

Summary
Rheumatoid arthritis (RA) remains a significant unmet medical need despite significant therapeutic advances. The pathogenesis of RA is complex and includes many cell types, including T cells, B cells, and macrophages. Fibroblast-like synoviocytes (FLS) in the synovial intimal lining also play a key role by producing cytokines that perpetuate inflammation and proteases that contribute to cartilage destruction. Rheumatoid FLS develop a unique aggressive phenotype that increases invasiveness into the extracellular matrix and further exacerbates joint damage. Recent advances in understanding the biology of FLS, including their regulation regulate innate immune responses and activation of intracellular signaling mechanisms that control their behavior, provide novel insights into disease mechanisms. New agents that target FLS could potentially complement the current therapies without major deleterious effect on adaptive immune responses.
Keywords: cytokines, rheumatoid arthritis, signaling protein, inflammation
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune joint disease that affects approximately 1% of the population. The disease characteristically involves the small joints of the hands and feet, although the larger joint inflammation is also frequent. In addition to disability and decreased quality of life, RA decreases life expectancy, most commonly from accelerated atherosclerosis (1). The precise etiology of RA is not known, but genetic and environmental influences clearly participate. Dysregulated adaptive immunity can precede the clinical manifestation joint disease for many years, and it is likely that repeated activation of innate immunity can contribute to a breakdown of tolerance. RA is a heterogeneous disease, varying from slowly progressive symptoms or waxing and waning symptoms to severely destructive unrelenting disease associated with nodules and systemic inflammation.
The introduction of novel biological therapies in the mid-1990s markedly improved clinical outcomes in RA. Cytokine antagonists, such as biologic agents that inhibit TNF, IL-6, or IL-1, decrease inflammation and joint destruction but impressive efficacy is only seen in about half of patients. Similarly, B cell depletion and T cell co-stimulation blockers are beneficial in non- or partially overlapping subsets of patients. These data suggest that the genetic background and the environmental stimuli that help initiate disease determine whether a particular patient has TNF-dependent, B cell-dependent, or T cell-dependent disease. Other subsets undoubtedly exist and await the development of therapies that target other pathways or cell lineages, such as macrophages or dendritic cells (2).
Of the potential cellular participants in RA, synovial lining fibroblasts [also called fibroblast-like synoviocytes (FLS)] contribute to the local production of cytokines, small molecule mediators of inflammation, and proteolytic enzymes that degrade the extracellular matrix. Our understanding of FLS biology has provided significant insights into the pathogenesis of RA, including novel ideas related to an unusual aggressive phenotype that results from exposure to the toxic rheumatoid synovial environment. Targeting FLS might improve clinical outcomes in inflammatory arthritis without suppressing systemic immunity.
Normal synovium
The architecture of diarthrodial joints provides the infrastructure that supports mobility. The segmental nature of the skeleton in combination with low friction cartilage surfaces allows movement and flexibility. The synovium encapsulates the joints and serves its function by providing structural support (with a fibrous capsule), lubricates the surfaces, and provides nutrients to the cartilage. The joint lining is normally is a delicate membrane that divided into two anatomical and functional compartments: the intimal lining layer and the sublining layer. The former is the superficial layer of that is in contact with the intra-articular cavity and produces lubricious synovial fluid. Normally, it is two to three cells deep and is composed of two cell types in relatively equal proportions: Type A or macrophage-like synovial cells and Type B or fibroblasts like synoviocytes. The synovial lining lacks epithelial cells, a basement membrane, tight junctions or desmosomes. Instead, it is a loose association cells in a bed of extracellular matrix interspersed with collagen fibrils and other matrix proteins. The porous organization of the synovial lining allows diffusion of the nutrients in serum to the avascular cartilage. This architecture probably contributes to the accumulation of immune complexes, bacterial cell walls, and other phlogistic material in the joint (3).
Type A, macrophage-like synovial cells in the intimal lining express markers of hematopoietic origin most consistent with the monocyte–macrophage lineage and are derived from the bone marrow (4). They migrate to the synovium and become resident cells, although it is not certain if differentiation occurs in situ or prior to arrival. Their phenotype is similar to other tissue resident macrophage populations, including CD11b, CD68, CD14, CD163, class II major histocompatability antigens, and Fc Rγ. Electron microscopy documented presence of vacuoles suggesting phagocytic activity. Like other tissue macrophages, Type A cells are terminally differentiated with little capacity to proliferate (Table 1).
Table 1.
Selected markers expressed by macrophage-like and fibroblast-like synoviocytes
| Marker |
Macrophage-like (type A) synovicytes |
Fibroblast-like (type B) synovicytes |
||
|---|---|---|---|---|
| Common | CD16/CD | Immunoglobulin G Fc receptor | Type IV and V collagens | Structural protein 64 |
| CD45 | Leukocyte common antigen | Vimentin | Intermediate filament | |
| CD14 | LPS/LBP receptor | Thy-1 | CD90 | |
| MHC class II | Major histocompatibility complex Class II | MHC class II | Major histocompatibility complex class II | |
| CD11b/C D18 | Integrin adhesion molecule and complement receptor ‘Mac-1’ | ICAM-1 | Intercellular adhesion molecule-1 | |
| CD68 | Lysosomal glycoprotein | |||
| Relatively specific | UDPGD | Uridine diphosphoglucose- dehydrogenase | ||
| VCAM-1 | Vascular cell adhesion molecule-1, CD106 | |||
| DAF | Decay accelerating- factor, CD55 | |||
| Very specific | Cadherin-11 | Calcium-dependent adhesion molecule-11 | ||
Type B synoviocytes, or FLS, are mesenchymal cells that display many characteristics of fibroblasts, including expression of type IV and V collagens, vimentin, and CD90 (Thy-1). In addition, the type B cells have some unique properties in situ that distinguishes them from many other fibroblast lineages, including sublining resident fibroblasts (5, 6). For instance, cadherin-11 is a specific adhesion molecule that plays a key role in homotypic aggregation of FLS in vitro and in vivo (7). Cadherin-11 deficient mice develop normally but lack a defined synovial intimal lining in diarthrodial joints. As will be described in more detail below, these mice are relatively resistant to joint inflammation and destruction in arthritis models (8). Uridine diphosphoglucose dehydrogenase (UDPGD) is preferentially expressed by the intimal lining fibroblasts and reflects ability to synthesize hyaluronan, an important constituent of synovial fluid and extracellular matrix (ECM) (5). Intimal lining fibroblasts also secrete lubricin, another critical protein for joint lubrication. Vascular adhesion molecule 1 (VCAM-1), which normally mediates adhesion of leukocytes to vascular endothelium, is also expressed on synoviocytes but is only rarely observed on non-intimal lining mesenchymal cells. Its role in the tissue is uncertain, but could participate by binding mononuclear cells in the tissue or anchoring FLS to alternatively spliced fibronectin in the matrix. Integrins, such as alpha4/beta1, and integrin receptors, like intercellular adhesion molecule 1 (ICAM-1), are also expressed by type B synoviocytes, as is CD55 (decay accelerating factor). The latter is often used to identify type B cells in the intimal lining using immunohistochemistry (6, 9, 10) (Table 1).
Rheumatoid arthritis synovium
RA is a chronic immune-mediated disease marked by inflammation in the lining of the joint (i.e. the synovium) and destruction of cartilage and bone. The synovium in RA transforms from a quiescent relatively acellular structure to a hyperplastic, invasive tissue teeming with immunocompetent cells (Fig. 1). Synovial tissue has two layers, i.e. the intimal lining and the sublining, both of which undergo major changes in RA. In the sublining region, T lymphocytes constitute 30–50% of all cell types and majority is CD4+ CD45RO+ memory cells. A smaller number of CD8+ T cells are scattered in the synovium. Lymphoid aggregates organized around follicular dendritic cells are seen in 15–20% of patients (6, 11). About 5% of sublining synovial cells are B lymphocytes, and their clonal expansion in the joint suggests antigen-driven maturation. Local autoantibody production occurs in many patients, including rheumatoid factors, anti-citrullinated peptide antibodies, and anti-collagen antibodies. Surprisingly, neutrophils are rare in the rheumatoid synovium even though they are abundant in synovial effusions. Blood vessel proliferation is common because of the influence of angiogenesis factors in the inflamed, hypoxic environment. All of these histologic changes are evident very early in RA and can be observed within days to weeks after the onset of symptoms. Based on animal model studies, it is likely that subclinical synovial inflammation occurs even before patients develop pain in the affected joint.
Fig. 1. Histopathologic appearance of rheumatoid arthritis(RA) synovial tissue.
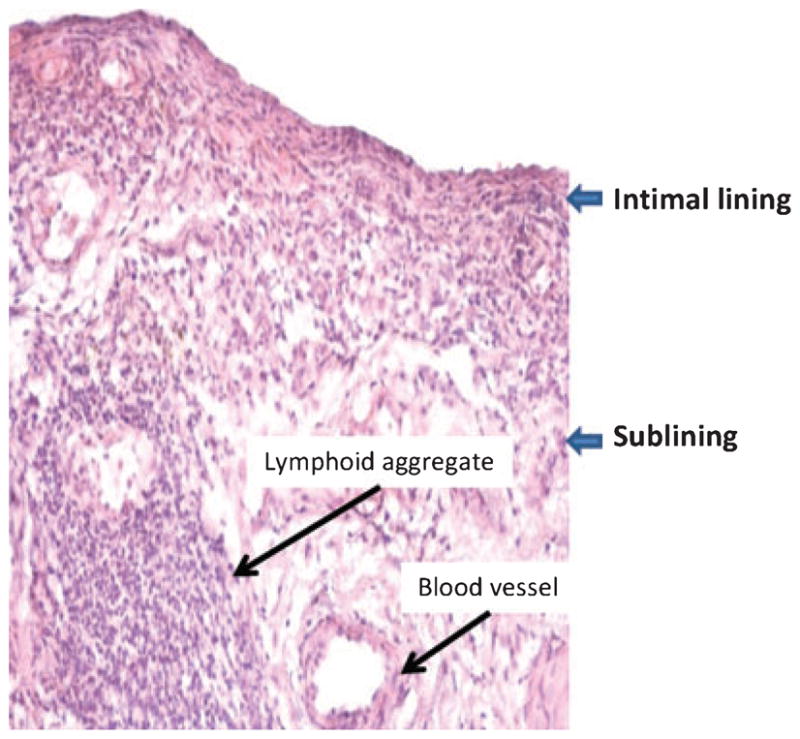
The synovium in RA is marked by intimal lining hyperplasia, infiltrating mononuclear cells in the sublining, and occasional lymphoid aggregates. From Reference (142).
The intimal lining of the synovium also displays remarkable changes in RA, with a marked increase in cellularity. The lining expands from 1–2 cells deep to a depth of up to 10–20 cells. Hypercellularity is due to an increase in both cell types present in this structure, namely type A macropahge-like cells and type B fibroblast-like cells (6). While studies vary considerably, many suggest that Type A cells predominate in RA because of migration of new cells from the bone marrow via the circulation. The macrophage-like cells display a highly activated phenotype and produce panoply of pro-inflammatory cytokines, chemokines, and growth factors. These products can activate local fibroblast-like synoviocytes (FLS) in the lining and induce the type B cells to produce their own array of mediators, especially IL-6, prostanoids, and matrix metallo-proteinases (MMPs). This process establishes a paracrine/autocrine network that can perpetuate synovitis, recruit new cells to the joint, and contribute to destruction of the extracellular matrix. Expansive synovial tissue called ‘pannus’ at the cartilage-bone interface cloaks the cartilage and erodes into bone. The pannus behaves like a locally invasive tumor; it is composed of macrophages, osteoclasts, and invasive FLS with relatively few lymphocytes.
The division of labor for destructive work of the synovium is now well delineated. Osteoclasts, which mature and are activated by macrophage colony stimulating factor (M-CSF), TNF, receptor activator of NFκB (RANKL) and other factors in the joint, erode into bone. FLS, on the other hand, are the primary effectors of cartilage destruction because of their unique invasive properties and the production of prodigious amount of proteases (6, 12). The concept that the cartilage and support structures of the joint are destroyed by FLS is supported by data in cadherin-11 knockout mice, which develop normally but lack an intimal lining. These synoviocyte-deficient mice are protected from arthritis-induced cartilage damage even though inflammation and bone damage progresses unabated (8).
Fibroblast-like synoviocytes and intimal lining hyperplasia
Origin of synoviocytes
The mechanisms of FLS accumulation in the intimal lining differ from the macrophage-like cells. The origin of the expanding synoviocyte population is uncertain, but could be due to migration of mesenchymal stem cells from the circulation or expansion of a stem cell pool in the synovium. FLS precursors could also migrate to the synovium through pores in cortical bone, as has been demonstrated in mice with collagen-induced arthritis (13). However, growth of the FLS population in RA is more likely because of an imbalance between cell proliferation, survival, and death. By and large, synovial intimal lining cell proliferation is difficult to demonstrate in RA. Limited numbers of mitotic figures or cells expressing cell cycle markers like Ki67 or proliferating cell nuclear antigen suggest that FLS DNA synthesis is not a major influence (6, 12).
Apoptosis in the synovial intimal lining
Instead, the synovial environment in RA promotes survival of FLS and discourages their deletion through apoptosis. Even though proliferation is difficult to demonstrate, completed apoptosis is extremely rare in the intimal lining. This interesting phenomenon occurs even though the rheumatoid synovium is highly genotoxic, with abundant reactive nitrogen and oxygen and cellular stress that normally kills cells. DNA strand breaks in the lining are plentiful, but this unexpectedly does not lead to removal of the damage synoviocytes as judged by TUNEL assays, histomorphometry, or electron microscopy (14, 15). Thus, considerable effort has been expended assessing potential mechanisms that protect RA synoviocytes from apoptosis in vivo.
