

Abstract
The function of transcription in dynamic gene expression programs has been extensively studied, but little is known about how it is integrated with RNA turnover at the genome-wide level. We investigated these questions using the meiotic gene expression program of Schizosaccharomyces pombe. We identified over 80 transcripts that co-purify with the meiotic-specific Meu5p RNA-binding protein. Their levels and half-lives were reduced in meu5 mutants, demonstrating that Meu5p stabilizes its targets. Most Meu5p-bound RNAs were also targets of the Mei4p transcription factor, which induces the transient expression of ∼500 meiotic genes. Although many Mei4p targets showed sharp expression peaks, Meu5p targets had broad expression profiles. In the absence of meu5, all Mei4p targets were expressed with similar kinetics, indicating that Meu5p alters the global features of the gene expression program. As Mei4p activates meu5 transcription, Mei4p, Meu5p and their common targets form a feed-forward loop, a motif common in transcriptional networks but not studied in the context of mRNA decay. Our data provide insight into the topology of regulatory networks integrating transcriptional and posttranscriptional controls.
Keywords: mRNA decay, RIp-chip, posttranscriptional control
Introduction
Many biological processes are accompanied by dynamic gene expression programs, in which the levels of hundreds or thousands of transcripts are regulated (Ferea and Brown, 1999). As RNA levels are determined by the balance between transcription and degradation, the contribution of both processes must be taken into account for a complete understanding of the regulation of gene expression (Perez-Ortin, 2007). However, most large-scale studies have focused on the function of transcription, and relatively little is known about the importance of posttranscriptional control and about how both levels of regulation are coordinated.
Global transcriptional processes have been extensively investigated by the systematic mapping of transcription factor binding sites (Farnham, 2009). These studies have revealed the existence of complex networks between transcription factors and the genes they regulate. Transcriptional networks are enriched in simple recurring connections called network motifs, which can confer specific kinetic properties to the regulatory network (Alon, 2007).
RNA turnover is regulated by RNA-binding proteins (RBPs) that recognize specific sequences in their targets and enhance or repress mRNA decay by modulating their interaction with the degradation machinery (Garneau et al, 2007). The genome-wide function of RBPs in the control of RNA turnover can be investigated by identifying RBP-associated transcripts and by analysing the functional consequences of inactivating the RBPs. RBP targets can be determined by purifying individual proteins together with bound RNAs, followed by identification of the transcripts using DNA microarrays (RIp-chip, for RBP immunoprecipitation followed by analysis with DNA chips) (Keene et al, 2006). RIp-chip has been applied to a large number of RBPs (see Mata et al, 2005; Halbeisen et al, 2008 for reviews and Hogan et al, 2008 for a recent example), including a few regulators of RNA decay (the Puf1, HuR, tristetraprolin and AUF1 proteins; Lopez de Silanes et al, 2004; Galgano et al, 2008; Mazan-Mamczarz et al, 2009; Morris et al, 2008; Stoecklin et al, 2008; Mukherjee et al, 2009). These studies have revealed that RBPs associate with specific RNA populations that often encode proteins with common features. These networks of RBP–RNA interactions are thought to coordinate the fates of RNA molecules through every step of posttranscriptional control (Keene and Tenenbaum, 2002). RIp-chip studies have been complemented by functional approaches, in which genes encoding RBPs have been silenced or mutated to reveal effects on transcript stability (Halbeisen et al, 2008). Moreover, transcript decay rates can be measured for whole genomes (most commonly by inactivating transcription and using microarrays to follow the reduction on RNA levels over time), thus allowing the distinction between direct effects on RNA stability and indirect consequences of the inactivation of the RBP (Perez-Ortin, 2007). This approach has been applied to yeast cells carrying mutations in genes encoding components of decay pathways (the deadenylases CCR4 and PAN2 and the specific RBPs PUB1 and PUF4 (reviewed in Halbeisen et al, 2008) and to higher eukaryotes (the tristetraprolin protein; Lai et al, 2006). A study that measured transcript levels and genome-wide decay rates during oxidative stress found complex relationships between changes in mRNA levels and turnover rates (Shalem et al, 2008), suggesting the existence of sophisticated coordination between mRNA production and degradation. Finally, a recent study showed that mathematical modelling incorporating genome-wide decay rates can be used to generate transcriptional activity profiles (which are different from gene expression profiles, which only reflect transcript levels). Activity profiles allow the identification of group of genes that are co-regulated by specific transcription factors (Barenco et al, 2009).
To analyse the contributions of transcription and mRNA turnover during dynamic gene expression programs, we have used sexual differentiation of the fission yeast Schizosaccharomyces pombe as a model. This developmental process culminates in meiosis and the formation of spores (Yamamoto et al, 1997), and involves a complex program of gene expression in which >40% of the genome (>2000 genes) is regulated (Mata et al, 2002). Although transcriptional control is critical for the regulation of a large part of the program (Mata and Bähler, 2006; Xue-Franzen et al, 2006; Mata et al, 2002, 2007), the control of mRNA stability also has a function (Harigaya et al, 2006; McPheeters et al, 2009). The genes upregulated during sexual differentiation have been classified into four main groups according to their expression profiles. In particular, a group of ∼555 genes (middle genes) is induced transiently at the time of the meiotic divisions by the activity of the Mei4p transcription factor, which itself peaks in expression during this period (Horie et al, 1998; Watanabe et al, 2001; Mata et al, 2007).
Several uncharacterized genes encoding RNA-binding domains are highly induced during sexual differentiation (Mata et al, 2002). We report here an investigation into the function of one of them, meu5, which is induced during the meiotic divisions. We used genome-wide approaches to characterize the function of Meu5p in the regulation of mRNA decay. Our results establish that Meu5p binds to and stabilizes a subset of meiotic genes, thus determining their correct temporal expression. Meu5p targets are transcriptionally induced by the Mei4p transcription factor, which also activates meu5 expression. Therefore, dynamic changes in mRNA levels during meiotic differentiation are determined by the coordinated regulation of transcription and mRNA decay.
