

Abstract
Background
Usefulness of iron chelation therapy in myelodysplastic patients is still under debate but many authors suggest its possible role in improving survival of low-risk myelodysplastic patients. Several reports have described an unexpected effect of iron chelators, such as an improvement in hemoglobin levels, in patients affected by myelodysplastic syndromes. Furthermore, the novel chelator deferasirox induces a similar improvement more rapidly. Nuclear factor-κB is a key regulator of many cellular processes and its impaired activity has been described in different myeloid malignancies including myelodysplastic syndromes.
Design and Methods
We evaluated deferasirox activity on nuclear factor-κB in myelodysplastic syndromes as a possible mechanism involved in hemoglobin improvement during in vivo treatment. Forty peripheral blood samples collected from myelodysplastic syndrome patients were incubated with 50 μM deferasirox for 18h.
Results
Nuclear factor-κB activity dramatically decreased in samples showing high basal activity as well as in cell lines, whereas no similar behavior was observed with other iron chelators despite a similar reduction in reactive oxygen species levels. Additionally, ferric hydroxyquinoline incubation did not decrease deferasirox activity in K562 cells suggesting the mechanism of action of the drug is independent from cell iron deprivation by chelation. Finally, incubation with both etoposide and deferasirox induced an increase in K562 apoptotic rate.
Conclusions
Nuclear factor-κB inhibition by deferasirox is not seen from other chelators and is iron and reactive oxygen species scavenging independent. This could explain the hemoglobin improvement after in vivo treatment, such that our hypothesis needs to be validated in further prospective studies.
Keywords: iron chelation therapy, nuclear factor-κB, myelodysplastic symdrome
Introduction
Myelodysplastic syndromes are a heterogeneous cluster of bone marrow disorders in which different grades of peripheral cytopenias are associated with bone marrow dysplasia1 and a percentage of bone marrow (BM) blasts of up to 20%. The International Prognostic Scoring System (IPSS), based on BM blast levels, the number of peripheral cytopenias, and cytogenetic abnormalities, allows clinicians to perform a prognostic stratification of patients.2 More recently, transfusion dependence has been added to the previous prognostic factors.3,4 Low- and intermediate-risk patients have a substantially longer life expectancy and often become transfusion-dependent. As such, these patients often develop secondary organ failure due to high iron intake.5 In order to avoid secondary iron overload, MDS patients are treated with iron chelation therapy (ICT) according to the MDS Foundation’s guidelines.6 Moreover, several studies have demonstrated an overall higher survival in MDS patients treated with ICT.4,7 Very recently, data on safety and efficacy in MDS patients of a once daily oral chelator (deferasirox) were published by Porter et al.8 In spite of this, the real usefulness of iron chelation therapy in MDS, and also which kind of patient can benefit more from the therapy, is still under debate9–11 and prospective studies are needed to address this. Several reports and our own experience have demonstrated an unexpected effect of deferasirox therapy on hemoglobin improvement and reductions in transfusion requirement even after only a few months of ICT.12–14 The effect seems to be shared by different diseases, including both MDS and primary myelofibrosis, but has never been observed in thalassemia patients. There are very few reports of similar effects with other iron chelators:15,16 In 1996, Jensen et al. described a hemoglobin improvement, but in some cases there were even trilinear responses in 11 MDS patients treated with deferioxamine for up to 60 months.17 It has also been reported that myelofibrotic patients have a similar response to deferiprone.18 In all of these reports, the hematologic response is strictly associated with both a sharp decrease in iron burden and a fairly long lasting therapy of at least one year.
Nuclear factor-κB is a transcriptional nuclear factor involved in the regulation of several fundamental cellular processes such as apoptosis, proliferation, differentiation and tumor migration.19 It consists of a small group of proteins that, if associated with the inhibitory complex IKK, maintains an inactive state of the p65 subunit and a cytoplasmic localization.20 After proteasomal degradation of this negative regulatory protein, NF-κB is able to enter the nucleus and regulate transcription of over 150 genes belonging to different pathways, essentially regulating proliferation and cell survival. NF-κB involvement in suppressing apoptosis gives it an undoubted role in the pathogenesis of various types of cancer21,22 and in hematologic malignancies. In 2001, Guzman et al. demonstrated that p65 nuclear localization and transcriptional activity occur in acute myeloid leukemia (AML) blasts but not in normal erythroid precursors.23 More recently, NF-κB activation in myelodysplastic patients has been reported.24 Increased activity is strictly related to the clonal population in the bone marrow, such that the IPSS risk is increased with increased percentages of nuclear p65-positive cells. The NF-κB pathway can be activated by a broad variety of different stimuli,25 one of the most important being the tumor necrosis factor (TNF) receptor signaling pathway, in which the involvement of the serine/threonine kinase RIP1 is crucial.26,27 However, TNF signaling in cells results in a subtle balance between survival and death. In fact, NF-κB activation mediated by TNF leads to an anti-apoptotic effect through both caspase and JNK (Jun N-terminal Kinase) cascade inhibition, while reactive oxygen species accumulation induced by the same stimulus leads to cell death through JNK activation.28–30 Moreover, NF-κB is a redox-sensitive transcription factor, and its activation is mediated by NIK (NF-κB-inducing kinase) in many cellular subsets,31–36 including different cancer cells.
In polytransfused MDS patients, NF-κB activation can be due to an increase in the amount of iron intake which leads to reactive oxygen species generation. But the fact that NF-κB activation can also be observed in non-transfused patients must be taken into account. Indeed, it is well known that free radical generation is not only a collateral event of a long transfusional history, but that reactive oxygen species are involved in the pathogenesis of the disease, as demonstrated by Padua et al. in a mouse model,37 and also play a crucial role in disease progression in myeloid malignancies38,39 and in MDS.
