

Abstract
The advent of high throughput genome-scale bioinformatics has led to an exponential increase in available cellular system data. Systems metabolic engineering attempts to use data-driven approaches – based on the data collected with high throughput technologies – to identify gene targets and optimize phenotypical properties on a systems level. Current systems metabolic engineering tools are limited for predicting and defining complex phenotypes such as chemical tolerances and other global, multigenic traits. The most pragmatic systems-based tool for metabolic engineering to arise is the in silico genome-scale metabolic reconstruction. This tool has seen wide adoption for modeling cell growth and predicting beneficial gene knockouts, and we examine here how this approach can be expanded for novel organisms. This review will highlight advances of the systems metabolic engineering approach with a focus on de novo development and use of genome-scale metabolic reconstructions for metabolic engineering applications. We will then discuss the challenges and prospects for this emerging field to enable model-based metabolic engineering. Specifically, we argue that current state-of-the-art systems metabolic engineering techniques represent a viable first step for improving product yield that still must be followed by combinatorial techniques or random strain mutagenesis to achieve optimal cellular systems.
Keywords: Genome-scale modeling, Metabolic engineering, Systems biology
1 Introduction
Cellular metabolism is a coordinated, interwoven collection of metabolites, enzymes, and regulatory factors. Metabolic engineering attempts to favor desired product formation by reconfiguring this interconnected network through the introduction of genetic controls and novel enzymes. In one respect, the complexity of these cellular networks makes engineering these systems a daunting task. However, at the same time, each of the sources of complexity within a cell provides an access point for improving cellular phenotype. In this regard, changes at the genetic, regulatory, enzymatic, and small molecule level can lead to desired phenotypes. However, it is usually difficult (and sometimes impossible) to naively select a genetic target among the wide array of potential candidate genes and components. To address this complexity, systematic approaches for redesigning cells have been formalized and explored. This basic premise founded the field of Metabolic Engineering – initially aimed at identifying pathway limitations through the systematic analysis and quantification of pathway fluxes.
A great deal has changed since the field of metabolic engineering was first described nearly 20 years ago [1]. Specifically, the advent of post-genomic technologies and high-throughput biology provides the ability to make cellular measurements and perturbations at vastly increased speeds and accuracy. The resulting explosion of data has enabled the newfound ability to take accurate snapshots of entire cellular function. Integrating and synthesizing this data forms the foundation for systems biology research. When the efforts of systems biology and metabolic engineering are combined, a systems metabolic engineering approach [2] emerges with promises to unlock cellular potential and describe cellular phenomena. This approach truly appreciates the cell as an integrated, global network, and attempts to use model-based and data-driven approaches to identify pathway bottlenecks and provide cellular reconstructions. However, the cost of producing these models (and the data required to create and validate them) is often high both financially and in terms of time consumption. Therefore, it is essential that a sound methodology and firm set of outcomes and expectations be defined prior to utilizing these approaches. The purpose of this review is to provide an overview of systems metabolic engineering and to highlight areas where more work must be done before realizing the potential of ground-up systems metabolic engineering. In this review, we will particularly recognize the contributions of genome-scale modeling as it remains the most tangible and applicable systems biology approach for metabolic engineering. Prior to discussing these approaches, we will review the area of metabolic engineering to build the context for the emerging systems metabolic engineering paradigm. We will then discuss the complete work throughput required to perform systems metabolic engineering for a newly discovered organism. Finally, we will conclude with prospects and challenges for the future of systems metabolic engineering as an enabling tool for improving cellular phenotypes.
2 Metabolic engineering defined and successes
Metabolic engineering embodies the manipulation of enzymatic, transport, and regulatory functions of a cell through recombinant DNA technologies with the goal of improving a cellular phenotype, often yield of a desired product. The traditional metabolic engineering toolbox comprises rationally selected deletions and overexpressions of native and heterologous genes [1]. More recently, this toolbox has been expanded to include many new tools for controlling gene expression, for modulating regulatory networks, for combinatorial genetics, and for employing synthetic biology approaches [3](Text Box 1). The current portfolio of advances in metabolic engineering is large for such a young field of study. A recent example of complex metabolic pathway engineering can be seen in work by the Keasling lab to produce artemisinic acid, a precursor to artemisinin, an anti-malarial drug. By relieving growth inhibition caused by a toxic pathway intermediate [4–7] and by tuning intergenic regions of polycistronic operons to alter expression levels of individual genes to balance flux [8], flux through the pathway to the intermediate amorphadiene was increased 1 000 000 fold and resulted in an artemisinic acid titer of 300 mg/L [6]. Similar heterologous pathway engineering approaches have been used to produce other complex products such as fosfomycin [9, 10] and novel polyketides [11–13].
Metabolic engineering has had additional success increasing the productivity of industrially relevant small molecules [14–17], alcohol-based biofuels [18–21], and biodiesel [22, 23]. Recently, this work has been expanded to hijack E. coli's amino acid biosynthetic pathway and divert 2-keto acid intermediates for the synthesis of 1-butanol, 2-methyl-1-butanol, 3-methyl-1-butanol, and 2-phenylethanol [24]. A similar approach was used to produce (S)-3-methyl-1-pentanol [25]. In contrast to these single pathway optimization projects, creating complex products or phenotypes places a larger demand on metabolic engineers often requiring novel approaches to modulate multiple gene targets at the same time. Combinatorial metabolic engineering approaches have attempted to overcome this problem. Examples include improving xylose metabolism [26, 27] and further probing metabolic landscapes [28] in E. coli. Genome shuffling and shotgun genomic approaches have been successful in inducing phenotypical improvements [29, 30] and has led to improved thermotolerance, ethanol tolerance, and ethanol production in Saccharomyces cerevisiae [31] and to improved acid tolerance and L-lactic acid production in Lactobacillus rhamnosus [32]. These are two industrially relevant strain improvements. A final example of combinatorial metabolic engineering is the use of global transcriptional machinery engineering (gTME) to increase ethanol tolerance and production in yeast [23, 33]. Most of these combinatorial tools were created to address a limited ability to systematically improve and model cellular phenotypes.
3 Systems metabolic engineering defined and successes
Systems metabolic engineering [2] embodies the incorporation and probing of large-scale datasets with the goal of improving a cellular phenotype and synthesizing cellular function in the form of models. Recent advances in the “omics” technologies enabled by high throughput biology techniques have expanded traditional metabolic engineering to further incorporate a systems-level view of cells [3]. These capabilities have ushered in the field of systems metabolic engineering [2, 34] (Fig. 1). However, this wealth of data has created new challenges. Specifically, a major goal of systems biology involves combining high throughput genomic, transcriptomic, proteomic, metabolomic, and fluxomic data to develop a robust and experimentally confirmable in silico cell model. This complete cell model could theoretically simulate (and ideally predict) cell and metabolic function. In this regard, this model would be invaluable for metabolic engineering by enabling rational predictions of phenotypical response for changes in media, gene knockouts, antibiotic effects, or incorporation of heterologous pathways.
Figure 1.

Systems Metabolic Engineering. The merging of systems biology with traditional metabolic engineering brings about the field of systems metabolic engineering. In this vision, a systems based approach is used for predicting phenotypical changes and selecting gene targets. This approach allows for a global metabolic engineering implementation.
