increase in fluorescence was measured using a Spectramax Gemini EM MAX (Molecular Devices Corp, Sunnyvale, CA) fluorometer and detected at excitation and emission wavelengths set to 365 nm and 450 nm, respectively. For inhibition studies using the enzymes other than GCase, the following buffer: substrate combinations were used: β-Gal: MUGal (1.6 mM in 20 mM CP 5.5), almond β-glucosidase: pNPGlc (1.6 mM in 20 mM CP pH 5.5:), human cytosolic β-glucosidase: pNPGlc (0.25 mM in 20 mM CP pH 5.5), α-MUαGlc Glc (0.5 mM in 20 mM CP pH 5.5), Hex: MUG (0.4 mM in 20 mM CP pH 4.5). All reactions were performed at 37 °C as an endpoint assay as described for GCase above.
To control for compounds that were either fluorescent or fluorescence quenchers near the emission maxima of MU, the inhibitory activity of compounds was also confirmed using the colourimetric substrate pNPGlc under conditions described for the MUGlc endpoint assay, except that absorbance was measured at 405 nm and for kinetic analyses 1.6 mM pNPGlc was used. IC50 values or kinetic parameters (KM, Vmax) were obtained by a nonlinear curve fitting of the data to the sigmoidal dose–response equation or Michaelis–Menten equation by using Prism 4.0 (Graphpad Software, La Jolla, CA, USA).
Heat inactivation assay
Heat inactivation experiments were performed using cerezyme powder (GCase) (13 mg mL−1) in CP buffer (20 mM), diluted a further 1/200–1/400 in CP buffer (20 mM, pH 5.5) containing TdC (0.2 %). Diluted samples of GCase containing inhibitors or DMSO, were split into two aliquots, one was left on ice, and the other heat-treated at 50°C. Heat-treated enzyme samples at each time point were cooled on ice until completion of the time series. For enzyme activity, samples were pre-equilibrated to room temperature for 10 min, followed by addition of MUGlc (0.8 mM final) substrate and incubated at room temperature for a further 20 min. Remaining activity was expressed as a ratio of GCase activity in the presence of the test compound following incubation at 50°C versus activity of the corresponding aliquot in the presence of the test compound held at 4°C.
Determination of pH dependence of inhibitory activity of 2 and 1 towards GCase
GCase (100 ng mL−1) was diluted into 20 mM CP buffer of pH ranging from 4.5–7 in steps of 0.5 units. Compound 2 (12 μM final), 1 (10 μM final) or DMSO (1 % final) was added to the enzyme mix and equilibrated for 15 minutes at room temperature. Following addition of an equal volume of MUGlc (0.8 mM final), an endpoint assay was performed at 37 °C as described above. Residual activity was expressed as a ratio of GCase activity at a given pH in the presence of the test compounds (1 or 2), versus activity at the corresponding pH in the presence of DMSO.
Evaluating chaperoning activity of compounds in cell culture
Gaucher patient fibroblasts (10 000–50 000 cells per well) were seeded onto 24 well plates at (about 50% confluence). The next day, the medium was replaced with fresh α-MEM-FBS with or without a test compound (1:100 dilution). Test compounds were dissolved in DMSO. Mock or compound-treated cells were evaluated in triplicate after growth for five days at 37 °C in a CO2-humidified incubator.
To measure GCase activity in treated Gaucher fibroblasts, media was removed, cells were washed twice with PBS and subsequently lysed by the addition of Triton X-100 (0.4 %) and TdC (0.4 %) in CP (20 mM, pH 5.5). An aliquot (25 μL) of the lysate was mixed with an equal volume of MUGlc (10 mM final) and assayed for total GCase activity. To control for variability in cell numbers between replicate wells, the remaining aliquot of the lysate, was used to assay for lysosomal Hex, with the substrate MUG using the endpoint assay described above.
Purification of iron-dextran-labeled lysosomes
An enriched lysosomal fraction was prepared from Gaucher patient N370S/N370S fibroblasts treated with either DMSO (0.1 %), 1 (12.5 μM) or 2 (12.5 μM) for five days, followed by labeling with iron-dextran colloid and subsequent purification by magnetic chromatography as previously described.[54] Lysosomal GCase was monitored fluorometrically using the substrate MUGlc.