We aimed to investigate the activity of the oral chelator deferasirox as an NF-κB inhibitor. We tested the drug in two types of leukemia cell line (K562 and HL60) characterized by high basal NF-κB activity. We then tested NF-κB activity in 40 peripheral blood (PB) samples of MDS and AML secondary to MDS (sAML) patients. In 28 of 40 peripheral blood samples with high basal NF-κB activity, deferasirox incubation induced a significant inhibition of NF-κB activity and a cytoplasmic sequestration of its active subunit p65 in an inactive form Finally, we investigated if other commercially available oral chelators share the same effect: neither deferiprone nor deferioxamine incubation with either the HL60 or K562 cell lines reduced NF-κB activation despite a similar reactive oxygen species clearance. Furthermore, no reduction on NF-κB inhibition has been observed after addition of a permeant iron carrier (ferric hydroxyquinoline) to K562 cells thus suggesting the mechanism of action of the drug is independent from cell iron deprivation by chelation. Our in vitro data could provide an explanation for the hemoglobin improvement observed in vivo in MDS patients treated with deferasirox.
Design and Methods
After written informed consent was obtained, 40 peripheral blood samples were collected from MDS patients (Table 1). The subtypes according to the WHO classification were as follows: 14 refractory anemia (RA), 2 refractory cytopenia with multilineage dysplasia (RCMD), 2 MDS unclassified (MDS-U), 14 refractory anemia with blast excess (RAEB) and 8 sAML. In 22 of 40 patients, serum ferritin levels were higher than 500 ng/mL and 12 presented a documented iron overload measured by SQUID (biomagnetic liver susceptometry). Fourteen samples were collected from untransfused patients. Twenty healthy volunteers were included in the study as controls.
Table 1.
Clinical features of the patients enrolled in the study.

Mononuclear cells collected from MDS patients and healthy subjects were cultured in RPMI supplemented with 5% fetal bovine serum and incubated with 50 μM deferasirox for 18h. The K562 and HL60 cell lines were used as controls and incubated with 50 μM deferasirox for 18h. Incubations with both deferioxamine and deferiprone were performed at 0.5 mM for 30 min as previously described.40 Deferiprone was also used at 100 μM for 18h. In order to better compare the different compounds in conditions mimicking the therapeutical concentrations reached in vivo during ICT, we also performed the same experiments using both deferioxamine and deferiprone at 50 μM for 18h obtaining similar results. FHQ (ferric hydroxyquinoline, Sigma-Aldrich) was added to the medium at either 2.5 μM or 5 μM as previously described.41 The NF-B inhibitor PS 1145 (Millenium, Cambridge, MA, USA) was added to the medium at 20 μM as previously described.42 Incubated and control cells were both evaluated for NF-κB activity. Unless stated otherwise, all the experiments were carried out independently three times.
Western blot was performed using 70 μg of both nuclear and cytoplasmic extracts. A p65 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used at a 1:1000 dilution. Antibodies against the unrelated proteins β-actin (Sigma Aldrich, St Louis, MO, USA) or lamin (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) were used both at 1:1000 dilutions as controls.42
Immunofluorescence assay was performed using a p65 polyclonal primary antibody (Santa Cruz Biotechnology) according to standard procedures. The signal was detected using a goat anti-rabbit Alexa Fluor 568 (FITC) secondary antibody (Molecular Probes) and propidium iodide to stain the nucleus. Images were analyzed with a confocal scanning microscope (LSM 5110; Carl Zeiss MicroImaging, Inc.) and captured using 100x objectives.
First 40 μg of nuclear extracts were incubated with a radioactive 32γ ATP-labeled oligonucleotide probe containing the DNA specific recognition sequence for p65 (5′-GGGACTTTCC-3′). The binding reaction and sample run were performed according to standard procedures. Gels were later dried and exposed to autoradiographic film at −70°C with enhancer foils.42
The DNA binding activity of NF-κB was determined according to the protocol of the TRANS AM ELISA Kit. Nuclear extracts (20 μg) were added to a 96-well plate coated with the DNA binding motif of NF-κB (5′-GGGACTTTCC-3′). For this, an HRP-conjugated secondary antibody provides a sensitive colorimetric reaction, which is quantified by spectrophotometry. Absorbances were read at 450 nm with a reference wave length of 655 nm.
2′-7′-dichlorofluorescin diacetate (DCF; Sigma) was added to K562 cells to a final concentration of 0.1 mM as previously described.36 Cells were incubated at 37°C for 15 min in a humidified atmosphere of 5% CO2 and were subsequently washed and resuspended in PBS. Samples were analyzed using a flow cytometer (FACS Calibur; Becton-Dickinson, Immunofluorometry Systems, Mountain View, CA, USA) with an argon laser beam of 488 nm. Mean fluorescence intensity (MFI) was calculated using CELLQUEST software (Becton-Dickinson).
Apoptosis was evaluated by flow cytometry for the detection of annexin V-positive cells. Cells incubated as previously described, as well as control cells, were labeled with annexin V conjugated with fluorescein isothiocyanate and propidium iodide. Briefly, cells were washed once in phosphate buffered saline and once in 1x-binding buffer, after which 5 μL of annexin V-fluorescein-S-iso-tyocianate (FITC) was added to the cells. Subsequently, 300 μL of 1x-binding buffer was added to the samples after a 15 min incubation at room temperature. The cells were then analyzed using flow cytometry, and the apoptotic fraction was defined as either annexin V-positive or propidium iodide-negative population.