Large-scale global measurements are favored as a means of assessing cellular and metabolic function. In some respects, the use of carbon-labeled substrates to reconstruct cell-wide flux maps represented the first attempt at applying a systems biology approach toward metabolic engineering. More recently, our capacities have expanded beyond this point to measure transcript levels, protein levels, interactions, concentrations, and even localizations. However, our ability to reconcile this data is not yet complete enough to build comprehensive cell models. Consequently, the most comprehensive and predictive models to come out of systems biology work are global metabolic network reconstructions. These models serve as a basic outgrowth of simple material balances and typically only account for stoichiometric reactions occurring within the cell. Despite being simplistic in their view of the cell, these models have successfully predicted various metabolic perturbations and can aid in designing improved cells (examples discussed below). These in silico genome-scale metabolic reconstructions form the backbone of future applications of systems metabolic engineering.
Novel Metabolic Engineering Tools.
Metabolic engineering has numerous successes to its credit, and new metabolic engineering tools have recently been discovered [3]. For instance, by mutating the genetic sequence of a constitutive promoter, Alper et al. [77] created a library of promoters with different strengths, allowing the transcription rate of a gene to be modulated to desired levels by inserting the correct promoter upstream. This allows for the possibility of engineering the transcription rates of all enzymes in a metabolic pathway to optimize pathway flux. In addition to modifying the promoters themselves, Alper et al. [23, 33] mutated sigma factors in prokaryotes and binding and associated transcription factors in eukaryotes to alter the global transcriptional machinery of the model organism. This global transcription machinery engineering (gTME) altered expression levels for hundreds of different compared to wildtype expression levels. Several groups have employed the use of shotgun genomics and combinatorial genetic screens to identify gene over-expressions in a high-throughput manner. Synthetic biologists have invented many devices that can be used by metabolic engineers for cellular programming. The development of an inducible “on” switch for cell pathways can allow for the production of toxic products when cell mass is maximized [84]. Furthering this, Gardner el al. [85] developed a novel genetic toggle switch, an inducible system that can switch between two states, with gene A on and gene B off or with gene B on and gene A off. These cellular “on/off” switches can easily be incorporated into metabolic models.
These novel metabolic engineering tools, promoter engineering, gTME, combinatorial shotgun overexpressions, and synthetic cell switches, will prove valuable additions to the metabolic engineer's toolbox.
4 Metabolic engineering applications of in silico analysis
While still an emergent field, systems metabolic engineering has already had significant successes. Park et al. [35, 36] and Lee et al. [2, 36] used similar approaches to improve the production of L-valine and L-threonine, respectively in E. coli. Their approaches diverged from the traditional means of creating industrial amino acid producing strains based on using random mutations and screening. Product yield was first increased using traditional pathway approaches (overexpression of rate limiting enzymes and deletion of genes to increase metabolic precursors). Then a systems metabolic engineering approach was used taking advantage of transcriptome analysis and in silico model-based metabolic reconstructions to identify gene knockouts. Sequential rounds of non-random mutations resulted in final product yields of 0.378 g L-valine per gram glucose and 0.393 g L-threonine per gram glucose. These yields are comparable to those values obtained from industrial strains, demonstrating that a rational approach can achieve yields similar to an unguided approach in shorter time-frames. Moreover, these strains may be further enhanced through in silico predictions because their genomes are still fully characterized, unlike in a randomly mutated strain. This outcome represents an important difference in systems metabolic engineering over strain improvement through random mutagenesis.
In a similar fashion, Alper et al. [37] used a genome-scale metabolic model of E. coli to identify single, double, and triple gene knockouts that improved lycopene production. A triple knockout system, which would have been intractable to discover without the genome-scale model using standard strain optimization search strategies [38], yielded lycopene production at nearly a 37% increase over an engineered parental strain. Furthermore, in 2002, Lee et al. [16, 39] used an early E. coli metabolic reconstruction to engineer the production of succinic acid, reaching 85% of the maximum theoretical yield.
More recently, these models have been employed to increase yields of complex products as in the case of recombinant human interleukin-2 (IL-2) production in E. coli [40]. A simplified stoichiometric model (a lumped model that ignored vitamins and minerals) was used to predict amino acid supplementations that would increase IL-2 production. Successful results included an increase from 81 to 195 mg IL-2/L in shake flask, and 403 to 594 mg IL-2/L in a fermenter, and 5150 to 10 010 mg IL-2/L in a fed-batch cultivated fermenter. Albeit simplified, this model was able to successfully predict changes in culturing conditions.
Beyond E. coli, this approach has seen significant adoption in the basic yeast, S. cerevisiae. Initial work was performed to improve succinic acid production [41] in yeast. Bro et al. [42] used an in silico genome-scale metabolic reconstruction of S. cerevisiae to engineer yeast reductive oxidative metabolism for decreased glycerol production and increased ethanol production on glucose as well as on a glucose/xylose carbon source.
A third industrially relevant organism suitable for systems metabolic engineering is the amino acid producing bacterium Corynebacterium glutamicum [43], responsible for producing nearly 1 500 000 tons/yr of L-glutamate and 550 000 tons/yr of L-lysine. Kieldsen et al. [44] recently published a genome-scale in silico metabolic reconstruction of C. glutamicum. In addition, work has already been done to aide in the reconstruction of transcriptional regulatory networks of Corynebacteria, easing potential future integration of metabolic and regulatory networks to increase model robustness [45]. Given the results above, this model may be used to engineer C. glutamicum for increased industrial amino acid titers.
The results above illustrate the power of in silico modeling as a complement to traditional metabolic engineering approaches for producing small molecules and biopharmaceuticals. The successful application of limited genome-scale models for metabolic engineering give reason to believe that further advances will be made when more comprehensive models are assembled. Beyond the limited scope of these models, a major potential drawback is the availability of a metabolic reconstruction. For standard, un-mutated strains of interest, network availability is often not an issue due to freely available genome-scale reconstructions for common organisms such as Escherichia coli [46], Saccharomyces cerevisiae [47], Aspergillus niger [48], Bacillus subtilus [49], and Corynebacterium glutamicum [44]. However, for industrial strains, mutant versions of common strains, or newly isolated organisms, the lack of an in silico model limits the applicability of a systems-based approach. Thus, researchers are challenged with the following dilemma: invest resources to create a model or use established, traditional approaches. In the next section, we go through the steps in network construction and highlight limitations and challenges that need to be overcome.
5 Enabling systems metabolic engineering
A critical decision for any metabolic engineering project is the selection of a platform organism [50]. Uncharacterized, non-model organisms may have innate biochemical pathways to produce desired products but can exhibit low growth rates or be limited by poorly developed genetic tools. Common industrial model organisms such as S. cerevisiae and E. coli have developed genetic tools, but may be lacking in necessary resistance or biosynthetic pathways.
When non-model organisms are chosen as the platform, building systems biology expertise and capacity from scratch is not a trivial task. High throughput biology measurements require high precision experiments. However, the price of these techniques is decreasing as they become more standardized. For the case of in silico genome-scale metabolic reconstructions, many steps are required if starting with an unsequenced organism (Fig. 2).
Figure 2.
