

Abstract
Motivation: A typical approach for the interpretation of high-throughput experiments, such as gene expression microarrays, is to produce groups of genes based on certain criteria (e.g. genes that are differentially expressed). To gain more mechanistic insights into the underlying biology, overrepresentation analysis (ORA) is often conducted to investigate whether gene sets associated with particular biological functions, for example, as represented by Gene Ontology (GO) annotations, are statistically overrepresented in the identified gene groups. However, the standard ORA, which is based on the hypergeometric test, analyzes each GO term in isolation and does not take into account the dependence structure of the GO-term hierarchy.
Results: We have developed a Bayesian approach (GO-Bayes) to measure overrepresentation of GO terms that incorporates the GO dependence structure by taking into account evidence not only from individual GO terms, but also from their related terms (i.e. parents, children, siblings, etc.). The Bayesian framework borrows information across related GO terms to strengthen the detection of overrepresentation signals. As a result, this method tends to identify sets of closely related GO terms rather than individual isolated GO terms. The advantage of the GO-Bayes approach is demonstrated with a simulation study and an application example.
Contact: song.zhang@utsouthwestern.edu; richard.scheuermann@utsouthwestern.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
In typical high-throughput experiments, such as gene expression microarrays, the first step in the analysis of the results is often to produce groups of genes based on certain criteria (e.g. genes that are differentially expressed). To gain more mechanistic insights into the underlying biology, overrepresentation analysis (ORA) is conducted to use the knowledge of the functional characteristics of the genes to investigate whether gene sets associated with particular biological functions are overrepresented in the identified gene groups. Drăghici et al. (2003) is the first paper to discuss the overrepresentation problem and propose different statistical methods that can be used in this area. ORA is based on the postulate that if a biological process has more identified genes than expected by chance alone, that biological process is probably linked to the experiment.
One of the most popular gene description databases used in ORA was developed by the Gene Ontology (GO) Consortium (Ashburner et al., 2000). Each GO term annotates a set of genes, indicating their known molecular functions, involvement in biological processes and cellular component locations. GO terms are structured in a directed acyclic graph (DAG) of parent–child relationship, where a child indicates a more specific biological classification than its parent(s). Based on the true-path rule the annotation of a gene to a GO term implies automatic annotation to all the ancestors of that term. Another feature is that a GO term is allowed to have more than one parent nodes, a feature known as multiple inheritance. For example, immune response is not only a specific form of organismal movement but also a part of defense response. Furthermore, some genes might be annotated by a parent GO node but not by any of its children because less is known about that gene's specific function. For a rigorous analysis, these dependency characteristics of the GO DAG need to be considered when developing statistical methods to detect overrepresentation of GO annotations.
In ORA, the most commonly used statistical test is based on the hypergeometric distribution or its binomial approximation (Cho et al., 2001; Khatri et al., 2002; Drăghici et al., 2003; Al-Shahrour et al., 2004; Beissbarth and Speed, 2004; Lee et al., 2005; Lee et al., 2006; Luo et al., 2007; among others). Let A denote a GO term or the set of genes annotated to A (with cardinality IA), and let S denote the set of genes (with cardinality IS) based on a certain criterion (i.e. differential expression) from a full gene list G (with cardinality I) in an experiment. The number of genes belonging to both S and A (S∩A), denoted by nA, indicates the representation of A in S. Under the null hypothesis that S and A are independent (i.e. the GO term is irrelevant to the gene cluster), nA follows a hypergeometric distribution. The P-value measuring the significance of association is the tail probability of observing nA or more genes annotated by A in S,
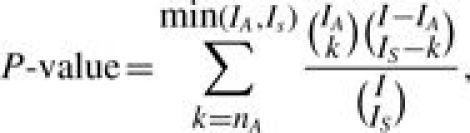 |
(1) |
where 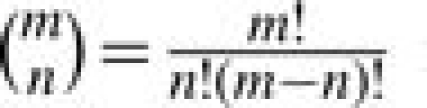 is the binomial coefficient. Many software and webtools (Onto-Express, CLASSIFI, GoMiner, EASEonline, GeneMerge, FuncAssociate, GOTree Machine, etc.) have been developed based on the hypergeometric P-value. Detailed review can be found in Khatri and Drăghici (2005).
is the binomial coefficient. Many software and webtools (Onto-Express, CLASSIFI, GoMiner, EASEonline, GeneMerge, FuncAssociate, GOTree Machine, etc.) have been developed based on the hypergeometric P-value. Detailed review can be found in Khatri and Drăghici (2005).
The hypergeometric P-value provides a straightforward measure of overrepresentation for each individual GO term. However, the major drawback of this approach is that it ignores the hierarchical structure in the GO DAG, which contains a substantial amount of information regarding the interactions among the GO terms.
We use an artificial DAG (Fig. 1) to illustrate this issue. It consists of 25 nodes {Aj, j=1,…, 25}, each denoted by a circle. Let G={a, b,…, z} denote the full list of genes, and 10 genes in S are marked in red. The rectangles contain the subset of genes annotated by each node, where (IA, nA) are listed under each rectangle. Figure 1 includes some important features of the GO DAG, such as multiple inheritance (e.g. A14 and A23 have two parent nodes) and that a gene might be annotated at different specific level (eg. gene k is annotated by A2, but not by any of its children: A5, A6 and A7). Note that in this example S is overrepresented in the regions under A2, while underrepresented in the regions under A3 and A4. We report the hypergeometric P-values estimated by (1) in blue.
Fig. 1.

