

Abstract
The par toxin−antitoxin system is required for the stable inheritance of the plasmid pAD1 in its native host Enterococcus faecalis. It codes for the toxin Fst and a small antisense RNA which inhibits translation of toxin mRNA, and it is the only known antisense regulated toxin−antitoxin system in Gram-positive bacteria. This study presents the structure of the par toxin Fst, the first atomic resolution structure of a component of an antisense regulated toxin−antitoxin system. The mode of membrane binding was determined by relaxation enhancements in a paramagnetic environment and molecular dynamics simulation. Fst forms a membrane-binding α-helix in the N-terminal part and contains an intrinsically disordered region near the C-terminus. It binds in a transmembrane orientation with the C-terminus likely pointing toward the cytosol. Membrane-bound, α-helical peptides are frequently found in higher organisms as components of the innate immune system. Despite similarities to these antimicrobial peptides, Fst shows neither hemolytic nor antimicrobial activity when applied externally to a series of bacteria, fungal cells, and erythrocytes. Moreover, its charge distribution, orientation in the membrane, and structure distinguish it from antimicrobial peptides.
Plasmids are extrachromosomal genetic entities carrying additional information for the host organism, and they rely mainly on the host machinery to be replicated. While the information encoded on a plasmid can be beneficial to the cell under various stress situations (e.g., exposure to antibiotics or heavy metals), plasmids also harbor a nutritional burden for bacteria under normal growth conditions. If the spread of the plasmid to daughter cells was only a random process, low copy number plasmids would get lost from bacterial cells within a short number of generations. Plasmidic toxin−antitoxin (TA)1 systems (also known as postsegregational killing or plasmid addiction systems) contribute to the stability of these mobile genetic elements in the growing host bacterial population by killing daughter cells which have not inherited a plasmid copy during cell division (1−3). TA systems consist of a stable toxin and an unstable antitoxin, which are both produced continuously. If the plasmid is lost, the pool of short-lived antitoxin is soon degraded due to the lack of de novo synthesis. The more stable toxin survives and thus kills plasmid-free cells. Besides being an integral component of various plasmids, TA systems were also found on bacterial chromosomes (4,5). While the function on plasmids is pretty clear, there is still no consensus about the function of chromosomal TAs. Current hypotheses range from growth regulation or even programmed cell death during stress conditions (e.g., starvation, high temperatures, oxidative stress, or exposure to antibiotics) to genomic junk (6−8).
Two types of TA systems are known: Type I systems consist of a short (<60 amino acid) toxin and a small antisense RNA acting as the antitoxin, which prohibits the translation of toxin mRNA (9,10). To exert its toxic function once the TA gene is lost or downregulated, the antisense RNA has to be less stable than the toxin-encoding mRNA. In type II systems both the toxin and antitoxin are small (∼70−135 residues) homodimeric proteins (1,5). A stable toxin and a short-lived antitoxin form a short operon whose transcription is autoregulated. The antitoxins contain intrinsically unstructured regions, which can be readily cleaved by proteases (Lon or Clp) (11). Much more information, especially as far as their structures is concerned, is currently available for type II toxin−antitoxin systems. Nine families of type II TA systems have been found so far (CcdAB, RelBE, MazEF, ParDE, phd/doc, VapBC, hipBA, HigBA, and epsilon/zeta/omega) (3,8). A variety of structural data is available for free toxin or antitoxins (CcdB (12,13), Kid (14), MazE (15), CcdA (16), ParD (17), VapC (18), omega (19), RelB (20), RelE (21)), a complex of a toxin with its target (CcdB-GyrA14) (22), toxin bound to antitoxin (MazE/MazF (23), RelB/RelE (24), YefM/YoeB (25), ParD/ParE (26), MqsR/MqsA (27), epsilon/zeta (28), PezA/PezT-DNA (29)), a DNA-bound antitoxin (CcdA-DNA) (16), and even a ternary toxin−antitoxin−DNA complex (FitA/FitB-DNA) (30). Neither sequences nor folds are conserved among toxins or antitoxins, and there is also great variability of combinations of toxin and antitoxin folds. For example, while the toxins CcdB and MazF share a similar fold (13,23), their corresponding antitoxins CcdA (16) and MazE (15) show completely unrelated structures. In contrast to type II TA systems, there has so far been no report on the structure of a type I toxin−antitoxin system component. Type I TA systems have been found on plasmids and chromosomes (10). The first type I systems found were the Hok/Sok pair of plasmid R1 (31) and the par locus of plasmid pAD1 (32). Homologues of Hok/Sok were also found on the chromosomes of Escherichia coli and other bacteria (33,34). Other chromosomal TA systems include the ldr (long direct repeat) sequences (35), the tisAB system (36), the OhsC/ShoB pair (37,38) (all in E. coli), and the TxpA/RatA system (39) of Bacillus subtilis. Although comparably few type I TA systems are known, there might be a much larger number which is just not yet being recognized by current protocols of genome searches as recently suggested (40). In all type I systems the toxins are small hydrophobic peptides. For none of them is the target known, but they seem to have some effect on bacterial membranes (10). Here we present the structure of the toxin Fst (faecalis plasmid stabilization toxin) (41) of the par addiction module of the plasmid pAD1 of Enterococcus faecalis, together with its mode of interaction with membrane-mimetics. The par toxin−antitoxin system, which was studied extensively by Weaver et al. (32,40−46), is the only one known in a Gram-positive bacterium. The par locus on pAD1 codes for two small RNAs, called RNA I (coding for the toxin) and RNA II (the antisense RNA) which are convergently transcribed in opposite directions across a pair of direct repeats toward a bidirectional terminator (42). RNA I and RNA II form a complex through various complementary interactions including ones in the translation initiation region for the Fst toxin. The par addiction system was found to have several homologues located both on plasmids and on chromosomes of Gram-positive bacteria (40). Translation of the toxin Fst m-RNA is inhibited through binding of a small antisense RNA. Translated Fst peptide was shown to affect cell membrane integrity and caused defects in cell division and chromosome segregation (46). Generally, atomic resolution structure determinations of small membrane-bound polypeptides are not possible by X-ray crystallography but can be carried out by NMR spectroscopy if the system under study is below the NMR size limit to prevent excessive line broadening. Thus, small membrane-mimetic systems (typically micelles) have to be used (47−49). Typically, the zwitterionic dodecylphosphocholine (DPC) is employed (47,48), since it structurally resembles the components of major eukaryotic biological membranes. It can well preserve the 3D structure of bound peptides and proteins (50) as well as the catalytic activity of membrane-bound enzymes (51,52). Here we present the structure and mode of membrane binding of the toxin Fst bound to DPC micelles, the first atomic resolution structure of a type I TA system component. Fst forms a transmembrane helix within the first ∼26 residues and contains a highly charged intrinsically unstructured C-terminus which sticks into the cytosol.