Various studies have evaluated the relative expression of Bcl-2 family proteins in RA. The ratio of these anti- and pro-apoptotic molecules constitutes a rheostat that sets the threshold for susceptibility to programmed cell death in the intrinsic apoptosis pathway. Immunohistochemical analysis reveals modestly increased expression of anti-apoptotic proteins Bcl-2 and Mcl-1 (Bcl-2 family member) in RA synovium compared with OA. In some cases, Bcl-2 expression is concentrated in the lymphoid aggregates suggesting that it might protect synovial T and B cells but not intimal lining synoviocytes (16, 17). The apoptotic Bcl-2 family member Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) is also widely expressed in RA synovium and OA synovium with minimal expression in normal tissue (18).
The extrinsic pathway of apoptosis is activated by ligand-bound death receptors, including tumor necrosis factor receptor 1(TNF-R1), Fas/CD95 and TNF-related apoptosis-inducing-ligand receptor (TRAIL-R1 and -R2). Fas is especially noteworthy and is expressed in the synovial lining. However, as will be discussed below, engaging this molecule by Fas ligand (FasL) on synoviocytes only kills a small percentage of cells (19). Several influences, on the other hand, protect FLS from apoptosis. For instance, NF-κB, which is highly activated in RA and lining cells, provides a strong pro-survival signal and provides a link between inflammation and decreased apoptosis (20). Post-translational modification of signaling molecules involved in apoptosis by sentrin-1 through sumoylation also protects synoviocytes. Sentrin-1 expression is quite high in the rheumatoid intimal lining, especially at sites of cartilage invasion (21). Expression of phosphatase and tensin homolog (PTEN), a well characterized tumor suppressor gene that dephosphorylates phosphatidylinositide 3-kinase (PI3K) substrates, is very low in the synovial intima. The relative absence of PTEN leads ultimately to higher Akt phosphorylation and NF-κB activation, thereby favoring cell survival (22).
Of the tumor suppressor genes implicated in RA, the p53 tumor suppressor is especially important. This protein, sometimes called ‘the guardian of the genome’, is induced by cell damage, thereby leading to cell cycle arrest through p21 and subsequent repair. If the damage is substantial, p53 directs cells toward apoptosis via genes like p53-upregulated modulator of apoptosis (PUMA) and NOXA. p53 expression is elevated in the rheumatoid intimal lining, perhaps as a response to DNA strand breaks in a genotoxic environment (23, 24) (Fig. 2). The relative lack of apoptosis despite p53 induction led to the discovery of somatic mutations in the p53 gene in RA synovium (25). Microdissection studies showed that the mutations are primarily located in the intimal lining (26). The mutations are functionally relevant as demonstrated by their dominant negative activity in cell transfection experiments and the fact that the same mutations are present in many tumors (27). Therefore, the lack of functional p53 in intimal lining synoviocytes in situ eliminates a key regulatory step that impairs cell survival. As will be discussed below, the relatively inability of wildtype p53 to induce PUMA in synoviocytes also contributes to cell survival, providing another explanation for the lack of p53-induced FLS apoptosis. Some abnormalities in DNA mismatch repair enzyme expression contribute to the accumulation of mutations (28).
Fig. 2. p53 protein expression in synovial tissue.
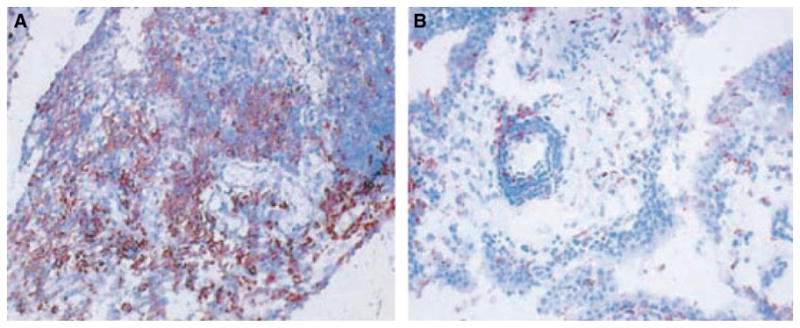
Immunohistochemistry was performed on synovial tissue from patients with (A) rheumatoid arthritis (RA) and (B) osteoarthritis (OA) to detect p53 protein. p53 was detected in the intimal lining and sublining infiltrating leukocytes. The p53 protein expression is significantly higher in the RA synovial tissue compared with OA synovium. From Reference (24).
Cultured fibroblast-like synoviocytes
FLS can be isolated from synovial tissue and grown in culture for prolonged periods of time. These cells, which are presumably derived from the intimal lining, serve as a useful tool for understanding the pathogenesis of RA and evaluating novel therapeutic targets. Enzymatic dispersion of synovial tissue yields a single cell suspension, and the cells are allowed to adhere to tissue culture dishes. The non-adherent cells are removed, leaving a mixture of two major cell populations that resemble the main cell populations found in the synovial lining. One cell type is macrophage-like, is phagocytic, and expresses DR antigens, Fc receptors and monocyte/macrophage markers CD14 and CD68. The second cell type is defined by the absence of phagocytic capability and macrophages markers and presence of antigens generally expressed by fibroblast (e.g. vimentin). The synovial macrophages are terminally differentiated with limited life span in vitro that rarely survive more than few weeks in culture. After three to four passages the proliferating FLS are the dominant cell type, resulting in a relatively homogenous population. These cells can be maintained for several months in vitro, with a doubling time of 5–7 days. After about 10–12 passages they senesce and proliferation gradually declines (29).
Origin of cultured FLS
The precise origin of FLS that overgrow synovial cell cultures is not certain, as they could potentially arise from the synovial lining, sublining or from other support structures of the joint included in the original sample. A significant percentage of the cultured fibroblasts express VCAM-1, CD55 and uridine diphosphoglucose dehydrogenase suggests that most of cells are derived from the intimal lining. In addition to traditional markers, cadherin-11 was identified as a relatively unique surface marker mainly present on intimal FLS (30). This adhesion molecule was identified as the major protein involved in homoaggregation of synoviocytes in vitro and in vivo. Moreover, caderin-11 can help recreate synovial architecture in vitro using artificial matrices to recapitulate a synovial intimal lining. After 3 weeks in ‘micromass’ cultures, FLS migrate to the surface and form a lining-layer-like structure at the interface between the matrix and fluid phase (Fig. 3). The structure resembles in vivo synovial lining, suggesting that FLS have intrinsically is capable to establish synovial architecture. This phenomenon is abrogated by specific antibodies and cadher-in-11 fusion proteins (31).
Fig. 3. Fibroblast-like synoviocytes (FLS) have intrinsic capacity to form a lining layer in vitro organ culture model.
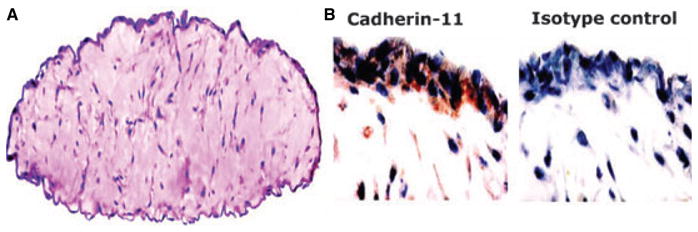
Cultured FLS were cultured in an artificial matrix and cultured for 3 weeks. The three-dimensional structure was fixed with 2% paraformaldehyde, embedded in paraffin and sections were stained with hematoxylin and eosin. FLS migrated to the surface of the matrix to establish a lining layer at the matrix-liquid interface (A). Immunohistochemistry was performed with cadherin-11 specific antibody and isotype control (B). Cadherin-11 specific staining was localized in the lining layer. From Reference (31).
Phenotype of FLS
FLS in cultures under light microscopy appear elongated, sometimes oval or polygonal with few branched cytoplasmic processes. When viewed by electron microscopy, FLS contain abundant rough endoplasmic reticulum and evidence of active secretory machinery. Cultured synovial fibroblasts spontaneously produce proteoglycans, cytokines, growth factors, MMPs, prostaglandins and other small molecule mediators during the first few weeks in culture. Production gradually decreases and FLS become fairly quiescent by the third passage. The RA phenotype can be restored by exposure to cytokines like IL-1 and TNF. Even long-term cultured FLS constitutively secrete some cytokines and growth factors, including IL-6, TGFβ, fibroblast growth factor without any additional stimuli. Adhesion molecules, including VCAM-1, ICAM-1 and integrins, are also constitutively expressed and display can be further enhanced by cytokines. IL-1, TNF, IFNγ, and IL-4 are the most potent stimuli for adhesion molecule induction (5, 6, 29).
FLS derived from RA synovium exhibit unique features with aggressive, invasive properties. They can grow under some anchorage free conditions and can escape contact inhibition like transformed cells (32). Unlike FLS from healthy individuals or patients with osteoarthritis, RA synoviocytes co-implanted with human cartilage into severe combined immune deficient (SCID) mice spontaneously invade and destroy the human cartilage (33). In terms of cell survival and growth RA synoviocytes appear more resistant than osteoarthritis FLS to apoptosis although their proliferation rates appear similar.
Aggressive phenotype of RA FLS
Rheumatoid synoviocytes display certain unique features that are reminiscent of transformed cells. Among these characteristics, the cells have inherent invasive qualities not observed in other fibroblasts. Initial descriptions of a tumor-like phenotype were made by Fassbender in the early 1980s, who noted the distinctive morphological features of RA FLS such as: abundant cytoplasm, large pale nuclei with several prominent nucleoli and a dense, rough endoplasmatic reticulum. He hypothesized that intimal fibroblast are able to invade articular cartilage even in the absence of immune cells due to these unusual properties (34). In vitro studies also suggest unusually aggressive behavior. However, it was not clear whether these features are inherent in RA FLS (transformed aggressors) or are somehow imprinted due to exposure to cytokines in the rheumatoid milieu in vivo (passive responders).
Cartilage implant model in SCID mice
The strongest evidence that the aggressive phenotype is not merely transient response to the environment comes from studies done in a SCID mouse model. As noted above, cultured human FLS devoid of other cell lineages after several passages were co-implanted with freshly isolated human cartilage into a SCID mouse under the renal capsule. RA FLS but not osteoarthritis (OA) or normal FLS (or dermal fibroblasts) adhere to and invade into the implanted cartilage after 2 months. The invading cells express several adhesion molecules, such as VCAM-1 and various integrins that facilitate attachment to cartilage and induce production of proteases that digest the extra-cellular matrix. The invading synoviocytes also influence adjacent chondrocytes in the implant to produce enzymes that degrade the local extracellular matrix (33). Thus, FLS are permanently altered and can orchestrate local destruction of the cartilage.
These observations lead to several studies to signaling pathways and the role of individual proteins responsible for the invasive properties. Using viral gene transfer, cultured RA FLS were transduced with a dominant negative mutant of c-raf to interfere with Ras-Raf-MAPK pathway. These transduced cells demonstrated decreased activation of the activator protein 1 (AP-1) pathway and MMP expression but not diminished invasiveness in the SCID model (35, 36). On the other hand, over-expression of IL-10 and IL-1Ra suppress invasion (37, 38). Only 40% of FLS express the tumor suppressor gene PTEN and expression in RA is mainly limited to the sublining regions, which could contribute to the invasive phenotype in SCID mice (12, 22).
Transformed phenotype
RA synoviocytes are not truly transformed, although they possess characteristics of tumor cells. For instance, a number of oncogenes involved in cell cycle regulation or that act as transcription factors such as c-fos, ras, raf, myc and myb are expressed at high levels in RA FLS. Abnormal cell growth probably occurs in vivo as well as suggested by presence of oligoclonality in synoviocytes derived from the invading pannus. These data suggest that certain FLS gain a growth advantage and can expand in the intimal lining. Increased telomerase activity, another feature of transformed cells, can also be observed in fibroblast growth factor stimulated RA synoviocytes (6, 19).
The mechanisms of permanently altered FLS function have also been explored. Some evidence implicates somatic mutations in key genes that regulate cell cycle, proliferation and apoptosis (27). The best characterized mutations have been documented in the p53 tumor suppressor gene, which have been described by several groups (39, 40). Inactivation of p53 by the human papilloma virus E6 protein in FLS also increases growth rate and suppresses apoptosis (41). Perhaps more important, p53 inactivation in normal FLS recapitulates the rheumatoid invasive phenotype in the cartilage-SCID mouse implantation model (42) (Fig. 4). Therefore, loss of p53 function can play a key role by permanently altering synoviocyte function.
Fig. 4. Effect of p53 deficiency on fibroblast-like synoviocytes (FLS) aggressive phenotype.

FLS derived from normal or rheumatoid synovial tissue were transduced with human papilloma virus type 18 (HPV-18) encoding E6 gene, which inactivates p53. The FLS were co-implanted with cartilage into SCID mice and harvested 60 days later. RA FLS (A) adhered to and invaded the matrix compared with normal FLS (C) which did not invade. Cartilage invasion was increased when RA FLS was transduced with HPV-18 encoding E6 gene (B). The normal FLS (C) acquired an RA-like invasive phenotype when p53 protein was inactivated with E6 (D). From Reference (42).
Function and mechanisms of somatic mutations
Abnormalities of the p53 gene have been long been implicated in numerous neoplastic diseases. The RA mutations observed in cultured rheumatoid synovicytes and synovial tissue are identical to the ones previously identified in tumors and tend to occur in the DNA binding domain of the protein. Most of the abnormalities are transition mutations, which are characteristic of damage induced by reactive oxygen or nitrogen oxide. Their functional relevance was confirmed in cell transfection studies in which rheumatoid mutant p53 genes were expressed in wildtype cells. These abnormal genes behaved in a dominant negative fashion by blocking the function of the normal allele and increasing production of MMPs and other genes associated with RA (27). Of interest, p53 deficiency increases disease severity and joint destruction in the collagen-induced arthritis model in mice but not in a passive model of arthritis that is independent of adaptive immunity. The effect in collagen-induced arthritis is associated with suppressed synovial apoptosis and increased cytokine gene expression. The lack of effect in the passive model suggests that influence of p53 on adaptive immune responses rather than innate immunity is more important in some models of RA (43–45).
