

Abstract
We have synthesized and purified silver nanoparticles (Ag NPs) (11.3 ± 2.3 nm) that are stable (non-aggregated) in cell culture medium and inside single living cells. We have developed new imaging methods to characterize sizes and number of single NPs in the medium and in single living cells in real-time and determine their stability (non-aggregation) in the medium and in single living cells at single NP resolution. These new approaches allow us to study toxic and therapeutic effects of single Ag NPs on tumor cells (L929, mouse fibroblast cells) with determined sizes and concentrations (doses) of NPs over time at single NP and single cell resolution. We found that Ag NPs inhibited the growth and division of tumor cells and their nuclei, at a dose and time dependent manner, showing significant inhibitory effects and abnormal cells with giant undivided nuclei or multiple nuclei beyond 12 h incubation. The results show that Ag NPs inhibited the segregation of chromosomes, but not their replications. Intracellular Ag NPs were well distributed in the cell population, and located in the nuclei and cytoplasm with higher number in the cytoplasm. This study demonstrates the possibility of using Ag NPs to inhibit the growth and division of the tumor cells and their cytotoxicity for potential therapeutic treatments, and offers a new method to count the number of single NPs in the medium for characterization their concentration and stability at single NP resolution over time.
Keywords: cytokinesis, karyokinesis, mouse fibroblast cells (L929), nanomedicine, silver nanoparticles, single cell imaging, single nanoparticle optics, toxicity, tumor cells
Introduction
Nanoparticles (NPs) exhibit unique physical, surface and chemical properties, offering the possibility of being used as optical and photonic probes for imaging and sensing, as carriers for smart drug delivery, and as medicines to treat a wide variety of diseases.1-5 For example, their small sizes allow them to penetrate into living cells and organisms.6-8 Their larger surface areas permit the surface modification with desired molecules for sensing,9-11 as well as for carrying larger payload of therapeutic molecules (drugs) for effective treatments.3, 4, 12 Their unusually high surface-area-to-volume ratios also make them highly unstable, subject to aggregation in suspension, and possibly chemical active. Even though mechanisms of NP reactivity (e.g., Ag) are still unknown, their interactions with living cells and organisms and their toxic effects have been reported.6, 7, 12-14 These effects offer the possibility of using NPs as unconventional medicines and spark great concern about their potential environmental impacts, which encourages a wide variety of research activities.6, 7, 12-16 In our previous studies, we focused on developing in vivo assays (zebrafish embryos), and study toxicity and biocompatibility of Ag and Au NPs in vivo.6, 17, 18 In this study, we investigate toxicity of Ag NPs on tumor cells and explore the possibility of using them as therapeutic agents.
Silver NPs have been used in diverse research fields, including optics, sensors, biological imaging, and catalysis.8-11, 19, 20 With such a wide range of applications and significant amounts of production, concerns about the potential impacts of Ag NPs in environment have led to the studies of their effects on eukaryotic cells (e.g., germ cells, rate liver cells, rat neuroendocrine cells, and rat alveolar macrophages), aiming to better assess and understand their cytotoxicity.21-23 Studies also reported that Ag NPs were germicidal and showed anti-viral properties against HIV, demonstrating that Ag NPs might act as potential therapeutic agents.2, 14, 15 Unfortunately, a majority of current studies used surface modified Ag NPs and/or unpurified Ag NPs, and did not develop and use effective means to characterize the stability, sizes and doses of NPs in the medium in situ, 2, 12-14, 16, 21-23 leading to inconclusive and sometimes contradictory results. It is unclear whether these effects are attributed to the reported doses and sizes of Ag NPs, aggregated NPs in the medium, or chemicals involved in synthesis of Ag NPs. To our knowledge, studies on the effects of purified and stable Ag NPs on tumor cells have not yet been widely reported. Typically, tumor cells can grow indefinitely. Therefore, we believe that they are excellent cellular models to study the potency of Ag NPs toward tumor cells and to better understand their inhibitory mechanisms.
In this study, we select one well-established tumor cell line, mouse fibrosarcoma cells (L929), which are derived from mouse cartilaginous tissue. This cell line has been widely used to study efficacy of chemical therapeutic agents and treatments (e.g., radiotherapy), and cell-cell communication among cancerous and immunological cells.24-32 We synthesize and characterize stable and purified Ag NPs, and study their effects on the growth, division, morphology, nuclei, intracellular DNA, and viability of the cells in a dose and time dependent manner at single NP and single cell resolution. The study of effects of Ag NPs on this particular cell line allows us to compare the efficacy of Ag NPs and the related mechanisms with other chemical therapeutic agents.