Comparison of the P-value and the B-score with an artificial DAG. The full list of genes (G) are denoted as lowercase letters; the genes in set S are marked in red. The rectangles contain the subset of genes annotated by each node, where (IA, nA) are listed under each rectangle. The hypergeometric P-values are listed in blue and the B-scores are listed in black underneath the P-values.
First, given I and IS, GO terms with the same (IA, nA) have exactly the same hypergeometric P-values. For example, with (IA, nA)=(2, 2), the P-values for A14 and A24 are both 0.139. Based on Figure 1, A14 is more likely to be linked with S because of the stronger evidence of overrepresentation in its neighboring nodes (related biological functions). Using evidence only from individual terms, the hypergeometric P-value does not differentiate between A14 and A24. Second, given I and IS, the hypergeometric P-value has a lower limit determined by IA, which is denoted as L(IA). Specifically, L(IA) is achieved when nA=IA, i.e. the P-value reaches its lower limit when all the genes annotated by A are in S. The smaller the IA, the larger the L(IA). For example, we have L(3)=0.046 as in A12 and L(2)=0.139 as in A14. This observation suggests that if we set the threshold for the P-value at L(k), then the hypergeometric test could not identify any GO terms with IA<k. In ORA, detecting more specific GO terms, which usually have a relatively smaller IA, might be more desirable because they provide more detailed biological information. However, the hypergeometric test tends to identify less specific GO terms because of the constraint of L(IA). In Figure 1, the most significant term selected by the P-value is A2, a term next to the root. The more specific terms that are associated with S (i.e. A12 and A14) are considered less significant compared with A2. All of these issues essentially stem from the limitation of the hypergeometric test in treating the GO terms as independent entities and ignoring their interrelated structure.
Recently some new methods have been proposed in ORA. Lewin and Grieve (2006) propose to group closely related GO nodes together and then compute a hypergeometric P-value for each group. In their approach, the graphical distance between nodes in the GO DAG is assumed to have some quantitative biological meaning. Alexa et al. (2006) calculate the overrepresentation of a GO term from leaf to root by downweighting the contribution of genes which are annotated by a child term that has been found to be significantly enriched. Grossmann et al. (2007) measure the overrepresentation of each GO term relative to its parent(s). From root to leaf, their method computes the significance of overrepresentation for a GO term conditional on the representation at the parent term(s). The last two methods purposely remove the dependence between parent and child terms.
In this article, we develop a Bayesian hierarchical model to incorporate the dependence structure of the DAG in assessing GO term overrepresentation. It takes into account evidence not only from individual GO terms, but also from their related terms (i.e. parents, children, siblings, etc.). The Bayesian framework enables borrowing information across related GO terms to strengthen the detection of overrepresentation signals. As a result, this method tends to identify sets of closely related GO terms rather than individual unrelated GO terms. The utility of the method is demonstrated using a gene expression microarray dataset from a human B cell stimulation experiment.
2 METHOD
2.1 The Bayesian model
The proposed method is called GO-Bayes: GO-based ORA using a Bayesian approach. In the model, each GO term has a relevance parameter measuring its association with the selected genes in S. The novelty of the model is that the complex dependence structure in the GO DAG is incorporated via a hierarchical prior on the relevance parameters.
To introduce GO-Bayes, we define the following notations. Let A={Aj, j=1,…, J} be the set of GO terms involved in the annotation of the full list of I genes (i.e. IAj>0, for j=1,…, J), among which IS genes are grouped in S in the experiment. We use Aj1→Aj2 to indicate that Aj1 is a parent of Aj2. We define Pj={Ak:Ak→Aj} and Cj={Ak:Aj→Ak} to be the sets of parent and child nodes of Aj, respectively. We use |U| to denote the cardinality of set U. Without loss of generality, let A1 denotes the root node and |P1|=0. For the non-root nodes, we have |Pj|≥1, where |Pj|>1 indicates multiple inheritance. We call Aj an end node if |Cj|=0 and an inner node otherwise. For example, in Figure 1, A1 is the root node, P10={A4}, C10={ A22, A23} and |P14|=|P23|=2 indicating multiple inheritance.
At the gene level, we use gi∈Aj to denote that gene i is annotated by Aj. We further use gi◃Aj to indicate that Aj is the most specific GO term that annotates gene i, with the formal definition being gi∈Aj and gi∉Ak for any Ak∈Cj. We define Bi={Aj:gi◃Aj} to be the set of the most specific annotations of gene i. The true-path rule implies that Bi contains all the annotation information about gene i. In Figure 1, we have Ba={A12} and Bk={A2, A18, A22}. Let yi(i =1,···, I) be the observed expression status of gene i, yi=1 if gene i is in S and yi=0 otherwise.