Western blotting
The enriched lysosomal fractions (1 μg) from treated and untreated Gaucher patient cells were subjected to SDS-PAGE on a bis-acrylamide gel (10 %), and the separated proteins were transferred to nitrocellulose. A rabbit polyclonal IgG against human GCase or mouse monoclonal Lamp-2 antibody were used as previously described.[55] Blots were developed using a chemiluminescent substrate according to the manufacturers protocol (Amersham, Bioscences, UK). Bands were visualized and optical density quantitated using a high sensitivity gel documentation system (Fluorchem 8000) consisting of a cooled CCD camera coupled with Alpha Innotech software (Alpha Innotech Corp., San Leandro, CA, USA).
Mass spectrometry
The mass of selected secondary hits was confirmed by the Advanced Proteomic Centre at Sickkids (Toronto, Canada) using a QToF mass spectrometer (Waters/Micromass, Manchester, UK)
Indirect immunofluorescence and confocal microscopy imaging
Indirect immunolabeling was performed using a previously described protocol[56] with small modifications. In brief, cells were seeded onto 18 mm diameter coverslips for 16–20 h, then washed and fixed with paraformaldehyde (2.5 %)(EMS) in PBS (pH 7.2), for 20 min at 37 °C. Blocking and permeabilization was performed for 1 h at room temperature with saponin (0.2 %; Sigma) and 10% of either goat or horse normal serum (Wisent Inc. St. Bruno, QC, Canada) in phosphate-buffered saline (SS-PBS). Primary antibodies were diluted in SS-PBS solution and incubated with the coverslips for 1 hour at room temperature. Secondary antibodies were diluted in SS-PBS solution and incubated with the coverslips for 1 hour, at room temperature in the dark. Extensive washes with PBS were performed after primary and secondary antibody incubations. Nuclear staining was done with DAPI (Molecular Probes) at 1/50000 in PBS. Coverslips were mounted onto glass slides by using fluorescent mounting medium (Dako Denmark A/S, Glostrup, Denmark). Primary antibodies used were rabbit polyclonal IgG anti-human GCase (raised by ourselves against purified recombinant GCase), mouse monoclonal IgG1 anti-human LAMP-1 (DHSB, Iowa) and anti-rat PDI (Stressgen Bioreagents, Ann Arbor, MI, USA). Secondary antibodies were Alexa Fluor 488 chicken anti-rabbit IgG and Alexa Fluor 594 chicken anti-mouse IgG (Invitrogen) at a 1/200 dilution in SS-PBS solution. Samples were analyzed using a Zeiss Axiovert confocal laser microscope equipped with a 63 01.4 numerical aperture Apochromat objective (Zeiss) and LSM 510 software; DAPI-stained nuclei were detected on the same system with a Chameleon two-photon laser. Confocal images were imported and contrast/brightness adjusted using Volocity 4 program (Improvision Inc., Waltham, MA, USA). Intensity settings were not changed when recording the images of GCase or PDI staining between the same treated and untreated cell lines.
Hydrogen/deuterium exchange mass spectrometry experiments
A GCase stock (80 μM) was prepared by dissolving cerezyme powder (31 mg) into H2O (500 μL). Stocks (47 mM) of IFG, 2 or 1 were prepared in DMSO. A 59:1 molar ratio of IFG, 2 or 1 to GCase was prepared by combining the GCase stock (50 μL) with the compound stock (5 μL). A DMSO containing “no-ligand” control was prepared by combining the GCase stock (50 μL) with DMSO (5 μL). An exchange reaction was initiated by diluting each mixture (5 μL) of with Tris (15 μL, 50 mM) to give a final pH of 7.8, and allowed to proceed at 23°C for a series of predetermined time periods (30, 100, 300, 1000 and 3000 s). The exchange was quenched by lowering the reaction temperature to 1 °C and by dropping the pH of the reaction to 2.5 by the addition of a pre-chilled solution (30 μL, 1 °C) containing urea (2 M) and tris(2-carboxyethyl)phosphine (TCEP) (1 M). The quenched solution was immediately pumped at 200 μLmin−1 over an immobilized porcine pepsin column (104 μL bed volume) with trifluoroacetic acid (TFA) (0.05 %) for three minutes with contemporaneous collection of proteolytic products by way of a trap column (4 μL bed volume). Pepsin was immobilized on Poros 20 AL media (30 mg mL−1, Applied Biosystems) as per the manufacturer’s instructions.