Mutations in other genes have also been described in RA, probably because of the same mechanisms of oxidative damage. Abnormal ras genes, HPRT, and mutation in mitochondrial DNA have been described in RA (46–48). However, no mutations have been described in the PTEN gene, and the prevalence of mutations in p53 is similar in erosion and non-erosion sites in RA (49). Most recently a high rate of microsatellite instability was identified in RA and was much higher than in OA synovium (28). This phenomenon has been associated in cancer with abnormalities of the DNA mismatch repair (MMR) mechanism. Western blot analysis of cultured FLS shows that the key repair enzymes are expressed by the cells constitutively, including human MutS homologue (hMSH) 2, 3, and 6. When FLS are stressed with a nitric oxide donor, the pattern of enzyme expression was altered, with increased hMSH3 and decreased hMSH6. This pattern of MSH expression was also observed in RA synovium (28, 50). The high hMSH3 and low hMSH6 phenotype permits single base pair errors and protects against large deletions and insertions (51), which is characteristic of mutations observed in RA tissue and synoviocytes. Therefore, oxidative stress can relax the DNA MMR system and contribute to the burden of mutations.
Overall, the aggressive behavior of FLS in RA is probably multifactorial. Imprinting due to chronic cytokine exposure probably contributes, and somatic mutations in key genes also play a role. It is important to understand that the mutations in FLS do not cause RA; rather, they are the result of a longstanding genotoxic environment. Once mutations occur, then they can potentially alter the natural history of disease due to increased synoviocyte MMP and IL-6 expression, decreased apoptosis, and increased invasive potential.
Cartilage destruction in RA
Cartilage and bone destruction are the hallmarks of rheumatoid arthritis. Osteoclasts are major effectors of bone destruction, but recent data suggests that FLS primarily responsible for cartilage damage. In situ hybridization studies confirm that the intimal lining, especially type B synoviocytes, are the source of MMPs in this process. The ability of FLS to erode cartilage is a multistep process that includes attachment to cartilage and synthesis of enzymes that degrade the ECM (6).
Adhesion molecules and cartilage damage
Adhesion molecules, especially integrins like α5/β1, facilitate anchoring of RA fibroblasts to components in the cartilage ECM. They preferentially interact with fibronectin, collagen and cartilage oligomeric matrix proteins components. Integrins constitute an important family of transmembrane cell adhesion receptors and are composed of two heterodimeric glycoproteins. Several members of the β1 integrins are highly expressed by cultured FLS and they function as fibronectin receptors. Anti-β1 integrin antibody partially inhibits attachment of RA FLS to bovine cartilage in vitro. Integrin engagement also modulates several intracellular signaling pathways relevant in RA, including mitogen-activated protein kinases (MAPKs) and Ras. Importantly, integrin engagement with fibronectin-derived peptides induces expression of MMPs, indicating that the matrix can regulate its own destruction (52).
In addition to integrins, VCAM-1 and ICAM-1 are highly expressed and inducible in cultured FLS. These immunoglobu-lin supergene family members bind to α4/β1 integrin and β2 integrins, respectively. α4/β1 integrin also binds to the CS1 component of alternatively spliced fibronectin. While ICAM-1 expression and induction by cytokines has been described for many types of fibroblasts, constitutive VCAM-1 display is relatively unique to FLS. VCAM-1 has been implicated in the adherence of FLS to cartilage in the SCID mouse model, where it can mediate binding and potentiate MMP expression. VCAM-1 also serves to help mononuclear cells to cultured FLS in in vitro adhesion assays. Because neutrophils lack α4/β1 integrin, they are able to pass through the intimal lining unimpeded by FLS that display VCAM-1 (53). Cadherin-11, the adhesion molecule responsible for synovial lining formation, is also relevant in cartilage destruction although the exact mechanism has not been elucidated. In vitro cadherin-11 expression facilitated synovicytes invasion into cartilage-like extra-cellular matrix (54).
FLS from the intimal lining are considered major effectors of cartilage destruction in RA based on their ability to produce massive amounts of degradative enzymes. Among the different classes of proteinases released by the inflamed synovium, MMPs and cathepsins are especially important. These enzymes are constitutively expressed at high levels by early passage RA FLS and their production gradually declines to basal levels Cytokines like IL-1 and TNF rapidly increase their expression in vitro. Cultured FLS also constitutively express a family of proteins that specifically block MMP activity, called tissue inhibitors of metalloproteinases (TIMPs). Unlike MMPs their production is not induced by IL-1 or TNF but instead appears to be regulated by IL-6 and TGFβ. Cartilage destruction can occur when the balance of TIMPs and MMPs favor the latter. The cytokine network in RA favors high MMP:TIMP ratios as shown by in situ hybridization studies on intact synovium.
Matrix metalloproteinases
MMPs are structurally and functionally related enzymes and subdivided into five groups depending on their matrix protein substrates. Collagenases (MMP-1, MMP-13) and stromelysins (MMP-3) are especially important in RA. Their synthesis and activation is induced by various factors including pro-inflammatory cytokines, growth factors, matrix proteins like fibronectin, Toll-like receptor (TLR) ligands, and reactive oxygen species. Exposure to IL-1 and TNF rapidly induces MMP gene expression in cultured FLS. The T cell cytokine IL-17 also can contribute directly or indirectly by synergizing with IL-1 and TNF to increase MMP production (55).
MMP gene expression in FLS is mainly regulated at the transcriptional level. The key promoter element that controls expression of most MMPs binds to activator protein-1 (AP-1). AP-1 is a family of transcription factors, composed of structurally and functionally related members of the Jun proteins (c-Jun, Jun B, Jun D) and Fos (c-fos, Fos B, Fra1, Fra2). Additionally, some members of the ATF (ATF-1, ATF-2, and ATF-3) and JDP (JDP-1 and JDP-2) families share structural similarities and form heterodimeric complexes. AP-1 is highly expressed in the RA synovium, mainly in the intimal layer where MMPs are mainly synthesized, and DNA binding is significantly higher in RA compared with OA. Components to the AP-1 complex, c-Jun and c-Fos are also expressed both in the lining and sublining (20, 56). Interference with AP-1 decoy oligonucleotides or using c-Fos/AP-1 inhibitors decreased severity of collagen-induced arthritis accompanied by decreased inflammatory cytokine and MMP synthesis, thus confirming the critical role of AP-1 in joint inflammation (57, 58).
Stimulation of FLS with IL-1 or TNF in vitro increases MMP mRNA accumulation and this is associated with markedly higher AP-1 binding and transcriptional activity. The latter was demonstrated using promoter constructs that include AP-1 and a reporter gene like luciferase. c-Jun and c-Fos are the major components of AP-1 in the stimulated FLS that mediate this function. The promoters in MMP genes also contain NF-κB-like binding sites. This transcription factor can also increase MMP production and is induced in cytokine-stimulated FLS.
AP-1 activation includes several key steps: (i) phosphorylation, especially of c-Jun to increase transcriptional activity of the complex; (ii) migration of the complex to the nucleus where it binds to the promoter; and (iii) increased expression of components like c-Jun to increase AP-1 binding even further. Cytokine stimulation induces each of these steps, including marked increases in c-Jun and c-Fos expression in cultured FLS in parallel with increased MMP production. This serves as a positive feedback loop to further increase MMP expression over several days in cultured cells. Overexpression of JunD, however, has the opposite effect, suppressed MMP production perhaps competing with the Jun or Fos containing heterodimers (6).
Mitogen-activated protein (MAP) kinases, especially c-Jun N-terminal kinase (JNK), are responsible for phosphorylation and activation of c-Jun in the AP-1 complex after FLS are stimulated with cytokines, especially IL-1 (59). Other MAP kinases, like p38 and extracellular regulating kinase (ERK) also can activate c-Jun and AP-1 in cultured FLS, although their contribution is less than JNK. The critical role of JNK was confirmed using small molecule inhibitors and JNK knockout synoviocytes where MMP3 expression is significantly decreased compared to control FLS (59). The same small molecule inhibitor (SP600125) also decreases joint destruction in the rat adjuvant arthritis model and demonstrates the critical role of JNK in matrix degradation in vivo. Two upstream kinases in the MAP kinase cascade, MAPK kinase (MKK) 4 and MKK7 modulate JNK function in FLS. RNA interference studies show that only MKK7 is needed for IL-1, TNF, TLR2 and TLR4 ligand-mediated JNK activation and MMP expression in cultured RA FLS, however TLR3 requires both MKK4 and MKK7 (60, 61).
TLR4 and TLR3 ligand stimulation also increase MMP production in FLS, although alternative signaling pathways that involve both JNK and the inhibitor of NF-κB (IKK)-related kinase IKKε are involved. After TLR3 stimulation by the synthetic ligand poly (I:C), IKKε is activated and can then directly phosphorylate c-Jun in cultured FLS. The function appears to be independent of the interferon regulatory factors and could synergize with JNK by providing two parallel pathways for AP-1 activation (62, 63). This also provides a novel link between antiviral responses, innate immunity, and destruction of the extracellular matrix in RA because TLR3 ligands have been identified in rheumatoid synovial effusions (64).
MMPs can also be post-transcriptionally regulated in FLS through modulation of mRNA stability. This process is mediated by adenine/uridine-rich elements located in the 3′-UTR region and promote mRNA decay and can be suppressed by regulatory mechanisms. MMP-1, 3 and 9 mRNA expression can be increased through enhanced stability in response to IL-1, epidermal growth factor (EGF), phorbol ester, or LPS (65). In addition, adenosine 2b receptor agonists destabilize RNA transcripts in FLS to suppress MMP gene expression after IL-1 stimulation. This process is mediated by increased cAMP in FLS and can be mimicked by cAMP mimetics or forskolin (66).
Other proteases: cathepsins and aggrecanases
Like MMPs, the cathepsins are proteases with broad specificity and are regulated by cytokines and proto-oncogenes. For instance, Ras, IL-1 and TNF induce cathepsin L expression in cultured FLS. Adenoviral delivery of ribozyme that specifically cleaves mRNA for either cathepsin L or K decreases FLS invasion and cartilage destruction in the SCID mouse model. Cathepsin K appears to be the main mediator of destruction in this model (35).
Aggrecanases are key mediators of cartilage destruction and have been implicated in OA. Two aggrecanases (1 and 2, also called ADAMTS-4 and -5) remove glycosaminoglycans between Glu373 and Ala374 (compared with MMPs like stromelysin that cleave at Asn341 and Phe342). ADAMTS-5 deficiency ameliorates destruction in degenerative arthritis models in mice. While generally considered chondrocyte products, both aggrecanases are constitutively expressed by cultured FLS. While not regulated by traditional pro-inflammatory cytokines, ADAMTS-4 expression is induced by TGFβ. This growth factor is present in rheumatoid synovial effusions and synoviocytes in the intimal lining could contribute to the loss of proteoglycan in cartilage (67, 68).
Synoviocyte apoptosis
Synovial lining hyperplasia can be due to alterations in several homeostatic mechanisms, including cell ingress, egress, proliferation, and death. As cell division is uncommon in the joint, deficient synoviocyte apoptosis could cause local FLS accumulation. Programmed cell death is tightly regulated by a complex regulatory network to maintain homeostasis. Every step in the apoptotic cascade is monitored and controlled by pro- and anti-survival signals. Pro-apoptotic factors counteract the inhibitory molecules when cell deletion is imperative. Several factors affect this balance in RA although the exact mechanism of the deficient apoptosis is not completely understood. Initial studies suggesting defective programmed cell death in the lining indicated that DNA damage was not accompanied by completed apoptosis. Subsequent experiments using in vitro culture FLS derived from RA synovium helped dissect the complex cellular interactions that participate in vivo.
Some of the key pathways are described in this section. As will be seen, information on the role of individual pathways has generated considerable date. However, a unifying hypothesis of how FLS apoptosis is regulated remains elusive. Instead, compendiums of stimulus-specific functions are reported in the literature.
Caspases
Caspases are a family of proteases that are main effectors of apoptosis and are activated by receptor-mediated and mitochondria-mediated pathways. Engagement of cell surface death receptors lead to death-inducing signaling complex (DISC) and include FADD, caspase 3 and 8, and cFLIP, which is a negative regulator of caspase 8. This mechanism functions well in FLS, and previous studies have shown that caspase activation accompanies Fas-mediated and p53 pathway-mediated cell death. The intrinsic mitochondrial pathway can be triggered also independently by various signals such as genotoxic stress and withdrawal from growth factors. Bcl-2 is a major regulator of this pathway that inhibits caspases and protects mitochondrial integrity (69, 70).
Bcl-2 family
Bcl-2 is expressed in cultured RA FLS and is induced by TNF and IL-1, thereby protecting them from cell death in an inflammatory milieu. IL-15, a cytokine with pleiotropic effects on innate and adaptive immunity, increases Bcl-2 and Bcl-xL mRNA levels. IL-15 blockade increases apoptosis in FLS along with suppression of Bcl-2 expression (69, 71). Mcl-1, an anti-apoptotic member of the Bcl-2 family is also induced in RA FLS after cytokine stimulation. Mcl-1 knockdown induces apoptosis through the expression of Bax, Bak, and Bim (72). The role of Bcl-2 proteins in apoptosis after exposure to hypoxia was recently analyzed. One member of this family, the pro-apoptotic BNIP3 protein, is induced in RA FLS in response to a low oxygen environment. Its pro-apoptotic action was inhibited by TNF or IL-1 (18). This provides an additional link between inflammation and resistance to apoptosis in RA.