The binary yi is assumed to follow a Bernoulli distribution, yi∣pi∼Bernoulli(pi), where pi is the probability that gene i belongs to S. Using the idea that if Aj is associated with S, the genes annotated by Aj have a higher chance of being grouped in S, we construct the following logistic model,
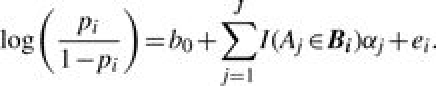 |
(2) |
We specify b0 as a constant and set b0=log[p0/(1−p0)] with p0=IS/I, where p0 is the background probability that gene i is grouped in S by chance. The random error ei is assumed to have a normal distribution with mean 0 and variance σ2, denoted by 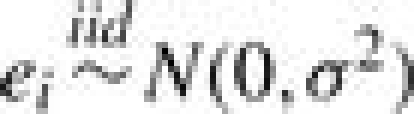 . Parameter αj characterizes the relevance of GO term Aj to the set of identified genes S, where it modifies the odds of gene i being grouped in S by a factor of exp(αj) if gi◃Aj. Thus, positive (negative) values of αj indicate over(under)-representation. Based on the true-path rule, we only include αj′s from Bi, the most specific annotations, in model (2) to avoid repeated use of information. The αj′s from less-specific annotations are assumed to affect the odds indirectly via a hierarchical prior on α={αj, j=1,…, J}, constructed according to the dependence structure in the GO DAG. We set α1=0 for the root node. Then the prior of αj(j=2,…, J) is specified conditionally given the relevance parameters of its parent nodes, denoted by αPj={αk:Ak∈Pj}. Specifically,
. Parameter αj characterizes the relevance of GO term Aj to the set of identified genes S, where it modifies the odds of gene i being grouped in S by a factor of exp(αj) if gi◃Aj. Thus, positive (negative) values of αj indicate over(under)-representation. Based on the true-path rule, we only include αj′s from Bi, the most specific annotations, in model (2) to avoid repeated use of information. The αj′s from less-specific annotations are assumed to affect the odds indirectly via a hierarchical prior on α={αj, j=1,…, J}, constructed according to the dependence structure in the GO DAG. We set α1=0 for the root node. Then the prior of αj(j=2,…, J) is specified conditionally given the relevance parameters of its parent nodes, denoted by αPj={αk:Ak∈Pj}. Specifically,
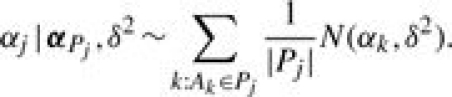 |
(3) |
Prior (3) assumes that αj arises from a mixture distribution of |Pj| components, each component being a normal distribution centered at the relevance parameter of one of its parents. The equal mixing probability 1/|Pj| in (3) assumes a priori that each parent has equal influence over αj. Parameter δ2 characterizes the variability among the children nodes. The joint prior of α is obtained by the product of (3) over j=2,…, J. This prior provides a mechanism to share information among the GO terms based on the DAG structure. It also naturally accommodates multiple inheritance.
We assign an inverse gamma prior, IG(aδ, bδ), on δ2, which has been used extensively in Bayesian models (Gelman et al., 2003). Similarly, an IG(aσ, bσ) prior is assumed for σ2. We infer the relevance of GO term Aj based on the posterior distribution of αj, denoted by [αj∣Y]. Here, Y={yi, i=1,…, I} is the collection of observations. Specifically, we use rj≡P(αj>0∣Y) (denoted as the B-score) to measure the relevance of a GO term to S. It is the posterior probability of Aj being positively associated with S. Making inferences based on posterior probabilities is a common practice in Bayesian analysis of microarray data (Newton et al. 2004; Do et al., 2005; Cao et al., 2009). Markov Chain Monte Carlo (MCMC) sampling algorithm is employed to simulate random samples from the joint posterior distribution. We implement the adaptive-rejection sampling method to take advantage of the log-concave property of the full conditional distributions (Gilks and Wild, 1992). The computation can be performed efficiently.
2.2 Demonstration of GO-Bayes with the artificial DAG
GO-Bayes is applied to the artificial DAG (Fig. 1) to detect overrepresentation of the terms. We present the B-score, rj, below the hypergeometric P-value. In the following discussion, we use association to refer to positive association between a GO term and S.
By incorporating the dependence structure of the GO DAG, GO-Bayes shows distinctive advantages over the hypergeometric test. First, GO-Bayes is capable of distinguishing terms with the same (IAj, nAj). For example, GO-Bayes produces r14=0.958 and r24=0.772, for A14 and A24, respectively, indicating that A14 is more likely to be associated with S based on the stronger evidence of overrepresentation of its neighboring nodes. Second, the B-score for all nodes has a range of 0–1 regardless of IA. Thus, GO-Bayes can identify more specific GO terms as long as their neighboring nodes are consistently overrepresented. For example, the top two terms selected by GO-Bayes are A12 and A14. Third, GO-Bayes also highlights underrepresentations as well as overrepresentations. In Figure 1, all the terms with nA=0 have the same P-value of 1.0. Under all the branches of A2, A3 and A4, there are terms with a P-value of 1.0. GO-Bayes suggests that it is the terms under A3, whose rj's are close to 0, that are most underrepresented in S.
We have also compared the GO-Bayes B-score with the elim P-value (Alexa et al., 2006) and the parent–child (union) P-value (Grossmann et al., 2007) based on the artificial DAG. Due to the space limit, the results are presented in Supplementary Figure 3. In general, both the elim method and the parent–child method address the ‘dependency problem’ caused by overlapping annotations between parent–child GO terms. The elim method removes (or downweights) all genes annotated to a significantly enriched node from all its ancestors. By doing this, the method tends to identify strongly overrepresented GO terms that remain significant even after discounting evidence from their offsprings. For illustrative purpose, we set the P-value cutoff at 0.05 for the elim method. Thus, A12 is considered significantly overrepresented and genes (a, b, c) are removed from its ancestor terms A5 and A2. Based on the elim P-value, the term A12 becomes the most significant, where A2 ranks second and A5 ranks sixth. The parent–child method computes the significance of a node conditional on the significance of its parents. It implements the idea by computing a hypergeometric P-value for each GO term in the context of its parent (treating the genes annotated by the parent term as the full gene list). This approach tends to identify GO terms that show stronger overrepresentation compared with their parents. Take A7 and A11, for example, which have the same (IA, nA). Based on the parent–child P−value, A11 has a higher rank (second) because its overrepresentation (2 out of 3) is stronger than its parent A4 (3 out of 11). In contrast, A7 has a lower rank (10th) because its overrepresentation (2 out of 3) is weaker compared with its parent A2 (8 out of 11). The GO-Bayes method accounts for the parent–child relationship through the hierarchical prior. The strategy of borrowing information from neighboring terms allows GO-Bayes to detect moderate but consistent signals from closely related GO terms. As a result, A7 has a higher rank than A11 (r7=0.802 and r11=0.397) because the overrepresentation in A7 is corroborated by related terms in its neighborhood. With the biological truth unknown, there is no gold standard to compare the methods in real studies (Grossmann et al., 2007). The results based on the artificial DAG, however, help to illustrate the distinctive characteristics of each method.