Results and Discussion
Synthesis and Characterization of Stable and Purified Ag NPs in Cell Culture Medium
We synthesized Ag NPs by reducing AgClO4 with NaBH4 and sodium citrate, and we purified the NPs by washing them with nanopure deionized (DI) water using centrifugation, as described in experimental section.6, 18 By removing chemicals from NP solution, the ionic strength of the solution was decreased, leading to an increase in thickness of electrical double layer on the surface of NPs and enhancement of the zeta potential of NPs. Therefore, the washed NPs are stable (non aggregated) in nanopure water for months, as we reported previously.6, 18
The sizes, shapes and optical properties of purified single NPs dispersed in the cell culture medium (MEM 1x with 1% PS-G and 10% FBS) were characterized using high-resolution transmission electron microscopy (HRTEM), dynamic light scattering (DLS) and dark-field optical microscopy and spectroscopy (DFOMS). The representative HRTEM images of single NPs in Figure 1A show nearly spherical shape NPs. The histogram of size distribution of single NPs (more than 100 NPs) determined using HRTEM in Figure 1B illustrates the average diameters of NPs at 11.3 ± 2.3 nm.
Figure 1.

Characterization of sizes, shapes and plasmonic optical properties of single Ag NPs dispersed in cell culture medium:
(A) HRTEM images of representative single Ag NPs show nearly spherical NPs. The scale bars are 5 nm.
(B) Histogram of more than one hundred of single Ag NPs measured by HRTEM in (A) shows the average diameter of NPs at 11.3 ± 2.3 nm.
(C) Representative dark-field image of 0.46 nM Ag NPs in the medium shows single blue, green and red NPs.
(D)Histogram of size distribution of Ag NPs in the medium measured by DLS shows the average diameter of NPs at 11.0 ± 2.1 nm.
(E) LSPR spectra of representative single Ag NPs show a peak wavelength (λmax) at 467, 531 and 616 nm, corresponding to blue, green and red NPs, respectively.
(F) Correlation of histogram of distribution of colors (λmax of LSPR spectra) of single NPs with their sizes determined using HRTEM in (A) shows 78% of blue (5-14 nm), 17% of green (14-20 nm) and 5% of red (> 20 nm) NPs, allowing us to determine the sizes of single NPs in solution in real-time using DFOMS.
Representative optical images of single Ag NPs characterized using DFOMS in Figure 1C show single blue, green and red NPs. Diameters of NPs dispersed in the medium measured using DLS are 11.07 ± 2.1 nm, as illustrated in Figure 1D. The localized surface plasmon resonance (LSPR) spectra of single NPs in Figure 1C are shown in Figure 1E, illustrating the blue, green and red NPs with the peak wavelength (λmax) at 467, 531 and 616 nm, respectively. The correlation of histogram of the LSPR spectra (colors) of single NPs measured using DFOMS with their sizes determined using TEM in Figure 1F allows us to use the LSPR spectra (colors) of single NPs to determine the sizes of single NPs in the medium in real time and to monitor their sizes over time.7, 14, 18 The result shows that the NP solution includes 78% of blue (5-14 nm in diameter), 17% of green (14-20 nm), and 5% of red (20-32 nm) NPs.
To study the stability of NPs in the medium, we determined the number of single NPs at single NPs using DFOMS. The number of single NPs per image versus time for 0.46 nM Ag NP solution remains essentially unchanged during 72 h of incubation (Figure 2A). Note that a minimal of 20 images at each given time for each experiment was acquired. In other words, a minimal of 20 volumes of NP solution at each given time for each experiment was sampled. The experiment was repeated three times. Therefore, a minimal of 360 NPs were determined at each given time to ensure the characterization of the bulk NP solution at single NP resolution.
Figure 2.