[34] Peptide fragments were eluted from the trap column and separated by C18 column (Magic C18, Michrom BioResources, Inc., Auburn, CA, USA) with a linear gradient of solvent B (13 %) to solvent B (40 %) over 23 min (solvent A, 0.05% TFA in water; solvent B, 95 % acetonitrile, 5% water, 0.0025 % TFA; flow rate 5 μL min−1–10 μL min−1). Mass spectrometric analyses were carried out with a Thermo Finnigan LCQ3 mass spectrometer (Thermo Fisher Scientific, San Jose, CA) with a capillary temperature of 200°C. Spectral data were acquired in data-dependent MS/MS mode with dynamic exclusion. The software program SEQUEST (Thermo Fisher Scientific, San Jose, CA) was used to tentatively identify the sequence of dynamically-selected parent-peptide ions. This tentative peptide identification was verified by visual confirmation of the parent ion charge state. These peptides were then further examined to determine if the quality of the measured isotopic envelope was of sufficient quality to allow an accurate geometric centroid determination. Centroid values were then determined using a proprietary program developed in collaboration with Sierra Analytics. Back-exchange corrections and deuteration level calculations were implemented as previously described elsewhere.[57,58]
Supplementary Material
Acknowledgments
We acknowledge the excellent technical assistance of Amy Leung, Daphne Benedict and Marie Anne Skomorowski, and thank John Callahan and M. Mylvaganam for their helpful discussions. Various aspects of this project were supported by grants from the Protein Engineering Network of Centres of Excellence of Canada (PENCE; to E.B. and D.M.), and a bequest from the Uger Estate (M.B.T. and D.M.), an NIH Grant (5R21NS051214-02; M.B.T. and D.M.) and a Canadian Institutes of Health Research (CIHR) “Team” grant (CTP-82944; E.B., M.B.T and D.M.). We thank Nathalie Juge (Biosciences FRE-3005-CNRS Université Paul Cézanne Aix-Marseille III, France) for a gift of human neutral β-glucosidase and F. Y. Choy (University of Victoria, BC, Canada) for the GD F213I/L444P cell line.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Beutler E, Grabowski GA. In: The Metabolic and Molecular Bases of Inherited Disease. 8. Scriver RC, Beaudet AL, Sly WS, Valle D, editors. III. McGraw–Hill; New York: 2001. p. 3635. [Google Scholar]
- 2.Butters TD. Expert Opin Pharmacother. 2007;8:427. doi: 10.1517/14656566.8.4.427. [DOI] [PubMed] [Google Scholar]
- 3.Knudson AG, Kaplan WD. Cerebral Sphingolipidoses. In: Volk BW, Aaronson SM, editors. A Symposium in Tay–Sachs Disease. Academic Press; New York: 1962. p. 395. [Google Scholar]
- 4.Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, Tayebi N, Sidransky E. Am J Hum Genet. 2000;66:1777. doi: 10.1086/302925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutler E, Gelbart T, Scott CR. Blood Cells Mol Dis. 2005;35:355. doi: 10.1016/j.bcmd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Schueler UH, Kolter T, Kaneski CR, Zirzow GC, Sandhoff K, Brady RO. J Inherited Metab Dis. 2004;27:649. doi: 10.1023/b:boli.0000042959.44318.7c. [DOI] [PubMed] [Google Scholar]
- 7.Grabowski GA, Leslie N, Wenstrup R. Blood Rev. 1998;12:115. doi: 10.1016/s0268-960x(98)90023-6. [DOI] [PubMed] [Google Scholar]
- 8.Amato D, Stachiw T, Clarke JT, Rivard GE. J Inherited Metab Dis. 2004;27:659. doi: 10.1023/b:boli.0000042983.60840.f3. [DOI] [PubMed] [Google Scholar]