3 APPLICATION
3.1 Dataset
In order to test the utility of the GO-Bayes approach, we selected a gene expression microarray dataset in which a B-cell lymphoma cell line (Ramos) was stimulated either through the B-cell antigen receptor (BCR), CD40 or a combination of the two (Basso et al., 2005). Specifically, we used an Affymetrix gene expression dataset selected from the GSE2350 series (GSM44051 to GSM44074) downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/projects/geo/). The expression values of all six replicates under four experimental conditions were normalized by rows and a list of 3952 differentially expressed genes (I=3952) were selected using the Significance Analysis of Microarray (SAM) approach (Tusher et al., 2001). The differentially expressed genes were subdivided into 20 groups based on their expression pattern by K-means clustering using Euclidean distance as the similarity metric.
Gene Cluster #7 (GC7) was chosen for detailed analysis because it promised to reveal some interesting biology about B-cell responses to receptor signaling. The 196 genes (IS=196) present in GC7 showed a particularly interesting expression pattern: these genes were upregulated in response to BCR signaling alone; however, this upregulation was suppressed when CD40 signaling was included (Fig. 2). These treatment conditions mimic important biological responses of immature B cells (Hsueh and Scheuermann, 2000). Immature B cells must learn to distinguish between signals delivered by authentic pathogen-derived antigens and signals delivered by self-antigens. In the former case, B cells need to respond by productive proliferation and differentiation into immune effector cells. In the later case, B-cell responses need to be suppressed either through the induction of a state of unresponsiveness or apoptotic cell death. The two-signal hypothesis is one mechanism proposed to elicit either responsiveness or non-responsiveness to antigen exposure, which states that B cells receiving only one signal, through the BCR, will proliferate and die, but B cells receiving two signals both through the BCR and a co-stimulatory receptor-like CD40 will proliferate and survive due to suppression of the cell death response. Thus, gene present in GC7 are those genes whose expression is suppressed with the addition of CD40 signaling and could thus be involved either in the cell death response or in the induction of unresponsiveness.
Fig. 2.
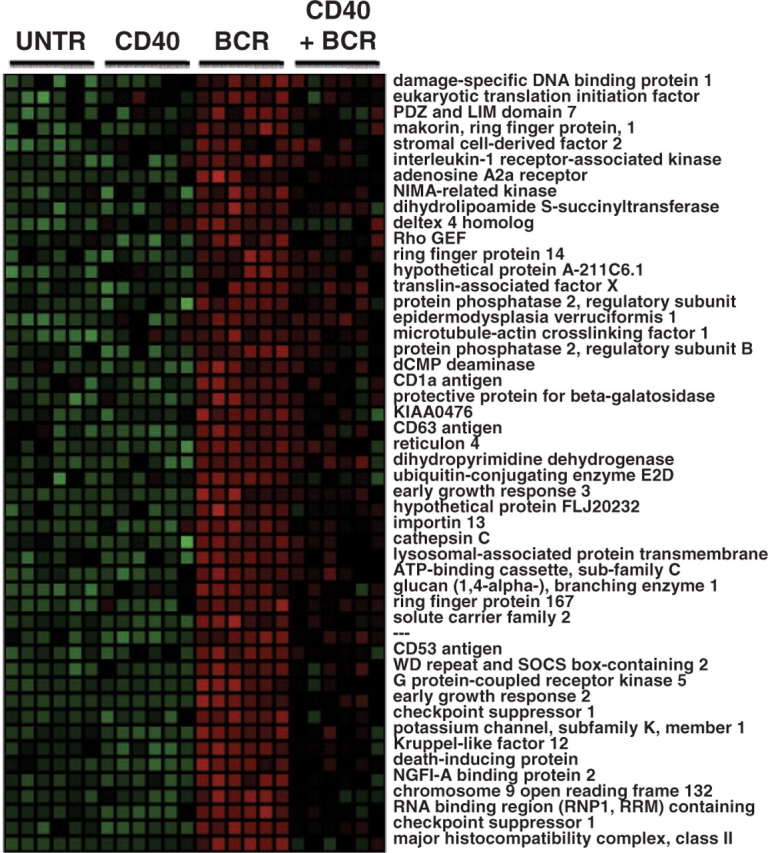
Gene expression pattern of Gene Cluster #7. A heat map of normalized expression values is shown in which green represents relatively low expression and red represents relatively high expression. Each column represents data from a single microarray. Six replicates from each of four experiment conditions were performed—untreated (UNTR), stimulation through the CD40 receptor (CD40), stimulation through the BCR and the combined stimulation. The expression pattern for a subset of 196 genes in GC#7 is shown.