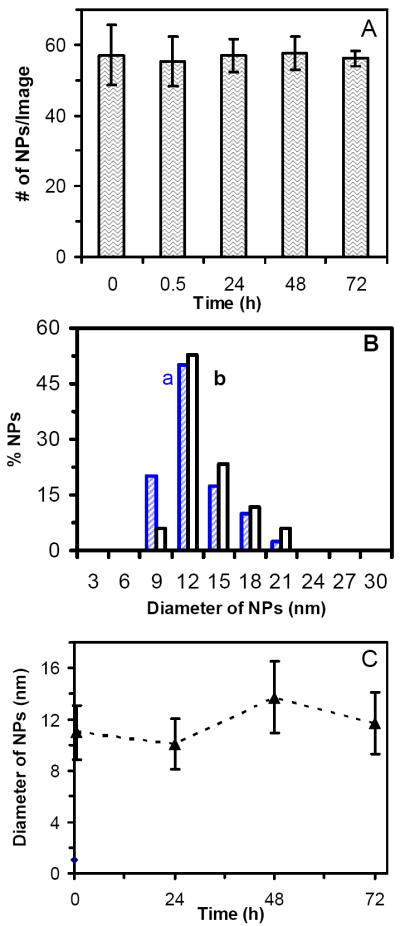
Characterization of stability of 0.46 nM Ag NPs incubated with cell culture medium at 37°C for 72 h at single NP resolution using DFOMS and in bulk solution using DLS:
(A) Plot of the average number of single Ag NPs per optical image versus incubation time shows that the total number of NPs (concentration of NPs) remains essentially constant over 72 h. For each experiment, at least 20 images similar to those in Figure 1C were acquired in 20 different locations (volumes) of NP solution at each given time using DFOMS. The average number of NPs per image is calculated by dividing total number of NPs with number of images.
(B) Histograms of size distributions of Ag NPs incubated with the medium at (a) 0 and (b) 72 h, measured by DLS, show the average diameters of NPs at 11.0 ± 2.1 nm and 11.7 ± 2.4 nm, respectively.
(C) Plot of average diameters of Ag NPs incubated in the medium measured by DLS versus incubation time shows that the average diameters of NPs remain essentially unchanged over 72 h. The average diameter of NPs in the medium over 72 h is 11.2 ± 2.8 nm.
We also determined the size distribution of bulk NPs in the medium using DLS (an ensemble method). The results in Figure 2B show that the average diameters of NPs in 0 and 72 h are at 11.0 ± 2.1 nm and 11.7 ± 2.4 nm , respectively. The average diameters of NPs in the medium measured over time in Figure 2C further indicate that the sizes of NPs remain essentially unchanged for 72 h. Taken together, the results in Figure 2 demonstrate that the NPs are stable (non-aggregated) in the medium for 72 h.
We also used the conventional ensemble method (Uv-vis spectroscopy) to determine the stability of NPs in the medium. UV-vis absorption spectra of 0.46 nM (22 μg/mL) NPs in nanopure DI water, in the medium, and the medium alone in Figure 1S-A in supporting information (SI) show the peak wavelength (λmax) of 395, 406 and 406 nm, respectively. The results suggest that the absorbance of medium attributes to the high baseline and shift of λmax of the absorption spectrum of NPs in the medium. Notably, the peak wavelength and absorbance of medium remain unchanged over time. The absorbance and peak wavelength of NPs in nanopure DI water also remain constant for 72 h (Figure 1S-B), showing that the NPs are stable in DI water, as we reported previously.6, 18
However, the absorption spectra of the NPs in the medium measured over time for 72 h show that the absorbance of the spectra decreases over time while its λmax remains unchanged (Figures 1S-C and D in SI). The unchanged λmax indicates that the sizes and shapes of NPs remain unchanged, showing that NPs are not aggregated in the medium for 72 h. The decreased absorbance over time suggests a possible decrease in absorptivity and/or reflectivity (scattering) of NPs in the medium due to the change of dielectric constant of embedded medium of NPs, and possible adsorption and exchange of surface molecules of NPs with the molecules of the medium. It takes time for exchange and adsorption of surface molecules to reach equilibrium, and for the electric double layers to re-establish its equilibrium on the surface of NPs in the medium. Therefore, we observed a gradual decrease in absorbance. Nonetheless, more studies are needed to determine detailed mechanisms of such interesting phenomena.
Note that the absorbance of NPs in the medium is attributed to the scattering and absorption of NPs, and medium. Therefore, this conventional ensemble method is not suited to determine the concentration of NPs in the medium over time, before it reaches equilibrium. By measuring the number of NPs at single NP resolution using DFOMS (Figure 2A), we ensured that number of NPs remained unchanged over time (72 h). Furthermore, we measured the sizes of NPs in the medium over time using DLS and the results in Figures 2B and C show that the sizes of NPs remain nearly constant. The slightly larger sizes of NPs at 72 may be attributed to the exchange of surface molecules of NPs with the molecules in the medium. These results further demonstrate the importance of characterization of stability of NPs at single NP resolution, and significance of the single NP imaging tool (DFOMS).