Results
A subset of meiotic middle genes is downregulated in the absence of Meu5p
The non-essential meu5 gene (also called crp79) encodes a 710-amino-acid RBP containing three RNA recognition motifs (Watanabe et al, 2001; Thakurta et al, 2002). Meu5p is similar to the S. pombe Mug28p RBP, but does not appear to have clear orthologs in other organisms. As meu5 is specifically induced during meiosis (Watanabe et al, 2001; Mata et al, 2002), we constructed a strain carrying a deletion in the meu5 gene to examine its ability to carry out sexual differentiation. meu5Δ cells were able to mate and proceed through the meiotic divisions with normal kinetics (Supplementary Figure S1A). However, meu5Δ cells produced few mature spores (10-fold less than wild-type), most of which presented an abnormal appearance (Figure 1A). In addition, the release of meu5Δ spores from the ascus was strongly delayed (around 3 days later than in wild-type cells). To investigate a possible function of Meu5p in the control of gene expression, we compared the transcriptome of wild-type and meu5Δ cells (Figure 1B). To achieve good synchrony and high reproducibility, we used thermosensitive mutants of the meiotic inhibitor Pat1p (Iino and Yamamoto, 1985; Nurse, 1985). Diploid pat1 cells (wild-type or meu5Δ) were arrested in G1 by removing nitrogen, and synchronous meiosis was induced by a temperature shift to inactivate Pat1p (Supplementary Figure S1A). Samples were taken at 6 h (1 h after the peak of expression of meu5 mRNA) and analysed using DNA microarrays. Although no transcripts were significantly upregulated in meu5Δ cells, 188 mRNAs were expressed at lower levels in the mutant (Figure 1B; Supplementary Table S1). This group of genes was strongly enriched in meiotic middle genes (Mata et al, 2002) (Figure 1C; P-value 10−68). Overall, 62% of these genes belonged to the middle group. Moreover, many of the non-middle genes showed ‘middle-like’ expression profiles, but had not been classified as such either because they did not reach the induction threshold used for the classification (only genes induced more than four-fold were considered) (Mata et al, 2002), or because they had more than one peak of expression (one of them at the time of induction of middle genes) and had been classified in another group because the other peak was prevalent (Supplementary Figure S1B). These data suggest that most of the affected genes behave as middle genes. However, the levels of a substantial fraction of middle gene transcripts (66%) did not vary in the absence of meu5.
Figure 1.

Meu5p has a function in sporulation and regulates the expression of a subset of meiotic middle genes. (A) Wild-type or meu5Δ h90 cells were incubated for 24 h in the absence of a nitrogen source to induce sexual differentiation and visualized using phase contrast microscopy. (B) Comparison of expression levels between wild-type and meu5Δ in pat1-synchronized meiotic diploid cells. Middle genes are shown in red and other genes in grey. Genes outside the dashed lines differ by more than two-fold in expression levels. (C) Overlap between genes downregulated in meu5Δ diploids (in pat1-synchronized meiosis) and middle genes. The numbers in brackets show the overlap between the two lists expected by chance given the sizes of the gene sets considered and the total number of genes. The P-value of the overlap is shown under the Venn diagram.
Although it has been shown that the regulation of middle genes is similar in pat1 and wild-type diploids (Mata et al, 2002), we sought to confirm our results by comparing meu5+ and meu5Δ diploids in a wild-type background. Despite meiosis being less synchronous in these cells, the same group of genes was underexpressed in the mutant (Supplementary Figure S1C; P-value 9 × 10−144).
Meu5p binds to RNAs encoded by middle genes
To discover direct targets of Meu5p, we purified TAP-tagged Meu5p and used DNA microarrays to identify the transcripts present in the immunoprecipitate. We carried out the experiment in synchronized pat1 diploids (at the same time point used for expression analysis) as well as in unsynchronized h90 haploid cells undergoing meiosis. Meu5p-TAP co-purified with 82 transcripts (Figure 2A; Supplementary Figure S2 and Table S2), which were not present in control immunoprecipitates (Amorim and Mata, 2009; data not shown). These mRNAs were strongly enriched in middle genes (Figure 2B; P-value 3 × 10−58) and in genes whose expression was reduced in meu5Δ meiotic cells (Figure 2C; P-value 10−112). Indeed, 87% of the Meu5p-bound transcripts were middle genes.
Figure 2.

Meu5p associates with transcripts encoded by middle genes and regulates their expression. (A) Meu5p targets are expressed at low levels in meu5Δ cells. Comparison of expression levels between wild-type and meu5Δ pat1-synchronized meiotic cells (data as in Figure 1B). Meu5p-associated transcripts identified by RIp-chip are shown in red and other genes are shown in grey. The position of the meu5 transcript is indicated with an arrow. (B) Overlap between Meu5p-associated transcripts and middle genes. (C) Overlap between Meu5p-associated transcripts and genes downregulated in meu5Δ cells (in pat1-synchronized meiosis). Labelling of the Venn diagrams is as in Figure 1C.
We carried out two additional experiments to confirm the specificity of the RIp-chip results: first, we analysed the results of a Meu5p immunoprecipitation experiment using a different microarray platform (see Materials and methods), and observed an extensive and highly significant overlap between the transcripts identified with both systems (Supplementary Figure S3; P-value 4 × 10−109). Second, we used quantitative PCR (qPCR) to examine the enrichment levels of several targets and negative controls, and found them to be comparable to those determined by RIp-chip (Supplementary Figure S4).
The nature and timing of expression of the Meu5p targets are consistent with the sporulation defect of meu5Δ cells, strongly suggesting that their low levels in the mutant are responsible for the phenotype. For example, Meu5p targets include genes involved in spore wall synthesis (mde10, which encodes an ADAM family protein; Nakamura et al, 2004) and in the degradation of the ascus wall (agn2 and eng2, encoding endoglucanases; Dekker et al, 2007; Encinar del Dedo et al, 2009) (Supplementary Table S2).
Meu5p stabilizes its mRNA targets
The results described above suggest that Meu5p stabilizes the transcripts it binds to. Therefore, Meu5p targets would be expected to have shortened half-lives in meu5Δ cells. To test this idea, we compared mRNA decay rates of wild-type and meu5Δ cells. Genome-wide decay rates are usually determined by following RNA levels with DNA microarrays after blocking transcription with drug treatments or heat-sensitive RNA polymerase mutants (Perez-Ortin, 2007). These methods present several disadvantages, because the treatments used to inactivate transcription may themselves influence mRNA stability, and because a general transcriptional shut-down is likely to affect the cellular physiology (Perez-Ortin, 2007).
To circumvent these problems, we adapted for fission yeast an existing technique to measure decay rates that does not rely on transcriptional inhibition (Cleary et al, 2005). This approach is based on the labelling of newly synthesized RNA during a short pulse, followed by its purification and quantification. During the labelling pulse, newly synthesized RNA replaces the pre-existing RNA. Under steady-state conditions, the fraction of newly synthesized RNA (compared with total RNA) at a given time can be used to calculate the rate of decay (Dolken et al, 2008). To implement this system, RNAs are labelled in vivo using a modified nucleoside, 4-thiouridine (4sU). After a labelling pulse, total RNA is purified, and 4sU-labelled RNA is biotinylated using a thio-specific reagent (biotin-HPDP). Finally, the biotinylated RNA is purified using streptavidin beads. Purified RNA is then compared with total RNA using DNA microarrays, allowing the determination of the fraction of labelled RNA and the estimation of decay rates.