- 9.Goker-Alpan O, Hruska KS, Orvisky E, Kishnani PS, Stubblefield BK, Schiffmann R, Sidransky E. J Med Genet. 2005;42:e37. doi: 10.1136/jmg.2004.028019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choy FY, Zhang W, Shi HP, Zay A, Campbell T, Tang N, Ferreira P. Blood Cells Mol Dis. 2007;38:287. doi: 10.1016/j.bcmd.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Beutler E. Mol Genet Metab. 2006;88:208. doi: 10.1016/j.ymgme.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Pastores GM, Barnett NL. Expert Opin Invest Drugs. 2003;12:273. doi: 10.1517/13543784.12.2.273. [DOI] [PubMed] [Google Scholar]
- 13.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Proc Natl Acad Sci USA. 2002;99:15428. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawkar AR, D’Haeze W, Kelly JW. Cell Mol Life Sci. 2006;63:1179. doi: 10.1007/s00018-005-5437-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vellodi A. Brit J Haematol. 2005;128:413. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- 16.Desnick RJ. J Inherited Metab Dis. 2004;27:385. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- 17.Fan JQ. Trends Pharmacol Sci. 2003;24:355. doi: 10.1016/S0165-6147(03)00158-5. [DOI] [PubMed] [Google Scholar]
- 18.Yu Z, Sawkar AR, Kelly JW. FEBS J. 2007;274:4944. doi: 10.1111/j.1742-4658.2007.06042.x. [DOI] [PubMed] [Google Scholar]
- 19.Fan JQ. Biol Chem. 2008;389:1. doi: 10.1515/BC.2008.009. [DOI] [PubMed] [Google Scholar]
- 20.Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA. Proc Natl Acad Sci USA. 2006;103:13813. doi: 10.1073/pnas.0605928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yam GH, Zuber C, Roth J. FASEB J. 2005;19:12. doi: 10.1096/fj.04-2375com. [DOI] [PubMed] [Google Scholar]
- 22.Fan JQ, Ishii S, Asano N, Suzuki Y. Nat Med. 1999;5:112. doi: 10.1038/4801. [DOI] [PubMed] [Google Scholar]
- 23.Yoshida H. FEBS J. 2007;274:630. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 24.Ron I, Horowitz M. Hum Mol Genet. 2005;14:2387. doi: 10.1093/hmg/ddi240. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz M, Alfalah M, Aerts JM, Naim HY, Zimmer KP. Int J Biochem Cell Biol. 2005;37:2310. doi: 10.1016/j.biocel.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Lin H, Sugimoto Y, Ohsaki Y, Ninomiya H, Oka A, Taniguchi M, Ida H, Eto Y, Ogawa S, Matsuzaki Y, Sawa M, Inoue T, Higaki K, Nanba E, Ohno K, Suzuki Y. Biochim Biophys Acta Mol Basis Dis. 2004;1689:219. doi: 10.1016/j.bbadis.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Sawkar AR, Adamski-Werner SL, Cheng WC, Wong CH, Beutler E, Zimmer KP, Kelly JW. Chem Biol. 2005;12:1235. doi: 10.1016/j.chembiol.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Lei K, Ninomiya H, Suzuki M, Inoue T, Sawa M, Iida M, Ida H, Eto Y, Ogawa S, Ohno K, Suzuki Y. Biochim Biophys Acta. 2007;1772:587. doi: 10.1016/j.bbadis.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Alfonso P, Pampin S, Estrada J, Rodriguez-Rey JC, Giraldo P, Sancho J, Pocovi M. Blood Cells Mol Dis. 2005;35:268. doi: 10.1016/j.bcmd.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 30.Compain P, Martin OR, Boucheron C, Godin G, Yu L, Ikeda K, Asano N. ChemBioChem. 2006;7:1356. doi: 10.1002/cbic.200600217. [DOI] [PubMed] [Google Scholar]
- 31.Zhu X, Sheth KA, Li S, Chang HH. J Q Fan, Angew Chem. 2005;117:7616. doi: 10.1002/anie.200502662. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:7450. [Google Scholar]
- 32.Lieberman RL, Wustman BA, Huertas P, Powe AC, Jr, Pine CW, Khanna R, Schlossmacher MG, Ringe D, Petsko GA. Nat Chem Biol. 2007;3:101. doi: 10.1038/nchembio850. [DOI] [PubMed] [Google Scholar]
- 33.Brumshtein B, Greenblatt HM, Butters TD, Shaaltiel Y, Aviezer D, Silman I, Futerman AH, Sussman JL. J Biol Chem. 2007;282:29052. doi: 10.1074/jbc.M705005200. [DOI] [PubMed] [Google Scholar]