Experimental Procedures
Materials
Fst (MKDLMSLVIAPIFVGLVLEMISRVLDEEDDSRK) and Fst1−27 (MKDLMSLVIAPIFVGLVLEMISRVLDE) were synthesized by FMOC-based solid-phase peptide synthesis and purchased from Peptide Specialty Laboratories GmbH (Heidelberg, Germany). Unlabeled and 98% deuterated dodecylphosphocholines (DPC) as well as tetradecylphosphocholine (TPC) were obtained from Anatrace Inc. (Maumee, OH) at a purity higher than 97%. Gd(DTPA-BMA) was purified from Omniscan (Nycomed, Oslo, Norway), a commercially available MRI contrast agent, as described (53). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) in the highest purity available.
NMR Spectroscopy
About 1 mg of peptide was dissolved in 600 μL of a 100 mM DPC-d38 solution, buffered by 50 mM KPi, pH 5.0, containing 0.02% NaN3 and 10% (v/v) D2O. All NMR spectra were acquired on a Varian Unity INOVA 600 MHz NMR spectrometer except the diffusion measurements which were carried out on a Bruker Avance DRX 500 MHz NMR spectrometer, both equipped with HCN triple-resonance probes and z-axis gradients. Chemical shifts were referenced relative to internal H2O. The H2O signal chemical shift was obtained according to δ(H2O) = 7.83 − T/96.9 ppm (54). Two-dimensional TOCSY spectra were acquired using different temperatures and mixing times (293 K/50 ms, 303 K/50 ms, 308 K/50 ms, 323 K/50 ms, and 308 K/30 ms, respectively). Two-dimensional NOESY spectra were acquired at the same temperatures and a mixing time of 150 ms. All spectra were processed using the programs nmrPipe (55) and TopSpin 2.1 (Bruker). Data analysis was done in NMRView 8.0.a22 (56). Self-diffusion coefficients were measured at 301 K by acquiring a series of nine pulsed-gradient stimulated echo spectra with variation of gradient strengths from 10% to 90% of maximum power (maximum ∼50 G/cm). Signals of interest were least-squares fitted to the equation
 |
where I and I0 correspond to the measured signal intensities at gradient strengths G and 0, τ2 is the delay between the first and second 90° pulse (1.05 ms) and τ1 the delay between the second and third 90° pulse (150 ms), T1 and T2 are the longitudinal and transverse relaxation rates, γ is the gyromagnetic ratio, δ is the length of the gradient (2 ms), Δ is the duration between the two gradients (151 ms), and D is the self-diffusion coefficient. Due to uncertainties of some parameters involved in the constants of eq 1, as for example the exact gradient strengths, all parameters were combined into the instrument-dependent constants “a” and “b” according to
and were obtained through calibration on an external water sample as described elsewhere (47). The experimental data gave linear fits with correlation coefficients typically above 0.99, proving the linearity up to 90% gradient strength. Proton T1 relaxation times were obtained from a series of 2D TOCSY spectra with a saturation recovery sequence at the beginning and using excitation sculpting solvent suppression before acquisition. Eight 2D data sets were acquired with recovery delays of 100, 200, 500, 700, 1000, 1500, 2000, and 3000 ms and 32 scans for each of the 512 increments. Due to the saturation period at the start of the sequence the interscan delay could be reduced to 0.2 s (57). Longitudinal relaxation times T1 were obtained by fitting the signal intensities to the equation
where I is the experimental signal intensity at time t, I0 the intensity at time 0. and T1 the longitudinal relaxation time. T1 values are calculated using the built-in Levenberg−Marquardt fitting algorithm in NMRView. PRE values were obtained by determination of the slopes of relaxation rates versus the concentration of added paramagnetic compound.
CD Spectroscopy
Far-UV CD experiments were recorded on a J-715 spectrapolarimeter with a PTC343 peltier unit (Jasco, Tokyo, Japan). Spectra were acquired at room temperature with a response time of 1 s and a step resolution of 0.2 nm. Five scans were averaged to obtain smooth spectra, and the background was corrected. The CD spectra were obtained on 100 μM Fst and 133 μM Fst1−27. A buffer consisting of 100 mM TPC, 50 mM KPi, pH 5.0, and 0.02% NaN3 was used.
Structure Determination
Peak intensities of manually assigned NOESY spectra were translated by the program NMRView into distance restraints using the built-in median method. The median intensity was set to a distance of 2.7 Å in the NOESY spectra of both Fst and Fst1−27. Additionally, ϕ and ψ angle restraints were obtained with the PREDITOR web server (58) using chemical shifts of amide and α-protons. A total of 279 NOEs and 25 ϕ-angle and 24 ψ-angle restraints were used as experimental input for the full simulated annealing structure calculation method using the CNS software package (59). Visualization of structures was carried out by the programs Pymol (60) and Molmol (61).
Hemolytic Activity
Human erythrocytes (donated by S. K.; healthy 29-year-old female) were used to explore the hemolytic activity of the peptide as previously described (62). The blood was centrifuged and washed four times with 0.9% NaCl solution to dispose of the plasma. Fst and Fst1−27 dissolved in PBS + 5% DMSO (pH 7.4) were added to erythrocytes (1 × 108 cells/mL in 0.9% NaCl). The suspensions were incubated for 2 h at 37 °C. The samples were centrifuged, and the optical density of the supernatant was measured at 451 nm to determine the extent of hemolysis. Hypotonically lyzed erythrocytes were used as a standard for 100% hemolysis. The experiment was repeated three times including positive and negative controls.