K562 cells cultured in RPMI supplemented with 5% fetal bovine serum were incubated with 10 μM etoposide for 72h alone or preceded by either deferasirox, deferioxamine and deferiprone 50 μM for 18h or deferioxamine and deferiprone 0.5 mM for 30 min. Subsequently cells were analyzed by flow cytometry to assess annexin V expression.
Results
Two leukemia cell lines (K562 and HL60) were incubated with 50 μM deferasirox for 18h and were subsequently evaluated for NF-κB activity and p65 localization. Immunofluorescence assays (Figure 1A) using a p65 antibody (green fluorescence) show both a cytoplasmic and nuclear localization of the active NF-κB subunit in basal conditions whereas, after drug incubation, p65 localization was mainly cytoplasmic (inactive form). Western blotting (Figure 1B) shows a significant decrease in the amount of p65 in nuclear extracts after deferasirox incubation. In addition, we performed an EMSA assay to evaluate the subunit’s activity, such that a reduction in DNA binding activity could be detected after deferasirox incubation (Figure 2A).
Figure 1.

Deferasirox inhibits NF-kB activity in K562 cells whereas deferioxamine and deferiprone do not. (A) Immunofluorescence assay using p65 antibody in control cells (a) and after incubation with deferasirox 50 mM for 18 h (b–c). Nuclei are stained in red, the green signal represents p65 subunit. In control cells the NF-κB subunit is mainly localized in the nucleus as indicated by the yellow signal whereas, after incubation with the drug, p65 subunit is localized in the cytoplasm in the inactive form as indicated by the green signal in the cytoplasm and the absence of yellow signal in the nucleus. The graph represents the signal intensity quantification in both the cellular compartments. Nuclear signal intensity is significantly different in control and incubated cells (P<0.001). (B) Western blotting using p65 antibody for the detection of proteins in either nuclear (N) or cytoplasmic (C) extracts in K562 cells. The upper line indicates the p65 antibody, while both the lower lines represent the internal controls for either cytoplasmic (actin antibody) or nuclear extracts (lamin antibody). p65 nuclear localization is decreased only after deferasirox incubation but neither deferioxamine nor deferiprone incubation. The graph shows the quantification of the signal intensity detected by Western blotting. (C) Immunofluorescence assay using p65 antibody in K562 cells before and after incubation with deferioxamine and deferiprone either at 0.5 mM for 30 min or 50 mM for 18 h. After all the different conditions of incubation, p65 remains localized in the nucleus in the active form as in control cells. The graph illustrates NF-κB signal intensity in all the different conditions of incubation. No statistically significant differences have been found between control values and K562 treated cells with both the drugs. (D) Western blotting using p65 antibody for the detection of proteins in either cytoplasmic or nuclear extracts in K562 cells. The upper line indicates the p65 antibody, while the lower line represents an internal control for either cytoplasmic (actin antibody) or nuclear extracts (lamin antibody). p65 nuclear localization is not decreased after deferioxamine incubation for 30 min.
Figure 2.
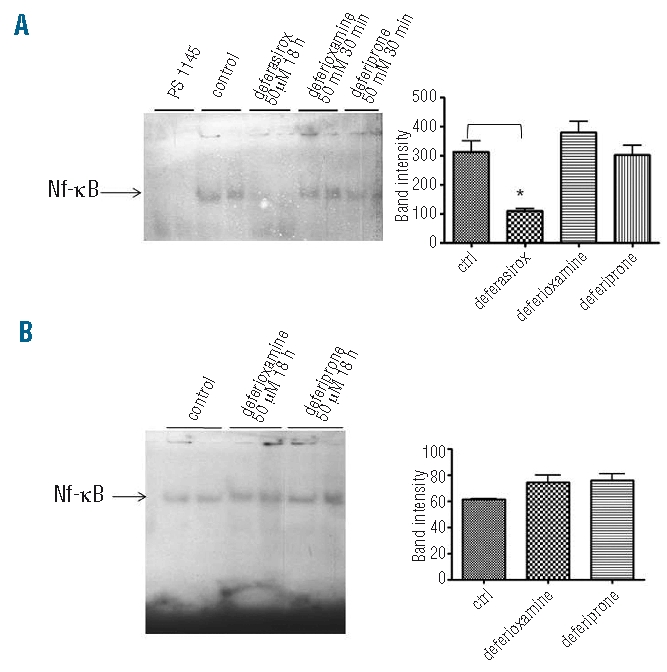
(A) NF-κB DNA binding activity (EMSA assay) in K562 cells treated either by deferasirox 50 mM for 18 h or by deferioxamine and deferiprone both at 0.5 mM for 30 min. Only after deferasirox incubation is the DNA binding activity of p65 subunit decreased. The first two lanes represent K562 cells incubated with the NF-κB inhibitor PS 1145 as negative control. The black arrow indicates specific binding to the consensus sequence. The graph on the right represents densitometry data. (B) EMSA assay for NF-κB DNA binding activity in K562 cells in basal conditions and after incubation with deferoxamine and deferiprone 50 mM both for 18 h. The DNA binding activity (indicated by the black arrow) is not decreased in both the different conditions of incubation with respect to the one detected in control cells. The graph on the right represents densitometry data.
We investigated whether NF-κB inhibition is a particular effect of deferasirox or if this is a shared feature of other chelators. K562and HL60 cell lines were incubated with deferasirox, deferiprone, deferioxamine and subsequently analyzed by Western blot and EMSA assays. An antioxidant compound (DTT, dithiothreitol) was used as a control. We observed that only deferasirox is able to reduce NF-κB activity as shown by immunofluorescence (Figure 1A and C), Western blot (Figure 1B and D) and EMSA assays (Figure 2A and B) with respect to the other chelators.