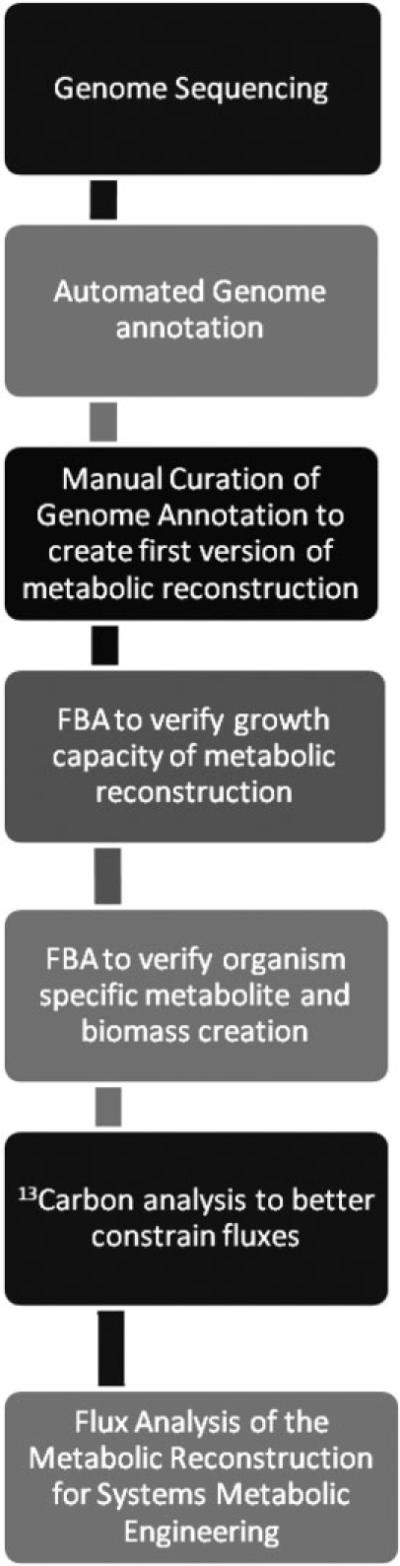
Flowchart for enabling systems metabolic engineering starting with an unsequenced organism.
A basic outline is provided for each of the steps required to create a successful genome-scale model for an unsequenced, novel organism.
5.1 Genome sequencing
The required input data for an in silico genome-scale metabolic reconstruction is access to the genome sequence for the organism to determine innate cellular capacities. The first genome sequenced was that of bacteriophage ΦX175, in 1977 [51]. A seemingly disproportionate amount of effort was necessary to discover the 5 kilobase sequence. By the early 1990s, Sanger's biochemistry had revolutionized sequencing technology [52], and the automation of Sanger's chain termination method would eventually allow for the shotgun sequencing of much larger genomes [53] with decreasing costs. However, recent advances in “next-generation” [54] sequencing (relying mainly on cheaper, shorter reads) have greatly reduced this cost. For example, the next-generation ABi SOLiD platform sequencer can sequence DNA for about $2 per megabase, compared to $500 per megabase with Sanger-based sequencing. Thus, next generation sequencers provide the means for rapid de novo sequencing, a required first step for model reconstruction.
These drastic decreases in sequencing cost have essentially brought major sequencing centers into the hands of individual investigators. This explosion of genomic data allows for comparative genomics, the systems-based study of genomes across different strains or species. Comparative genomics can be used to rationally engineer metabolic pathways by uncovering essential enzymes that can increase product yield. As an example, a comparative genomic analysis between the E. coli and Mannheimia succiniciproducens genomes, combined with in silico flux analysis, allowed for a seven-fold improvement in succinic acid yield in E. coli [55]. In the near future, these costs will be reduced further, removing this step as a financial or time limitation in the systems metabolic engineering approach. We expect that sequencing diverse collections of organisms will lead to systems-based pathway design and will help advance the field of metabolic engineering by facilitating pathway construction and design. Currently however, the cost of sequencing for more than a handful of organisms cannot be overlooked as a trivial cost.
5.2 Genome annotation and automatic metabolic reconstruction
Following genome sequencing, the next step in the metabolic network reconstruction process requires the bioinformatic discovery of all unique ORFs coding for enzymes in the metabolic network. Once identified, ORFs are assigned an enzyme functionality based on database information that includes activity, substrate specificity, cofactor dependence, and location within the cell (for compartmentalized models) [56]. Challenges to this process include assigning function to enzymes that may catalyze several reactions, enzymes with broad substrate specificity, or enzymes unique to the organism of study.
Genome annotation and metabolic reconstructions can be automated through a coupling with metabolic databases (Table 1), such as KEGG [57], LIGAND [58], BioCYC [59], EcoCYC [60], MetaCYC [61], PathBLAST [62], FMM [63], SEED [64], and BRENDA [65]. These databases collect bioinformatic and systems biology knowledge sets, serving as a repository for new or well-characterized pathways and reconstructions. As this repository grows in size and continues to be characterized, it essentially amounts to available biological catalysts that can be “pulled off the shelf” and imported into cells via synthetic biology constructs. Thus, the prospect for de novo pathway design is being advanced in these endeavors.
Table 1.
Online tools and databases for in silico model construction. A variety of tools are available to aid the automated construction, validation and verification of in silico models. These include tools for genome annotation, ORF function assessment, pathway design, and optimization routines.
| Systems metabolic engineering online databases and computational tools | |
|---|---|
| Databases for automated and manual genome annotation | TRANSPORT DB [86], WIT [87], SGD [88], KEGG [57], LIGAND [58], BioCYC [59], EcoCYC [60], MetaCYC [61], and BRENDA [65], UniProt [89], SWISS-PROT [90] |
| Elucidating gene function for unknown ORF | Gene Finders: GLIMMER [91], GlimmerM, Exonomy and Unveil [92] or GENSCAN [93] |
| Homology based – BLAST [94, 95], FASTA [96], or HMMER (http://hmmer.janelia.org/) | |
| Non-homology based - Prolinks database [97] | |
| Pathway completion tools | REBIT [98], BNICE[99, 100], FMM [63], PathBLAST [62] |
| In silico modeling tools | MOMA[70], ROOM[71], OptKnock [72], OptStrain [73], OptReg [74], OMNI [101], COBRA Toolbox [102], COPASI [103], GrowMatch [69] |
For uncommon genes, function can be assigned using gene finding algorithms, sequence homology searches, and non-homology based algorithms (Table 1). The drawback of these approaches is that they rely upon databases and commonalities of studied organisms, but seemingly similar enzymes can have different functionalities in different organisms. Furthermore, many unsequenced organisms contain pathways, enzymes, or metabolites that have not been characterized. In particular, the biochemical verification of even very closely related enzymes is important because minor differences in sequence have been known to drastically alter enzyme function. Thus, a great deal of hand curation is necessary to ensure the accuracy of the genome annotation, making this step the limiting factor for employing a systems metabolic engineering approach to unsequenced organisms.
Once organism annotation is completed, it is necessary to create the metabolic network and component interactions [56]. For a metabolic network enzyme, reaction stoichiometry, cofactor specificity, substrate specificity, directionality or reversibility, and cellular location all need to be included in the model [66]. The combination of verified automatically generated network reactions and manually curated reactions yields the first version of the genome-scale metabolic reconstruction.
5.3 Iteratively improving upon the metabolic reconstruction using flux analysis
The first metabolic reconstruction is often neither sufficiently accurate nor complete, thus an approach to improve the model is necessary. At this stage, computer programming-based flux analysis can be performed using the in silico metabolic reconstruction to assess its accuracy (Table 1). In this regard, flux balance analysis (FBA) is a constraint-based optimization approach toward quantifying flux distribution inside a genome-scale in silico metabolic reconstruction under the assumption of steady state conditions. An informative review of many flux analysis techniques has previously been published by Park et al. [67]. Initially, these in silico models are used to assess whether the model can predict biomass formation in general. These biomass formation tests are used to address the absence of key metabolic enzymes.