Antimicrobial Activity
The antimicrobial activity was tested with an inhibition zone assay on agar plates according to Hultmark et al. (63). The peptide was serially diluted in 100 mM TPC solution, containing 50 mM potassium phosphate at pH 5.5. Inhibition zones were measured from wells filled with 5 μL of peptide solutions and in parallel from sterile paper disks (Sigma-Aldrich) loaded with 20 μL of peptide solution. Each experiment was repeated three times, and disk diameters were considered in the calculations. Bacterial and fungal strains used in this assay were the Gram-negative E. coli (BL 21), the Gram-positive strains B. subtilis (DSM 1089) and Enterococcus caccae (DSM 19114), and the fungal strains Candida parapsilosis (NBCC 0707) and Saccharomyces cerevisiae (BCY 1020). E. caccae is phylogenetically closely related to the Fst producing E, faecalis(64) but less pathogenic. Bacteria and fungi were grown to an OD600 of 0.7 in Luria−Bertani broth (LB; Sigma-Aldrich) and universal medium (YM), respectively. In midlogarithmic phase bacteria were diluted in LB medium and approximately 2 × 105 colony forming units (CFU) plated on Petri dishes with LB + 1.5% agar (Sigma-Aldrich). Fungal cells were diluted in YM and plated on Petri dishes with YM + 1.5% agar.
Molecular Dynamics Simulations
The simulation system was constructed from an equilibrated POPC bilayer (65) consisting of 200 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) molecules and 35 water molecules per lipid. Two full-length Fst toxin peptides were inserted using g_membed (66). Thereby, two peptides were sequentially first overlaid with the preequilibrated membrane at the largest possible distance from one another and then scaled by 0.1 in the membrane plane. Still overlapping lipid (cutoff radius 0.22 nm) and water molecules were removed. The peptides were subsequently expanded to their original size while pushing on water and lipid molecules. The system was electroneutralized by adding four K+ ions. Further K+ and Cl− ions were added to obtain a 100 mM concentration. The system was equilibrated for 5 ns while restraining the position of the Fst peptides with 1000 kJ/mol. Subsequently, 50 ns production runs were carried out. Berger lipids (67), converted into the format of the OPLS all-atom force field (40), were used to describe the POPC molecules. The OPLS all-atom force field was applied for the Fst peptide. The SPC (68) water model was used. The simulation was performed using the Gromacs 4.0.7 MD package (69,70) using an integration time step of 2 fs and a neighbor search list updated at every 10th step. The temperature was maintained at 310 K using the velocity rescale (v-rescale) algorithm (71) applying a coupling time of 0.1 ps, independently coupling water plus ions and lipids plus Fst peptides to an external bath. Pressure was maintained at 1 bar both in the membrane plane and in the membrane normal using the semiisotropic Berendsen pressure coupling scheme (72) with a time constant of 4 ps. Bond lengths were constrained using the LINCS method (73). A cutoff of 1.0 nm was applied for vdW interactions. Long-range electrostatic interactions were calculated applying the particle mesh Ewald method (74) using a cutoff of 1.0 nm, and the reciprocal space interactions were evaluated on a 0.14 nm grid using fourth-order B-splines. Periodic boundary conditions were applied in all dimensions.
Results and Discussion
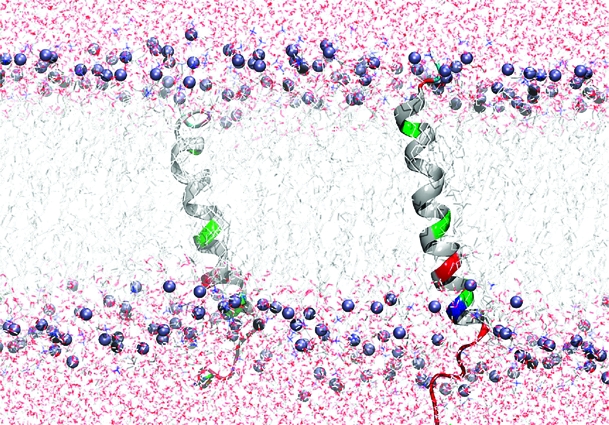
Fst was insoluble in water but can be easily solubilized by adding membrane mimetics. Well-resolved 1D NMR spectra could be acquired even at relatively low DPC concentrations of 50 mM. No significant changes in the NMR spectra were observed beyond 100 mM DPC-d38. Unlabeled peptides in micelles can be structurally characterized typically up to ∼30 residues by solution NMR. The spectra of Fst showed a few very strong and slowly relaxing peaks (based on their narrow line width) while most signals are very weak and broad. The strong signals could be fully sequentially assigned using 2D TOCSY and NOESY spectra. They all belong to the last 7 C-terminal residues and are characterized by chemical shifts close to random coil values, and they show only trivial NOEs. Thus the C-terminus of Fst is intrinsically unstructured. Only a few of the other residues could be assigned. Besides being much broader and therefore weaker, these amino acids have chemical shifts closer to values observed in α-helical peptide and protein regions (75). To enable the assignment and structure determination also of the structured part of Fst, a shorter version of the peptide was synthesized which consists of the 27 N-terminal residues (Fst1−27). Fst1−27 is also insoluble in water but can be easily dissolved in 100 mM DPC-d38. The TOCSY and NOESY spectra of Fst1−27 show a much more uniform distribution of signal intensities and line widths. The chemical shifts of the signals stemming from the N-terminal 27 residues of Fst are very similar to those of Fst1−27 (see Figure 1). Therefore, the structure and probably also mode of membrane binding of Fst1−27 represent quite well the one for these residues in full-length Fst. Additionally, CD spectra of Fst and Fst1−27 are very similar (see Figure 2). Both show typical α-helical secondary structure formation in phosphocholine micelles with minima at 207 and 222 nm. The molar ellipticity per residue is higher for Fst1−27 as expected for an overall better structured peptide (Figure 2). To enable the complete assignment of Fst1−27, several two-dimensional TOCSY and NOESY spectra at different temperatures between 293 and 325 K had to be acquired to resolve overlapping signals. All but the very first N-terminal residue could be assigned, and the chemical shifts are deposited in the BMRB database under accession number 16769. No NOEs between the last 6 C-terminal residues of Fst with the first 27 residues were found, and all NOEs within the first 27 residues