Mononuclear peripheral blood samples from 20 healthy subjects and 40 myelodysplastic patients were tested for NF-κB activity using ELISAs. We found an increase in activity in 6 of 14 RA, one of 2 RCMD, one of 2 MDS-U, 12 of 14 RAEB, and in all the cases of s-AML but not in healthy subjects.
No significant difference was detected in NF-κB activity comparing patients with or without iron overload (P=0.5). The levels of NF-κB activity increased during disease progression, being higher in RAEB and s-AML as compared to RA (P<0.001). A positive correlation was found between NF-κB activity and both blast percentage (r=0.75, P<0.0001)) and reactive oxygen species levels in mononuclear cells (r= 0.82, P<0.0001), but not between NF-κB and serum ferritin amount (r=0.12, P=0.42).
Among patients with increased basal NF-κB activity (n=28), the incubation with deferasirox induced a significant NF-κB inhibition (P=0.0002) as shown in Figure 3A and B (EMSA and ELISA assays, respectively), and confirmed by immunofluorescence images (Figure 3C). This effect has been observed both in patients with or without iron overload. A significant inhibition of NF-κB was also detected after in vivo deferasirox treatment for iron overload in a patient who experienced hemoglobin improvement during ICT (data not shown).
Figure 3.
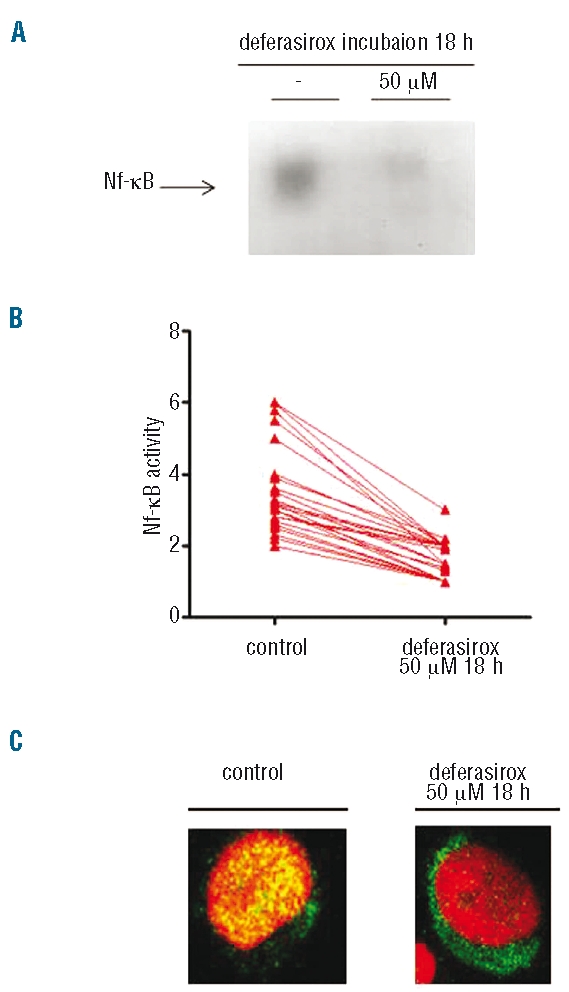
Deferasirox inhibits NF-κB activity in MDS patients. (A) NF-kB DNA binding activity (EMSA assay) in a MDS patient: NF-κB activity can be detected in control cells and its amount decreases after 18 h of deferasirox incubation in vitro at 50 mM. The black arrow indicates specific binding to the consensus sequence. The second lane of each condition was loaded with an unspecific cold probe as negative control. (B) Inhibition of NF-κB activity after in vitro incubation with 50 mM deferasirox of mononuclear PB cells of MDS patients for 18 h, as measured by ELISA assay. In all patients with high basal NF-κB activity (28 patients out of 40 tested), it is possible to detect transcription factor (TF) inhibition after drug incubation. (C) Immunofluorescence assay of PB cells from an MDS patient. At basal conditions on the left, p65 is completely localized in the nucleus, but after deferasirox incubation, the subunit is largely located in the cytoplasm, as demonstrated by the presence of the green signal.
We investigated whether the action of deferasirox on NF-κB could be related to a different reactive oxygen species scavenging activity of the chelator. We, therefore, tested our samples for reactive oxygen species levels before and after the different conditions of incubation previously described, adding DCF to a final concentration of 0.1 mM. As shown in Figure 4A, incubation with the three different compounds leads to a statistically significant reduction in reactive oxygen species concentration (expressed as MFI) with respect to the value obtained in basal conditions, but no statistically significant difference can be shown when comparing MFI after deferasirox incubation with respect to the values after deferiprone or deferioxamine treatment. Our data strongly support the conclusion that NF-κB inhibition by deferasirox is independent of its reactive oxygen species scavenging activity, and could be due to a different mechanism.
Figure 4.