3.2 Result
For the full list of 3952 differentially expressed genes (I=3952), 6768 GO terms (J=6768) have been used to annotate their functions. Four groups of GO terms are found in the top 20 list of most significant terms based on the GO-Bayes approach (Table 1). The GO term with the highest B-score (0.9004) is ‘transferase activity, transferring hexosyl groups’ (GO:0016758). Nine of the top 20 GO terms based on the B-score are related to this group. Five of the top 20 GO terms are related to ‘G-protein signaling, coupled to cAMP nucleotide second messenger’ (GO:0007188); six are related to ‘lysosome’ (GO:0005764); and two are in the group of ‘negative regulation of transcription, DNA-dependent’ (GO:0045892). In the case of the ‘transferase activity, transferring hexosyl groups’, all nine related terms (GO:0016758, GO:0016757, GO:0000030, GO:0003844, GO:0008375, GO:0015020, GO:0042328, GO:0004703, GO:0004674) can be found near GO:0016758 in the GO hierarchy (see Supplementary Fig. 4). Even though some of these terms are poorly represented in this dataset (e.g. GO:0003844 represented by only a single gene in the dataset), all of these terms have relatively high B-scores due to the overrepresentation of other terms that are close relatives in the hierarchy.
Table 1.
The top 20 lists GO terms associated with Gene Cluster #7 by CLASSIFI and GO-Bayes
| GO ID | IA | nA | P-value | RankP | B-score | RankB | Group | GO name |
|---|---|---|---|---|---|---|---|---|
| GO:0019933 | 21 | 6 | 4.00E-04 | 1 | 0.7652 | 149 | a | cAMP-mediated signaling |
| GO:0005773 | 91 | 13 | 4.61E-04 | 2 | 0.7544 | 187 | b | Vacuole |
| GO:0001726 | 23 | 6 | 6.85E-04 | 3 | 0.7724 | 118 | b | Ruffle |
| GO:0007188 | 16 | 5 | 7.95E-04 | 4 | 0.8688 | 7 | a | G-protein signaling, coupled to cAMP nucleotide second messenger |
| GO:0007190 | 10 | 4 | 9.73E-04 | 5 | 0.6928 | 508 | a | Activation of adenylate cyclase activity |
| GO:0045762 | 10 | 4 | 9.73E-04 | 5 | 0.6582 | 778 | a | Positive regulation of adenylate cyclase activity |
| GO:0031281 | 10 | 4 | 9.73E-04 | 5 | 0.4976 | 3148 | a | Positive regulation of cyclase activity |
| GO:0051349 | 10 | 4 | 9.73E-04 | 5 | 0.4712 | 3545 | a | Positive regulation of lyase activity |
| GO:0019935 | 25 | 6 | 1.11E-03 | 9 | 0.7568 | 177 | a | Cyclic-nucleotide-mediated signaling |
| GO:0007189 | 5 | 3 | 1.12E-03 | 10 | 0.8346 | 27 | a | G-protein signaling, adenylate cyclase activating pathway |
| GO:0016757 | 57 | 9 | 1.71E-03 | 11 | 0.8716 | 6 | c | Transferase activity, transferring glycosyl groups |
| GO:0000323 | 80 | 11 | 1.74E-03 | 12 | 0.8276 | 33 | b | Lytic vacuole |
| GO:0005764 | 80 | 11 | 1.74E-03 | 12 | 0.8762 | 5 | b | Lysosome |
| GO:0010324 | 69 | 10 | 1.87E-03 | 14 | 0.5084 | 2957 | b | Membrane invagination |
| GO:0006897 | 69 | 10 | 1.87E-03 | 14 | 0.5596 | 2015 | b | Endocytosis |
| GO:0007187 | 20 | 5 | 2.40E-03 | 16 | 0.8220 | 43 | a | G-protein signaling, coupled to cyclic nucleotide second messenger |
| GO:0006898 | 20 | 5 | 2.40E-03 | 16 | 0.5870 | 1601 | b | Receptor -mediated endocytosis |
| GO:0001609 | 2 | 2 | 2.45E-03 | 18 | 0.2706 | 6218 | a | Adenosine receptor activity, G-protein coupled |
| GO:0032230 | 2 | 2 | 2.45E-03 | 18 | 0.7562 | 178 | f | Positive regulation of synaptic transmission, GABAergic |
| GO:0048285 | 2 | 2 | 2.45E-03 | 18 | 0.5246 | 2625 | f | Organelle fission |
| GO:0007217 | 2 | 2 | 2.45E-03 | 18 | 0.8452 | 17 | a | Tachykinin signaling pathway |
| GO:0051319 | 2 | 2 | 2.45E-03 | 18 | 0.5124 | 2878 | f | G2-phase |
| GO:0015851 | 2 | 2 | 2.45E-03 | 18 | 0.3764 | 5103 | f | Nucleobase transport |
| GO:0016519 | 2 | 2 | 2.45E-03 | 18 | 0.1972 | 6600 | f | Gastric inhibitory peptide receptor activity |
| GO:0000085 | 2 | 2 | 2.45E-03 | 18 | 0.5650 | 1945 | f | G2-phase of mitotic cell cycle |
| GO:0016021 | 874 | 59 | 4.68E-03 | 31 | 0.8924 | 3 | a, b | Integral to membrane |
| GO:0005886 | 727 | 50 | 6.95E-03 | 36 | 0.8562 | 9 | a, b | Plasma membrane |
| GO:0004703 | 3 | 2 | 7.10E-03 | 40 | 0.8536 | 15 | c | G-protein-coupled receptor kinase activity |
| GO:0000030 | 4 | 2 | 1.37E-02 | 61 | 0.8556 | 11 | c | Mannosyltransferase activity |
| GO:0016758 | 41 | 6 | 1.45E-02 | 70 | 0.9004 | 1 | c | Transferase activity, transferring hexosyl groups |
| GO:0015020 | 5 | 2 | 2.22E-02 | 92 | 0.8538 | 14 | c | glucuronosyltransferase activity |
| GO:0045892 | 89 | 9 | 3.09E-02 | 104 | 0.8996 | 2 | d | Negative regulation of transcription, DNA dependent |