of Fst were also found in Fst1−27. Therefore, we combined the NOEs of Fst1−27 with the ones found for the last 6 residues of Fst for the structure calculation of full-length Fst. It should be noted that due to the different dynamical behavior between the first 27 and last 6 residues the translation of NOEs into distances is probably not uniform along the peptide chain. However, since only trivial NOEs were found in the C-terminal region and to indicate the increased flexibility in this part of the peptide, we included these additional NOEs from full-length Fst into the structure calculation. The NOE connectivity pattern of Fst (Figure 3) shows typical α-helix NOEs (dαN(i,i + 4), dαN(i,i + 3), and dαβ(i,i + 3)) (75) for residues ∼4−27. For the structure calculation a total of 279 NOEs from both Fst and Fst1−27 were used together with 49 ϕ and ψ angle restraints derived from chemical shifts using the PREDITOR webserver. A total of 100 structures were calculated in CNS 1.1, and the 20 lowest energies which gave no NOE and dihedral angle violations are shown in Figure 4 and are deposited in the PDB database under accession number 2kv5 (Table 1). Fst forms an α-helical structure between amino acids 5 and 26. A proline known as a potential destabilizer of secondary structure is found at position 11. In Fst, the α-helix is well-defined at the proline residue, showing clear and well-resolved NOEs to amino acids 7, 8, 10, 12, and 14. A glycine residue at position 15 gives NOEs to amino acids 12, 14, 16, and 17, but the number of NOE connectivities of glycines is generally low as a result of the lack of side chain atoms. Although still α-helical, small distortions in the final structures around glycine 15 result in a slightly bent α-helix. As expected based on the narrow line widths and low numbers of (only trivial) NOEs found, the last 7 C-terminal residues are completely disordered. Besides the structure, knowing the orientation and location inside a membrane are also important to understand the function of membrane-bound peptides and proteins. The positioning of a peptide in a small membrane-mimetic can be determined using relaxation enhancements induced by the inert and soluble paramagnetic probe gadolinium diethylenetriaminepentaacetic acid bismethylamide (Gd(DTPA-BMA)) (53,76−78). Using this complex the orientation of the peptide in the micelle can be studied without chemical modification of the peptide or micelle. Since the orientation of Fst and Fst1−27 in the micelle might be different due to the changed charge of the C-terminus, we did not attempt to determine the orientation of Fst1−27 but only full-length Fst. Paramagnetic relaxation enhancements (PREs) were acquired by addition of Gd(DTPA-BMA) to concentrations of 0.98 and 1.95 mM. Due to extensive line broadening of most residues within the structured region of Fst, PREs could only be determined for amino acids 2, 14, 17, 29, and 31−33. The PRE values (see Figure 5) are high near the termini and much lower for residues 14 and 17 and thus indicate that Fst penetrates directly through the micelle. It obviously forms a transmembrane helix with the first ∼2 and last ∼7 residues sticking out of the membrane. Side chain protons 2.Hε3 and 32.Hε show higher values than backbone protons. These polar lysine (residue 2) and arginine (residue 32) side chains obviously snorkel into the aqueous solution surrounding the micelle. Further insight into the mode of membrane-binding of Fst was obtained by molecular dynamics simulations. Simulation of two Fst peptides, inserted in a preequilibrated membrane by overlapping the hydrophobic regions of the POPC membrane and the Fst peptide, was carried out for 50 ns. The use of two peptides allows for duplication of conformational phase space sampling and for an internal consistency control. Both peptide molecules were found to behave similarly. In Figure 6 a representative snapshot of the simulation is shown. The polar headgroup region of the membrane is highlighted by the blue spheres of the phosphate atoms. Both peptides show a similar conformation with the hydrophobic part well within the hydrophobic region of the membrane. At the N-terminus the peptides reach the edge of the membrane, while at the C-terminus, in perfect accordance with our experimental results, the polar residues are water exposed. Glu19 and Arg23 form a salt bridge and/or reach to the polar headgroup region of the membrane. The mobility of the individual residues is shown as root-mean-square fluctuations (RMSF) in Figure 7, averaged over both peptides. Residues within the membrane show a low level of fluctuation, while especially at the C-terminus, beginning with residue Asp26, the mobility of the residues strongly increases. An analysis of the secondary structure (79) is shown in Figure 8. The transmembrane part, including residues Asp3−Asp26 forms a stable helical structure, which can be observed throughout the 50 ns time window in both peptides. Consistent with the NMR data, due to increased dynamics, no stable secondary structure is observed at the C-terminal region.
Figure 1.

Overlay of the fingerprint region of 2D TOCSY spectra of Fst (red) and Fst1−27 (black) at 301 K. Correlations between amide protons and α-protons are indicated by the residue numbers. Cross-peaks between NH and other (non-α) protons are indicated by the position in the side chain.
Figure 2.

CD spectra of Fst (filled circles) and Fst1−27 (empty circles) in 100 mM TPC solution showing the mean residue ellipticity (mre) per residue versus the wavelength. The reduced mean residual ellipticity of Fst is a result of the intrinsically disordered C-terminus.
Figure 3.

Sequence plot of Fst and Fst1−27. The strengths of the NOEs are indicated by the thicknesses of the bars.
Figure 4.

Least-squares superposition of the 20 lowest energy structures of Fst showing all atoms (a) or the backbone only (b). The backbone atoms of residues 5−25 were used for the fitting.