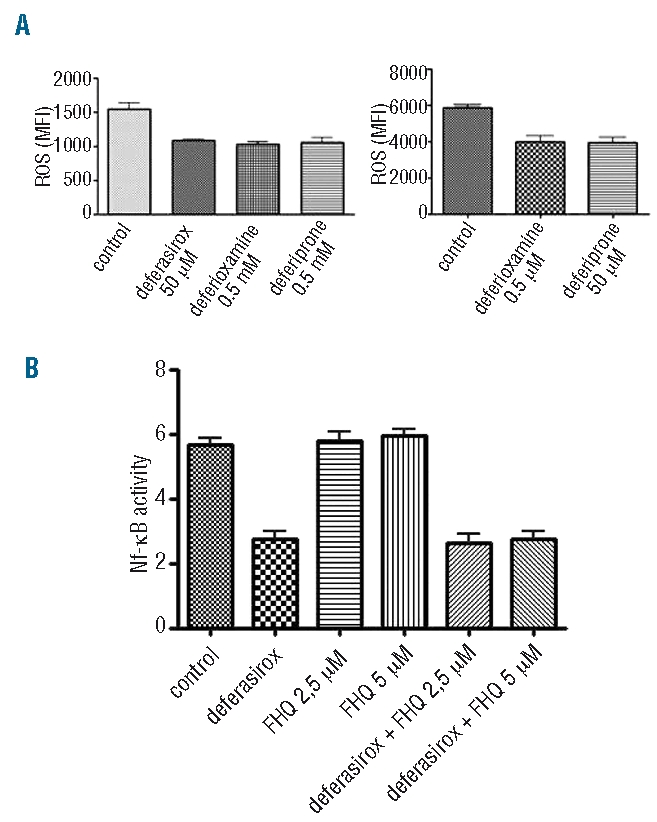
Deferasirox inhibits NF-κB activity independently from cell iron deprivation by chelation and reactive oxygen species scavenging. (A) ROS assay by DCF staining in K562 cells before and after incubation with three chelators at the conditions previously described. Mean fluorescence intensities (MFI) of K562 untreated cells and after incubation with deferioxamine, deferiprone and deferasirox are shown in the two graphs. There is a statistically significant difference comparing MFI samples before and after incubation with the three different drugs. Conversely, no statistically significant difference can be detected comparing MFI after deferasirox incubation with respect to either deferioxamine or deferiprone incubation. (B) The graph shows NF-κB activity measured by ELISA assay in K562 cells incubated respectively with deferasirox 50 mM alone, ferric hydroxyquinoline (FHQ) at 2.5 mM and 5 mM and with both deferasirox and FHQ. No statistically significant difference in NF-κB activity reduction has been detected in samples incubated by both deferasirox and FHQ with respect to those treated by deferasirox alone, suggesting a mechanism of action of the drug whch is independent from cell iron deprivation by chelation.
In order to test whether deferasirox inhibition of NF-κB activity is dependent or not on cell iron deprivation by chelation, we added the highly membrane-permeant iron carrier FHQ41 to the cells. K562 cells were incubated with deferasirox 50 μM alone or in combination with FHQ either 2.5 μM or 5μM. After 18h of incubation, the samples were tested for NF-κB activity by ELISA assay. No statistically significant difference in the reduction in NF-κB activity has been observed in cells incubated with both deferasirox and FHQ with respect to cells treated by deferasirox alone (Figure 4B) thus suggesting the action of the drug is independent from cell iron deprivation.
We performed a chemosensitivity assay in order to demonstrate that NF-κB inhibition by deferasirox enhances the activity of chemotherapeutic agents. First, no increase in the apoptotic rate of K562 cells could be detected by annexin V FACS analysis after incubation with deferioxamine, deferiprone or deferasirox alone (Figure 5A). Subsequently, K562 cells were incubated with either 10 μM etoposide alone or preceded by deferasirox 50 μM for 18h and analyzed by FACS for annexin V expression. As shown in Figure 5B, the percentage of apoptotic cells increased in a statistically significant manner in the sample incubated simultaneously with both drugs (P=0.003). No increase in the apoptotic rate has been observed in cells preincubated with deferioxamine whereas a slight increase has been induced by incubation with deferiprone.
Figure 5.

Apoptosis evaluated by fluorescence-activated cell sorting for the detection of annexin V-positive K562 cells after incubation with the three chelators and with etoposide and deferasirox. The apoptotic rate, represented by the percentage of cells both positive for Annexin V and negative for propidium iodide by FACS is indicated in each graph and represented on the right. (A) The apoptotic rate is not increased with respect to basal conditions after incubation with deferioxamine and deferiprone 0.5 mM for 30 min and deferasirox 50 mM for 18 h. The same results have been obtained after deferioxamine 50 mM incubation and deferiprone 50 and 100 mM incubation for 18 h. (B) Apoptosis assay of K562 cells in control cells and after etoposide 10 mM for 72 h alone or preceded by 50 mM deferasirox incubation for 18 h. After etoposide 10 mM for 72 h incubation an increase in the apoptotic cells amount can be detected (mean value 4.68%±0.46) and this is even higher if etoposide is preceded by deferasirox 50 mM for 18 h incubation (mean value 9.13±0.2). K562 cells were also incubated with etoposide preceeded by deferioxamine and deferiprone 50 mM for 18 h and 0,5 mM for 30 min without any statistically significant increase in the apoptotic rate as shown also by the graph on the right. No changes in apoaptotic rate can be detected after deferioxamine incubation (mean 5.1±0.64 at 50 mM and 4.3±0.49 at 50 mM), whereas deferiprone induces a slight increase in apoptotic rate (mean 6.7±0.45 at 0.5 mM and 6.8±0.7 at 50 mM).