Receptor-mediated apoptosis: Fas, TNF, TNF-related apoptosis-inducing factor (TRAIL), and others
Fas, one of the best characterized member of death domain containing receptors, is constitutive expressed by cultured FLS. Anti-Fas antibody leads to RA FLS apoptosis, albeit only in about 15–20% of cells. Engaging this pathway is associated with JNK activation in FLS, with a concomitant increase in AP-1 binding. Some data suggest that OA FLS are even less susceptible to Fas induced death than RA cells. This difference is associated with lower JNK activation in OA FLS. However, pre-incubation of OA synoviocytes with TNF increases JNK activation and sensitizes the cells to Fas-induced death, recapitulating the RA phenotype (73, 74). When cultured RA FLS are exposed to recombinant LIGHT (another member of the TNF superfamily), Fas-mediated death is further reduced without affecting cell proliferation. LIGHT also increases MMP-9, IL-6 and ICAM-1 expression. Although the source of LIGHT in RA is not certain, its receptors HVEM and LTβ R are expressed in resting FLS (75). The decoy receptor DcR3 prevents RA FLS from Fas Ligand-induced cell death (76).
TNF can elicit a death-inducing signal through its receptor TNF-R1 but this function is masked in FLS by the strong survival signal from accompanying NF-κB activation. However, TNF can induce apoptosis in RA FLS when NF-κB is inhibited. The mechanism is thought to be through activation of the PI3K/Akt pathway, because PI3K inhibition potentiates TNF-induced apoptosis. TNF can also interfere with Fas/Fas ligand-mediated apoptosis by increasing release of soluble Fas.
Attention has recently been focused on potential therapeutic benefits of TRAIL because is preferentially toxic to cancer cells and does not affect normal cells at therapeutic doses. TRAIL interacts with five different receptors: TRAIL-R1/DR4, R2/DR5, R3/DcR1, R4/DcR2 and soluble osteoprotegrin. R1, R2 are main death mediating receptors while the others behave as decoy receptors. TRAIL or anti-DR5 antibody but not anti-DR4 induces apoptosis of a subset of RA FLS followed by proliferation of the survivors. This pleiotropic effect has been also described for other TNF superfamily members in cancer cells.
The molecular basis of TRAIL-triggered signaling pathway in RA FLS involves inhibiting PI3K/Akt signaling pathway. PI3K inhibition was associated with decreased expression of anti-apoptotic proteins Mcl-1, XIAP, and RIP and increased expression of the cell cycle inhibitor p21. Interestingly, caspase 8 is involved in TRAIL induced apoptosis and proliferation, as was demonstrated with siRNA knock down. Inhibition of apoptosis is not a prerequisite for TRAIL induced FLS proliferation. Caspases synergize with PI3K and/or MAPK mediated TRAIL-induced proliferation in RA FLS (77–81).
Adhesion molecule and FLS survival
Signaling pathways initiated by adhesion molecules faciliate cartilage destruction and also modulate apoptosis in vitro. Focal adhesion kinase (FAK) is a key kinase that participates in integrin signaling is a major substrate for the caspases implicated in cell death. A potential role of FAK in apoptosis is supported by the observation that RA FLS grown on fibronectin are more resistant to Fas Ligand-induced apoptosis then cells grown on poly-lysine. This activity requires α5/β1 integrin. The link between matrix degradation and the resistance of RA FLS to receptor-mediated apoptosis was further demonstrated by overexpression on tissue inhibitor of metalloproteinases TIMP3 in FLS. TIMP3 adenovirus gene transfer sensitizes RA FLS to Fas-induced apoptosis (12).
Intracellular signaling and oncogenes in FLS
In addition to external signals, modulations of downstream pathways have also been studied in RA FLS. Pro-caspase 8/FLICE like inhibitory protein (FLIP) is one component of the death-inducing signaling complex and can interfere with pro-caspase 8. FLIP expression is induced by TNF and requires NF-κB. Suppression of FLIP with siRNA sensitizes FLS to Fas-induced apoptosis (82). Post-translational modification of signaling proteins also plays a role in FLS apoptosis. SUMO-1/sentrin 1 is a small ubiquitin-like protein that changes the protein conformation of the affected targets instead of marking cells for degradation. SUMO-1 mediated modification protects cells from Fas and TNF R1 induced apoptosis. It is highly expressed in RA FLS and appears to protect from cell death. Recently data suggests that SUMO-1 does not directly interact with the death domain, instead modifies the nuclear promyelocytic leukemia (PML) protein, which traps the pro-apoptotic molecules such as DAXX in PML nuclear bodies (21, 83).
The relative paucity of apoptosis in RA also can be partially explained by the pattern of oncogene expression. Various studies have revealed high expression of immediate early genes in RA FLS, such as egr-1 and fos, as well as proto-oncogenes such as c-jun and c-myc. The high expression of Fos and Jun, which are involved in the formation of the AP-1 transcription factor, appears to be mediated through upstream oncogenes like ras, scr and raf. These oncogenes in turn activate MAP kinases and regulate expression of other proteins involved in apoptosis. Expression of tumor-suppressor genes like PTEN is also altered in RA. Only 40% of cultured FLS expressed PTEN, a phosphatase that is a negative regulator of PI3K/Akt, and its relative absence favors survival (22).
p53-mediated cell death
Aside from the presence of somatic mutations in RA FLS that improve cell survival, p53 also has surprisingly limited ability to induce apoptosis when the wildtype protein is overexpressed. Instead of inducing cell death, p53 preferentially induces p21 in FLS and directs cells to cell cycle arrest. The mechanism for this survival versus apoptosis decision is based on the limited ability of p53 to induce its primary mediator of apoptosis in FLS, namely PUMA (p53 upregulated mediator of apoptosis). Expression of PUMA is surprisingly low in RA FLS. PUMA overexpression, however, leads to rapid death of the majority of FLS through caspase 3 activation (Fig. 5). Therefore, gene transfer approaches to delete FLS should focus on PUMA or other p53 effectors rather than p53 itself (84, 85).
Fig. 5. Apoptosis of RA FLS induced by PUMA overexpression.

(A) RA FLS were transfected with pCEP4, hemagglutinin-tagged, full-length PUMA expression vector (HA-PUMA), or HA-tagged PUMA expression vector with a deletion of the Bcl-2 homology 3 domain (HA-PUMAdBH3). Data are presented as the percentage of nonviable cells. HA-PUMA-transfected FLS showed significantly more dead cells compared with pCEP4- or HA-PUM-AdBH3-transfected cells (*P < 0.01). (B) Comparison of apoptosis induced by pCEP4, HA-PUMA, or HA-PUMAdBH3 in RA and OA FLS lines. The extent of PUMA-induced apoptosis was the same in both cell lines. *P < 0.05 compared with controls. (C) RA FLS were transfected with pCEP4, HA-PUMA, or HA-PUMAdBH3 and DNA fragmentation, another measure of apoptosis, was measured by ELISA. Significant induction of DNA fragmentation was noted in HA-PUMA-transfected cells compared with pCEP4-transfected cells (*P < 0.05). (D) FLS transfected with pCEP4, HA-PUMA, or HA-PUMAdBH3 were cultured in chamber slides. The cells were then immunostained for activated caspase 3. HA-PUMA-transfected FLS showed significantly more activated caspase 3-positive cells compared with pCEP4- or HA-PUMAdBH3-transfected cells (P < 0.01). From reference (84).
Effect of FLS on survival of other cells
RA FLS also contribute to the accumulation of infiltrating T and B cells by regulating their apoptotic response through cell–cell interaction and/or soluble factors. For instance, T-cell resistance to apoptosis in vitro is prolonged in presence of cultured FLS. SDF-1α, the ligand of the chemokine receptor CXCR4, is produced by FLS and inhibits T-cell apoptosis through PI3K and MAPK pathways. In addition, RA FLS can function like follicular dendritic cells to support B-cell survival. B cells are protected from cell death when incubated with FLS through a mechanism that requires VCAM-1 and α4/β1 integrin. Reduced apoptosis is associated with increased expression of apoptosis inhibitors like Bcl-X (L). The B-cell survival factor BAFF is also produced by FLS in RA after engagement of α5 and β1 integrins on the cell surface (69, 86–88).
Synoviocytes as effectors of innate immunity
FLS are not just passive responders in the rheumatoid joint but also actively participate in synovial inflammation. They can contribute to initiation, propagation, and maintenance of chronic inflammation through cell–cell contact and through elaboration of soluble products. In response to environmental stimuli and interactions with various cell types in the inflamed synovium, FLS secrete a broad array of mediators: cytokines, chemokines, growth factors and several other proinflammatory molecules like prostaglandins and leukotrienes. The mechanisms of synovicyte activation are many, but include stimulation of Toll-like receptors (TLRs), exposure of cytokines and ligation of integrins by matrix molecules. TLRs are conserved pattern recognition molecules that recognize preserved structures of various pathogens and trigger an immediate inflammatory response, activate antigen-presenting cells and enhance adaptive immune response (89).
Toll-like receptors
FLS emerged as innate immunity effectors when several groups confirmed that they express TLRs and that ligation of these receptors increases expression of adhesion molecules, cytokines, and MMPs. For instance, RNA transcripts for TLRs 1 through 6 are expressed by cultured FLS but not TLRs 7, 8, 9, and 10. RA FLS express especially high levels of functional TLR2, TLR3, and TLR4 (90–92). In addition to exogenous bacterial peptidoglycan, several endogenous ligands such as heat shock protein, fibrinogen, and hyaluronan are commonly found in inflamed joint and can potentially activate cells through TLR2 and 4 (93). Double stranded RNA released from necrotic synovial fluid cells can activate RA FLS via TLR3. While TLRs are constitutively expressed, TLR2 expression can be increased by IL-1 and TNF (64).
Three key TLR ligands, namely LPS, peptidoglycan and double stranded RNA (poly (I:C), readily induce production of proinflammatory cytokines like IL-6 and MMPs by cultured FLS. Surprisingly, TLR3 stimulation, which is normally associated with interferon responses, is the potent stimulator of IL-6, MMP1, and MMP3. TLR2 activation in response to bacterial peptidoglycan also induces VEGF as well as various chemokines like GRL-2, MCP-2, RANTES, IL-8, and GCP-2. Stimulation of cultured synoviocytes with the TLR3 ligand poly (I:C) increases production of type I IFNs, IL-6, and MMPs (63). The TLR3 mechanisms are complex in FLS, but involve activation of the IKK-related kinase IKKε and TBK1 (see below), which then activate the interferon regulatory factors (IRFs), especially IRF3 (61).
Interferon regulatory factors and the IKK–related kinases
The relevance of the IRF family in RA is only been recently recognized in the setting of TLR-mediated responses. IRF3 is constitutively expressed in both RA and OA synovium. The active, phosphorylated form is predominantly expressed in RA along with phosphorylated IKKε (63, 64). IRF3 serves as a substrate for two upstream kinases TANK-binding kinase 1 (TBK1) and IKKε, which from a complex with NAK-associated protein 1 (NAP1). Upon phosphorylation by the IKK-related kinases (or JNK) and dimerization IRF3 translocates to the nucleus and in cooperation with other transcription factors like activating transcription factor 2 (ATF2)/c-Jun regulates type I IFN expression (Fig. 6).
Fig. 6. TLR-3 mediated MMPs and type I IFN gene expression in fibroblast-like synoviocytes (FLS).

MMPs gene expression: After TLR-3 receptor stimulation by synthetic ligand poly (I:C), two independent pathways are activated, MKK7/JNK and IKKε/TBK1. Both of these complexes phosphorylate c-Jun, leading to increased AP-1 activation. JNK-mediated c-Jun activation and AP-1-mediated gene expression of MMPs require MKK7. IKKε, independent of JNK can also phosphorylate c-Jun after forming a complex with TBK1 and NAP1. Type I IFN gene expression: Transcriptional activation of type I IFN genes require formation of an enhanceosome, an interaction between transcription factors ATF2/c-Jun and IRF3. In FLS, in response to TLR-3 activation, IRF3 serves as a substrate for both JNK and IKKε, followed by phosphorylation, dimerization and translocation to the nucleus. JNK-mediated IRF3 activation and IFN gene expression requires MKK7. MKK4 in complex with JNK also contributes to type I IFN gene expression through ATF2 phosphorylation and formation of ATF2/c-Jun complex. TLR3, Toll-like receptor; MKK4, MAPK kinase 4; MKK7, MAPK kinase 7; JNK, c-Jun N-terminal kinase; TBK1, TANK-binding kinase; NAP1, NAK-associated protein-1; IKKe, IKK-related kinase; IRF3, interferon regulatory factor 3; c-JUN, component of AP-1 transcription factor; ATF2, activated transcription factor 2; MMPs, matrix metalloproteinases; Type I IFNs, interferon α, β and interferon-stimulated genes.
In FLS, IKKε inhibition partially decreases IRF phosphorylation and IFNβ expression, suggesting that this kinase has a dominant role in some culture conditions. Additional data show that TBK1 also regulates a distinct set of genes (Deepa Hammaker and Gary S Firestein, unpublished data). In cultured synoviocytes, the TLR3 ligands activate IRF3 and increase production of IFNβ and other genes in the interferon response repertoire. LPS also stimulates the pathway through TLR4, but to a lesser degree. Surprisingly, TNF increases IRF3 phosphorylation and IRF3-mediated gene expression in FLS, a function that appears to be unique in synoviocytes. TRL3 signaling in FLS also activates ATF2/c-Jun via JNK and IKKε to increase MMP expression.
The pathways activated by TLRs intersect with other signaling cascades, including the MAP kinases. Peptidoglycan and LPS stimulation increase MMP expression through a JNK-dependent mechanism. Of the two JNK kinases (MKK4 and MKK7), only MKK7 is required for this activity. In contrast, TLR3 ligation depends on both MKK4 and MKK7 for induction of IP-10, RANTES, and IFNβ. There is also cross talk with the MAPKs, especially JNK that expands the cluster of genes regulated by IRF3 (61, 63).