Table 1. Structural Statistics of Fst in 100 mM DPC Micelles.
| restraints | |
| NOE restraints | |
| intraresidue NOEs | 140 |
| interresidue NOEs (i, i+1) | 73 |
| medium-range NOEs (i, i+2,3,4) | 66 |
| total NOEs | 279 |
| angle restraints | |
| ϕ angles (2−26) | 25 |
| ψ angle (2−25) | 24 |
| energies (kcal/mol) | |
| total | 56.4 ± 2.4 |
| bond | 1.6 ± 0.4 |
| angle | 23.7 ± 0.8 |
| improper | 1.4 ± 0.2 |
| van der Waals | 18.6 ± 1.5 |
| NOE | 10.7 ± 0.7 |
| dihedral | 0.27 ± 0.03 |
| statistics (5−25) | |
| rmsd for backbone atoms | 0.41 |
| rmsd for all atoms | 1.27 |
| number of NOE violations >0.5 Å | 0 |
| number of dihedral angle violations >5° | 0 |
| Ramachandran plot details | |
| residues in most favored regions | 91.1% |
| residues in additionally allowed regions | 8.9% |
| residues in generously allowed regions | 0.0% |
| residues in disallowed regions | 0.0% |
| statistics (1−33) | |
| rmsd for backbone atoms | 2.03 |
| rmsd for all atoms | 3.63 |
| number of NOE violations >0.5 Å | 0 |
| number of dihedral angle violations >5° | 0 |
| Ramachandran plot details | |
| residues in most favored regions | 75.7% |
| residues in additionally allowed regions | 22.9% |
| residues in generously allowed regions | 1.0% |
| residues in disallowed regions | 0.3% |
Figure 5.

Longitudinal (R1) paramagnetic relaxation enhancements (PREs) of Fst obtained by adding Gd(DTPA-BMA). Only values of well-resolved peaks with correlation coefficients of least-squares fits higher than 0.98 were used. A low PRE is indicative of a larger distance from the micelle surface.
Figure 6.

Representative structures of Fst of a 50 ns MD simulation run. The peptide is shown as a color-coded ribbon (red = negatively charged, blue = positively charged, green = polar, including glycine, and white = unpolar). The phosphate headgroups are indicated by blue spheres.
Figure 7.

Residue root-mean-square fluctuation: average RMSF of each residue, averaged over the entire simulation, and both peptides are shown together with the standard deviation.
Figure 8.

Time evolution of the secondary structure of the two Fst toxin peptides.
The binding of well-immersed peptides of comparable size of Fst1−27 to micelles results in hydrodynamic radii not remarkably different from pure micelles (47). A radius of 23.4 ± 2.0 Å was obtained for Fst1−27 bound to DPC micelles using self-diffusion NMR experiments, whereas 23.3 ± 1.4 Å was found previously for a pure DPC solution (47). An unusually large proportion of charged residues is found near the C-terminus. A strongly negatively charged stretch (D26−D30) is found right where the peptide leaves the membrane. Since the outside of Gram-positive membranes is negatively charged, Fst likely binds to the cell membrane with the C-terminus pointing into the cytosol. This orientation (outside to inside) is also the strongly preferred model obtained with the transmembrane helix tool TMpred (80).
All type I TA system toxins are small hydrophobic peptides and at least Fst is mainly α-helical. Overexpression of several type I toxins has been shown to lead to membrane depolarization or disruption. Based on these observations it has been speculated that type I toxins might act extracellularly, similar to antimicrobial peptides (9). Antimicrobial peptides occur in a wide variety of hosts, including bacteria (81), and show a strong activity against microorganisms (82,83). They are part of the innate immune system that protects the host in an unspecific manner. Interestingly, some of the first bacterial antimicrobial peptides were found in Enterococci (84) and even the pAD1 plasmid of E. faecalis(85). The host organism itself is immune through the coexpression of immunity proteins (81). According to the commonly accepted Shai−Matsuzaki−Huang model, antimicrobial peptides bind to the membrane surface and, after reaching a critical concentration, penetrate the membrane and lyze it through pore formation (86−89). This leads to collapse of the transmembrane electrochemical gradient and, finally, to cell death (88,89). To examine any potential antibiotic properties, we investigated the hemolytic and antimicrobial activity of Fst. Neither Fst nor Fst1−27 lyzed red blood cells. Both peptides were not able to lyze the membranes of erythrocytes up to a concentration of 100 μM peptide. Also, both peptides did not inhibit the growth of bacterial or fungal strains when tested in inhibition zone assays. The toxin and its truncated form were assayed against the yeast strains C. parapsilosis and S. cerevisiae against which they did not show any toxic activity. The same negative result was obtained for the Gram-negative E. coli and the Gram-positive B. subtilis. The growth of the species E. caccae which is closely related to the Fst producing E. faecalis(64) was also not inhibited by any of the two peptides. This is consistent with results of a study by Weaver et al. (46), where Fst was added to liquid cultures of E. faecalis without showing any influence on the growth of the bacterium. Although they found a negative effect on membrane integrity when overexpressing Fst in E. faecalis cells, the mechanism of action of Fst is obviously different from antimicrobial peptides. Despite some apparent similarities between toxins of type I TA systems and antimicrobial peptides, one has to keep in mind that the biological roles are very different. Antimicrobial peptides serve as a first defense mechanism against a broad range of microbes and are produced by organisms which themselves are not affected by them. Type I toxins, on the other hand, ensure the maintenance of a plasmid in a bacterial population by killing cells that did not inherit these genes. Thus they have to exert their toxic effect right in the cell which produces them, and it would make no sense to secrete them into the environment and also kill other cells which harbor the TA coding plasmid. In contrast to antimicrobial peptides, Fst works supposedly on a specific target inside the cell. While antimicrobial peptides are usually aligned parallel to the membrane surface prior to pore formation, Fst is in a transmembrane orientation. In addition, antimicrobial peptides are usually completely immersed in the membrane (62), and Fst contains a C-terminus which sticks into the cytosol. Weaver et al. also found that the primary function of Fst is likely an inhibition of macromolecule synthesis in the cell (46). While a perturbation in membrane integrity and topology was observed, the main effects of Fst are related to chromosome segregation and cell division. We believe that Fst is located in the membrane to facilitate interactions with a specific membrane-bound target rather than being directed against the membrane itself. Insertion into the membrane reduces the problem of finding its target from a three-dimensional (freely soluble) to a two-dimensional (gliding in the membrane) search. The C-terminus might become structured upon binding to its specific target. Intrinsically disordered proteins (90) or domains have a reduced energy barrier when binding to their targets as they can more readily adapt to the structure in the bound state without the need to unfold beforehand.