Incubation of S. pombe wild-type cells with 4sU for up to 30 min did not affect growth and did not cause strong changes in gene expression (Supplementary Figure S5A). We then applied this method to vegetatively growing cells, and determined half-lives for almost 90% of all fission yeast transcripts, ranging from 2 min to several hours (Supplementary Figure S5B). The results were not strongly affected by the length of the pulse and correlated well with earlier genome-wide estimates of half-lives obtained by blocking transcription with 1,10-phenantroline (Lackner et al, 2007) (Supplementary Figure S5C).
A fundamental requirement of this approach is that the system must be in steady state (i.e. mRNA levels must not change over time). Given the dynamic nature of the sexual differentiation gene expression program, this condition is unlikely to apply for meiotic cells. Therefore, we decided to identify conditions under which Meu5p targets are expressed at constant levels. To achieve this, we exploited the facts that the transcription of most Meu5p targets is under the control of the Mei4p transcription factor, and that overexpression of Mei4p in vegetative cells leads to ectopic expression of its targets (Horie et al, 1998; Mata et al, 2007). We overexpressed Mei4p and verified that the levels of Mei4p targets did not change during an incubation with 4sU (Supplementary Figure S5A), thus confirming the steady-state assumption. We then compared the transcriptome of wild-type and meu5Δ vegetative cells overexpressing Mei4p. We found a significant correlation between genes expressed at lower levels in meu5Δ meiotic cells and in meu5Δ vegetative cells overexpressing Mei4p (Supplementary Figure S6; P-value 10−92). Therefore, the ‘vegetative system’ recapitulates the meiotic phenotype of meu5Δ, while expressing Mei4p targets at constant levels. We therefore applied the 4sU labelling method to wild-type and meu5Δ vegetative cells overexpressing Mei4p, and identified 90 transcripts with significantly lower half-lives in the mutant (Figure 3A; Supplementary Table S3, note that a higher labelled fraction corresponds to a shorter half-life). The changes in expression levels correlated well with the differences in half-lives (Supplementary Figure S7), further validating the method. The group of genes with shortened half-lives in meu5Δ was significantly enriched in Meu5p-bound transcripts (Figure 3B; P-value 2 × 10−83) and in genes expressed at lower levels in meu5Δ cells (Figure 3B; P-value 9 × 10−110). These results show that Meu5p directly stabilizes its targets.
Figure 3.

Meu5p stabilizes its targets. (A) Comparison of the fraction of newly synthesized RNA (4sU-labelled) between wild-type and meu5Δ pat1-synchronized meiotic cells. Meu5p-associated transcripts are shown in red and other mRNAs are shown in grey. Genes outside the dashed lines differ by more than two-fold in labelling levels. Note that a higher fraction corresponds to a shorter half-life. (B) Overlap between genes destabilized in meu5Δ cells, genes downregulated in meu5Δ diploid cells (in pat1-synchronized meiosis) and Meu5p-associated transcripts. The numbers outside the Venn diagrams show the P-values of the pairwise comparisons.
Although the vegetative system recapitulates the behaviour of meu5 mutants, we sought to verify that Meu5p activity in meiotic cells is also posttranscriptional. To this end, we used global RNA Polymerase II (Pol II) occupancy across coding regions as a surrogate for transcriptional activity (Lackner et al, 2007). We carried out chromatin immunoprecipitation experiments using antibodies against Pol II in both meu5Δ and wild-type diploid cells (pat1-induced meiosis), and analysed the results with DNA microarrays (Supplementary Figure S8). Most genes underexpressed in meu5Δ cells showed similar Pol II occupancy in meu5Δ and wild-type cells, confirming that the effects on Meu5p on its target genes are posttranscriptional in both vegetative and meiotic cells.
Meu5p targets have distinct meiotic expression patterns
Middle genes have been defined by their expression profiles in a meiotic time course (Mata et al, 2002) and are induced by the action of the Mei4p transcription factor (Mata et al, 2007). We noticed that many Meu5p targets were expressed with a specific pattern during meiotic time courses, with a broad expression period between 5 and 7 h after induction of meiosis (Figure 4A; data are from Mata et al, 2002). By contrast, the other middle genes were expressed more transiently, with a peak of expression at 5 h followed by a sharp decrease (Mata et al, 2002). These observations prompted us to re-examine the classification of middle genes. We separated the middle genes into two groups according to their expression profiles in published meiotic time courses (Mata et al, 2002) (Figure 4B and C; Supplementary Table S4). One group (‘early-decrease’) showed a sharp peak of expression, and was enriched in genes involved in cell-cycle regulation, proteolysis and spindle function (Table I). The other group (‘late-decrease’) was expressed in a wider window and was enriched in genes encoding proteins localized to the endoplasmic reticulum (Table I). The fact that both sets were enriched in different categories suggested that the classification was biologically relevant. We then examined whether these two groups were functionally related to meu5. Although ‘late-decrease’ genes overlapped significantly with Meu5p-bound transcripts (P-value 7 × 10−76), ‘early-decrease’ genes were not enriched in Meu5p targets (P-value 0.6) (Figure 4D). Similar results were obtained when comparing the two groups with genes being underexpressed (Supplementary Figure S9) or showing increased decay (Supplementary Figure S10) in meu5Δ mutants. We conclude that Meu5p targets show distinct expression profiles during meiosis that are different from those of other Mei4p targets, consistent with a function of Meu5p in the stabilization of its targets.
Figure 4.

Meu5p targets have distinct meiotic expression profiles. (A) Meiotic gene expression profiles of Meu5p targets. Vegetatively growing cells (V) are synchronized in G1 by nitrogen removal and enter meiosis by inactivation of pat1 at time 0 (expression data are from Mata et al, 2002). The expression profiles of mei4 (orange) and meu5 (green) are also shown. Note that neither meu5 nor mei4 transcripts are bound by Meu5p. (B, C) Classification of middle genes according to the kinetics of their downregulation (expression data are from Mata et al, 2002). ‘Early decrease’ genes are shown in red and ‘late-decrease’ are shown in blue. Average expression profiles for each group are shown in (B) and profiles for all genes are shown in (C). (D) Overlap between Meu5p-associated transcripts and ‘early-decrease’ genes (left) or ‘late decrease’ (right). Labelling of the Venn diagrams is as in Figure 1C.