| GO:0015075 | 135 | 12 | 3.41E-02 | 115 | 0.8568 | 8 | b | Ion transmembrane transporter activity |
| GO:0003844 | 1 | 1 | 4.96E-02 | 281 | 0.8506 | 16 | c | 1,4-Alpha-glucan branching enzyme activity |
| GO:0022891 | 160 | 13 | 5.22E-02 | 289 | 0.8562 | 10 | b | Substrate -specific transmembrane transporter activity |
| GO:0042328 | 2 | 1 | 9.67E-02 | 471 | 0.8414 | 20 | c | Heparan sulfate N-acetylglucosaminyltransferase activity |
| GO:0000122 | 66 | 6 | 1.07E-01 | 491 | 0.8556 | 12 | d | Negative regulation of transcription from RNA polymerase II promoter |
| GO:0004674 | 200 | 14 | 1.18E-01 | 512 | 0.8550 | 13 | c | Protein serine/threonine kinase activity |
| GO:0008375 | 14 | 2 | 1.51E-01 | 620 | 0.8770 | 4 | c | Acetylglucosaminyltransferase activity |
| GO:0007186 | 101 | 7 | 2.33E-01 | 814 | 0.8430 | 19 | a | G-protein-coupled receptor protein signaling pathway |
| GO:0042629 | 2 | 0 | 1.0 | NA | 0.8432 | 18 | b | Mast cell granule |
The columns IA and nA represent the cardinality of the relevant GO term and the number of genes in the GO term that also appear in Gene Cluster #7; P-value and RankP represent the hypergeometric P-value computed by CLASSIFI, and the rank of the GO terms based on the P-value; and B-score and RankB represent the GO-Bayes measure and the corresponding rank of the GO terms. Under the column ‘Group’, ‘a’ represents GO terms closely related to GO:0007188 (G-protein signaling, coupled to cAMP nucleotide second messenger) in the GO hierarchy; ‘b’ represents GO terms closely related to GO:0005764 (lysosome); ‘c’ represents GO terms closely related to GO:0016757 (transferase activity, transferring glycosyl groups); ‘d’ represents GO terms closely related to GO:0045892 (negative regulation of transcription, DNA-dependent); and ‘f’ represents GO terms unrelated to the above four groups. NA, not applicable.
In some cases, the association of the group of GO terms highlighted by the GO-Bayes approach matches our expectations. It is well known that activation of B cells through the BCR induces endocytosis and the fusion of endocytic vesicles with lysosomes as a mechanism to capture antigen for presentation to T cells in order to stimulate the helper immune response (Lee et al., 2006). Thus, the presence of lysosome-related GO terms in the cluster of genes upregulated in response to BRC stimulation might be expected. In other cases, the association is not immediately expected, but subsequent investigations revealed that the association is supported by previous experiment data. For example, the majority of genes giving rise to the ‘negative regulation of transcription, DNA-dependent’ association, including ID3 (Pan et al., 1999), CTCF (Qi et al., 2003), SMAD3 (Ramesh et al., 2009), KLF12 (Roth et al., 2000), E2F6 (Xu et al., 2007), FosB (Yin et al., 2007) and PA2G4 (Zhang et al., 2008), have been found to be upregulated in response to BCR stimulation in B cells or somehow involved in B-cell signaling responses. In the case of CTCF, Qi et al. (2003) showed that this upregulation is associated with the induction of apoptosis in B cells and can be suppressed by co-stimulation through CD40 in agreement with the findings reported here. In still other cases, no direct corroborative evidence could be found (e.g. for ‘transferase activity, transferring hexosyl groups’). Thus, this finding serves as a hypothesis for future testing.
In order to compare the results of the GO-Bayes approach with the standard ORA based on the hypergeometric test, we processed the gene set in GC7 with the CLASSIFI algorithm (Lee et al., 2006) and selected the top 20 GO terms. The top 20 lists by CLASSIFI and GO-Bayes, respectively, are presented in Table 1. While four GO terms were ranked in the top 20 by both methods, the remaining top 20 terms from each method were distinct. Based on this comparison, several distinctions between the two approaches can be made.
First, three of the four GO term groups are found in both top 20 term lists. Two of the groups, G protein signaling and lysosome, have multiple terms in both top 20 lists. One group, transferase activity, is only represented once in the CLASSIFI list, but multiple times in the GO-Bayes list. Given that little could be found in the literature about these genes in B cell biology, this association would likely be ignored from the hypergeometric analysis. One group, the negative regulation of transcription group was not found in the hypergeometric top 20, and yet is potentially the most interesting given the strong literature support described above.