After successfully modeling cell growth, the reconstruction should be checked to confirm that it generates all known metabolites produced by the organism. This step requires either prior knowledge of the strain or global systems biology measurements of cellular components, including metabolites. The inability of the metabolic reconstruction simulation to produce a known cellular component infers a gap in the network that must be resolved using database or experimental methods. Furthermore, Manichaikul et al. [68] developed a methodology based on RT-PCR and RACE to verify hypothetical enzymes in order to refine genome annotations.
When the metabolic reconstruction can replicate cellular metabolites synthesis, further iterative improvement of the model is possible using comparative gene knockout data. Experimental data about growth or lack of growth of gene knockout strains can be compared to the in silico model. If the in silico model predicts no growth, while the experimental knockout grows, the discrepancy is likely due to an isozyme or an unsuspected metabolic pathway. If the in silico predicts growth, but the experimental knock out will not grow, then most likely there are enzyme functionalities in the metabolic reconstruction that are not actually present in the organism. GrowMatch [69] is a novel program that can automatically search for reactions to add to or to suppress from the network to help fix these growth versus no growth discrepancies.
5.4 Using flux analysis of the metabolic reconstruction for systems metabolic engineering
To the present date, most successful systems metabolic engineering approaches have used flux analysis to model gene knockouts in metabolic reconstructions in order to identify otherwise intractable deletions to increase product yield. These gene knockouts are modeled in silico by constraining the flux through the deleted reaction to be zero. Often, these reconstructions also introduce synthetic enzyme pathways to model non-native products.
A number of optimization algorithms have been developed to better reflect flux redistributions in response to a gene deletion. In addition to the linear-programming of FBA, minimization algorithms such as [70] minimization of metabolic adjustment (MOMA) and [71] regulatory on/off minimization (ROOM) have been developed to model gene knockouts. MOMA minimizes the flux redistributions in knockout models compared to wildtype fluxes while ROOM minimizes the number of significant flux changes in knockout flux models compared to wildtype fluxes. FBA, MOMA, and ROOM all only have a single objective function when calculating cellular flux distributions, normally to optimize cell growth. However, the common objective of a systems metabolic engineer is to optimize production of a given metabolite without decreasing cell growth. Therefore, the ability to specify multiple objectives, as in OptKnock [72], is very useful for the dual optimization of cell growth and product yield. Additional modeling constructions such as OptStrain [73], OptReg [74], and OptGene [75] have potential systems metabolic engineering application. These modeling approaches attempt to find multiple targets for flux improvement through gene overexpressions, gene knockdowns, gene deletions, and heterologous protein incorporations. These approaches are expanding the in silico metabolic engineering toolbox to complement our experimental capacities.
These flux analysis algorithms can be used to model gene knockouts and insertion of heterologous enzymes for metabolic pathway engineering applications, however, they do have inherent limitations. Heterologous enzymes are simply inserted into the reconstruction matrix, assuming that they are going to be actively expressed in their new host. This is often not the case due to solubility problems, unknown regulators, or problems with unoptimized codons. In addition, these flux analyses are not particularly adept at modeling overexpressions of enzymes. This limitation results from the fact that flux analyses based on stoichiometric matrices tend to represent gene overexpression by adjusting the flux constraint. When looking at the basic genome to fluxome cellular data pathway: Genome → transcriptome → proteome → metabolome → fluxome, it can be seen that the enzyme overexpression occurs at the genomic level. Due to cellular complexity, there is no direct, linear relation between any of the levels of cellular data [76]. In addition, the enzyme may not be the rate-limiting step or may be part of a tightly regulated pathway, so overexpression will have no phenotypical effect. Thus, these models are more predictive for the type of activity change required in the cell rather than the way in which to deliver this change. In this respect, these systems metabolic engineering tools provide guidance for strain engineering, but still rely on the availability of a powerful metabolic engineering toolbox (Textbox 1) capable of inducing these changes.
A general advantage of the systems metabolic engineering approach to strain improvement is complete cataloguing of all genetic modifications, allowing for future rational pathway or systems based engineering. To utilize traditional metabolic engineering techniques, only knowledge of the basic pathway of interest is prerequisite, and all engineering endeavors exploit only reactions closely related to the basic pathway. On the contrary, the systems metabolic engineering approach can target enzymes for gene knockout that are seemingly not related to the pathway or phenotype of interest. The cumulative effects of these knockouts combined with traditional metabolic engineering approaches generate an increased product titer or an enhanced phenotype. Even after several rounds of rational engineering, it is possible to adapt the metabolic reconstruction to model the engineered strain by constraining it to accurately reflect experimental 13Carbon flux data. This reconstruction can be used for further in silico analysis to search for potential genetic modifications [2, 35, 36]. Thus, an iterative systems metabolic engineering approach may be used to engineer already altered cells.
6 Improving the capabilities of systems metabolic engineering
Systems metabolic engineering is dependent on the availability and accuracy of high throughput data to incorporate into in silico models. The omics revolution has vastly increased our knowledge base, but our basic understanding of the cell's complexity is rudimentary. Therefore, our in silico cell models are also comparatively simple. Systems metabolic engineering's effectiveness at phenotypical predictions will theoretically increase as in silico reconstructions further reflect the complexity of the cell.
Reconstruction complexity can be increased in three ways in the near future. Firstly, in vivo enzyme kinetic data are lacking for many metabolic reactions, and incorporating this data into the metabolic model will increase its predictive prowess. Secondly, a suitable in silico representation of traditional enzyme overexpression and novel enzyme underexpression [8, 77] will permit the modeling of essential metabolic engineering techniques, increasing the capacity of the reconstruction to model genetic perturbations. By simulating gene over-expression, knockdown, and knockout, instead of only gene knockout, the power of the reconstruction will have increased three fold. Finally, the incorporation of transcriptional regulatory networks and signaling networks into the metabolic network reconstruction will greatly increase the predictive efficacy of the model and allow for dynamic cellular representation.
Transcriptional regulatory network reconstructions can be created following the same basic outline as for metabolic reconstructions [66]. Recent work by Faith et al. [78] using network inference algorithms may automate the future development of regulatory networks. In 2002, Covert and Palsson developed an experimentally confirmable integrated metabolic/regulatory model for the central carbon metabolism of E. coli that could model growth, substrate uptake, product secretion, and gene expression [79]. Furthermore, a very complete genome-scale reconstruction of E. coli's transcriptional and translational machinery was recently completed by Thiele et al. [80], but it has not been integrated into the metabolic reconstruction. Advanced integrated transcriptional regulatory/metabolic reconstructions could be used to model global transcriptional modulation, a key facet missing from existing metabolic reconstructions. Dynamic analysis of integrated metabolic, transcriptional regulatory, and signaling networks is in the initial stages. Lee et al. [81] proposed an integrative, dynamic FBA to solve a stoichiometric reconstruction containing metabolic, regulatory, and signaling processes of a S. cerevisiae pathway. Likewise, Covert et al. [82] modeled the dynamic behavior of the three networks in E. coli central carbon metabolism using an altered FBA approach. When the integration of the metabolic, transcriptional regulatory, and signaling transduction cellular networks is scaled up to the genome level, it will vastly improve the predictive power of in silico cell models.