Intracellular sensors of the innate immune system
Other pattern recognition receptors located in the cytoplasm rather than on the cell surface are involved in synoviocyte innate immune responses. These proteins belong to two major subfamilies: called NODs and NALPs. The NALPs form a structure called ‘inflammasome’ that responds to various danger signals like uric acid crystals or pathogen components with production of IL-1 by engaging caspase 1. Mutations in these proteins have been associated with various autoinflammatory disorders. Cryopyrin, a key member of the NALP family, is expressed in RA synovium and cultured FLS. Expression of cryopyrin in cultured FLS is highly inducible by TNF (94).
NOD2 recognizes muramyl dipeptide and peptidoglycan derived peptide from Gram-negative and Gram-positive bacteria. NODs have been implicated in inflammatory bowel disease and they are expressed on intestinal antigen presenting and epithelial cells. NOD-2 is also expressed in the RA synovium, predominantly at sites of cartilage invasion. In cultured FLS, NOD-2 is induced by poly (I:C), LPS or TNF. After NOD2 expression is increased, stimulation with its ligand muramyl dipeptide increases production of IL-6, IL-8, and MMPs. Although the role of these mechanisms in RA is not well defined, their ability to induce cytokines and chemokines suggests that they might contribute to the proinflammatory environment (95).
Synoviocytes and cell recruitment
Recruitment and retention of inflammatory cells is another fundamental feature of synovitis. This process is coordinated by presence of chemoattractant proteins at the site of inflammation assisted by expression of adhesion molecules. Chemokines and other small chemoattractant molecules are abundant in the RA synovium and can be produced by macrophages and synovial fibroblasts in the intimal lining.
Chemokine production by synoviocytes
The chemokine system includes numerous chemoattractants and G-protein coupled receptors. Although there is redundancy in the system, the specific chemokine–chemokine receptor interaction can define the phenotype of the cells recruited to the site of inflammation. Regulation of chemokine synthesis in synoviocytes contributes to the cellular composition of the rheumatoid synovium. For instance, T cell that constitute about 30–50% of cells in RA synovium are most commonly CD4+ with Th1 phenotype (6). This is, in part is related to highly expressed CXCR3 and CCR5 on T cell and presence of MIP-1α and RANTES in the synovium (both of which are produced by cultured synoviocytes). SDF-1 and CXCL16 produced by synovial fibroblasts also help retain CD4+ T memory T cells in the inflamed joint. CXCR4, the receptor specific for SDF-1 is mainly present on memory CD4+ T cells and expression is enhanced by inflammatory cytokine IL-15 (96–100).
Synoviocytes produce an array of chemokines that signal cells to migrate into the joint and can, in some cases, direct angiogenesis. IL-8 is a C-X-C chemokine originally characterized as a potent neutrophil attractant that also stimulates blood vessel growth. Cultured macrophages constitutively produce IL-8, but IL-1 and TNF markedly increase its production by cultured FLS. This, along with numerous other chemokines, including RANTES, IP-10, ENA-78, and MCP-1, MIP-1α, and MIP-1β are major monocyte/macrophage recruiting proteins that are produced by FLS in response to IL-1 or TNF stimulation. Release of these chemoattractants contributes to the cell mix that is subsequently recruited to the inflamed joint (12). Their biological relevance has been confirmed by in vitro studies using synovial fluid to induce cell migration. Antibodies to these and other chemokines neutralize much of this activity (101). The chemokine fractalkine expressed on FLS can also activate CD4+ CD28− memory T cells, which are expanded in RA blood and in the synovium. Fractalkine can also serve an autocrine function and stimulate synoviocyte chemotaxis (101–103).
In addition to cytokine-mediated chemokine production, TLR stimulation increases chemokine expression by FLS. TLR2 and 4 signaling are mediated through the MyD88 pathway and activation of NF-κB, which is especially important for IL-8 (104). However, other chemokines are largely driven by the interferon pathway. As noted above, the IKK-related kinases and IRF3 can contribute to cytokine and MMP expression. Chemokine expression is also an important part of this repertoire. Thus, TLR3 ligands, which are potent inducers of ‘anti-viral’ responses, also provide signals to draw cells into the synovium such as production of IP-10 and RANTES (61, 63).
While the chemokine system is an attractive target, specific agents that block their function, such as anti-MCP-1 antibody and small molecule inhibitors of the MCP-1 receptor (CCR2) or the MIP-1/RANTES receptor (CCR1) have been unsuccessful in RA. Thus, the role of these chemokines in the pathogenesis of disease remains uncertain. Alternative strategies, such as interfering with numerous chemokines by virtue of their intracellular signaling mechanisms (e.g. PI3 kinase γ) might be a solution if the lack of efficacy is due to a high level of redundancy in the system (105–107).
New blood vessel formation
Functions mediated by the chemokine system are not limited to cell trafficking. The distinct signaling pathways like PI3K/Akt coupled to G-protein coupled receptors are involved in cell proliferation, survival, and regulation of apoptosis. In addition, some of the chemokines present in the RA are also involved in synovial vasculogenesis and angiogenesis. Neovascularization is required to support the proliferating synovium. In conjunction with cytokines and various growth factors, chemokines contribute to both the angiogenic and angiostatic side of the balance. For instance VEGF, IL-8, and SDF-1 produced by FLS are inducers of angiogenesis while IP-10 inhibits neovascularization. Soluble VCAM, which is released by activated synoviocytes, also has pro-angiogenic properties. Therefore, the combination of increased blood vessel proliferation, greater adhesion molecule expression, and chemokine production serve to attract new cells to the inflamed joint (108).
Synoviocytes and cytokine networks
Characterization of the cytokine profile in RA and defining the contribution of cytokine networks to disease perpetuation were major breakthroughs in our understanding of disease pathogenesis. Detailed studies of the rheumatoid cytokine milieu shows that it is dominated by macrophage and fibroblast derived proinflammatory cytokines. Activated macrophages and fibroblast in the intimal layer interact with each other through secretion of mediators in a paracrine and autocrine fashion. This positive feedback loop leads to further activation of intimal lining cells, recruitment of new cells to the joint and increased production of inflammatory mediators. Cytokines like IFNγ produced by Th1 cells in the joint were identified in synovial effusions albeit at low concentration compared with TNF, IL-1, IL-6 and others derived from macrophages and synoviocytes. Cytokines were initially proposed as self-perpetuating feedback loops independent of the T cell. Later, this model was updated as the role of T cells re-emerged as major regulator of the innate immune response. The recognition that IL-17 and Th17 cells can contribute to synovitis and the demonstration that abatacept (which inhibits T-cell co-stimulation) is effective in RA provides new evidence of the complexity of adaptive immune responses in this disease (2, 109).
Synoviocyte-derived cytokines
Of the classical cytokines implicated in RA, most are derived from macrophages. However, FLS in the intimal lining are the primary source of IL-6 as shown by in situ hybridization and immunohistochemistry studies. Thus, the critical role of synoviocytes in the pathogenesis of disease is underscored by recent studies showing that IL-6 inhibition dramatically decreases signs and symptoms of RA. Cultured FLS spontaneously produce IL-6, and its production is markedly increased by IL-1 or TNF through an NF-κB-dependent pathway. MAP kinases also contribute to IL-6 production by cytokine-activated FLS, mainly through post-transcriptional mechanisms. Other newly described cytokines implicated in RA, the IL-1 family members IL-18 and IL-33 and the novel cytokine IL-32, can also be produced by cytokine stimulated FLS (109).
Colony-stimulating factors in the joint also appear to be derived largely from FLS in RA. These growth factors were originally identified by virtue of their ability to increase lineage specific maturation of hematopoietic stem cells in the bone marrow. However, it is now clear that they play a key role in dendritic cell and macrophage activation. Granulocyte macrophage colony-stimulating factor (GM-CSF) and M-CSF are especially abundant in RA synovium and synovial effusions. In situ hybridization studies showed that they are produced mainly by intimal lining cells. Cultured FLS that have been activated by IL-1 or TNF markedly increase production of GM-CSF, which could contribute to the local expansion and activation of macrophages. GM-CSF, rather than IFNγ, is the primary macrophage activating factor in the rheumatoid joint and is responsible for induction of class II MHC on macrophage lineage cells (110).
Cultured FLS also are a major source of type I interferons in RA synovium. The role of type I IFNs in this disease is dichotomous. On the one hand, the pathway is pro-inflammatory due to the release of chemokines and metalloproteinases in cultured synoviocytes. On the other hand, IFNβ itself is generally anti-inflammatory in models of arthritis. Clinical trials using IFNβ in RA did not demonstrate efficacy, but the dose was limited because of side effects (111). An alternative strategy in animal models showed that IKKε deficiency in combination with low dose ‘replacement’ IFNβ was as effective as high dose IFNβ in wildtype mice. The benefit of IFNβ was associated with increased IL-1Ra production in mice. Thus, dissecting the cytokine pathway in FLS led to the discovery that the pro- and anti-inflammatory effects of interferon could potentially be resolved with a combination therapy approach (112).
FLS can also modulate the synovial cytokine profile through direct cell–cell contact. Cultured macrophages and fibroblasts produce abundant inflammatory mediators when are stimulated by fixed T cells or membrane derived from activated T cells. The exact cell surface proteins responsible for this mechanism are not known but integrins have been implicated. Pretreatment of FLS with IFNγ to induce MHC II expression confers the ability to stimulate T cells in the presence of superantigens. The activated lymphocytes can then produce cytokines that contribute to the pro-inflammatory milieu (113).
Anti-inflammatory cytokines
Synoviocytes also have the capacity to produce anti-inflammatory cytokines and factors that can potentially suppress synovitis. As noted above, cultured FLS produce IFNβ especially after they are stimulated with TLR3 ligands. They also can serve as a source of TGFβ, which suppresses protease and pro-inflammatory cytokine production. IL-1Ra is a natural protein that competes with IL-1 for the IL-1 receptors. IL-1Ra has two splice forms: one is secreted and one remains intracellular. Cultured FLS express substantial amounts of IL-1Ra; however, it is predominantly the intracellular form. Therefore, it is unable to interfere with IL-1 in the synovial extracellular space, a phenomenon that is permissive for a more robust IL-1-mediated effect in RA (114).
Microarray analysis of gene expression patterns
A global approach to cytokine and growth factor gene expression using microarray analysis identified two main subgroups of FLS grown out of synovial tissue based on their gene expression profiles. One group showed TGFβ/activin A-inducible gene signature characteristic of myofibroblasts, and the other reminiscent of insulin-like growth factor regulated genes. There was a correlation between the cytokine expression profile of the cultured cells and the level of synovial inflammation. FLS cell lines with the TGFβ/myofibroblast phenotype were mainly derived from synovial tissue with characterized as highly inflammatory while the other group originated from tissue with low inflammation (115). These data strongly suggest that the phenotype of cells in the synovium can persist for prolonged periods of time in culture, reflect the cytokine network for individual patients, and can potentially predict clinical outcomes.
Signal transduction in synoviocytes
Targeted anti-cytokine therapies have a major impact on clinical symptoms of RA as well as destruction of the extracellular matrix. Nevertheless, there is still a significant unmet medical need because of the cost of these agents and the fact that many patients do not have a robust response. As an alternative to biologics that inhibit individual cytokines, understanding the mechanisms that regulate their production and function might be useful because they affect pathways in concert. Dissecting the signaling mechanisms that participate in this process has identified a cornucopia of targets important to synoviocyte function and the pathogenesis of RA. A variety of signaling molecules relevant to RA and synoviocyte biology have been discussed in the previous sections, such as IRFs and AP-1. Some additional key pathways implicated in the mechanisms of RA and can potentially serve as therapeutic targets are described below.
Mitogen-activated protein kinases
MAPKs are family of evolutionary conserved signaling molecules that control cellular functions from metabolism, proliferation, apoptosis, cell differentiation to various stress responses and immune function. They include a group of kinases that govern many aspects of host defense, including innate and adaptive immunity. Role of MAPKs has been explored extensively in RA because they participate in cellular responses to cytokines, TLR ligands and control expression of various genes relevant in chronic inflammation, including cytokines, chemokines, and MMPs.
There are three well-defined MAPK pathways: the extra-cellular signal-regulated kinase, the c-Jun N-terminal kinase (JNK), and the p38. The activation pathway of MAPKs is relatively conserved and involves several key steps. Following receptor activation at the membrane, members of the small GTPase of the ρ family are recruited followed by interaction with MAPK kinase kinases (MAP3Ks), which leads to activation of the next tier of kinases, namely MAP2Ks (also called MKKs). The MAPKs themselves are then activated by the MKKs and complete the cascade by phosphorylating their substrates, such as c-Jun, ATF2, and many others. Signal propagation is accomplished by serial phosphorylation events and terminated by protein phosphatases that remove phosphates from the kinases. The individual kinase modules include the MAP3K, MKK, and MAPK along with regulatory proteins. Each MAPK has several unique and also overlapping functions. ERK has a predominant role in regulating mitotic responses and cell growth and differentiation while p38 and JNK are the main responders to various stress signals, including inflammation, heat, and genotoxic events (116).
Because MAPKs regulate expression of inflammatory mediators like IL-1, TNF, IL-6, and MMPs, the concept that they play a pivotal role in RA pathogenesis is not surprising. ERK, JNK and p38 are constitutively expressed in RA and OA synovium but expression of the activated phosphorylated forms is much higher in RA. Similarly ERK, JNK and p38 are also constitutively expressed in cultured synoviocytes, although in low serum medium that mainly exist in the unphosphorylated state. After stimulation with TNF, other pro-inflammatory cytokines and TLR ligands, the dual phosphorylated kinases can be translocated to the nucleus and can alter gene transcription or mRNA stability (117, 118).
p38 MAPK has generated considerable interest as a potential therapeutic target because (119, 120):
It is activated in the joints of patients with RA; p38 inhibitors are effective in animal models of arthritis; p38 blockade decreases cytokine production by cultured RA synovial tissue cells and cultured synoviocytes in vitro; and p38 blockade decreases in vivo cytokine responses in humans exposed to LPS.