Table 1. Overlap between ‘early-decrease’ and ‘late-decrease genes’ and Gene Ontology categories.
| GO category (GO code) | GO type | % In group | % In genome | P-value |
|---|---|---|---|---|
| ‘Early-decrease’ middle genes | ||||
| M phase (0000279) | Process | 23.0 | 5.2 | 6.88 × 10−33 |
| Cell cycle (0007049) | Process | 28.5 | 9.7 | 3.13 × 10−25 |
| Meiotic cell cycle (0051321) | Process | 13.7 | 3.4 | 4.81 × 10−16 |
| APC-dependent activity (0031145) | Process | 3.4 | 0.3 | 2.24 × 10−10 |
| Protein ubiquitination (0016567) | Process | 6.6 | 1.6 | 9.32 × 10−7 |
| Microtubule cytoskeleton organization (0000226) | Process | 5.3 | 1.1 | 6.92 × 10−6 |
| Chromosome segregation (0007059) | Process | 8.7 | 2.7 | 1.40 × 10−6 |
| Anaphase-promoting complex (0005680) | Component | 3.2 | 0.2 | 1.01 × 10−10 |
| Microtubule cytoskeleton (0015630) | Component | 13.2 | 4.7 | 1.05 × 10−8 |
| Spindle (0005819) | Component | 11.9 | 4.3 | 1.94 × 10−7 |
| Spindle pole body (0005816) | Component | 9.2 | 3.5 | 1.5 × 10−4 |
| Condensin complex (0000796) | Component | 1.3 | 0.1 | 3.8 × 10−3 |
| ‘Late-decrease’ middle genes | ||||
| Membrane lipid metabolism (0006643) | Process | 8.7 | 1.46 | 4.1 × 10−3 |
| Endoplasmic reticulum (0005783) | Component | 25.4 | 9.3 | 1.4 × 10−4 |
Meu5p activity determines the temporal expression pattern of its targets
The results presented above suggest a model in which Meu5p stabilizes the RNAs of its targets, thus allowing them to persist for a longer period of time. A prediction of this hypothesis is that in the absence of meu5, ‘late-decrease’ genes should behave as ‘early-decrease’ genes. To test this hypothesis, we carried out meiotic time courses in pat1-synchronized wild-type and meu5Δ diploids (Figure 5A, average profiles are shown). As expected, the expression profiles of ‘early-decrease’ genes were almost identical in wild-type and meu5Δ cells. ‘Late-decrease’ genes displayed a wide expression profile in wild-type cells, similar to published results (Mata et al, 2002). By contrast, they reached a lower peak in the absence of meu5, and their levels decreased sharply after 5 h. Indeed, their profiles in meu5Δ cells became indistinguishable from those of ‘early-decrease’ genes. Meu5p-mediated stabilization of its RNA targets would be expected to influence their protein levels. To verify this prediction, we monitored protein levels of several Mei4p targets over meiotic time courses by western blot, in both wild-type and meu5 mutant cells (Supplementary Figure S11). As predicted, the levels of proteins encoded by Meu5p targets were clearly reduced in meu5Δ cells, whereas those of other Mei4p targets were not affected.
Figure 5.

Meu5p regulates the dynamics of expression of its targets. (A) Average expression profiles of ‘early-decrease’ (left) or ‘late-decrease’ (right) genes in pat1-induced meiotic time courses. Labelling of the graphs is as in Figure 4A, except that expression ratios were normalized to the levels at 3 h after the induction of meiosis in the corresponding experiment. Data from wild-type meiosis are shown in purple and from meu5Δ are shown in green. (B) Diagram summarizing the regulation of the expression of middle genes. Proteins are shown as ovals and transcripts are shown as curvy lines. All middle genes are induced transcriptionally by Mei4p. Meu5p stabilizes its targets allowing them to be expressed for longer, whereas middle genes not bound by Meu5p are induced more transiently. Mei4p induces the expression of meu5, and thus Mei4p, Meu5p and their common targets form a feed-forward network motif.
The meu5 mRNA is expressed transiently during meiosis (Figure 4A), suggesting that the downregulation of Meu5p allows the eventual degradation of its targets. To test this idea, we followed the expression pattern of Meu5-TAP in pat1-syncrhonized cells. Meu5p was expressed between 4 and 7 h after induction of meiosis, with a peak between 5 and 6 h (Supplementary Figure S12).
Discussion
We present a comprehensive analysis of the function and targets of the Meu5p RBP. Our results underscore how the cooperation between transcriptional and posttranscriptional controls is essential for the temporal regulation of gene expression: The Mei4p transcription factor is induced transiently at the time of the meiotic divisions (Figure 4A), leading to the temporary expression of the middle genes. One of the targets of Mei4p is meu5, whose product in turn binds to and stabilizes a subset of the Mei4p targets, allowing them to be expressed for a longer period of time. As Meu5p expression is also transient (Supplementary Figure S12), its subsequent downregulation would allow the turnover of its targets. By contrast, Mei4p targets that are not bound by Meu5p are expressed during a shorter window. Therefore, not only does Mei4p induce the expression of its targets, but it also triggers a system that determines for how long they will be expressed (Figure 5B).
It has been reported that the complexes between RBPs and their targets can be altered during cell lysis, potentially leading to artefactual results in RIp-chip experiments (Mili and Steitz, 2004). This is very unlikely to be the case here, given that the results of RIp-chip have been extensively validated by our analysis of Meu5p function. Notably, the Meu5p-bound mRNAs were identified independently in expression profiling of meu5Δ mutants, determination of decay rates in meu5Δ cells and classification of expression profiles in meiotic time courses.
We attempted to find putative Meu5p-binding sites by searching for overrepresented motifs in the untranslated regions (UTRs) and coding sequences of Meu5p targets (see Materials and methods). Although we failed to identify any clear motifs, both the 5′ and 3′ UTRs of Meu5p targets were enriched in poorly defined U-rich sequences (data not shown). The failure to identify these sequences could be explained in several ways: First, Meu5p could recognize structural motifs (rather than simple linear sequences). Second, different RRMs in Meu5p might recognize different target sequences. Third, Meu5p could bind some or all of its targets indirectly, through the association with additional RBPs. A recent study, in which the targets of ∼30 RBPs were determined using RIp-chip, identified potential binding sites for more than half of them (Hogan et al, 2008). This success rate highlights both the power and the limitations of bioinformatic approaches to identify RBP-binding sites from RIp-chip data.
We have identified fewer transcripts with shortened half-lives in meu5Δ cells than mRNAs with reduced levels (Figure 3B), and not every ‘late-decrease’ gene seems to have a reduced half-life in meu5Δ (Supplementary Figure S10B). We believe that this difference reflects a larger number of false negatives in the identification of transcripts with reduced decay rates. Measuring RNA levels using microarrays is very straightforward, and the results are highly reproducible. By contrast, the determination of decay rates is a more complex experiment, which involves in vivo and in vitro labelling of transcripts as well as an affinity purification step. These issues make the estimation of decay rates noisier and less sensitive, and probably lead to a large fraction of false negatives. Therefore, we expect that most ‘late-decrease’ transcripts also have shorter half-lives, explaining their behaviour in the time courses (Figure 5). Consistent with this view, most mRNAs with decreased levels in meu5Δ show a typical ‘late-decrease’ profile, regardless of whether their transcripts are destabilized in meu5Δ or not. Similarly, RIp-chip identified only 40% of the mRNAs whose levels are affected by meu5Δ (Figure 2C). Given the complexity of the RIp-chip experiments, it is likely that they are also less sensitive than expression profiling, and that most RNAs expressed at low levels in meu5Δ cells are directly bound and stabilized by Meu5p.