It is important to note that as network or integrated network reconstructions increase in complexity, the computational power necessary to model flux analyses increases. Modeling multiple gene knockouts by systematically searching the reconstruction for single, double, triple, or more knockouts leads very quickly to combinatorial explosion. Therefore, a sequential, iterative approach is typically employed whereby the reconstruction is searched for a certain number of potential single knockouts, and then these single knockouts are searched for potential double knockouts, and so on. Alternatively, the optimization function can be augmented to simultaneously guarantee product yields and maximal growth. Regardless, increased computational power and refined optimization techniques would allow for a more thorough search.
7 The drawbacks of systems metabolic engineering
Assuming that reconstruction and modeling capabilities continue to be improved, there are still two potential difficulties blocking the proliferation of the systems metabolic engineering approach. Most importantly, even though the cost of generating the necessary high throughput data for a systems approach has been decreasingly rapidly, the price tag can still be prohibitively high, especially in an industrial setting where many mutant strains are under study. These high throughput costs include sequencing the genome and generating microarrays for analyzing transcript and protein levels, and 13Carbon flux data to verify the flux model. For instance, if multiple randomly mutated strains have improved phenotype, all of their genomes would have to be sequenced and analyzed in order to find the beneficial mutations. It may be more cost-effective to first shuffle the genomes together to combine the phenotypical improvements, and then sequence only the most improved strain. The second difficulty slowing the incorporation of systems-based approaches is that high throughput data in effect takes a “snap shot” of certain cellular component levels, transcript levels, metabolite levels, or protein levels. This snap shot provides a very descriptive view of the cell status but inherently does not have any information about any past or future cell states. Therefore, when a reconstruction is fitted to this high throughput data, it runs the risk of becoming a descriptive model, instead of the desired predictive model. As a result, systems metabolic engineering must address both cost and model predictivity to compete with traditional strategies for strain improvement.
8 The future of systems approaches to metabolic Engineering
In the near future, the most successful approaches utilizing systems metabolic engineering will continue to employ genome-scale metabolic reconstructions to model gene knockouts. Currently, there are metabolic reconstructions for over 30 organisms [56, 83], but of these, it has been mainly the metabolic reconstructions of commonly modeled organisms such as E. coli and S. cerevisiae that have been used for systems approaches. As metabolic reconstructions are expanded and genetic tools are developed for non-model organisms, systems approaches toward engineering these organisms will thrive. The capability to model gene overexpression and underexpression will be difficult due to the unquantified relation between transcription levels and network flux modulation. However, these impacts will certainly advance the field.
To maximize the benefit of using systems approaches, a metabolic engineer must ask the following questions. First, what organism should be engineered to optimize yield? Second, in what stage of the strain development process should a systems biology approach be applied? Third, is a systems metabolic engineering approach more cost-effective than traditional pathway engineering or industry-styled random mutagenesis and screening?
What organism should be engineered to optimize yield? Before attempting to begin any modeling or experimental work, literature should be referenced to determine how easily potential organisms grow under standard laboratory or industrial conditions. Traditional pathway engineering and systems metabolic engineering are possible only for organisms that have developed genetic tools, such as the transformative insertion of heterologous DNA and protocols for the deletions of unwanted genes. In certain cases, properties of an uncommon organism make it seemingly ideal for the desired application. If this potential organism does not have developed genetic tools, phenotype improvement is often limited to random mutagenesis. If the organism has developed genetic tools, it can be altered using traditional metabolic engineering techniques. If, in addition, the organism has a developed metabolic reconstruction, in silico modeling can be utilized to test for desired phenotypes in a systems-based setting. If the metabolic reconstruction does not exist, it can be created by following the procedure discussed above. However, the time demand and costs associated with the annotation and reconstruction process can be prohibitive, so it may be more cost-effective to engineer the organism using non-systems-based methods. After weighing the benefits and shortcomings of using model or non-model platforms, the optimal organism identified can be used for experimental work.
In what stage of the strain development process should a systems biology approach be applied? The largest quantity of bioinformatic data is available for wildtype strains because they have not been genetically modified or randomly mutated. Therefore, most genome-scale metabolic reconstructions are likely to be available for non-modified wildtype strains, and a systems metabolic engineering approach should be employed in the beginning stages of strain development. The largest degree of success has been seen in improving the metabolic phenotypes of cells, rather than more complex phenotypes. Even if the metabolic reconstruction is being produced from scratch, it is still best to begin with a systems approach, both to improve phenotype, and to check the reconstruction for accuracy. Additionally, all systems metabolic engineering-based genetic alterations are fully characterized and traceable, unlike combinatorial and evolutionary approaches. Thus, sequential, iterative approaches toward improving phenotypes are easier when employing a systems-based approach. Following this approach, the use of combinatorial approaches for further improving phenotypes should be employed since suitable systems approaches for global cellular engineering are not currently available. Moreover, these techniques can create genomic mutations that are difficult to track and model using systems approaches.
Is a systems metabolic engineering approach more cost-effective than traditional pathway engineering or industry-styled random mutagenesis and screening? The cost-effectiveness of systems metabolic engineering depends heavily on two factors: if the platform organism has developed genetic tools and if the organism has a previously existing metabolic reconstruction. Developing genetic tools for uncommon organisms can be costly and prone to failure. Therefore, random mutagenesis is most likely the only viable method to improve phenotype for these organisms. If the organism has genetics tools but lacks a metabolic reconstruction, the cost of producing such a reconstruction must be weighed against the potential phenotypical improvement through systems metabolic engineering. Finally, there is the scenario in which the organism has both genetic tools and an existing reconstruction; the field of systems metabolic engineering has had the most successes under these conditions. In this case, the metabolic reconstruction can be used to find gene knockouts to improve product yield at little to no charge. Updating the reconstruction after successful knockouts requires newly acquired fluxomic data. This process, which can be repeatedly iteratively and inexpensively, can be considered a first leap in improving product yield. After potential gene perturbations have been exhausted, combinatorial techniques (gTME or genome shuffling) and random mutagenesis can be employed to obtain another leap in product yield. Combinatorial approaches are often necessary because the multigenic nature of many phenotypes cannot be captured by a simplified model.
In conclusion, the emerging field of systems metabolic engineering is rife with prospects and capabilities. Previous systems metabolic engineering successes have used model organisms with existing metabolic reconstructions to discover gene knockouts to improve product yield. At the present time, strains improved in this manner can be furthered optimized using novel combinatorial techniques and random mutagenesis to further increase product yield. As more non-model organisms or industrial strains are sequenced and annotated, the library of organisms (and enzymes) available to a systems metabolic engineer will increase. Furthermore, reconstruction predictive power can be improved by incorporating other cellular processes, allowing for the improvement of multigenic traits and non-metabolic phenotypes. At present, systems biology approach for metabolic engineering work well for predicting changes for metabolic pathway engineering. The computational support of the field must also be advanced to match our current capabilities of high-throughput biology measurements. In addition, this approach has significant limitations in the engineering of complex, multigenic phenotypes such as tolerance and thus still requires follow-up using combinatorial metabolic and cellular engineering tools. As these approaches evolve, the cost of systems metabolic engineering will continue to decrease allowing for widespread adoption as an iterative first step in phenotype improvement.