Despite these observations, numerous p38 inhibitors have been tested in RA and the benefits are modest (see below).
Upstream activators of p38, such as MKK3 and MKK6, are being explored as an alternative to p38. Cultured synoviocytes constitutively express MKK3 and MKK6, and both are phosphorylated in IL-1 and TNF stimulated synoviocytes (121). Studies transfecting FLS with dominant negative MKK3 or MKK6 constructs show that MKK3 blockade has a greater suppressive effect on IL-6, IL-8, and MMP3 production (122). The potential utility of MKK blockade in vivo is supported by studies in MKK3−/− and MKK6−/− mice, where passive K/BxN arthritis severity is suppressed if either kinase is deficient (121, 123).
MKK3 and MKK6 are not redundant and regulate distinct genes. For instance, TNF and LPS induced expression of MIP1α and KC (IL-8 homolog) is MKK6 dependent. However, TNF-induced RANTES is more dependent on MKK3 (123).
Each of the MAPKs exists as multiple isoforms. Among the three different JNK isoforms, JNK2 expression is the highest in cultured FLS. In vivo the other isoforms might be relevant because JNK2 deficient mice had only modest decreased cartilage destruction. More recently, studies in knockout mice show that deficiency of the JNK1 isoform decreases severity in some passive arthritis models. In contrast, lack of JNK1 has no effect in TNF transgenic mice (124). Therefore, the relative importance of JNK1 and JNK2 in RA and in synoviocytes remains uncertain.
JNK is differentially regulated by its upstream kinases MKK4 and MKK7, both of which are activated in RA synovium depending on the stimulus (125). Overall, MKK7 is responsible for most of the pro-inflammatory activities of JNK in FLS (60). Of the MAP3Ks that regulate JNK, MEKK1, MEKK2, and TAK1 are the most abundant in RA synovium and RA synoviocytes. MEKK2 was initially identified as a major MAP3K regulating IL-1 stimulated JNK based on in vitro kinase assays and expression levels (126). However, siRNA knock down unexpectedly showed that MEKK1, MEKK2, MEKK3 deficiency does not alter IL-1 induced JNK, p38 or ERK activation. The MAP3K TGFβ-activated kinase (TAK1) is the major regulator of IL-1 induced JNK activation in FLS (127). TAK1 siRNA inhibits JNK, MKK4, MKK7 phosphorylation which leads to decreased AP-1 binding. As expected MMP3 expression was significantly decreased in TAK1 deficient cells in response to IL-1.
Endogenous MAPK inhibitors
Mechanisms contributing to sustained activation of MAPK in RA are less understood. There are several known negative regulators and regulatory mechanisms of unclear relevance to RA. In the recent years, a large family of dual-specificity phosphatases has been described as regulators of the MAPK. MKP-1, which is one of these phosphatases, is induced IL-1 and its expression is further enhanced in presence of glucocorticoids (128). Another important negative regulator of the JNK pathway, Growth arrest and DNA-damage-inducible, beta protein (Gadd45β), was recently characterized both in animal model of RA and in cultured synoviocytes. Gadd45β is a NF-κB-inducible gene, but its expression is surprisingly low in RA synovium despite abundant NF-κB levels. Similarly TNF and IL-1 stimulation, which activate the NF-κB pathway, only induces a modest transient expression of Gadd45β cultured synoviocytes (129).
These observations raised the possibility that deficient Gadd45β expression might contribute to persistent JNK activation in RA. Recent studies show that Gadd45β can bind to and inhibit MKK7 function and thereby prevents JNK phosphorylation. As MKK7 is the most important upstream kinase in the JNK pathway, its relative absence could have significant deleterious effects. To test this hypothesis, cultured FLS were transfected with the Gadd45β cDNA, which then suppressed JNK/AP-1 regulated MMP production in MKK7 dependent manner. In vivo studies in Gadd45β knockout mice confirmed that the lack of this regulatory gene increased arthritis severity in the passive K/BxN model and increased JNK-dependent joint destruction. These data suggest that increased JNK activation in RA synovium could be due to insufficient Gadd45β induction in synoviocytes.
Nuclear factor κB
The transcription factor NF-κB is ubiquitously expressed and is involved in inflammation, cell survival, proliferation and differentiation. It plays a central role in inflammation by regulating expression of several proinflammatory gene clusters, such as cytokines, chemokines, adhesion molecules, prostaglandin synthases, and nitric oxide synthases. Increased NF-κB activity has been linked to many chronic inflammatory disorders including RA.
NF-κB is a family of five proteins: RelA (p65), RelB, c-REl, p50/p105, and p52/p100 that form homo or heterodimers. They normally reside as a cytoplasmic complex with IκB, which masks the nuclear localization sequence, thus inhibiting its entry into the nucleus. Phosphorylation of IκB is an important step in NF-κB activation and is mediated by two kinases IKKα and IKKβ. Phosphorylated IκB is marked by ubiquitination for proteosome degradation. This process releases NF-κB dimers allowing migration to the nucleus where they bind to the promoter regions of the target genes. These molecular events are referred to as ‘classical’ or ‘canonical’ NF-κB pathway. The IKK complex represents the convergence point for the signals that are transmitted in response to different cellular stimuli, such as proinflammatory cytokines and TLR agonists (130, 131).
NF-κB activation is observed in RA synovium and immunohistochemical analysis shows expression of p50 and p65 in the synovial intimal lining cells. Electromobility shift assays demonstrate that NF-κB binding is significantly higher in RA synovium compared with osteoarthritis. Nuclear translocation of NF-κB occurs rapidly in cultured FLS after IL-1 or TNF stimulation through activation of IKK signaling complex and leads to consequent induction of genes like IL-6, IL-8, ICAM-1, and MMPs.
IKKα and IKKβ are constitutively expressed in synoviocytes and their kinase activity is increased over 10-fold within minutes of cytokine exposure (132). IKKβ is the primary signaling molecule under these circumstances, as small molecule inhibitors and dominant negative IKKβ constructs block the activation of NF-κB and synoviocyte production of pro-inflammatory cytokines, MMPs, and adhesion molecules. The p50 and p65 subunits of NF-kB are the most relevant to synoviocytes. siRNA knockdown of p65 in particular suppresses production of pro-inflammatory cytokines and combined knockdown of both is even more effective (131).
IKKβ is also required for TLR4-induced cytokine production by synoviocytes and provides an important survival signal that suppresses apoptosis. In vitro and in vivo studies suggest that synoviocyte death is decreased when NF-κB activation is suppressed, such as with decoy oligonucleotides that interfere with its ability to bind to gene promoter sites. Proteosome inhibitors that interfere with IκB degradation or gene therapy with NF-κB inhibitors like the IκB super-repressor or XIAP (X-linked inhibitor of apoptosis) also decrease programmed cell death in cultured synoviocytes (130).
Animal models of RA also support the role of NF-κB in synovial inflammation. Increased NF-κB binding occurs very early in the course of collagen-induced arthritis in mice and adjuvant arthritis in rats. IKKβ emerged as an attractive target based on its ability to regulate proinflammatory cytokine production in several cell types. Proof of its pivotal role was demonstrated in adjuvant arthritis where gene therapy using a dominant negative IKKβ construct suppressed (133) inflammatory joint disease. Additionally, small molecule IKKβ inhibitors are effective in animal models of RA. The beneficial effects in vivo are probably complex and include effects on synoviocytes, lymphocytes, and macrophages (134, 135).
Progress targeting signaling molecules in RA
As previously noted, p38 MAPK fulfills many characteristics of an ideal target for treatment of RA. It regulates the production of inflammatory mediators not only in FLS but also in immune cells in response to cytokines, TLR ligands, and stress. Over 20 p38 inhibitors that demonstrated efficacy in animal models have entered clinical trials over the last few years. Early generation compounds like BIRB-794 and SCIO-469 had, at best, limited clinical benefit in RA and were associated with significant toxicity. Recently, new generation, more selective p38 inhibitors like VX-702 and pamapimod were tested in phase II clinical trials. The results of these studies are similarly disappointing, demonstrating only transient reduction of inflammatory markers and minimal clinical efficacy (136–138). ARRY-162, a MAPK inhibitor that blocks the upstream kinases that regulate ERK, also did not improve signs and symptoms in RA.
Despite these initial failures, targeting other signaling pathways, such as Janus kinases (JAK) and spleen tyrosine kinase (Syk), has been more promising (139, 140). The JAK kinases (Jak1, Jak2, Jak3, and Tyk2) are utilized by several cytokines and growth factors relevant to RA pathogenesis. Syk is a key regulator of immune receptor signaling in macrophages, neutrophils, B cells, and mast cells, but is also implicated in synoviocytes regulation of IL-6 and MMPs (141).
A JAK inhibitor (CP-690 550) that inhibits multiple JAK isoforms demonstrated efficacy in a phase II study when used in combination with methotrexate (140). Of interest, this response was achieved in patients who already failed other TNF blockers. Fostaminib, a Syk inhibitor, also improves signs and symptoms in patients with RA (139). Recent data suggest that this compound might not be effective in TNF failures, suggesting that its mechanism might be related to TNF production or signaling. For each of these compounds, efficacy was relatively rapid (weeks rather than months) and approached the level observed with biologics. The key questions relate to safety, as these pathways play a major role in host defense and homeostasis. Additional studies will be needed to assess safety.
Conclusion
Over five decades of research have shown that the pathogenesis of RA is enormously complex. Variable clinical responses to targeted therapies, such as TNF blockers, T-cell costimulation inhibitors, and B-cell depletors, demonstrate that the disease is heterogeneous and probably lacks a single mechanism that applies to all patients. Fibroblast-like synoviocytes in the synovial intimal lining undoubtedly contribute to the disease, and recent data using cadherin-11 deficient mice confirm their role in cartilage destruction and synovial inflammation. Therefore, attempts to target this interesting and unique cell type could potentially complement current therapies. The contribution of synoviocytes to RA is especially interesting because their sojourn through rheumatoid joint confers a unique aggressive phenotype that can perpetuate disease and increase joint destruction. The mechanisms are still uncertain, but imprinting due to chronic cytokine exposure, aberrant signaling, and somatic mutations might participate. Understanding this facet of RA will not only help identify novel therapeutic targets but also understand the pathogenesis of a disease that remains a major unmet medical need.