The meu5 gene was originally isolated as a multi-copy suppressor of mutants involved in mRNA export, although the meu5 mutant did not display any defects in mRNA export (Thakurta et al, 2002). Given the function of Meu5p in mRNA stabilization described in this work, it is possible that the ectopic expression of Meu5p in vegetative cells suppresses the nuclear export defects indirectly, by increasing the amounts of either mRNAs encoding nuclear export components, or of transcripts whose transport is impaired in the mutants. An alternative possibility is that Meu5p regulates the stability of its targets by facilitating their export to the cytoplasm, where they would be stabilized by translation.
Mei4p also activates the expression of several other genes encoding transcription factors and RBPs of unknown function (Mata et al, 2002). Moreover, several genes encoding components of general decay pathways are induced during meiosis. For example, the ccr4 gene (encoding an exonuclease from the CCR4-NOT complex, involved in deadenylation) and exo2 (a homologue of budding yeast XRN1, which encodes an exonuclease involved in 5′->3′ degradation of mRNAs) are both induced during sexual differentiation in a Mei4p-dependent manner. These observations suggest that the correct implementation of the gene expression program of sexual differentiation is governed by regulatory interactions between transcription factors and regulators of RNA stability.
As meu5 is a transcriptional target of Mei4p, Meu5p forms a coherent feed-forward loop (FFL) with Mei4p and their common targets (Figure 5B). FFLs are among the most frequent motifs in transcriptional networks, where they can confer specific kinetic properties to the transcription of target genes (Alon, 2007). FFLs are also common in posttranslational networks (Csikasz-Nagy et al, 2009), and have recently been observed in the interaction networks between micro-RNAs and transcription factors (Tsang et al, 2007). However, the structure of the regulatory networks formed by the combination of transcription factors and RBPs has not been analysed exhaustively (Zhu et al, 2007). It is likely that these networks will be enriched only in certain types of motifs, some of which may differ from those present in transcriptional networks. Addressing this question will require the systematic analysis of the function and targets of more RBPs, and the integration of these data with transcriptional networks.
Materials and methods
Yeast methods and experimental design
Standard methods were used for fission yeast growth and manipulation (Forsburg and Rhind, 2006). meu5 was deleted and TAP-tagged using a one-step PCR method in a h90 background (Bähler et al, 1998; Tasto et al, 2001). Successful tagging was verified by western blot as described earlier (Amorim and Mata, 2009). Meu5-TAP-tagged strains grew normally, displayed normal cell shape and produced normal spores, showing that the tagged proteins were functional. Meu5-TAP and meu5Δ-derived strains were generated by crossing or protoplast fusion. Other TAP-tagged strains (except mug110) were made as follows: PCR products containing fragments of the coding and the full 3′ untranslated sequence were amplified by PCR from genomic DNA and cloned into pFA6a-2x-TAP::kanMX6 using the restriction sites indicated in Supplementary Table S6 (Tasto et al, 2001), and subsequently subcloned into pFA6a-NatMX6 (Hentges et al, 2005). mug110 ORF and UTR were cloned directly into pFA6a-4x-TAP::NatMX6 (Van Driessche et al, 2005). The plasmids were linearized using the following restriction enzymes that cut in the coding sequences of the corresponding genes: XhoI (meu14), BclI (mug110), MfeI (mpf1) and BbsI (mug103). The linearized fragments were directly transformed into pat1 or pat1 meu5Δ diploids. The correct integration of the plasmid was verified by PCR, and the expression of the tagged protein by western blot (see below). This strategy ensures that mRNAs encoding the tagged proteins are transcribed from the endogenous promoters and contain wild-type 5′ and 3′ UTRs. A complete list of the strains used in this work is presented in Supplementary Table S7.
To induce meiosis in h90 or in wild-type diploid backgrounds, cells were grown in Edinburgh minimal medium containing 2% glucose (EMM) plus 0.5% NH4Cl, washed with EMM containing 0.5% glucose without NH4Cl (EMM-N low glucose) and resuspended in EMM-N low glucose. h90 cells were incubated at 25°C for 18 h and wild-type diploids at 30°C for 6.5 h. Induction of meiosis using pat1 mutations was carried out exactly as in our earlier studies (Mata et al, 2002, 2007). Briefly, pat1-114/pat1-114 ade6-M210/ade6-M216 h+/h+ diploid cells were grown in EMM plus 0.5% NH4Cl, resuspended in EMM without NH4Cl (EMM-N) and incubated for 14 h at 25°C. Meiosis was started by shifting the cells to 34°C in the presence of 0.05% NH4Cl. For assessment of meiotic synchronization, cells were collected from each time point, stained with DAPI, and the number of nuclei counted (n=100). For the time course, RNA extracted from each time point was compared with a reference RNA prepared from pat1-114/pat1-114 cells treated as described above to induce meiosis. The reference consisted of equal amounts of RNA extracted from cells at 0, 1, 2, 3, 4, 5, 6, 7, 8 and 9 h after the temperature shift. The expression ratios at each time point were normalized to those of the 3-h time point of the corresponding strain. For other experiments, RNA extracted from wild-type and meu5Δ cells was compared directly.
For time courses to monitor protein levels, pat1 diploids were synchronized as described above. Samples were taken at 3, 4, 5, 6, 7, 8 and 9 h after meiotic induction. Cell extracts were prepared as for RIp-chip experiments (see below). TAP-tagged proteins were detected using peroxidase–anti-peroxidase-soluble complexes (Sigma). Pcp1p was detected with a polyclonal antibody (provided by Takashi Toda). Cig2p and tubulin were detected with monoclonal antibodies (anti-Cig2p 3A11 from Abcam and anti-tubulin B-5-1-2 from Sigma).
For overexpression experiments using the nmt1 promoter, cells were grown in EMM containing 0.5% NH4Cl and 15 μM thiamine, washed three times in EMM with 0.5% NH4Cl, and incubated at 32°C for 18 h. In every experiment, RNA extracted from cells overexpressing the Mei4p transcription factor was compared with RNA from cells transformed with empty vectors that were treated in exactly the same way to induce the nmt1 promoter.