In conclusion, the structure and mode of membrane binding of Fst, the first atomic resolution structure of a type I toxin−antitoxin system component, are presented. Fst binds to membranes via the insertion of an α-helix between residues ∼4−26 and contains an intrinsically disordered stretch near the C-terminus. The N- and C-termini stick out of the membrane, likely with the highly charged C-terminal region pointing into the cytosol. The transmembrane orientation and charge distribution as well as the unstructured, protruding C-terminus distinguish it from antimicrobial peptides. It shows no hemolytic or toxic effects when Fst was applied externally to a series of microbes and is obviously directed against a specific membrane-bound target inside E. faecalis cells.
Financial support by the Austrian Science Foundation (Fonds zur Förderung der wissenschaftlichen Forschung, FWF) under project number 20020 to K.Z. and through the “Doktoratskolleg Molecular Enzymology” is gratefully acknowledged. C.G. thanks the Austrian Academy of Sciences (ÖAW) for a DOC fellowship.
Footnotes
Abbreviations: DPC, dodecylphosphocholine; Fst, faecalis plasmid stabilization toxin; Fst1−27, N-terminal 27 residues of Fst; Gd(DTPA-BMA), gadolinium diethylenetriaminepentaacetic acid bismethylamide; MD, molecular dynamics simulations; NOESY, nuclear Overhauser effect spectroscopy; POPC, 1-palmitoyl-2-oleoylphosphatidylcholine; PRE, paramagnetic relaxation enhancement; RMSF, root-mean-square fluctuations; TA, toxin−antitoxin; TOCSY, total correlation spectroscopy; TPC, tetradecylphosphocholine.
References
- Buts L.; Lah J.; Dao-Thi M. H.; Wyns L.; Loris R. (2005) Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 30, 672–679. [DOI] [PubMed] [Google Scholar]
- Gerdes K.; Christensen S. K.; Lobner-Olesen A. (2005) Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382. [DOI] [PubMed] [Google Scholar]
- Pandey D. P.; Gerdes K. (2005) Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelberg-Kulka H.; Glaser G. (1999) Addiction modules and programmed cell death and antideath in bacterial cultures. Annu. Rev. Microbiol. 53, 43–70. [DOI] [PubMed] [Google Scholar]
- Hayes F. (2003) Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301, 1496–1499. [DOI] [PubMed] [Google Scholar]
- Christensen S. K.; Mikkelsen M.; Pedersen K.; Gerdes K. (2001) RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U.S.A. 98, 14328–14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson R. D. (2007) Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189, 6089–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Melderen L.; Saavedra De Bast M. (2009) Bacterial toxin-antitoxin systems: more than selfish entities?. PLoS Genet. 5, e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozo E. M.; Hemm M. R.; Storz G. (2008) Small toxic proteins and the antisense RNAs that repress them. Microbiol. Mol. Biol. Rev. 72, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K.; Wagner E. G. H. (2007) RNA antitoxins. Curr. Opin. Microbiol. 10, 117–124. [DOI] [PubMed] [Google Scholar]
- Van Melderen L.; Thi M. H.; Lecchi P.; Gottesman S.; Couturier M.; Maurizi M. R. (1996) ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 271, 27730–27738. [DOI] [PubMed] [Google Scholar]
- De Jonge N.; Hohlweg W.; Garcia-Pino A.; Respondek M.; Buts L.; Haesaerts S.; Lah J.; Zangger K.; Loris R. (2010) Structural and thermodynamic characterization of Vibrio fischeri CcdB. J. Biol. Chem. 285, 5606–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loris R.; Dao-Thi M. H.; Bahassi E. M.; Van Melderen L.; Poortmans F.; Liddington R.; Couturier M.; Wyns L. (1999) Crystal structure of CcdB, a topoisomerase poison from E. coli. J. Mol. Biol. 285, 1667–1677. [DOI] [PubMed] [Google Scholar]
- Hargreaves D.; Santos-Sierra S.; Giraldo R.; Sabariegos-Jareno R.; de la Cueva-Mendez G.; Boelens R.; Diaz-Orejas R.; Rafferty J. B. (2002) Structural and functional analysis of the kid toxin protein from E. coli plasmid R1. Structure 10, 1425–1433. [DOI] [PubMed] [Google Scholar]
- Loris R.; Marianovsky I.; Lah J.; Laeremans T.; Engelberg-Kulka H.; Glaser G.; Muyldermans S.; Wyns L. (2003) Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 278, 28252–28257. [DOI] [PubMed] [Google Scholar]
- Madl T.; Van Melderen L.; Mine N.; Respondek M.; Oberer M.; Keller W.; Khatai L.; Zangger K. (2006) Structural basis for nucleic acid and toxin recognition of the bacterial antitoxin CcdA. J. Mol. Biol. 364, 170–185. [DOI] [PubMed] [Google Scholar]
- Oberer M.; Zangger K.; Gruber K.; Keller W. (2007) The solution structure of ParD, the antidote of the ParDE toxin antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 16, 1676–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard D. B.; Joanna L. M.; Edward N. B.; Vickery L. A. (2008) Crystal structure of PAE0151 from Pyrobaculum aerophilum, a PIN-domain (VapC) protein from a toxin-antitoxin operon. Proteins: Struct., Funct., Bioinf. 72, 510–518. [DOI] [PubMed] [Google Scholar]
- Murayama K.; Orth P.; de la Hoz A. B.; Alonso J. C.; Saenger W. (2001) Crystal structure of [omega] transcriptional repressor encoded by Streptococcus pyogenes plasmid pSM19035 at 1.5 Å resolution. J. Mol. Biol. 314, 789–796. [DOI] [PubMed] [Google Scholar]
- Li G.-Y.; Zhang Y.; Inouye M.; Ikura M. (2008) Structural mechanism of transcriptional autorepression of the Escherichia coli RelB/RelE antitoxin/toxin module. J. Mol. Biol. 380, 107–119. [DOI] [PubMed] [Google Scholar]
- Neubauer C.; Gao Y. G.; Andersen K. R.; Dunham C. M.; Kelley A. C.; Hentschel J.; Gerdes K.; Ramakrishnan V.; Brodersen D. E. (2009) The structural basis for mRNA recognition and cleavage by the ribosome-dependent endonuclease RelE. Cell 139, 1084–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao-Thi M. H.; Van Melderen L.; De Genst E.; Afif H.; Buts L.; Wyns L.; Loris R. (2005) Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol. 348, 1091–1102. [DOI] [PubMed] [Google Scholar]