References
- 1.Gabriel SE, Michaud K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res Ther. 2009;11:229. doi: 10.1186/ar2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.Bresnihan B, Flanagan AM. Synovium. In: Firestein GS, Budd RC, Harris T, McInnes IB, Ruddy S, Sergent JS, editors. Kelly’s Textbook of Rheumatology. 8. Philadelphia, PA: Saunders Elsevier; 2009. pp. 23–37. [Google Scholar]
- 4.Edwards JC, Willoughby DA. Demonstration of bone marrow derived cells in synovial lining by means of giant intracellular granules as genetic markers. Ann Rheum Dis. 1982;41:177–182. doi: 10.1136/ard.41.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- 6.Firestein GS. Etiology and pathogenesis of rheumatoid arthritis. In: Firestein GS, Budd RC, Harris T, McInnes IB, Ruddy S, Sergent JS, editors. Kelly’s Textbook of Rheumatology. 8. Philadelphia, PA: Saunders Elsevier; 2009. pp. 1035–1086. [Google Scholar]
- 7.Valencia X, et al. Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004;200:1673–1679. doi: 10.1084/jem.20041545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DM, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315:1006–10010. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]
- 9.Szekanecz Z, et al. Differential distribution of intercellular adhesion molecules (ICAM-1, ICAM-2, and ICAM-3) and the MS-1 antigen in normal and diseased human synovia. Their possible pathogenetic and clinical significance in rheumatoid arthritis. Arthritis Rheum. 1994;3:221–231. doi: 10.1002/art.1780370211. [DOI] [PubMed] [Google Scholar]
- 10.Morales-Ducret J, Wayner E, Elices MJ, Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Alpha 4/beta 1 integrin (VLA-4) ligands in arthritis. Vascular cell adhesion molecule-1 expression in synovium and on fibroblast-like synoviocytes. J Immunol. 1992;149:1424–1431. [PubMed] [Google Scholar]
- 11.Takemura S, et al. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167:1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 12.Pap T, Gay S. Fibroblasts and fibroblast-like synoviocytes. In: Firestein GS, Budd RC, Harris T, McInnes IB, Ruddy S, Sergent JS, editors. Kelly’s Textbook of Rheumatology. 8. Philadelphia, PA: Saunders Elsevier; 2009. pp. 201–214. [Google Scholar]
- 13.Marinova-Mutafchieva L, Williams RO, Funa K, Maini RN, Zvaifler NJ. Inflammation is preceded by tumor necrosis factor-dependent infiltration of mesenchymal cells in experimental arthritis. Arthritis Rheum. 2002;46:507–513. doi: 10.1002/art.10126. [DOI] [PubMed] [Google Scholar]
- 14.Nakajima T, et al. Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. Arthritis Rheum. 1995;38:485–491. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- 15.Firestein GS, Yeo M, Zvaifler NJ. Apoptosis in rheumatoid arthritis synovium. J Clin Invest. 1995;96:1631–1638. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto S, Müller-Ladner U, Gay RE, Nishioka K, Gay S. Ultrastructural demonstration of apoptosis, Fas and Bcl-2 expression of rheumatoid synovial fibroblasts. J Rheumatol. 1996;23:1345–1352. [PubMed] [Google Scholar]
- 17.Perlman H, Georganas C, Pagliari LJ, Koch AE, Haines K, 3rd, Pope RM. Bcl-2 expression in synovial fibroblasts is essential for maintaining mitochondrial homeostasis and cell viability. J Immunol. 2000;164:5227–5235. doi: 10.4049/jimmunol.164.10.5227. [DOI] [PubMed] [Google Scholar]
- 18.Kammouni W, Wong K, Ma G, Firestein GS, Gibson SB, El-Gabalawy HS. Regulation of apoptosis in fibroblast-like synoviocytes by the hypoxia-induced Bcl-2 family member Bcl-2/adenovirus E1B 19-kd protein-interacting protein 3. Arthritis Rheum. 2007;56:2854–2863. doi: 10.1002/art.22853. [DOI] [PubMed] [Google Scholar]
- 19.Imamura F, et al. Monoclonal expansion of synoviocytes in rheumatoid arthritis. Arthritis Rheum. 1998;41:1979–1986. doi: 10.1002/1529-0131(199811)41:11<1979::AID-ART13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 20.Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity. 1998;28:197–208. doi: 10.3109/08916939808995367. [DOI] [PubMed] [Google Scholar]
- 21.Franz JK, et al. Expression of sentrin, a novel antiapoptotic molecule, at sites of synovial invasion in rheumatoid arthritis. Arthritis Rheum. 2000;43:599–607. doi: 10.1002/1529-0131(200003)43:3<599::AID-ANR17>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 22.Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S. Activation of synovial fibroblasts in rheumatoid arthritis: lack of expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Firestein GS, Nguyen K, Aupperle KR, Yeo M, Boyle DL, Zvaifler NJ. Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am J Pathol. 1996;149:2143–2151. [PMC free article] [PubMed] [Google Scholar]
- 24.Tak PP, et al. p53 overexpression in synovial tissue from patients with early and longstanding rheumatoid arthritis compared with patients with reactive arthritis and osteoarthritis. Arthritis Rheum. 1999;42:948–953. doi: 10.1002/1529-0131(199905)42:5<948::AID-ANR13>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 25.Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997;94:10895–10900. doi: 10.1073/pnas.94.20.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamanishi Y, Boyle DL, Rosengren S, Green DR, Zvaifler NJ, Firestein GS. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 2002;99:10025–10030. doi: 10.1073/pnas.152333199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han Z, Boyle DL, Shi Y, Green DR, Firestein GS. Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999;42:1088–1092. doi: 10.1002/1529-0131(199906)42:6<1088::AID-ANR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 28.Lee SH, et al. Microsatellite instability and suppressed DNA repair enzyme expression in rheumatoid arthritis. J Immunol. 2003;170:2214–2220. doi: 10.4049/jimmunol.170.4.2214. [DOI] [PubMed] [Google Scholar]
- 29.Rosengren S, Boyle DL, Firestein GS. Acquisition, culture, and phenotyping of synovial fibroblasts. Methods Mol Med. 2007;135:365–375. doi: 10.1007/978-1-59745-401-8_24. [DOI] [PubMed] [Google Scholar]
- 30.Kiener HP, Brenner MB. Building the synovium: cadherin-11 mediates fibroblast-like synoviocyte cell-to-cell adhesion. Arthritis Res Ther. 2005;7:49–54. doi: 10.1186/ar1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiener HP, Lee DM, Agarwal SK, Brenner MB. Cadherin-11 induces rheumatoid arthritis fibroblast-like synoviocytes to form lining layers in vitro. Am J Pathol. 2006;168:1486–1499. doi: 10.2353/ajpath.2006.050999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. J Clin Invest. 1989;83:1267–1276. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Müller-Ladner U, et al. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am J Pathol. 1996;149:1607–1615. [PMC free article] [PubMed] [Google Scholar]
- 34.Fassbender HG, Simmling-Annefeld M. The potential aggressiveness of synovial tissue in rheumatoid arthritis. J Pathol. 1983;139:399–406. doi: 10.1002/path.1711390314. [DOI] [PubMed] [Google Scholar]
- 35.Pierer M, Müller-Ladner U, Pap T, Neidhart M, Gay RE, Gay S. The SCID mouse model: novel therapeutic targets - lessons from gene transfer. Springer Semin Immunopathol. 2003;25:65–78. doi: 10.1007/s00281-003-0126-2. [DOI] [PubMed] [Google Scholar]
- 36.Pap T, et al. Cooperation of Ras- and c-Myc-dependent pathways in regulating the growth and invasiveness of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2004;50:2794–2802. doi: 10.1002/art.20461. [DOI] [PubMed] [Google Scholar]
- 37.Müller-Ladner U, et al. Gene transfer of cytokine inhibitors into human synovial fibroblasts in the SCID mouse model. Arthritis Rheum. 1999;42:490–497. doi: 10.1002/1529-0131(199904)42:3<490::AID-ANR14>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 38.Müller-Ladner U, et al. Human IL-1Ra gene transfer into human synovial fibroblasts is chondroprotective. J Immunol. 1997;158:3492–3498. [PubMed] [Google Scholar]
- 39.Rème T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998;111:353–358. doi: 10.1046/j.1365-2249.1998.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inazuka M, et al. Analysis of p53 tumour suppressor gene somatic mutations in rheumatoid arthritis synovium. Rheumatology (Oxford) 2000;39:262–266. doi: 10.1093/rheumatology/39.3.262. [DOI] [PubMed] [Google Scholar]
- 41.Aupperle KR, et al. Regulation of synoviocyte proliferation, apoptosis, and invasion by the p53 tumor suppressor gene. Am J Pathol. 1998;152:1091–1098. [PMC free article] [PubMed] [Google Scholar]
- 42.Pap T, Aupperle KR, Gay S, Firestein GS, Gay RE. Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001;44:676–681. doi: 10.1002/1529-0131(200103)44:3<676::AID-ANR117>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 43.Simelyte E, Rosengren S, Boyle DL, Corr M, Green DR, Firestein GS. Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum. 2005;52:1876–1884. doi: 10.1002/art.21099. [DOI] [PubMed] [Google Scholar]
- 44.Yamanishi Y, et al. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am J Pathol. 2002;160:123–130. doi: 10.1016/S0002-9440(10)64356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tak PP, Klapwijk MS, Broersen SF, van de Geest DA, Overbeek M, Firestein GS. Apoptosis and p53 expression in rat adjuvant arthritis. Arthritis Res. 2000;2:229–235. doi: 10.1186/ar92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roivainen A, Jalava J, Pirilä L, Yli-Jama T, Tiusanen H, Toivanen P. H-ras oncogene point mutations in arthritic synovium. Arthritis Rheum. 1997;40:1636–1643. doi: 10.1002/art.1780400913. [DOI] [PubMed] [Google Scholar]
- 47.Cannons JL, Karsh J, Birnboim HC, Goldstein R. HPRT- mutant T cells in the peripheral blood and synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 1998;41:1772–1782. doi: 10.1002/1529-0131(199810)41:10<1772::AID-ART9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 48.Da Sylva TR, Connor A, Mburu Y, Keystone E, Wu GE. Somatic mutations in the mitochondria of rheumatoid arthritis synoviocytes. Arthritis Res Ther. 2005;7:R844–R851. doi: 10.1186/ar1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamanishi Y, et al. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R12–R18. doi: 10.1186/ar1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simelyte E, Boyle DL, Firestein GS. DNA mismatch repair enzyme expression in synovial tissue. Ann Rheum Dis. 2004;63:1695–1699. doi: 10.1136/ard.2003.017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jascur T, Boland CR. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006;119:2030–2035. doi: 10.1002/ijc.22023. [DOI] [PubMed] [Google Scholar]
- 52.Werb Z, Tremble PM, Behrendtsen O, Crowley E, Damsky CH. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J Cell Biol. 1989;109:877–889. doi: 10.1083/jcb.109.2.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Müller-Ladner U, et al. Alternatively spliced CS-1 fibronectin isoform and its receptor VLA-4 in rheumatoid arthritis synovium. J Rheumatol. 1997;24:1873–1880. [PubMed] [Google Scholar]
- 54.Kiener HP, Niederreiter B, Lee DM, Jimenez-Boj E, Smolen JS, Brenner MB. Cadherin 11 promotes invasive behavior of fibroblast-like synoviocytes. Arthritis Rheum. 2009;60:1305–1310. doi: 10.1002/art.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- 56.Asahara H, et al. Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum. 1997;40:912–918. doi: 10.1002/art.1780400520. [DOI] [PubMed] [Google Scholar]
- 57.Shiozawa S, Shimizu K, Tanaka K, Hino K. Studies on the contribution of c-fos/AP-1 to arthritic joint destruction. J Clin Invest. 1997;99:1210–1216. doi: 10.1172/JCI119277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aikawa Y, et al. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat Biotechnol. 2008;26:817–823. doi: 10.1038/nbt1412. [DOI] [PubMed] [Google Scholar]
- 59.Han Z, et al. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of JNK by MKK-7 in fibroblast-like synoviocytes. Arthritis Rheum. 2006;54:2127–2135. doi: 10.1002/art.21919. [DOI] [PubMed] [Google Scholar]
- 61.Yoshizawa T, Hammaker D, Sweeney SE, Boyle DL, Firestein GS. Synoviocyte innate immune responses: I. differential regulation of interferon responses and the JNK pathway by MAPK kinases. J Immunol. 2008;181:3252–3258. doi: 10.4049/jimmunol.181.5.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sweeney SE, Hammaker D, Boyle DL, Firestein GS. Regulation of c-Jun phosphorylation by the I kappa B kinase-epsilon complex in fibroblast-like synoviocytes. J Immunol. 2005;174:6424–6430. doi: 10.4049/jimmunol.174.10.6424. [DOI] [PubMed] [Google Scholar]
- 63.Sweeney SE, Mo L, Firestein GS. Antiviral gene expression in rheumatoid arthritis: role of IKKepsilon and interferon regulatory factor 3. Arthritis Rheum. 2007;56:743–752. doi: 10.1002/art.22421. [DOI] [PubMed] [Google Scholar]
- 64.Brentano F, Schorr O, Gay RE, Gay S, Kyburz D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005;52:2656–2665. doi: 10.1002/art.21273. [DOI] [PubMed] [Google Scholar]
- 65.Makkonen KM, Pasonen-Seppänen S, Törrönen K, Tammi MI, Carlberg C. Regulation of the hyaluronan synthase 2 gene by convergence in cyclic AMP response element-binding protein and retinoid acid receptor signaling. J Biol Chem. 2009;284:18270–18281. doi: 10.1074/jbc.M109.012492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boyle DL, Sajjadi FG, Firestein GS. Inhibition of synoviocyte collagenase gene expression by adenosine receptor stimulation. Arthritis Rheum. 1996;39:923–930. doi: 10.1002/art.1780390608. [DOI] [PubMed] [Google Scholar]
- 67.Yamanishi Y, et al. Expression and regulation of aggrecanase in arthritis: the role of TGF-beta. J Immunol. 2002;168:1405–1412. doi: 10.4049/jimmunol.168.3.1405. [DOI] [PubMed] [Google Scholar]
- 68.Stanton H, et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005;434:648–652. doi: 10.1038/nature03417. [DOI] [PubMed] [Google Scholar]
- 69.Korb A, Pavenstädt H, Pap T. Cell death in rheumatoid arthritis. Apoptosis. 2009;14:447–454. doi: 10.1007/s10495-009-0317-y. [DOI] [PubMed] [Google Scholar]
- 70.Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 71.Kurowska M, et al. Fibroblast-like synoviocytes from rheumatoid arthritis patients express functional IL-15 receptor complex: endogenous IL-15 in autocrine fashion enhances cell proliferation and expression of Bcl-x(L) and Bcl-2. J Immunol. 2002;169:1760–1767. doi: 10.4049/jimmunol.169.4.1760. [DOI] [PubMed] [Google Scholar]
- 72.Liu H, et al. Mcl-1 is essential for the survival of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2005;175:8337–8345. doi: 10.4049/jimmunol.175.12.8337. [DOI] [PubMed] [Google Scholar]