RIp-chip experiments
We used the protocol described in Amorim and Mata (2009), with the following modifications: (1) 25% of the extract was used for total RNA purification; (2) the immunoprecipitation was performed for 1 h and (3) magnetic beads containing the immunoprecipitate were resuspended in 100 μl of wash buffer containing 1 mM DTT, 0.2 units/μl of SuperaseIN (Ambion) and 3 units/μl of AcTev protease (Invitrogen). The solution with the beads was incubated for 1 h at 19°C, the supernatant recovered and RNA extracted using RNAqueous micro columns (Ambion) according to the manufacturer's instructions. The RNA was eluted from the column in 12 μl and used for labelling without amplification. To minimize protein degradation during the preparation of the cell extracts, cultures were incubated with 1 mM PMSF before collection of the cells.
Real-time qPCR
For qPCR experiments, total and Meu5p-bound RNAs were purified as for RIp-chip experiments, and cDNA was prepared from them using Superscript III (Invitrogen). The qPCR reactions were performed in 20 μl of Power Sybr Green Master Mix (Applied Biosytems) containing 1 μl of cDNA (diluted 1:10) and 0.5 μM of each primer. The reactions were analysed with an Applied Biosystems 7300 Real-Time PCR system.
The ratio between transcript amounts in the IP and total RNA samples was calculated as 2−ΔCT, where ΔCT is the difference between the threshold cycle of the IP RNA and that of the total RNA. For each experiment, the data were normalized by dividing the enrichment ratio of each transcript by that of myo1 (a non-target of Meu5p).
Selection of RIp-chip targets
To define an enrichment cutoff in the RIp-chip experiments that identifies real Meu5p targets, we initially ranked all mRNAs present in the immunoprecipitates according to their enrichment. We then made lists of increasing size, and quantified the number of genes in each list whose expression was affected in meu5Δ mutants (functional targets). The number of validated targets in each of the lists of Meu5p-bound mRNAs was plotted against the total number of genes in the list (Supplementary Figure S2A). A similar analysis was performed with randomized enrichment ranks (Supplementary Figure S2A). The initial slope in the ranked RIp-chip is higher than that in the randomized data, indicating that the target lists are enriched in functional targets. The point at which the slope changes (arrow in Supplementary Figure S2A) indicates when the RIp-chip experiment ceases to identify functional targets at a rate higher than random, and was chosen as a threshold for the definition of Meu5p targets. The list presented in Supplementary Table S2 shows all targets that were identified in at least two independent experiments. A false-positive rate can be estimated from the data on Figure 2C. Assuming that all binding events affect gene expression and that we have identified all changes in gene expression caused by the deletion of meu5Δ, the false-positive rate would be 5/83 (0.06). However, these assumptions lead to a very conservative estimate (e.g. it is unlikely that all changes in RNA levels in meu5 mutants have been detected). Therefore, the real false-positive rate is likely to be lower.
RNA Pol II ChIP-chip
The experiment was performed with vegetative cells overexpressing Mei4p, as well as in wild-type diploids undergoing meiosis (in wild-type and meu5Δ backgrounds). Chromatin immunoprecipitation and DNA labelling were performed exactly as described in Aligianni et al (2009) using an anti Pol II antibody (Abcam 5408). Labelled material was hybridized to the 4x44K Chip-on-chip whole genome DNA microarray platform (Agilent).
Microarray experiments (all except ChIP-chip)
All microarray experiments except the ChIP-chip and a single RIp-chip (see below) were performed with custom-made PCR-based microarrays using previously published protocols (Lyne et al, 2003; Amorim and Mata, 2009). A custom-designed oligonucleotide-based microarray was manufactured by Agilent (ArrayExpress accession A-MEXP-1801). For this microarray, labelled probes were prepared using SuperScript Plus Direct cDNA Labeling System (Invitrogen), and the microarrays were processed according to the manufacturer's instructions. Microarrays were scanned with a Genepix 4000B scanner and analysed with GenePix software (Axon Instruments). Data clustering and visualization were done with GeneSpring (Agilent). Transcriptome analysis of meu5Δ in pat1 cells was carried out three times. Determination of decay rates in cells overexpressing Mei4p was done three times, and from each experiment a transcriptome analysis comparing meu5Δ and wild-type strains was performed before 4sU incorporation. An additional transcriptome analysis of Mei4p-overexpressing cells was done (a total of four repeats). Meu5-TAP RIp-chip experiments were done three times with the PCR-based microarray platform (twice in h90 cells and once in pat1 diploids) and once with Agilent microarrays (pat1 diploids). The wild-type and meu5Δ pat1-synchronized time course were performed once. All experiments were independent biological replicates. Dyes were swapped between experiments. The complete normalized data are presented in Data set S1 (all expression arrays except 4sU-labelling experiments and pat1 time courses), Data set S2 (RIp-chip experiments), Data set S3 (all 4sU experiments) and Data set S4 (pat1 time courses). Microarray raw data have been deposited in the Array Express database with the following accession numbers: E-TABM-775 (expression data except time course), E-TABM-774 (meu5Δ time course expression arrays), E-TABM-776 (decay rate determinations), E-TABM-777 (RIp-chip with PCR-product microarrays), E-TABM-932 (RIp-chip with Agilent microarray) and E-TABM-993 (RNA Pol II ChIP-chip).
Genome-wide determination of decay rates by in vivo metabolic labelling
4sU was added to the cells at a final concentration of 75 μg/ml, and cells were incubated for 30 min at 32°C, harvested and frozen. Total RNA was extracted using hot phenol (Lyne et al, 2003). Half of the sample was kept for use as reference, and 100 μg of RNA were used for biotinylation. Biotinylation of 4sU-labelled RNA was performed as follows: a solution containing 100 μg/ml of total RNA, 0.2 mg/ml EZ-link Biotin-HPDP (Pierce), 1 mM EDTA and 10 mM Tris–HCl pH 7.6 was incubated at room temperature for 90 min. RNA was purified by extraction with chloroform:isoamyl alcohol (24:1) and precipitated by adding 1 volume of isopropanol and 1/10th volume of 5 M NaCl. Finally, biotinylated RNA was resuspended in nuclease-free H2O, denatured at 65°C for 5 min, and incubated with Dynabeads® M-280 Streptavidin (Invitrogen) for 40 min at room temperature. Streptavidin beads were blocked for 30 min before use with 0.5 mg/ml yeast tRNA (Ambion) in MPG buffer (100 mM Tris–HCl pH 7.6, 1 M NaCl, 10 mM EDTA), washed and equilibrated in MPG buffer. After the incubation with biotinylated RNA, beads were washed three times with MPG buffer at 65°C and three times at room temperature. RNAs were eluted from the beads by reduction of the linker with 5% (v/v) β-mercaptoethanol for 5 min at room temperature. The eluted RNA was further purified using an RNAqueous Micro columns (Ambion) following the manufacturer's instructions.