Discussion
Iron chelation therapy in myelodysplastic syndromes is still controversial9–11 and there is a lack of evidence linking tissue iron overload to reduced survival. In spite of this, several international guidelines6 based on expert panel agreements strongly support iron chelation therapy for myelodys-plastic patients undergoing chronic transfusional therapy with a sufficiently long life expectancy in order to improve patient survival and clinical conditions. Some reports in recent years have described a hematopoiesis improvement under highly effective chelation therapy almost exclusively with deferioxamine.16,17 More recently, a similar effect was observed also during deferasirox treatment within only a few months of therapy.14 In the present work, we investigated if this particular effect could be due to a specific action of deferasirox on the malignant clone. Our attention was focused on the NF-κB pathway based on the important role of the transcription factor in the pathogenesis of MDS as demonstrated in different studies23,24 and on the observation that the efficacy of chemotherapeutic agents in AML cells is magnified adding specific NF-κB inhibitors in the in vitro model.41 Moreover, the transcription factor activation is a particular characteristic of the blast cells, so the various compounds with NF-κB inhibitory activity are selectively targeted on the neoplastic clone. Due to such a selective property, inhibiting NF-κB, indeed, has no consequence in normal cells; thus, the pathway is not hyperactivated. This is consistent with an absence of normal cell suppression induced by different NF-κB inhibitors tested in vivo. We investigated whether deferasirox acts as a specific NF-κB inhibitor in leukemic cell lines and in mononuclear cells of MDS patients. Our data demonstrate that deferasirox acts in vitro as a NF-κB inhibitor both in cell lines and in patients’ cells. In order to explore whether this effect is unique to this drug, we also tested NF-κB activity after incubation with other commercially available chelators. In our study, no other drugs showed similar inhibition of the transcription factor. It is well known that NF-κB can be activated by a broad range of stimuli and that one of these is mediated by reactive oxygen species. These compounds are present in polytransfused patients at a very high level and could be responsible for NF-κB activation. But this is only one of the different pathways that triggers its activation considering that in AML blast cells a high basal NF-κB activity can be detected despite normal iron parameters. We demonstrated that reactive oxygen species scavenging activity is not the crucial mechanism by which deferasirox induces p65 delocalization, considering that all drugs tested showed comparable reactive oxygen species reduction in our in vitro model. Furthermore, we excluded the possibility that the different iron chelators could induce an increase in apoptosis in the cells incubated and tested for annexin V expression by flow cytometry. We also ruled out the possibility that deferasirox activity is dependent on cell iron deprivation by chelation, as demonstrated by the persistence of the drug activity also after addition of a permeant iron-organic complex (FHQ) to the medium. Our data suggest that the deferasirox effect is not due to an intracellular iron chelation per se (all the chelators act in this way) but could be related to a specific ability of the drug to sequester iron in a particular target not accessible to deferioxamine and deferiprone. A recent study by Ruddell et al.43 demonstrates that in hepatic stellate cells, tissue ferritin and both H- and L-ferritin subsets are able to activate NF-κB signaling in an iron-independent mechanism involving PI-3 kinase, PKC-ζ and p44/p42-mitogen activated protein kinase, suggesting a cytokine-like effect of ferritin. Our data are in agreement with these findings providing evidence that NF-κB is activated in an iron independent manner, although the specific target of deferasirox is still under investigation. We cannot rule out the possibility that, also in our setting, PI-3 kinase could be responsible for the activation of this pathway as already demonstrated by Follo et al.44
The data regarding hematopoiesis improvement in patients treated with deferasirox in vivo are currently mainly based on case reports collected in a retrospective manner, so it is unknown which mechanism could be responsible for or which kind of patient would develop such a response during iron chelation therapy. Our hypothesis is that deferasirox acts in vivo as a specific inhibitor of blast cells because it selectively targets the NF-κB pathway when hyperactivated only in this cell population, inducing highly specific apoptosis. This effect in low-risk MDS patients leads to a reduction in ineffective erythropoiesis induced by a malignant clone, while in sAML or high-risk MDS, the drug reduces the percentage of blast cells through an increase in apoptosis. Otherwise, we cannot exclude a direct effect of the drug on erythroid precursors similar to the one seen in inherited sideroblastic anemia presenting GLRX5 deficiency, as previously described by Camaschella et al.45 in which after removing red cell precursors, the iron excess could re-establish a more effective erythropoiesis. Moreover, due to its specific NF-κB inhibitory properties, deferasirox is able to increase the antineoplastic activity of a chemotherapeutic agent, such as etoposide, in our in vitro model. A similar effect has been demonstrated previously with other NF-κB inhibitors in different in vitro models.46,47
In summary, in our study, deferasirox acts as a potent NF-κB inhibitor, so we can hypothesize that it could act directly on the malignant clone also during in vivo therapy, inducing a hematopoietic improvement in many patients. NF-κB inhibition is a particular and iron independent effect of deferasirox, not shared by other chelators and not relying on the reactive oxygen species scavenging properties of the drug. This observation offers new insights into iron chelation therapy in myelodysplastic patients, which is not only essential for secondary hemosiderosis prevention, but also acts as a targeted therapy on malignant clones. This could be associated with other therapeutic options.
Acknowledgments
we thank Prof. Ioav Cabantchik (Hebrew University of Jerusalem) for suggestions and discussion.
Footnotes
Funding: this work has been supported by grants from AIRC (Associazione Italiana per la Ricerca sul Cancro), MURST-COFIN, AIL (Associazione Italiana contro le Leucemie) and Regione Piemonte. CZ is a fellow of Josè-Carreras Foundation.
Authorship and Disclosures
EM wrote the manuscript and designed the study; DC designed the study and revised the manuscript; CZ and EG collected clinical data; EB and CM performed the EMSA assays; ARot collected samples; ID, SC, VR and RMP performed the in vitro incubations and western blotting; ARoe, FM and FA performed the immunofluorescence assays; MP and SC performed FACS analysis; DA provided the drug; GS gave final approval for the manuscript.
EM, SC, CM, MP, EB, ARoe, FM, FA, ID, VR, CZ, ARot, EG, RMP and DC have no conflict of interest to disclose; GS is on the advisory board and speaker bureau of Novartis and Bristol Meyers Squibb, and received research grants from Novartis. DA is an employee of Novartis.