Acknowledgments
We acknowledge support from NIH (Grant number: 1R01GM090221-01) and the Camille and Henry Dreyfus New Faculty Award.
Abbreviations
- FBA
flux balance analysis
- gTME
global transcription machinery engineering
- MOMA
minimization of metabolic adjustment
- ROOM
Regulatory On/Off Minimization
Biographies

Dr. Hal Alper is an Assistant Professor in the Department of Chemical Engineering at The University of Texas at Austin. He earned his Ph.D. in chemical engineering from the Massachusetts Institute of Technology in 2006 and was a postdoctoral research associate at the Whitehead Institute for Biomedical Research from 2006–2008, and at Shire Human Genetic Therapies from 2007–2008. Dr. Alper's research is in the area of cellular and metabolic engineering. His research focuses on metabolic and cellular engineering in the context of biofuel, biochemical, and biopharmaceutical production in an array of model host organisms.

John Blazeck received his BS in chemical engineering from the University of Florida, and is a graduate student in the Laboratory for Cellular and Metabolic Engineering at the University of Texas at Austin. His current research utilizes oleaginous yeast for the production of biofuels.
Footnotes
The authors have declared no conflict of interest.
9 References
- [1].Bailey JE. Toward a science of metabolic engineering. Science. 1991;252:1668–1675. doi: 10.1126/science.2047876. [DOI] [PubMed] [Google Scholar]
- [2].Lee KH, Park JH, Kim TY, Kim HU, et al. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol. Syst. Biol. 2007;3:149. doi: 10.1038/msb4100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tyo KE, Alper HS, Stephanopoulos GN. expanding the metabolic engineering toolbox: more options to engineer cells. Trends Biotechnol. 2007;25:132–137. doi: 10.1016/j.tibtech.2007.01.003. [DOI] [PubMed] [Google Scholar]
- [4].Kizer L, D. Pitera J, Pfleger BF, Keasling JD. Application of functional genomics to pathway optimization for increased isoprenoid production. Appl. Environ. Microbiol. 2008;74:3229–3241. doi: 10.1128/AEM.02750-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pitera DJ, Paddon CJ, Newman JD, Keasling JD. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab. Eng. 2007;9:193–207. doi: 10.1016/j.ymben.2006.11.002. [DOI] [PubMed] [Google Scholar]
- [6].Keasling JD. Synthetic biology for synthetic chemistry. ACS Chem. Biol. 2008;3:64–76. doi: 10.1021/cb7002434. [DOI] [PubMed] [Google Scholar]
- [7].Keasling JD, Newman JD, Pitera DJ. Method for enhancing production of isoprenoid compounds. 2007. US 07183089. [Google Scholar]
- [8].Pfleger BF, Pitera DJ, Smolke CD, Keasling JD. Combinatorial engineering of intergenic regions in operons tunes expression of multiple genes. Nat. Biotechnol. 2006;24:1027–1032. doi: 10.1038/nbt1226. [DOI] [PubMed] [Google Scholar]
- [9].Woodyer RD, Li GY, Zhao HM, van der Donk WA. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem. Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- [10].Woodyer RD, Shao ZY, Thomas PM, Kelleher NL, et al. Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem. Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- [11].Khosla C, Kapur S, Cane DE. Revisiting the modularity of modular polyketide synthases. Curr. Opin. Chem. Biol. 2009;13:135–143. doi: 10.1016/j.cbpa.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ridley CP, Khosla C. Synthesis and biological activity of novel pyranopyrones derived from engineered aromatic polyketides. ACS Chem. Biol. 2007;2:104–108. doi: 10.1021/cb600382j. [DOI] [PubMed] [Google Scholar]
- [13].McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel `unnatural' natural products. Proc. Natl. Acad. Sci. USA. 2000;97:5011–5011. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Nakamura CE, Whited GM. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 2003;14:454–459. doi: 10.1016/j.copbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- [15].Suthers PF, Cameron DC. Production of 3-hydroxypropionic acid in recombinant organisms. 2005. US 06852517. [Google Scholar]
- [16].Lee SY, Hong SH. Engineering of Escherichia coli central metabolic pathways for the production of succinic acid. Am. Chem. Soc. 2002:30–38. [Google Scholar]
- [17].Alper H, Miyaoku K, Stephanopoulos G. Characterization of lycopene-overproducing E. coli strains in high cell density fermentations. Appl. Microbiol. Biotechnol. 2006;72:968–974. doi: 10.1007/s00253-006-0357-y. [DOI] [PubMed] [Google Scholar]
- [18].Inui M, Suda M, Kimura S, Yasuda K, et al. Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl. Microbiol. Biotechnol. 2008;77:1305–1316. doi: 10.1007/s00253-007-1257-5. [DOI] [PubMed] [Google Scholar]
- [19].Steen EJ, Chan R, Prasad N, Myers S, et al. Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb. Cell Fact. 2008;7:1–8. doi: 10.1186/1475-2859-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Atsumi S, Cann AF, Connor MR, Shen CR, et al. Metabolic engineering of Escherichia coli for 1-butanol production. Metab. Eng. 2008;10:305–311. doi: 10.1016/j.ymben.2007.08.003. [DOI] [PubMed] [Google Scholar]
- [21].Shen CR, Liao JC. Metabolic engineering of Escherichia coli for 1-butanol and 1-propanol production via the keto-acid pathways. Metab. Eng. 2008;10:312–320. doi: 10.1016/j.ymben.2008.08.001. [DOI] [PubMed] [Google Scholar]
- [22].Kajiwara S, Suga K, Sone H, Nakamura K. Improved ethanol tolerance of Saccharomyces cerevisiae strains by increases in fatty acid unsaturation via metabolic engineering. Biotechnol. Lett. 2000;22:1839–1843. [Google Scholar]
- [23].Alper H, Moxley J, Nevoigt E, Fink GR, et al. Engineering yeast transcription machinery for improved ethanol tolerance and production. Science. 2006;314:1565–1568. doi: 10.1126/science.1131969. [DOI] [PubMed] [Google Scholar]
- [24].Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451:86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- [25].Zhang K, Sawaya MR, Eisenberg DS, Liao JC. Expanding metabolism for biosynthesis of nonnatural alcohols. Proc. Natl. Acad. Sci. USA. 2008;105:20653–20658. doi: 10.1073/pnas.0807157106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jin YS, Alper H, Yang YT, Stephanopoulos G. Improvement of xylose uptake and ethanol production in recombinant Saccharomyces cerevisiae through an inverse metabolic engineering approach. Appl. Environ. Microbiol. 2005;71:8249–8256. doi: 10.1128/AEM.71.12.8249-8256.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma Q, Li X, Vale-Cruz D, Brown ML, et al. Activating transcription factor 2 controls Bcl-2 promoter activity in growth plate chondrocytes. J. Cell. Biochem. 2007;101:477–487. doi: 10.1002/jcb.21198. [DOI] [PubMed] [Google Scholar]
- [28].Jin YS, Stephanopoulos G. Multi-dimensional gene target search for improving lycopene biosynthesis in Escherichia coli. Metab. Eng. 2007;9:337–347. doi: 10.1016/j.ymben.2007.03.003. [DOI] [PubMed] [Google Scholar]
- [29].Stephanopoulos G. Metabolic engineering by genome shuffling - Two reports on whole-genome shuffling demonstrate the application of combinatorial methods for phenotypic improvement in bacteria. Nat. Biotechnol. 2002;20:666–668. doi: 10.1038/nbt0702-666. [DOI] [PubMed] [Google Scholar]