- 34.Hamuro Y, Coales SJ, Southern MR, Nemeth-Cawley JF, Stranz DD, Griffin PR. J Biomol Tech. 2003;14:171. [PMC free article] [PubMed] [Google Scholar]
- 35.Kornhaber GJ, Tropak MB, Maegawa G, Tuske SJ, Coales SJ, Mahuran DJ, Hamuro Y. ChemBioChem. 2008 doi: 10.1002/cbic.200800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tropak MB, Blanchard J, Withers SG, Brown E, Mahuran D. Chem Biol. 2007;14:153. doi: 10.1016/j.chembiol.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tropak MB, Mahuran D. FEBS J. 2007;274:4951. doi: 10.1111/j.1742-4658.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng W, Padia J, Urban DJ, Jadhav A, Goker-Alpan O, Simeonov A, Goldin E, Auld D, Lamarca ME, Inglese J, Austin CP, Sidransky E. Proc Natl Acad Sci USA. 2007;104:13192. doi: 10.1073/pnas.0705637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sawkar AR, Schmitz M, Zimmer KP, Reczek D, Edmunds T, Balch WE, Kelly JW. ACS Chem Biol. 2006;1:235. doi: 10.1021/cb600187q. [DOI] [PubMed] [Google Scholar]
- 40.Li YK, Byers LD. Biochim Biophys Acta Protein Struct Mol Enzymol. 1989;999:227. doi: 10.1016/0167-4838(89)90001-0. [DOI] [PubMed] [Google Scholar]
- 41.Hayashi Y, Okino N, Kakuta Y, Shikanai T, Tani M, Narimatsu H, Ito M. J Biol Chem. 2007;282:30889. doi: 10.1074/jbc.M700832200. [DOI] [PubMed] [Google Scholar]
- 42.Tribolo S, Berrin JG, Kroon PA, Czjzek M, Juge N. J Mol Biol. 2007;370:964. doi: 10.1016/j.jmb.2007.05.034. [DOI] [PubMed] [Google Scholar]
- 43.Rosowsky A, Mota CE, Queener SF, Waltham M, Ercikan-Abali E, Bertino JR. J Med Chem. 1995;38:745. doi: 10.1021/jm00005a002. [DOI] [PubMed] [Google Scholar]
- 44.Zolli-Juran M, Cechetto JD, Hartlen R, Daigle DM, Brown ED. Bioorg Med Chem Lett. 2003;13:2493. doi: 10.1016/s0960-894x(03)00480-3. [DOI] [PubMed] [Google Scholar]
- 45.Maegawa GHB, Tropak M, Butner J, Stockley T, Kok F, Clarke JTR, Mahuran DJ. J Biol Chem. 2007;282:9150. doi: 10.1074/jbc.M609304200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brumshtein B, Wormald MR, Silman I, Futerman AH, Sussman JL. Acta Crystallogr Sect D Biol Crystallogr. 2006;62:1458. doi: 10.1107/S0907444906038303. [DOI] [PubMed] [Google Scholar]
- 47.Krissinel E, Henrick K. J Mol Biol. 2007;372:774. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 48.Liou B, Kazimierczuk A, Zhang M, Scott CR, Hegde RS, Grabowski GA. J Biol Chem. 2006;281:4242. doi: 10.1074/jbc.M511110200. [DOI] [PubMed] [Google Scholar]
- 49.Qi X, Grabowski GA. Biochemistry. 1998;37:11544. doi: 10.1021/bi980785+. [DOI] [PubMed] [Google Scholar]
- 50.Baker JG, Hill SJ. Trends Pharmacol Sci. 2007;28:374. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Petäjä-Repo UE, Hogue M, Bhalla S, Laperrière A, Morello JP, Bouvier M. EMBO J. 2002;21:1628. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huyer G, Piluek WF, Fansler Z, Kreft SG, Hochstrasser M, Brodsky JL, Michaelis S. J Biol Chem. 2004;279:38369. doi: 10.1074/jbc.M402468200. [DOI] [PubMed] [Google Scholar]
- 53.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screening. 1999;4:67. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 54.Tropak MB, Reid S, Guiral M, Withers SG, Mahuran DJ. J Biol Chem. 2004;279:13478. doi: 10.1074/jbc.M308523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hou Y, McInnes B, Hinek A, Karpati G, Mahuran D. J Biol Chem. 1998;273:21386. doi: 10.1074/jbc.273.33.21386. [DOI] [PubMed] [Google Scholar]
- 56.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. J Biol Chem. 2006;281:11374. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Z, Smith DL. Protein Sci. 1993;2:522. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamuro Y, Coales SJ, Morrow JA, Molnar KS, Tuske SJ, Southern MR, Griffin PR. Protein Sci. 2006;15:1883. doi: 10.1110/ps.062103006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.