References
- 1.Aul C, Bowen DT, Yoshida Y. Pathogenesis, etiology and epidemiology of myelodysplastic syndromes. Haematologica. 1998;83(1):71–86. [PubMed] [Google Scholar]
- 2.Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–88. [PubMed] [Google Scholar]
- 3.Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005;23(30):7594–603. doi: 10.1200/JCO.2005.01.7038. [DOI] [PubMed] [Google Scholar]
- 4.Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R, et al. Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol. 2007;25(23):3503–10. doi: 10.1200/JCO.2006.08.5696. [DOI] [PubMed] [Google Scholar]
- 5.Leitch HA. Improving clinical outcome in patients with myelodysplastic syndrome and iron overload using iron chelation therapy. Leuk Res. 2007;31S3:S7–9. doi: 10.1016/S0145-2126(07)70460-5. [DOI] [PubMed] [Google Scholar]
- 6.Gattermann N. Guidelines on iron chelation therapy in patients with myelodysplastic syndromes and transfusional iron overload. Leuk Res. 2007;31S3:S10–5. doi: 10.1016/S0145-2126(07)70461-7. [DOI] [PubMed] [Google Scholar]
- 7.Rose C, Brechignac S, Vassilief D, Beyne-Rauzy O, Stamatoullas A, Larbaa D, et al. Positive Impact of Iron Chelation Therapy (CT) on Survival in Regularly Transfused MDS Patients. A Prospective Analysis by the GFM. Blood. 2007;110 doi: 10.1016/j.leukres.2009.12.004. abstract [249] [DOI] [PubMed] [Google Scholar]
- 8.Porter J, Galanello R, Saglio G, Neufeld EJ, Vichinsky E, Cappellini MD, et al. Relative response of patients with myelodysplastic syndromes and other transfusion-dependent anaemias to deferasirox (ICL670): a 1-yr prospective study. Eur J Haematol. 2008;80:168–76. doi: 10.1111/j.1600-0609.2007.00985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pullarkat V. Objectives of iron chelation therapy in myelodysplastyc syndromes: more than meets the eye? Blood. 2009;114(26):5251–5. doi: 10.1182/blood-2009-07-234062. [DOI] [PubMed] [Google Scholar]
- 10.Tefferi A, Stone RM. Iron chelation therapy in myelodysplastic syndrome - Cui bono? Leukemia. 2009;23(8):1373. doi: 10.1038/leu.2009.39. [DOI] [PubMed] [Google Scholar]
- 11.Steensma DP. Myelodysplasia paranoia: iron as the new radon. Leuk Res. 2009;33(9):1158–63. doi: 10.1016/j.leukres.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 12.List AF, Baer MR, Steensma D, Raza A, Esposito J, Virkus J, et al. Iron chelation with deferasirox (Exjade) improves iron burden in patients with myelodysplastic syndromes (MDS) Blood. 2008;112 abstract[634] [Google Scholar]
- 13.Di Tucci AA, Murru R, Alberti D, Rabault B, Deplano S, Angelucci E. Correction of anemia in a transfusion-dependent patient with primary myelofibrosis receiving iron chelation therapy wih deferasirox (Exjade, ICL670) Eur J Haematol. 2007;78(6):540–2. doi: 10.1111/j.1600-0609.2007.00840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messa E, Cilloni D, Messa F, Arruga F, Roetto A, Saglio G. Deferasirox treatment improved the hemoglobin level and decreased transfusion requirements in four patients with the myelodysplastic syndrome and primary myelofibrosis. Acta Haematol. 2008;120(2):70–4. doi: 10.1159/000158631. [DOI] [PubMed] [Google Scholar]
- 15.Marsh JH, Hundert M, Schulman P. Deferoxamine-induced restoration of haematopoiesis in myelofibrosis secondary to myelodysplasia. Br J Haematol. 1990;76(1):148–9. doi: 10.1111/j.1365-2141.1990.tb07851.x. [DOI] [PubMed] [Google Scholar]
- 16.Jensen PD, Jensen IM, Ellegaard J. Desferrioxamine treatment reduces blood transfusion requirements in patients with myelodysplastic syndrome. Br J Haematol. 1992;80(1):121–4. doi: 10.1111/j.1365-2141.1992.tb06411.x. [DOI] [PubMed] [Google Scholar]
- 17.Jensen PD, Heickendorff L, Pedersen B. The effet of iron chelation on haemopoiesis in MDS patients with transfusional iron overload. Br J Haematol. 1996;94(2):288–99. doi: 10.1046/j.1365-2141.1996.d01-1795.x. [DOI] [PubMed] [Google Scholar]
- 18.Smeets ME, Vreugdenhil G, Holdrinet RS. Improvement of erythropoiesis during treatment with deferiprone in a patient with myelofibrosis and transfusional hemosiderosis. Am J Hematol. 1996;51(3):243–4. doi: 10.1002/(SICI)1096-8652(199603)51:3<243::AID-AJH12>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal BB. Nuclear factor-kB: the enemy within. Cancer Cell. 2004;6(3):203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 21.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 22.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 2004;14(2):64–9. doi: 10.1016/j.tcb.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Guzman M, Neering SJ, Upchurch D, Grimes B, Howard DS, Rizzieri DA, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98(8):2301–7. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 24.Braun T, Carvalho G, Coquelle A, Vozenin MC, Lepelley P, Kiladjian JJ, et al. NF-kappaB constitutes a potential therapeutic target in high-risk myelodysplastic syndrome. Blood. 2006;107(3):1156–65. doi: 10.1182/blood-2005-05-1989. [DOI] [PubMed] [Google Scholar]