- [30].Lynch MD, Warnecke T, Gill RT. SCALEs: multiscale analysis of library enrichment. Nat. Methods. 2007;4:87–93. doi: 10.1038/nmeth946. [DOI] [PubMed] [Google Scholar]
- [31].Shi DJ, Wang CL, Wang KM. Genome shuffling to improve thermotolerance, ethanol tolerance and ethanol productivity of Saccharomyces cerevisiae. J. Ind. Microbiol. Biotechnol. 2009;36:139–147. doi: 10.1007/s10295-008-0481-z. [DOI] [PubMed] [Google Scholar]
- [32].Wang YH, Li Y, Pei XL, Yu L, et al. Genome-shuffling improved acid tolerance and L-lactic acid volumetric productivity in Lactobacillus rhamnosus. J. Biotechnol. 2007;129:510–515. doi: 10.1016/j.jbiotec.2007.01.011. [DOI] [PubMed] [Google Scholar]
- [33].Alper H, Stephanopoulos G. Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab. Eng. 2007;9:258–267. doi: 10.1016/j.ymben.2006.12.002. [DOI] [PubMed] [Google Scholar]
- [34].Park JH, Lee SY, Kim TY, Kim HU. Application of systems biology for bioprocess development. Trends Biotechnol. 2008;26:404–412. doi: 10.1016/j.tibtech.2008.05.001. [DOI] [PubMed] [Google Scholar]
- [35].Park JH, Lee KH, Kim TY, Lee SY. Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc. Natl. Acad. Sci. USA. 2007;104:7797–7802. doi: 10.1073/pnas.0702609104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Park JH, Lee SY. Towards systems metabolic engineering of microorganisms for amino acid production. Curr. Opin. Biotechnol. 2008;19:454–460. doi: 10.1016/j.copbio.2008.08.007. [DOI] [PubMed] [Google Scholar]
- [37].Alper H, Jin YS, Moxley JF, Stephanopoulos G. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab. Eng. 2005;7:155–164. doi: 10.1016/j.ymben.2004.12.003. [DOI] [PubMed] [Google Scholar]
- [38].Alper H, Stephanopoulos G. Uncovering the gene knockout landscape for improved lycopene production in E. coli. Appl. Microbiol. Biotechnol. 2008;78:801–810. doi: 10.1007/s00253-008-1373-x. [DOI] [PubMed] [Google Scholar]
- [39].Lee SY, Hong SH, Moon SY. In silico metabolic pathway analysis and design: succinic acid production by metabolically engineered Escherichia coli as an example. Genome Inf. 2002;13:214–223. [PubMed] [Google Scholar]
- [40].Yegane-Sarkandy S, Farnoud AM, Shojaosadati SA, Khalilzadeh R, et al. Overproduction of human interleukin-2 in recombinant Escherichia coli BL21 high-cell-density culture by the determination and optimization of essential amino acids using a simple stoichiometric model. Biotechnol. Appl. Biochem. 2009;54:31–39. doi: 10.1042/BA20080300. [DOI] [PubMed] [Google Scholar]
- [41].Otero JM, Olsson L, Nielsen J. Metabolic engineering of Saccharomyces cerevisiae microbial cell factories for succinic acid production. Biotechnol. J. 2007:S205–S205. [Google Scholar]
- [42].Bro C, Regenberg B, Forster J, Nielsen J. In silico aided metabolic engineering of Saccharomyces cerevisiae for improved bioethanol production. Metab. Eng. 2006;8:102–111. doi: 10.1016/j.ymben.2005.09.007. [DOI] [PubMed] [Google Scholar]
- [43].Wendisch VF, Bott M, Kalinowski J, Oldiges M, et al. Emerging Corynebacterium glutamicum systems biology. Biotechnol. J. 2006:74–92. doi: 10.1016/j.jbiotec.2005.12.002. [DOI] [PubMed] [Google Scholar]
- [44].Kieldsen KR, Nielsen J. In silico genome-scale reconstruction and validation of the Corynebacterium glutamicum metabolic network. Biotechnol. Bioeng. 2009;102:583–597. doi: 10.1002/bit.22067. [DOI] [PubMed] [Google Scholar]
- [45].Baumbach J, Brinkrolf K, Czaja LF, Rahmann S, et al. CoryneRegNet: An ontology-based data warehouse of corynebacterial transcription factors and regulatory networks. BMC. Genomics. 2006;7:12. doi: 10.1186/1471-2164-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Feist AM, Henry CS, Reed JL, Krummenacker M, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007;3:1–18. doi: 10.1038/msb4100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Herrgard MJ, Swainston N, Dobson P, Dunn WB, et al. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol. 2008;26:1155–1160. doi: 10.1038/nbt1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Andersen MR, Nielsen ML, Nielsen J. Metabolic model integration of the bibliome, genome, metabolome and reactome of Aspergillus niger. Mol. Syst. Biol. 2008;4:1–13. doi: 10.1038/msb.2008.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Oh YK, Palsson BO, Park SM, Schilling CH, et al. Genome-scale reconstruction of metabolic network in Bacillus subtilis based on high-throughput phenotyping and gene essentiality data. J. Biol. Chem. 2007;282:28791–28799. doi: 10.1074/jbc.M703759200. [DOI] [PubMed] [Google Scholar]
- [50].Alper H, Stephanopoulos G. Engineering for biofuels: exploiting innate microbial capacity or importing biosynthetic potential? Nat. Rev. Microbiol. 2009;7:715–723. doi: 10.1038/nrmicro2186. [DOI] [PubMed] [Google Scholar]
- [51].Sanger F, Air GM, Barrell BG, Brown NL, et al. Nucleotide-sequence of bacteriophage PhiChi174 DNA. Nature. 1977;265:687–695. doi: 10.1038/265687a0. [DOI] [PubMed] [Google Scholar]
- [52].Sanger F. Sequences, Sequences, and Sequences. Annu. Rev. Biochem. 1988;57:1–28. doi: 10.1146/annurev.bi.57.070188.000245. [DOI] [PubMed] [Google Scholar]
- [53].Hunkapiller T, Kaiser RJ, Koop BF, Hood L. Large-scale and automated DNA-sequence determination. Science. 1991;254:59–67. doi: 10.1126/science.1925562. [DOI] [PubMed] [Google Scholar]
- [54].Shendure J, Ji HL. Next-generation DNA sequencing. Nat. Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- [55].Lee SJ, Lee DY, Kim TY, Kim BH, et al. Metabolic engineering of Escherichia coli for enhanced production of succinic acid, based on genome comparison and in silico gene knockout simulation. Appl. Environ. Microbiol. 2005;71:7880–7887. doi: 10.1128/AEM.71.12.7880-7887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Reed JL, Famili I, Thiele I, Palsson BO. Towards multidimensional genome annotation. Nat. Rev. Genet. 2006;7:130–141. doi: 10.1038/nrg1769. [DOI] [PubMed] [Google Scholar]
- [57].Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Goto S, Nishioka T, Kanehisa M. LIGAND: chemical database of enzyme reactions. Nucleic Acids Res. 2000;28:380–382. doi: 10.1093/nar/28.1.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Karp PD, Ouzounis CA, Moore-Kochlacs C, Goldovsky L, et al. Expansion of the BioCyc collection of pathway/genome databases to 160 genomes. Nucleic Acids Res. 2005;33:6083–6089. doi: 10.1093/nar/gki892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Keseler IM, Collado-Vides J, Gama-Castro S, Ingraham J, et al. EcoCyc: a comprehensive database resource for Escherichia coli. Nucleic Acids Res. 2005;33:D334–D337. doi: 10.1093/nar/gki108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Caspi R, Foerster H, Fulcher CA, Kaipa P, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008;36:D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kelley BP, Yuan BB, Lewitter F, Sharan R, et al. Path-BLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res. 2004;32:W83–W88. doi: 10.1093/nar/gkh411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chou CH, Chang WC, Chiu CM, Huang CC, et al. FMM: a web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009;37:W129–W134. doi: 10.1093/nar/gkp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].DeJongh M, Formsma K, Boillot P, Gould J, et al. Toward the automated generation of genome-scale metabolic networks in the SEED. BMC Bioinf. 2007;8:1–17. doi: 10.1186/1471-2105-8-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chang A, Scheer M, Grote A, Schomburg I, et al. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res. 2009;37:D588–D592. doi: 10.1093/nar/gkn820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Herrgard MJ, Thiele I, Reed JL, et al. Reconstruction of biochemical networks in microorganisms. Na. Rev. Microbiol. 2009;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Park JM, Kim TY, Lee SY. Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol. Adv. 2009 doi: 10.1016/j.biotechadv.2009.05.019. in press. DOI: 10.1016/j.biotechadv.2009.05.019, 2009. [DOI] [PubMed] [Google Scholar]