- 73.Okamoto K, et al. Fas-associated death domain protein is a Fas-mediated apoptosis modulator in synoviocytes. Rheumatology (Oxford) 2000;39:471–480. doi: 10.1093/rheumatology/39.5.471. [DOI] [PubMed] [Google Scholar]
- 74.Mountz JD, Zhang HG. Regulation of apoptosis of synovial fibroblasts. Curr Dir Autoimmun. 2001;3:216–239. doi: 10.1159/000060524. [DOI] [PubMed] [Google Scholar]
- 75.Pierer M, et al. The TNF superfamily member LIGHT contributes to survival and activation of synovial fibroblasts in rheumatoid arthritis. Rheumatology (Oxford) 2007;46:1063–1070. doi: 10.1093/rheumatology/kem063. [DOI] [PubMed] [Google Scholar]
- 76.Hayashi S, et al. Decoy receptor 3 expressed in rheumatoid synovial fibroblasts protects the cells against Fas-induced apoptosis. Arthritis Rheum. 2007;56:1067–1075. doi: 10.1002/art.22494. [DOI] [PubMed] [Google Scholar]
- 77.Meyer LH, Franssen L, Pap T. The role of mesenchymal cells in the pathophysiology of inflammatory arthritis. Best Pract Res Clin Rheumatol. 2006;20:969–981. doi: 10.1016/j.berh.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 78.Ichikawa K, et al. TRAIL-R2 (DR5) mediates apoptosis of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2003;171:1061–1069. doi: 10.4049/jimmunol.171.2.1061. [DOI] [PubMed] [Google Scholar]
- 79.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–15718. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 80.Audo R, Combe B, Coulet B, Morel J, Hahne M. The pleiotropic effect of TRAIL on tumor-like synovial fibroblasts from rheumatoid arthritis patients is mediated by caspases. Cell Death. 2009;16:1227–1237. doi: 10.1038/cdd.2009.38. [DOI] [PubMed] [Google Scholar]
- 81.Bai S, et al. NF-kappaB-regulated expression of cellular FLIP protects rheumatoid arthritis synovial fibroblasts from tumor necrosis factor alpha-mediated apoptosis. Arthritis Rheum. 2004;50:3844–3855. doi: 10.1002/art.20680. [DOI] [PubMed] [Google Scholar]
- 82.Palao G, et al. Fas activation of a proinflammatory program in rheumatoid synoviocytes and its regulation by FLIP and caspase 8 signaling. Arthritis Rheum. 2006;54:1473–1481. doi: 10.1002/art.21768. [DOI] [PubMed] [Google Scholar]
- 83.Meinecke I, et al. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc Natl Acad Sci USA. 2007;104:5073–5078. doi: 10.1073/pnas.0608773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cha HS, Rosengren S, Boyle DL, Firestein GS. PUMA regulation and proapoptotic effects in fibroblast-like synoviocytes. Arthritis Rheum. 2006;54:587–592. doi: 10.1002/art.21631. [DOI] [PubMed] [Google Scholar]
- 85.You X, Boyle DL, Hammaker D, Firestein GS. PUMA-mediated apoptosis in fibroblast-like synoviocytes does not require p53. Arthritis Res Ther. 2006;8:R157. doi: 10.1186/ar2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lindhout E, van Eijk M, van Pel M, Lindeman J, Dinant HJ, de Groot C. Fibroblast-like synoviocytes from rheumatoid arthritis patients have intrinsic properties of follicular dendritic cells. J Immunol. 1999;162:5949–5956. [PubMed] [Google Scholar]
- 87.Hayashida K, Shimaoka Y, Ochi T, Lipsky PE. Rheumatoid arthritis synovial stromal cells inhibit apoptosis and up-regulate Bcl-xL expression by B cells in a CD49/CD29-CD106-dependent mechanism. J Immunol. 2000;164:1110–1116. doi: 10.4049/jimmunol.164.2.1110. [DOI] [PubMed] [Google Scholar]
- 88.Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest. 2001;107:305–315. doi: 10.1172/JCI11092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1–16. doi: 10.1042/BJ20090272. [DOI] [PubMed] [Google Scholar]
- 90.Radstake TR, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum. 2004;50:3856–3865. doi: 10.1002/art.20678. [DOI] [PubMed] [Google Scholar]
- 91.Seibl R, et al. Expression and regulation of Toll-like receptor 2 in rheumatoid arthritis synovium. Am J Pathol. 2003;162:1221–1227. doi: 10.1016/S0002-9440(10)63918-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis. Cell Immunol. 2005;233:90–96. doi: 10.1016/j.cellimm.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 93.van der Heijden IM, et al. Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000;43:593–598. doi: 10.1002/1529-0131(200003)43:3<593::AID-ANR16>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 94.Rosengren S, Hoffman HM, Bugbee W, Boyle DL. Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis. 2007;64:708–714. doi: 10.1136/ard.2004.025577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ospelt C, et al. Expression, regulation, and signaling of the pattern-recognition receptor nucleotide-binding oligomerization domain 2 in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:355–363. doi: 10.1002/art.24226. [DOI] [PubMed] [Google Scholar]
- 96.Patel DD, Haynes BF. Leukocyte homing to synovium. Curr Dir Autoimmun. 2001;3:133–167. doi: 10.1159/000060517. [DOI] [PubMed] [Google Scholar]
- 97.Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol. 2001;98:39–45. doi: 10.1006/clim.2000.4957. [DOI] [PubMed] [Google Scholar]
- 98.Nanki T, et al. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4 + T cell accumulation in rheumatoid arthritis synovium. J Immunol. 2000;165:6590–6598. doi: 10.4049/jimmunol.165.11.6590. [DOI] [PubMed] [Google Scholar]
- 99.Tsubaki T, et al. Accumulation of plasma cells expressing CXCR3 in the synovial sublining regions of early rheumatoid arthritis in association with production of Mig/CXCL9 by synovial fibroblasts. Clin Exp Immunol. 2005;141:363–371. doi: 10.1111/j.1365-2249.2005.02850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmutz C, et al. Chemokine receptors in the rheumatoid synovium: upregulation of CXCR5. Arthritis Res Ther. 2005;7:R217–R229. doi: 10.1186/ar1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sawai H, Park YW, Roberson J, Imai T, Goronzy JJ, Weyand CM. T cell costimulation by fractalkine-expressing synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2005;52:1392–1401. doi: 10.1002/art.21140. [DOI] [PubMed] [Google Scholar]
- 102.van der Voort R, et al. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. Arthritis Rheum. 2005;52:1381–1391. doi: 10.1002/art.21004. [DOI] [PubMed] [Google Scholar]
- 103.Murphy G, Caplice N, Molloy M. Fractalkine in rheumatoid arthritis: a review to date. Rheumatology (Oxford) 2008;47:1446–1451. doi: 10.1093/rheumatology/ken197. [DOI] [PubMed] [Google Scholar]
- 104.Pierer M, et al. Chemokine secretion of rheumatoid arthritis synovial fibroblasts stimulated by Toll-like receptor 2 ligands. J Immunol. 2004;172:1256–1265. doi: 10.4049/jimmunol.172.2.1256. [DOI] [PubMed] [Google Scholar]
- 105.Wells TN, Power CA, Shaw JP, Proudfoot AE. Chemokine blockers–therapeutics in the making? Trends Pharmacol Sci. 2006;27:41–47. doi: 10.1016/j.tips.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 106.Proudfoot AE. Is CCR2 the right chemokine receptor to target in rheumatoid arthritis? Arthritis Rheum. 2008;58:1889–1891. doi: 10.1002/art.23590. [DOI] [PubMed] [Google Scholar]
- 107.Waldburger JM, Firestein GS. Garden of therapeutic delights: new targets in rheumatic diseases. Arthritis Res Ther. 2009;11:206. doi: 10.1186/ar2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bodolay E, Koch AE, Kim J, Szegedi G, Szekanecz Z. Angiogenesis and chemokines in rheumatoid arthritis and other systemic inflammatory rheumatic diseases. J Cell Mol Med. 2002;6:357–376. doi: 10.1111/j.1582-4934.2002.tb00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–3545. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Cytokines in chronic inflammatory arthritis. IV. Granulocyte/macrophage colony-stimulating factor-mediated induction of class II MHC antigen on human monocytes: a possible role in rheumatoid arthritis. J Exp Med. 1989;170:865–875. doi: 10.1084/jem.170.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Genovese MC, Chakravarty EF, Krishnan E, Moreland LW. A randomized, controlled trial of interferon-beta-1a in patients with rheumatoid arthritis: a pilot study [ISRCTN03626626] Arthritis Res Ther. 2004;6:R73–R77. doi: 10.1186/ar1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Corr M, Boyle DL, Ronacher L, Flores N, Firestein GS. Synergistic benefit in inflammatory arthritis by targeting I kappaB kinase epsilon and interferon beta. Ann Rheum Dis. 2009;68:257–263. doi: 10.1136/ard.2008.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis T lymphocytes. Arthritis Res Ther. 2007;9:202. doi: 10.1186/ar2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Firestein GS, et al. Synovial interleukin-1 receptor antagonist and interleukin-1 balance in rheumatoid arthritis. Arthritis Rheum. 1994;37:644–652. doi: 10.1002/art.1780370507. [DOI] [PubMed] [Google Scholar]
- 115.Kasperkovitz PV, et al. Fibroblast-like synoviocytes derived from patients with rheumatoid arthritis show the imprint of synovial tissue heterogeneity: evidence of a link between an increased myofibroblast-like phenotype and high-inflammation synovitis. Arthritis Rheum. 2005;52:430–441. doi: 10.1002/art.20811. [DOI] [PubMed] [Google Scholar]
- 116.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 117.Han Z, Boyle DL, Aupperle KR, Bennett B, Manning AM, Firestein GS. Jun N-terminal kinase in rheumatoid arthritis. J Pharmacol Exp Ther. 1999;291:124–130. [PubMed] [Google Scholar]
- 118.Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67:909–1016. doi: 10.1136/ard.2007.074278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:409–414. doi: 10.1093/rheumatology/kem297. [DOI] [PubMed] [Google Scholar]
- 120.Schett G, et al. Activation, differential localization, and regulation of the stress-activated protein kinases, extracellular signal-regulated kinase, c-JUN N-terminal kinase, and p38 mitogen-activated protein kinase, in synovial tissue and cells in rheumatoid arthritis. Arthritis Rheum. 2000;43:2501–2512. doi: 10.1002/1529-0131(200011)43:11<2501::AID-ANR18>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 121.Inoue T, et al. Mitogen-activated protein kinase kinase 3 is a pivotal pathway regulating p38 activation in inflammatory arthritis. Proc Natl Acad Sci USA. 2006;103:5484–5489. doi: 10.1073/pnas.0509188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Inoue T, Hammaker D, Boyle DL, Firestein GS. Regulation of p38 MAPK by MAPK kinases 3 and 6 in fibroblast-like synoviocytes. J Immunol. 2005;174:4301–4306. doi: 10.4049/jimmunol.174.7.4301. [DOI] [PubMed] [Google Scholar]
- 123.Yoshizawa T, et al. Role of MAPK kinase 6 in arthritis: distinct mechanism of action in inflammation and cytokine expression. J Immunol. 2009;183:1360–1367. doi: 10.4049/jimmunol.0900483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Köller M, et al. JNK1 is not essential for TNF-mediated joint disease. Arthritis Res Ther. 2005;7:R166–R173. doi: 10.1186/ar1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sundarrajan M, Boyle DL, Chabaud-Riou M, Hammaker D, Firestein GS. Expression of the MAPK kinases MKK-4 and MKK-7 in rheumatoid arthritis and their role as key regulators of JNK. Arthritis Rheum. 2003;48:2450–2460. doi: 10.1002/art.11228. [DOI] [PubMed] [Google Scholar]
- 126.Hammaker DR, Boyle DL, Chabaud-Riou M, Firestein GS. Regulation of c-Jun N-terminal kinase by MEKK-2 and mitogen-activated protein kinase kinase kinases in rheumatoid arthritis. J Immunol. 2004;172:1612–1618. doi: 10.4049/jimmunol.172.3.1612. [DOI] [PubMed] [Google Scholar]
- 127.Hammaker DR, Boyle DL, Inoue T, Firestein GS. Regulation of the JNK pathway by TGF-beta activated kinase 1 in rheumatoid arthritis synoviocytes. Arthritis Res Ther. 2007;9:R57. doi: 10.1186/ar2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Toh ML, Yang Y, Leech M, Santos L, Morand EF. Expression of mitogen-activated protein kinase phosphatase 1, a negative regulator of the mitogen-activated protein kinases, in rheumatoid arthritis: up-regulation by interleukin-1beta and glucocorticoids. Arthritis Rheum. 2004;50:3118–3128. doi: 10.1002/art.20580. [DOI] [PubMed] [Google Scholar]
- 129.Svensson CI, et al. Gadd45beta deficiency in rheumatoid arthritis: enhanced synovitis through JNK signaling. Arthritis Rheum. 2009;60:3229–3240. doi: 10.1002/art.24887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Firestein GS. NF-kappaB: holy Grail for rheumatoid arthritis? Arthritis Rheum. 2004;50:2381–2386. doi: 10.1002/art.20468. [DOI] [PubMed] [Google Scholar]
- 131.Simmonds RE, Foxwell BM. Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology (Oxford) 2008;47:584–590. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- 132.Aupperle K, Bennett B, Han Z, Boyle D, Manning A, Firestein G. NF-kappa B regulation by I kappa B kinase-2 in rheumatoid arthritis synoviocytes. J Immunol. 2001;166:2705–2711. doi: 10.4049/jimmunol.166.4.2705. [DOI] [PubMed] [Google Scholar]
- 133.Tak PP, et al. Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001;44:1897–1907. doi: 10.1002/1529-0131(200108)44:8<1897::AID-ART328>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 134.Ichikawa H, Takada Y, Murakami A, Aggarwal BB. Identification of a novel blocker of I kappa B alpha kinase that enhances cellular apoptosis and inhibits cellular invasion through suppression of NF-kappa B-regulated gene products. J Immunol. 2005;174:7383–7392. doi: 10.4049/jimmunol.174.11.7383. [DOI] [PubMed] [Google Scholar]
- 135.Tas SW, et al. Amelioration of arthritis by intraarticular dominant negative Ikk beta gene therapy using adeno-associated virus type 5. Hum Gene Ther. 2006;17:821–832. doi: 10.1089/hum.2006.17.821. [DOI] [PubMed] [Google Scholar]
- 136.Alten RE, et al. Efficacy and safety of pamapimod in patients with active rheumatoid arthritis receiving stable methotrexate therapy. Ann Rheum Dis. 2009 doi: 10.1136/ard.2008.104802. [DOI] [PubMed] [Google Scholar]
- 137.Cohen SB, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- 138.Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009;60:1232–1241. doi: 10.1002/art.24485. [DOI] [PubMed] [Google Scholar]
- 139.Weinblatt ME, et al. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. 2008;58:3309–3318. doi: 10.1002/art.23992. [DOI] [PubMed] [Google Scholar]
- 140.Kremer JM, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo. Arthritis Rheum. 2009;60:1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- 141.Cha HS, et al. A novel spleen tyrosine kinase inhibitor blocks c-Jun N-terminal kinase-mediated gene expression in synoviocytes. J Pharmacol Exp Ther. 2006;317:571–578. doi: 10.1124/jpet.105.097436. [DOI] [PubMed] [Google Scholar]
- 142.Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. 2004;36:372–378. doi: 10.1016/s1357-2725(03)00259-0. [DOI] [PubMed] [Google Scholar]