Calculation of mRNA half-lives
The analysis of data from the 4sU labelling experiments is described in detail in Dolken et al (2008), where the derivation of all the equations can be found. Briefly, under steady-state conditions, mRNAs half-lives can be calculated following the equation:
where t is the length of the labelling period and F is the fraction of mRNA that has been labelled with 4sU. The microarray experiments produce an unnormalized measure of the relative abundance of labelled mRNA compared with total mRNA, which needs to be multiplied by a correction factor to calculate F:
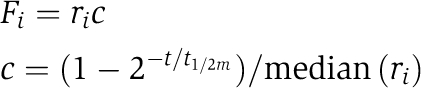 |
where ri is the ratio measured in the microarray experiment, c is the correction factor, t1/2m is the median half-life of all mRNAs and median (ri) is the median of all ratios measured in the experiment.
To estimate t1/2m we carried out an experiment in which we compared pre-existing RNA (the supernatant after purification of 4sU-labelled RNA) with total RNA, as well as 4sU-labelled RNA with total RNA. The ratios of pre-existing RNA/total RNA and newly synthesized RNA/total RNA allow the calculation of the fraction of labelled mRNA (and thus half-lives) by a linear regression (see Dolken et al, 2008 for details). This estimate of the median half-life (30.2 min) was used to calculate the correction factors for all the experiments described in this article. Supplementary Table S5 displays the calculated wild-type half-lives from this experiment.
Classification of middle genes into ‘early’ and ‘late-decrease’
Middle genes (555) were classified into two groups according to how their levels decreased after reaching a peak at 5 h into meiosis (in pat1 synchronous meiosis; Mata et al, 2002). Some middle genes are regulated by the late acting transcriptions factors Atf21p and Atf31p, leading to an extended period of expression. To avoid any confounding effects, Atf21p and Atf31p targets (defined as genes whose expression was induced by co-expression of both transcription factors; Mata et al, 2007) were excluded from the analysis, leaving 502 genes. Nine of the 53 genes removed from the data set were bound by Meu5p. Genes whose expression levels at 6 h were 0.75-fold or lower than those at 5 h (376) were classified as ‘early-decrease’, whereas the rest (126) were defined as ‘late-decrease’. These groups are presented in Supplementary Table S4. Similar results were obtained by using k-means to cluster middle genes into two groups (data not shown).
Statistical analysis
Statistical significance of differential gene expression was determined using significance analysis of microarrays (Tusher et al, 2001), with the false-discovery rate adjusted to 0.1% (for expression analysis) or 5% (for comparison of fractions of 4sU-labelled mRNA). No minimal fold-difference was required for selection. The significance of the overlaps between gene lists was determined assuming that the overlap between random groups follows a hypergeometric distribution.
Identification of potential regulatory sequences
We extracted sequences from 5′ and 3′ UTRs from RIp-chip targets and a control set of similar number of genes from the ‘early-decrease’ group. The information on the UTR length in meiotic cells (pat1 synchronized between 5 and 6 h after meiosis induction) was obtained from Wilhelm et al (2008). The minimal UTR length was set to 100 nucleotides. The four sets of sequences had similar compositions (data not shown) and were scanned for potential regulatory sequences using multiple expectation maximization for motif elicitation (Bailey et al, 2009) with default parameters. As a control, upstream sequences of 500 nucleotides upstream of the coding sequences were scanned for the Meu5p targets and the control set. In both cases, sequences related to the Mei4p-binding site were readily detected (data not shown).
Accession numbers
Microarray data have been deposited in the ArrayExpress database (see Materials and methods).
Supplementary Material
Genes underexpressed in meu5 mutants.
Meu5p-bound transcripts.
Transcripts with shortened half-lives in meu5p mutants.
Classification of meiotic middle genes according to their downregulation profiles.
Estimated mRNA half-lives in wild type cells.
Transcriptome analysis of meu5p cells.
Meu5p RIp-chip experiments.
Determination of half-lives using 4sU labelling.
Wild type and meu5 pat1-synchronised time courses.
Supplementary figures S1–12, Supplementary table S6 and S7, index to supplementary materials.
Acknowledgments
We thank Jürg Bähler for his support throughout this project, Ana Matia for help with the development of the 4sU method, Sofia Aligianni for assistance with ChIP-chip experiments, and Jürg Bähler and Samuel Marguerat for comments on the article. We also thank Takashi Toda for the generous gift of antibodies. This work was supported by a Medical Research Council New Investigator Award (G0501168) and a Biological Sciences and Biotechnology Research Council (BBSRC) grant (BB/G011869/1).
Footnotes
The authors declare that they have no conflict of interest.
References
- Aligianni S, Lackner DH, Klier S, Rustici G, Wilhelm BT, Marguerat S, Codlin S, Brazma A, de Bruin RA, Bähler J (2009) The fission yeast homeodomain protein Yox1p binds to MBF and confines MBF-dependent cell-cycle transcription to G1-S via negative feedback. PLoS Genet 5: e1000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- Amorim MJ, Mata J (2009) Rng3, a member of the UCS family of myosin co-chaperones, associates with myosin heavy chains cotranslationally. EMBO Rep 10: 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A III, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14: 943–951 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenco M, Brewer D, Papouli E, Tomescu D, Callard R, Stark J, Hubank M (2009) Dissection of a complex transcriptional response using genome-wide transcriptional modelling. Mol Syst Biol 5: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary MD, Meiering CD, Jan E, Guymon R, Boothroyd JC (2005) Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat Biotechnol 23: 232–237 [DOI] [PubMed] [Google Scholar]
- Csikasz-Nagy A, Kapuy O, Toth A, Pal C, Jensen LJ, Uhlmann F, Tyson JJ, Novak B (2009) Cell cycle regulation by feed-forward loops coupling transcription and phosphorylation. Mol Syst Biol 5: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker N, van Rijssel J, Distel B, Hochstenbach F (2007) Role of the alpha-glucanase Agn2p in ascus-wall endolysis following sporulation in fission yeast. Yeast 24: 279–288 [DOI] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, Koszinowski UH (2008) High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar del Dedo J, Duenas E, Arnaiz Y, del Rey F, Vazquez de Aldana CR (2009) beta-glucanase Eng2 is required for ascus wall endolysis after sporulation in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell 8: 1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ (2009) Insights from genomic profiling of transcription factors. Nat Rev Genet 10: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferea TL, Brown PO (1999) Observing the living genome. Curr Opin Genet Dev 9: 715–722 [DOI] [PubMed] [Google Scholar]
- Forsburg SL, Rhind N (2006) Basic methods for fission yeast. Yeast 23: 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, Gerber AP (2008) Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One 3: e3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ (2007) The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126 [DOI] [PubMed] [Google Scholar]