- [68].Manichaikul A, Ghamsari L, Hom EFY, Lin CW, et al. Metabolic network analysis integrated with transcript verification for sequenced genomes. Nat. Methods. 2009;6:589–U553. doi: 10.1038/nmeth.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kumar VS, Maranas CD. GrowMatch: an automated method for reconciling in silico/in vivo growth predictions. PLoS. Comput. Biol. 2009;5 doi: 10.1371/journal.pcbi.1000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Shlomi T, Berkman O, Ruppin E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. USA. 2005;102:7695–7700. doi: 10.1073/pnas.0406346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Burgard AP, Pharkya P, Maranas CD. OptKnock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- [73].Pharkya P, Burgard AP, Maranas CD. OptStrain: A computational framework for redesign of microbial production systems. Genome Res. 2004;14:2367–2376. doi: 10.1101/gr.2872004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Pharkya P, Maranas CD. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 2006;8:1–13. doi: 10.1016/j.ymben.2005.08.003. [DOI] [PubMed] [Google Scholar]
- [75].Patil KR, Rocha I, Forster J, Nielsen J. Evolutionary programming as a platform for in silico metabolic engineering. BMC Bioinf. 2005;6:12. doi: 10.1186/1471-2105-6-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Moxley JF, Jewett MC, Antoniewicz MR, Villas-Boas SG, et al. Linking high-resolution metabolic flux phenotypes and transcriptional regulation in yeast modulated by the global regulator Gcn4p. Proc. Natl. Acad. Sci. USA. 2009;106:6477–6482. doi: 10.1073/pnas.0811091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA. 2005;102:12678–12683. doi: 10.1073/pnas.0504604102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Faith JJ, Hayete B, Thaden JT, Mogno I, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS. Biol. 2007;5:54–66. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Covert MW, Palsson BO. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J. Biol. Chem. 2002;277:28058–28064. doi: 10.1074/jbc.M201691200. [DOI] [PubMed] [Google Scholar]
- [80].Thiele I, Jamshidi N, Fleming RMT, Palsson BO. Genome-scale reconstruction of Escherichia coli's transcriptional and translational machinery: a knowledge base, its mathematical formulation, and its functional characterization. PLoS. Comput. Biol. 2009;5:13. doi: 10.1371/journal.pcbi.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lee JM, Gianchandani EP, Eddy JA, Papin JA. Dynamic analysis of integrated signaling, metabolic, and regulatory networks. PLoS. Comput. Biol. 2008;4:20. doi: 10.1371/journal.pcbi.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Covert MW, Xiao N, Chen TJ, Karr JR. Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics. 2008;24:2044–2050. doi: 10.1093/bioinformatics/btn352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Oberhardt MA, Palsson BO, Papin JA. Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol. 2009;5:320. doi: 10.1038/msb.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Ham TS, Lee SK, Keasling JD, Arkin AP. A tightly regulated inducible expression system utilizing the fim inversion recombination switch. Biotechnol. Bioeng. 2006;94:1–4. doi: 10.1002/bit.20916. [DOI] [PubMed] [Google Scholar]
- [85].Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- [86].Ren QH, Kang KH, Paulsen IT. TransportDB: a relational database of cellular membrane transport systems. Nucleic Acids Res. 2004;32:D284–D288. doi: 10.1093/nar/gkh016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Overbeek R, Larsen N, Pusch GD, D'Souza M, et al. WIT: integrated system for high-throughput genome sequence analysis and metabolic reconstruction. Nucleic Acids Res. 2000;28:123–125. doi: 10.1093/nar/28.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Christie KR, Weng S, Balakrishnan R, Costanzo MC, et al. Saccharomyces Genome Database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 2004;32:D311–D314. doi: 10.1093/nar/gkh033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Apweiler R, Bairoch A, Wu CH, Barker WC, et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Boeckmann B, Bairoch A, Apweiler R, Blatter MC, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Delcher AL, Harmon D, Kasif S, White O, et al. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Majoros WH, Pertea M, Antonescu C, Salzberg SL. GlimmerM, Exonomy and Unveil: three ab initio eukaryotic genefinders. Nucleic Acids Res. 2003;31:3601–3604. doi: 10.1093/nar/gkg527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- [94].Altschul SF, Gish W, Miller W, Myers EW, et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- [95].Altschul SF, Madden TL, Schaffer AA, Zhang JH, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bowers PM, Pellegrini M, Thompson MJ, Fierro J, et al. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 2004;5:13. doi: 10.1186/gb-2004-5-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Prather KLJ, Martin CH. De novo biosynthetic pathways: rational design of microbial chemical factories. Curr. Opin. Biotechnol. 2008;19:468–474. doi: 10.1016/j.copbio.2008.07.009. [DOI] [PubMed] [Google Scholar]
- [99].Li CH, Henry CS, Jankowski MD, Ionita JA, et al. Computational discovery of biochemical routes to specialty chemicals. Chem. Eng. Sci. 2004;59:5051–5060. [Google Scholar]
- [100].Hatzimanikatis V, Li CH, Ionita JA, Henry CS, et al. Exploring the diversity of complex metabolic networks. Bioinformatics. 2005;21:1603–1609. doi: 10.1093/bioinformatics/bti213. [DOI] [PubMed] [Google Scholar]
- [101].Herrgard MJ, Fong SS, Palsson BO. Identification of genome-scale metabolic network models using experimentally measured flux profiles. PLoS. Comput. Biol. 2006;2:676–686. doi: 10.1371/journal.pcbi.0020072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Becker SA, Feist AM, Mo ML, Hannum G, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox. Nat. Protoc. 2007;2:727–738. doi: 10.1038/nprot.2007.99. [DOI] [PubMed] [Google Scholar]
- [103].Hoops S, Sahle S, Gauges R, Lee C, et al. COPASI- A COmplex PAthway SImulator. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485. [DOI] [PubMed] [Google Scholar]