Second, eight GO terms in GC7 with (IA, nA)=(2, 2) have the same hypergeometric P-value of 0.0024, and all of them are in the hypergeometric top 20. The GO-Bayes measure suggests that they are very different in their association with the cluster. For example, GO:0007217 (tachykinin signaling pathway) has a B-score of 0.8452 and it ranks in the GO-Bayes top 20. In contrast, GO:0016519 (gastric inhibitory peptide receptor activity) has a B-score of 0.1972 and its GO-Bayes rank is 6600. To shed light on the difference in the B-scores between these two terms, we compare their regional DAGs, which are shown in Supplementary Figures 5 and 6. The three direct ancestors of GO:0007217 have a stronger association with the cluster than those of GO:0016519. In addition, GO:0007217 has nine siblings, five of which have genes represented in the cluster (nA>0). By comparison, GO:0016519 has 13 siblings, of which only one sibling has genes represented in the cluster. The support from the related GO terms is substantially higher for GO:0007217 than for GO:0016519, and thus GO:0007217 is judged to be more likely associated with the cluster based on the GO-Bayes measure. While there is no evidence for the involvement of the gastric inhibitory peptide receptor activity in the regulation of B-cell function in the literature, tachykinin (also known as hemokinin-1) has been found to be secreted during the differentiation of B-cell precursors thereby regulating their own development (Milne et al., 2004). Thus, GO:0007217 does appear to be biologically associated with GC7.
Among the top 20 GO terms identified by GO-Bayes, 8 GO terms have two genes or fewer belonging to GC7 (GO: 0007217, GO:0004703, GO:0000030, GO:0015020, GO:0003844, GO:42328, GO:0008375 and GO:0042629). Researchers may have concern over these findings, suspecting that they may be false positives. Based on the GO hierarchy, GO: 0007217 is a child of GO:0007186, which ranked seventh in the top 20 GO-Bayes list. The term is also found to be biologically associated with the experiment (Milne et al., 2004). Six of the GO terms (GO:0004703, GO:0000030, GO:0015020, GO:0003844, GO:42328 and GO:0008375) are all related to ‘transferase activity’, and all of them are in a close neighborhood in the GO DAG as shown in Supplementary Figure 4. As for GO:0042629, the fact that none of the two annotated genes were found in GC7 requires in-depth discussion on the term. Note that GO:0042629 is a direct child of ‘lysosome’ (GO:0005764), which ranked fifth in the top 20 GO-Bayes list. The two genes annotated with GO:0042629 are IL8 receptor beta (IL8RB) and serglycin (SRGN). IL8RB has been found to be involved in B-cell chemotaxis across the blood-brain barrier (Alter et al., 2003). Mice deficient for IL8RB show lymphadenopathy due to a specific expansion of the B-cell compartment (Cacalano et al., 1994). Importantly, IL8RB was found to be dramatically upregulated (∼9-fold) in response to BCR stimulation of primary splenic B cells by microarray analysis (Sato et al., 2005) (see IL8RB expression profile in http://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS1467). Although less is known about the function of SRGN in B cells, it was also found to be upregulated following BCR stimulation of B cells from TAK1-deficient mice in the same microarray experiment. Taken together, these findings suggest that the identification of GO:0042629 by GO-Bayes is significant and represents an example where the GO-Bayes approach was able to overcome false negative results generated by deficiencies in microarray data generation or processing. Thus, GO-Bayes will only identify a GO term with relatively few annotated genes belonging to S when there is strong evidence of overrepresentation from its neighboring GO terms.
4 DISCUSSION
In this article, we proposed GO-Bayes, a Bayesian approach for GO-based OR. The model has a relatively simple format, with the first level as a logistic regression model and straightforward prior specification. The key innovation is that it can incorporate the dependence structure of the GO DAG on a global scale. The resulting measure on the association between GO terms and selected genes borrows information across related GO terms to strengthen the detection of overrepresentation signals. We have given detailed comparison between the GO-Bayes approach and the hypergeometric test, which is widely used in ORA. Our analysis using an artificial dataset and a real microarray dataset suggests that the GO-Bayes approach can produce more biologically meaningful results than the hypergeometric test.
Relying on individual GO terms, the hypergeometric P-value has a closed form, which only requires the information on I, IS and (IA, nA) for each GO term. GO-Bayes, on the other hand, does not have a closed form and it needs the information on the structure of the GO DAG. We have developed a program in C to implement the GO-Bayes approach. For the B-cell lymphoma cell line example in the application section, the B-score calculations were completed in ∼10 min on a MAC (OS X 10.4.11, 1.83 GHz Intel Core Duo processor) computer. The program is available upon request from S.Z.
The GO-Bayes measure, rj≡P(αj>0∣Y), is the posterior probability of a GO term being positively associated with S. As a reviewer pointed out, rj behaves like a frequentist P-value under the null hypothesis (Bochkina and Richardson, 2007), and a frequentist-type estimator of false discovery rate (Storey, 2002) can be employed to assess the statistical significance.
Currently, the proposed method is based on binary outcomes (membership of genes in S). We are working on generalizing the GO-Bayes approach to cases with ordinal or continuous outcomes. Although we have focused on the use of this approach for the interpretation of gene expression microarray data and GO annotation, the general strategy can be applied to any circumstance in which groups of entities are annotated with terms derived from any ontology hierarchy or similar dependency structure.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the three reviewers for their constructive comments and suggestions.
Funding: U.S. National Institutes of Health (N01AI40076 and UL1 RR024982, in part).
Conflict of Interest: none declared.