- 25.Hayden MS, Ghosh S. Shared principles in NF-kappaB signalling. Cell. 2008;132(3):344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 26.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the YNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 27.Zhang SQ, Kovalenko A, Cantarella G, Wallach D. Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity. 2000;12(3):301–11. doi: 10.1016/s1074-7613(00)80183-1. [DOI] [PubMed] [Google Scholar]
- 28.Bubici C, Papa S, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB and JNK: an intricate affair. Cell Cycle. 2004;3(12):1524–9. doi: 10.4161/cc.3.12.1321. [DOI] [PubMed] [Google Scholar]
- 29.Bubici C, Papa S, Dean K, Franzoso D. Mutual cross-talk between reactive oxygen species and nuclear factor-kappa B: molecular basis and biological significance. Oncogene. 2006;25(51):6731–48. doi: 10.1038/sj.onc.1209936. [DOI] [PubMed] [Google Scholar]
- 30.Papa S, Bubici C, Pham CG, Zazzeroni F, Franzoso G. NF-kappaB meets ROS: an “ironic” encounter. Cell Death Differ. 2005;12(10):1259–62. doi: 10.1038/sj.cdd.4401694. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Huang X, Cang H, Gao F, Yamamoto T, Osaki T, et al. The endogenous reactive oxygen species promote NF-kappaB activation by targeting on activation of NF-kappaB-inducing kinase in oral squamous carcinoma cells. Free Radic Res. 2007;41(9):963–71. doi: 10.1080/10715760701445045. [DOI] [PubMed] [Google Scholar]
- 32.Lluis J, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mithocondrial reactive oxygen species in hypoxia signalling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007;67(15):7368–77. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- 33.Jimenez LA, Thompson J, Brown DA, Rahman I, Antonicelli F, Duffin R, et al. Activation of NF-kappaB by PM(10) occurs via an iron-mediated mechanism in the absence of IkappaB degradation. Toxicol Appl Pharmacol. 2000;166(2):101–10. doi: 10.1006/taap.2000.8957. [DOI] [PubMed] [Google Scholar]
- 34.Xiong S, She H, Tsukamoto H. Signaling role of iron in NF-kappa B activation in hepatic macrophages. Comp Hepatol. 2004;3 (Suppl 1):S36. doi: 10.1186/1476-5926-2-S1-S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto H. Iron regulation of hepatic macrophage TNFalpha expression. Free Radic Biol Med. 2002;32(4):309–13. doi: 10.1016/s0891-5849(01)00772-9. [DOI] [PubMed] [Google Scholar]
- 36.Kim KM, Kim PK, Kwon YG, Bai SK, Nam WD, Kim YM. Regulation of apoptosis by nitrosative stress. J Biochem Mol Biol. 2002;35(1):127–33. doi: 10.5483/bmbrep.2002.35.1.127. [DOI] [PubMed] [Google Scholar]
- 37.Omidvar N, Kogan S, Beurlet S, le Pogam C, Janin A, West R, et al. BCL-2 and mutant NRAS interact physically and functionally in a mouse model of progressive myelodysplasia. Cancer Res. 2007;67(24):11657–67. doi: 10.1158/0008-5472.CAN-07-0196. [DOI] [PubMed] [Google Scholar]
- 38.Rassool FV, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M, et al. Reactive oxygen species, DNA damage, and error-prone repair: a model for genomic instability with progression in myeloid leukemia? Cancer Res. 2007;67(18):8762–71. doi: 10.1158/0008-5472.CAN-06-4807. [DOI] [PubMed] [Google Scholar]
- 39.Koptyra M, Falinski R, Nowicki, Stoklosa T, Majsterek I, Nieborowska-Skorska M, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108(1):319–27. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghoti H, Amer J, Winder A, Rachmilewitz E, Fibach E. Oxidative stress in red blood cells, platelets and polymorphonuclear leukocytes from patients with myelodysplastic syndrome. Eur J Haematol. 2007;79(6):463–7. doi: 10.1111/j.1600-0609.2007.00972.x. [DOI] [PubMed] [Google Scholar]
- 41.Shvartsman M, Kikkeri R, Shanzer A, Cabantchik ZI. Non-transferrin-bound iron reaches mitochondria by a chelator-inaccessible mechanism: biological and clinical implications. Am J Physiol Cell Physiol. 2007;293(4):1383–94. doi: 10.1152/ajpcell.00054.2007. [DOI] [PubMed] [Google Scholar]
- 42.Cilloni D, Messa F, Arruga F, Defilippi I, Morotti A, Messa E, et al. The NF-kappaB pathway blockade by the IKK inhibitor PS1145 can overcome imatinib resistance. Leukemia. 2006;20(1):61–7. doi: 10.1038/sj.leu.2403998. [DOI] [PubMed] [Google Scholar]
- 43.Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, et al. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology. 2009;49(3):887–900. doi: 10.1002/hep.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Follo MY, Finelli C, Mongiorgi S, Clissa C, Bosi C, Martinelli G, et al. PKR is activated in MDS patients and its subcellular localization depends on disease severity. Leukemia. 2008;22(12):2267–9. doi: 10.1038/leu.2008.122. [DOI] [PubMed] [Google Scholar]
- 45.Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110(4):1353–8. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 46.Frelin C, Imbert V, Griessinger E, Peyron AC, Rochet N, Philip P, et al. Targeting NF-kappaB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood. 2005;105(2):804–11. doi: 10.1182/blood-2004-04-1463. [DOI] [PubMed] [Google Scholar]
- 47.Cilloni D, Messa F, Rosso V, Arruga F, Defilippi I, Carturan S, et al. Increase sensitivity to chemotherapeutical agents and cytoplasmatic interaction between NPM leukemic mutant and NF-kappaB in AML carrying NPM1 mutations. Leukemia. 2008;22(6):1234–40. doi: 10.1038/leu.2008.68. [DOI] [PubMed] [Google Scholar]