- Kamada K.; Hanaoka F.; Burley S. K. (2003) Crystal structure of the MazE/MazF complex: molecular bases of antidote-toxin recognition. Mol. Cell 11, 875–884. [DOI] [PubMed] [Google Scholar]
- Takagi H.; Kakuta Y.; Okada T.; Yao M.; Tanaka I.; Kimura M. (2005) Crystal structure of archaeal toxin-antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat. Struct. Mol. Biol. 12, 327–331. [DOI] [PubMed] [Google Scholar]
- Kamada K.; Hanaoka F. (2005) Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 19, 497–509. [DOI] [PubMed] [Google Scholar]
- Dalton K. M., and Crosson S. A. (2010) Conserved mode of protein recognition and binding in a ParD-ParE toxin-antitoxin complex, Biochemistry (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown B. L.; Grigoriu S.; Kim Y.; Arruda J. M.; Davenport A.; Wood T. K.; Peti W.; Page R. (2009) Three dimensional structure of the MqsR:MqsA complex: a novel TA Pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 5, e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhart A.; Alonso J. C.; Strater N.; Saenger W. (2003) Crystal structure of the plasmid maintenance system epsilon/zeta: functional mechanism of toxin zeta and inactivation by epsilon 2 zeta 2 complex formation. Proc. Natl. Acad. Sci. U.S.A. 100, 1661–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo S. K.; Loll B.; Chan W. T.; Shoeman R. L.; Ngoo L.; Yeo C. C.; Meinhart A. (2007) Molecular and structural characterization of the PezAT chromosomal toxin-antitoxin system of the human pathogen Streptococcus pneumoniae. J. Biol. Chem. 282, 19606–19618. [DOI] [PubMed] [Google Scholar]
- Mattison K.; Wilbur J. S.; So M.; Brennan R. G. (2006) Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 281, 37942–37951. [DOI] [PubMed] [Google Scholar]
- Gerdes K.; Thisted T.; Martinussen J. (1990) Mechanism of post-segregational killing by the hok/sok system of plasmid R1: sok antisense RNA regulates formation of a hok mRNA species correlated with killing of plasmid-free cells. Mol. Microbiol. 4, 1807–1818. [DOI] [PubMed] [Google Scholar]
- Weaver K. E.; Tritle D. J. (1994) Identification and characterization of an Enterococcus faecalis plasmid pAD1-encoded stability determinant which produces two small rna molecules necessary for its function. Plasmid 32, 168–181. [DOI] [PubMed] [Google Scholar]
- Faridani O. R.; Nikravesh A.; Pandey D. P.; Gerdes K.; Good L. (2006) Competitive inhibition of natural antisense Sok-RNA interactions activates Hok-mediated cell killing in Escherichia coli. Nucleic Acids Res. 34, 5915–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen K.; Gerdes K. (1999) Multiple hok genes on the chromosome of Escherichia coli. Mol. Microbiol. 32, 1090–1102. [DOI] [PubMed] [Google Scholar]
- Kawano M.; Oshima T.; Kasai H.; Mori H. (2002) Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol. Microbiol. 45, 333–349. [DOI] [PubMed] [Google Scholar]
- Vogel J.; Argaman L.; Wagner E. G.; Altuvia S. (2004) The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr. Biol. 14, 2271–2276. [DOI] [PubMed] [Google Scholar]
- Fozo E. M.; Kawano M.; Fontaine F.; Kaya Y.; Mendieta K. S.; Jones K. L.; Ocampo A.; Rudd K. E.; Storz G. (2008) Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol. Microbiol. 70, 1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M.; Reynolds A. A.; Miranda-Rios J.; Storz G. (2005) Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 33, 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvaggi J. M.; Perkins J. B.; Losick R. (2005) Small untranslated RNA antitoxin in Bacillus subtilis. J. Bacteriol. 187, 6641–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver K. E.; Reddy S. G.; Brinkman C. L.; Patel S.; Bayles K. W.; Endres J. L. (2009) Identification and characterization of a family of toxin-antitoxin systems related to the Enterococcus faecalis plasmid pAD1 par addiction module. Microbiology 155, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield T. J.; Ehli E.; Kirshenmann T.; Franch T.; Gerdes K.; Weaver K. E. (2000) The antisense RNA of the par locus of pAD1 regulates the expression of a 33-amino-acid toxic peptide by an unusual mechanism. Mol. Microbiol. 37, 652–660. [DOI] [PubMed] [Google Scholar]
- Greenfield T. J.; Weaver K. E. (2000) Antisense RNA regulation of the pAD1 par post-segregational killing system requires interaction at the 5′ and 3′ ends of the RNAs. Mol. Microbiol. 37, 661–670. [DOI] [PubMed] [Google Scholar]
- Weaver K. E. (2007) Emerging plasmid-encoded antisense RNA regulated systems. Curr. Opin. Microbiol. 10, 110–116. [DOI] [PubMed] [Google Scholar]
- Weaver K. E.; Clewell D. B.; An F. (1993) Identification, characterization, and nucleotide sequence of a region of Enterococcus faecalis pheromone-responsive plasmid pAD1 capable of autonomous replication. J. Bacteriol. 175, 1900–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver K. E.; Walz K. D.; Heine M. S. (1998) Isolation of a derivative of Escherichia coli-Enterococcus faecalis shuttle vector pAM401 temperature sensitive for maintenance in E. faecalis and its use in evaluating the mechanism of pAD1par-dependent plasmid stabilization. Plasmid 40, 225–232. [DOI] [PubMed] [Google Scholar]
- Weaver K. E.; Weaver D. M.; Wells C. L.; Waters C. M.; Gardner M. E.; Ehli E. A. (2003) Enterococcus faecalis plasmid pAD1-encoded Fst toxin affects membrane permeability and alters cellular responses to lantibiotics. J. Bacteriol. 185, 2169–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göbl C.; Dulle M.; Hohlweg W.; Grossauer J.; Falsone S. F.; Glatter O.; Zangger K. (2010) Influence of phosphocholine alkyl chain length on peptide-micelle interactions and micellar size and shape. J. Phys. Chem. B 114, 4717–4724. [DOI] [PubMed] [Google Scholar]