REFERENCES
- Alexa A, et al. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. doi: 10.1093/bioinformatics/btl140. [DOI] [PubMed] [Google Scholar]
- Alter A, et al. Determinants of human B cell migration across brain endothelial cells. J. Immunol. 2003;170:4497–4505. doi: 10.4049/jimmunol.170.9.4497. [DOI] [PubMed] [Google Scholar]
- Al-Shahrour F, et al. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. doi: 10.1093/bioinformatics/btg455. [DOI] [PubMed] [Google Scholar]
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, et al. Reverse engineering of regulatory networks in human B cells. Nat. Genet. 2005;17:182–190. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Beissbarth T, Speed TP. GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics. 2004;20:1464–1465. doi: 10.1093/bioinformatics/bth088. [DOI] [PubMed] [Google Scholar]
- Bochkina N, Richardson S. Tail posterior probability for inference in pairwise and multiclass gene expression data. Biometrics. 2007;63:1117–1125. doi: 10.1111/j.1541-0420.2007.00807.x. [DOI] [PubMed] [Google Scholar]
- Cacalano G, et al. Neutrophil and B cell expansion in mice that lack the murine IL-8 receptor homolog. Science. 1994;265:682–684. doi: 10.1126/science.8036519. [DOI] [PubMed] [Google Scholar]
- Cao J, et al. Bayesian optimal discovery procedure for simultaneous significance testing. BMC Bioinformatics. 2009;10:5. doi: 10.1186/1471-2105-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, et al. Transcriptional regulation and function during the human cell cycle. Nat. Genet. 2001;27:48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- Do K, et al. A Bayesian mixture model for differential gene expression. Appl. Stat. 2005;54:627–644. [Google Scholar]
- Drăghici S, et al. Global functional profiling of gene expression. Genomics. 2003;81:98–104. doi: 10.1016/s0888-7543(02)00021-6. [DOI] [PubMed] [Google Scholar]
- Grossmann S, et al. Improved detection of overrepresentation of Gene-Ontology annotations with parent-child analysis. Bioinformatics. 2007;23:3024–3031. doi: 10.1093/bioinformatics/btm440. [DOI] [PubMed] [Google Scholar]
- Gelman A, et al. Bayesian Data Analysis. London: CRC Press; 2003. [Google Scholar]
- Gilks W, Wild P. Adaptive rejection sampling for Gibbs sampling. Appl. Stat. 1992;41:337–348. [Google Scholar]
- Hsueh R, Scheuermann RH. Tyrosine kinase activation in the growth, differentiation and death responses initiated from the B cell antigen receptor. Adv. Immunol. 2000;75:283–316. doi: 10.1016/s0065-2776(00)75007-3. [DOI] [PubMed] [Google Scholar]
- Khatri P, et al. Profiling gene expression using Onto-Express. Genomics. 2002;79:266–270. doi: 10.1006/geno.2002.6698. [DOI] [PubMed] [Google Scholar]
- Khatri P, Drăghici S. Ontological analysis of gene expression data: current tools, limitations, and open problems. Bioinformatics. 2005;21:3587–3595. doi: 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, et al. ErmineJ: tool for functional analysis of gene expression data sets. BMC Bioinformatics. 2005;6:269. doi: 10.1186/1471-2105-6-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JA, et al. Components of the antigen processing and presentation pathway revealed by gene expression microarray analysis following B cell antigen receptor (BCR) stimulation. BMC Bioinformatics. 2006;7:237. doi: 10.1186/1471-2105-7-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, et al. Modular organization of protein Interaction networks. Bioinformatics. 2007;23:207–214. doi: 10.1093/bioinformatics/btl562. [DOI] [PubMed] [Google Scholar]
- Lewin AM, Grieve IC. Grouping Gene Ontology terms to improve the assessment of gene set enrichment in microarray data. BMC Bioinformatics. 2006;7:426. doi: 10.1186/1471-2105-7-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne CD, et al. Mechanisms of selection mediated by interleukin-7, the preBCR, and hemokinin-1 during B-cell development. Immunol. Rev. 2004;197:75–88. doi: 10.1111/j.0105-2896.2004.0103.x. [DOI] [PubMed] [Google Scholar]
- Newton MA, et al. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biostatistics. 2004;4:155–176. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- Pan L, et al. Impaired Immune Responses and B-Cell Proliferation in Mice Lacking the Id3 Gene. Mol. Cell. Biol. 1999;19:5969–5980. doi: 10.1128/mcb.19.9.5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, et al. CTCF functions as a critical regulator of cell-cycle arrest and death after ligation of the B cell receptor on immature B cells. Proc. Natl Acad. Sci. USA. 2003;100:633–638. doi: 10.1073/pnas.0237127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh S, et al. Transforming growth factor β (TGFβ)-induced apoptosis. Cell Cycle. 2009;8:11–17. doi: 10.4161/cc.8.1.7291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth C, et al. Genomic structure and DNA binding properties of the human zinc finger transcriptional repressor AP-2rep (KLF12) Genomics. 2000;63:384–390. doi: 10.1006/geno.1999.6084. [DOI] [PubMed] [Google Scholar]
- Sato S, et al. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rate. J. R. Stat. Soc. Ser. B. 2002;64:479–498. [Google Scholar]
- Tusher VG, et al. Significance analysis of microarrays applied to transcriptional responses to ionizing radiation. Proc. Natl Acad. Sci. USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, et al. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals interchangeable roles of E2F family members. Genome Res. 2000;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q, et al. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2007;283:2654–2662. doi: 10.1074/jbc.M708218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. Alterations in cell growth and signaling in ErbB3 binding protein-1 (Ebp1) deficient mice. BMC Cell Biol. 2008;9:69. doi: 10.1186/1471-2121-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.