- Halbeisen RE, Galgano A, Scherrer T, Gerber AP (2008) Post-transcriptional gene regulation: from genome-wide studies to principles. Cell Mol Life Sci 65: 798–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y, Tanaka H, Yamanaka S, Tanaka K, Watanabe Y, Tsutsumi C, Chikashige Y, Hiraoka Y, Yamashita A, Yamamoto M (2006) Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 442: 45–50 [DOI] [PubMed] [Google Scholar]
- Hentges P, Van Driessche B, Tafforeau L, Vandenhaute J, Carr AM (2005) Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe. Yeast 22: 1013–1019 [DOI] [PubMed] [Google Scholar]
- Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO (2008) Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol 6: e255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie S, Watanabe Y, Tanaka K, Nishiwaki S, Fujioka H, Abe H, Yamamoto M, Shimoda C (1998) The Schizosaccharomyces pombe mei4+ gene encodes a meiosis-specific transcription factor containing a forkhead DNA-binding domain. Mol Cell Biol 18: 2118–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino Y, Yamamoto M (1985) Mutants of Schizosaccharomyces pombe which sporulate in the haploid state. Mol Gen Genet 198: 416–421 [DOI] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, Friedersdorf MB (2006) RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nature Protocols 1: 302–307 [DOI] [PubMed] [Google Scholar]
- Keene JD, Tenenbaum SA (2002) Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell 9: 1161–1167 [DOI] [PubMed] [Google Scholar]
- Lackner DH, Beilharz TH, Marguerat S, Mata J, Watt S, Schubert F, Preiss T, Bähler J (2007) A network of multiple regulatory layers shapes gene expression in fission yeast. Mol Cell 26: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ (2006) Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol Cell Biol 26: 9196–9208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M (2004) Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci USA 101: 2987–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyne R, Burns G, Mata J, Penkett CJ, Rustici G, Chen D, Langford C, Vetrie D, Bähler J (2003) Whole-genome microarrays of fission yeast: characteristics, accuracy, reproducibility, and processing of array data. BMC Genomics 4: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Bähler J (2006) Global roles of Ste11p, cell type, and pheromone in the control of gene expression during early sexual differentiation in fission yeast. Proc Natl Acad Sci USA 103: 15517–15522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Lyne R, Burns G, Bähler J (2002) The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet 32: 143–147 [DOI] [PubMed] [Google Scholar]
- Mata J, Marguerat S, Bähler J (2005) Post-transcriptional control of gene expression: a genome-wide perspective. Trends Biochem Sci 30: 506–514 [DOI] [PubMed] [Google Scholar]
- Mata J, Wilbrey A, Bähler J (2007) Transcriptional regulatory network for sexual differentiation in fission yeast. Genome Biol 8: R217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazan-Mamczarz K, Kuwano Y, Zhan M, White EJ, Martindale JL, Lal A, Gorospe M (2009) Identification of a signature motif in target mRNAs of RNA-binding protein AUF1. Nucleic Acids Res 37: 204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPheeters DS, Cremona N, Sunder S, Chen HM, Averbeck N, Leatherwood J, Wise JA (2009) A complex gene regulatory mechanism that operates at the nexus of multiple RNA processing decisions. Nat Struct Mol Biol 16: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, Steitz JA (2004) Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA 10: 1692–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, Keene JD (2008) Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol Cell Biol 28: 4093–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Lager PJ, Friedersdorf MB, Thompson MA, Keene JD (2009) Coordinated posttranscriptional mRNA population dynamics during T-cell activation. Mol Syst Biol 5: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Abe H, Hirata A, Shimoda C (2004) ADAM family protein Mde10 is essential for development of spore envelopes in the fission yeast Schizosaccharomyces pombe. Eukaryot Cell 3: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P (1985) Mutants of the fission yeast Schizosacharomyces pombe which alter the shift between cell proliferation and sporulation. Mol Gen Genet 198: 497 [Google Scholar]
- Perez-Ortin JE (2007) Genomics of mRNA turnover. Brief Funct Genomic Proteomic 6: 282–291 [DOI] [PubMed] [Google Scholar]
- Shalem O, Dahan O, Levo M, Martinez MR, Furman I, Segal E, Pilpel Y (2008) Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol Syst Biol 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoecklin G, Tenenbaum SA, Mayo T, Chittur SV, George AD, Baroni TE, Blackshear PJ, Anderson P (2008) Genome-wide analysis identifies interleukin-10 mRNA as target of tristetraprolin. J Biol Chem 283: 11689–11699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasto JJ, Carnahan RH, McDonald WH, Gould KL (2001) Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast 18: 657–662 [DOI] [PubMed] [Google Scholar]
- Thakurta AG, Whalen WA, Yoon JH, Bharathi A, Kozak L, Whiteford C, Love DC, Hanover JA, Dhar R (2002) Crp79p, like Mex67p, is an auxiliary mRNA export factor in Schizosaccharomyces pombe. Mol Biol Cell 13: 2571–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A (2007) MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 26: 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driessche B, Tafforeau L, Hentges P, Carr AM, Vandenhaute J (2005) Additional vectors for PCR-based gene tagging in Saccharomyces cerevisiae and Schizosaccharomyces pombe using nourseothricin resistance. Yeast 22: 1061–1068 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Miyashita K, Saito TT, Yoneki T, Kakihara Y, Nabeshima K, Kishi YA, Shimoda C, Nojima H (2001) Comprehensive isolation of meiosis-specific genes identifies novel proteins and unusual non-coding transcripts in Schizosaccharomyces pombe. Nucleic Acids Res 29: 2327–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bähler J (2008) Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453: 1239–1243 [DOI] [PubMed] [Google Scholar]
- Xue-Franzen Y, Kjaerulff S, Holmberg C, Wright A, Nielsen O (2006) Genomewide identification of pheromone-targeted transcription in fission yeast. BMC Genomics 7: 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Imai I, Watanabe Y (1997) S. pombe mating and sporulation. In The Molecular and Cellular Biology of the Yeast Saccharomyces: Life Cycle and Cell Biology, Pringle JR, Broach JR, Jones EW (eds.), pp 1035–1106. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory [Google Scholar]
- Zhu X, Gerstein M, Snyder M (2007) Getting connected: analysis and principles of biological networks. Genes Dev 21: 1010–1024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes underexpressed in meu5 mutants.
Meu5p-bound transcripts.
Transcripts with shortened half-lives in meu5p mutants.
Classification of meiotic middle genes according to their downregulation profiles.
Estimated mRNA half-lives in wild type cells.
Transcriptome analysis of meu5p cells.
Meu5p RIp-chip experiments.
Determination of half-lives using 4sU labelling.
Wild type and meu5 pat1-synchronised time courses.
Supplementary figures S1–12, Supplementary table S6 and S7, index to supplementary materials.