- Kallick D. A.; Tessmer M. R.; Watts C. R.; Li C. Y. (1995) The use of dodecylphosphocholine micelles in solution NMR. J. Magn. Reson. B 109, 60–65. [DOI] [PubMed] [Google Scholar]
- Lauterwein J.; Boesch C.; Brown L. R.; Wuethrich K. (1979) Physicochemical studies of the protein-lipid interactions in melittin-containing micelles. Biochim. Biophys. Acta 556, 244–264. [DOI] [PubMed] [Google Scholar]
- Arora A.; Abildgaard F.; Bushweller J. H.; Tamm L. K. (2001) Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 8, 334–338. [DOI] [PubMed] [Google Scholar]
- Czerski L.; Sanders C. R. (2000) Functionality of a membrane protein in bicelles. Anal. Biochem. 284, 327–333. [DOI] [PubMed] [Google Scholar]
- Vinogradova O.; Sönnichsen F.; Sanders C. R. (1998) On choosing a detergent for solution NMR studies of membrane proteins. J. Biomol. NMR 11, 381–386. [DOI] [PubMed] [Google Scholar]
- Respondek M.; Madl T.; Göbl C.; Golser R.; Zangger K. (2007) Mapping the orientation of helices in micelle-bound peptides by paramagnetic relaxation waves. J. Am. Chem. Soc. 129, 5228–5234. [DOI] [PubMed] [Google Scholar]
- Cavanagh J., Fairbrother W. J., Palmer A. G., and Skelton N. J. (1996) Protein NMR Spectroscopy, Academic Press, San Diego, CA. [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Johnson B. A.; Blevins R. A. (1994) A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614. [DOI] [PubMed] [Google Scholar]
- Madl T.; Bermel W.; Zangger K. (2009) Use of relaxation enhancements in a paramagnetic environment for the structure determination of proteins using NMR spectroscopy. Angew. Chem., Int. Ed. Engl. 48, 8259–8262. [DOI] [PubMed] [Google Scholar]
- Berjanskii M. V.; Neal S.; Wishart D. S. (2006) PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 34, W63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 54, 905–921. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA.
- Koradi R.; Billeter M.; Wuthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graphics 14, 51–55. [DOI] [PubMed] [Google Scholar]
- Kosol S.; Zangger K. (2010) Dynamics and orientation of a cationic antimicrobial peptide in two membrane-mimetic systems. J. Struct. Biol. 170, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultmark D.; Engstrom A.; Bennich H.; Kapur R.; Boman H. G. (1982) Insect immunity: isolation and structure of cecropin D and four minor antibacterial components from Cecropia pupae. Eur. J. Biochem. 127, 207–217. [DOI] [PubMed] [Google Scholar]
- Carvalho M. d. G. S.; Shewmaker P. L.; Steigerwalt A. G.; Morey R. E.; Sampson A. J.; Joyce K.; Barrett T. J.; Teixeira L. M.; Facklam R. R. (2006) Enterococcus caccae sp. nov., isolated from human stools. Int. J. Syst. Evol. Microbiol. 56, 1505–1508. [DOI] [PubMed] [Google Scholar]
- Jerabek H.; Pabst G.; Rappolt M.; Stockner T. (2010) Membrane-mediated effect on ion channels induced by the anesthetic drug ketamine. J. Am. Chem. Soc. 132, 7990–7997. [DOI] [PubMed] [Google Scholar]
- Wolf M. G.; Hoefling M.; Aponte-Santamaria C.; Grubmuller H.; Groenhof G. (2010) g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J. Comput. Chem. 31, 2169–2174. [DOI] [PubMed] [Google Scholar]
- Berger O.; Edholm O.; Jahnig F. (1997) Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J.; Postma J. P. M.; van Gunsteren W. F.; Hermans J. (1981) Intermol. Forces, 331–342. [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theor. Comput. 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Vangunsteren W. F.; Dinola A.; Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Darden T.; York D.; Pedersen L. (1993) Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, Wiley, New York. [Google Scholar]
- Franzmann M.; Otzen D.; Wimmer R. (2009) Quantitative use of paramagnetic relaxation enhancements for determining orientations and insertion depths of peptides in micelles. ChemBioChem 10, 2339–2347. [DOI] [PubMed] [Google Scholar]
- Pintacuda G.; Otting G. (2002) Identification of protein surfaces by NMR measurements with a pramagnetic Gd(III) chelate. J. Am. Chem. Soc. 124, 372–373. [DOI] [PubMed] [Google Scholar]
- Zangger K.; Respondek M.; Gobl C.; Hohlweg W.; Rasmussen K.; Grampp G.; Madl T. (2009) Positioning of micelle-bound peptides by paramagnetic relaxation enhancements. J. Phys. Chem. B 113, 4400–4406. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Hofmann K.; Stoffel W. (1993) TMbase—a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 374, 166. [Google Scholar]
- Drider D.; Fimland G.; Hechard Y.; McMullen L. M.; Prevost H. (2006) The continuing story of class IIa bacteriocins. Microbiol. Mol. Biol. Rev. 70, 564–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman H. G. (2000) Innate immunity and the normal microflora. Immunol. Rev. 173, 5–16. [DOI] [PubMed] [Google Scholar]
- Hancock R. E.; Lehrer R. (1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16, 82–88. [DOI] [PubMed] [Google Scholar]
- Casaus P.; Nilsen T.; Cintas L. M.; Nes I. F.; Hernandez P. E.; Holo H. (1997) Enterocin B, a new bacteriocin from Enterococcus faecium T136 which can act synergistically with enterocin A. Microbiology 143, Part 72287–2294. [DOI] [PubMed] [Google Scholar]
- Clewell D. B. (2007) Properties of Enterococcus faecalis plasmid pAD1, a member of a widely disseminated family of pheromone-responding, conjugative, virulence elements encoding cytolysin. Plasmid 58, 205–227. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 3, 238–250. [DOI] [PubMed] [Google Scholar]
- Jenssen H.; Hamill P.; Hancock R. E. (2006) Peptide antimicrobial agents. Clin. Microbiol. Rev. 19, 491–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai Y. (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell. Biol. 6, 197–208. [DOI] [PubMed] [Google Scholar]