Notably, it is essential to characterize the stability of NPs in the medium over time in order to study the effect of NPs with determined sizes and concentrations (doses) on the growth and division of tumor cells. This is because cells need to be cultured in the medium with NPs over time and instability of the NPs in the medium will affect the doses and sizes of NPs that are incubated with the cells in the medium, providing misleading results.
Inhibitory Effects of Ag NPs on Cell Growth and Division
Tumor cells cultured in the absence and presence of various doses (concentrations) of stable and purified Ag NPs (0.23 and 0.46 nM) (11 and 22 μg/mL) were imaged over time (Figure 3), showing that the morphologies and number of cells that were attached on the surface of flasks depended upon the doses of NPs and incubation time. Note that mouse fibroblast cells (L929) are adherent cells and only those cells attached on the surface can grow. Thus, we used the amount of cells on the desired surface area of the flasks to follow their growth. No cells were attached on the surface of the flasks at the very beginning of incubation time (Figure 3a), and the number of cells on the surface increased with time at the first 12 h, which was due to the time needed for the cells to attach onto the surface of the flasks. Interestingly, the number of the cells cultured in the medium with 0 and 0.23 nM Ag NPs continued to increase with time, while the number of the cells cultured in the medium with 0.46 nM Ag NPs remained essentially unchanged.
Figure 3.

Study of inhibitory effects of Ag NPs on the growth of single tumor cells in a dose and time dependent manner. Representative optical images of cells on the surface of flasks, which contain the medium with (A) 0, (B) 0.23 and (C) 0.46 nM Ag NPs, at (a) 0, (b) 12, (c) 24, (d) 48, and (e) 72 h. The scale bar = 50 μm
The cell growth curves (number of the cells versus time) in the medium containing 0, 0.23 and 0.46 nM Ag NPs (Figure 4A) show that the cell growth rates were significantly affected by Ag NPs in a dose and time dependent manner. After 12 h of incubation, Ag NPs showed significant inhibitory effects on the cell growth. The inhibitory effect of Ag NP increases with time at a dose dependent manner. Notably, the inhibitory effects of 0.46 nM Ag NPs are higher than that of 0.23 nM Ag NPs. The results suggest that Ag NPs inhibit the cell growth after its first cell cycle and the higher dose (0.46 nM) of Ag NPs inhibits the cell growth much more effectively than the lower dose (0.23 nM) of NPs.
Figure 4.

Study of cytotoxic and genotoxic effects of Ag NPs on the tumor cells:
Plots of (A) number of cells and (B) number of nuclei per mm2, versus incubation time, for cells that are cultured in the medium containing Ag NPs of: (a) 0, (b) 0.23, and (c) 0.46 nM, show that Ag NPs significantly affect (A) the growth and (B) division of the cells, on a dose and time dependent manner, after 12 h of culture time.
(C) Fluorescence images of EtBr-DNA of single cells characterized using comet assay. The cells are incubated with the medium containing (a) 0, (b) 0.23 and (c) 0.46 nM of Ag NPs for 72 h, showing that the intracellular DNA of single cells is intact. (d) The positive control experiment carried out by using the cells that were treated with 880 μM of H2O2 for 15 min, shows a long trail (comet) of intracellular DNA of single cells under gel electrophoresis, which suggests the fragmentation of intracellular DNA by H2O2. The scale bar = 20 μm
In Figures 3B and C, the cells with multiple nuclei or single larger nuclei were observed after 12 h of cell culture in the presence of 0.23 and 0.46 nM Ag NPs, respectively. To investigate whether Ag NPs inhibit the division of the cells, we plot the number of their nuclei versus time (Figure 4B). The curves in Figure 4B show the similar trend as those in Figure 4A, indicating that the Ag NPs affect the replication and division of nuclei which leads to the inhibitory effect of the cell growth. Interestingly, the number of nuclei for the cells cultured in the medium with 0.23 and 0.46 nM Ag NPs (Figure 4B) is slightly higher than the number of their cells (Figure 4A) at each given time, after 12 h incubation, correspondingly. The results show some cells with multiple nuclei, suggesting that the Ag NPs also inhibit the cell division.
To study whether Ag NPs affected the intracellular DNA, we investigated the DNA of the cells cultured in the medium with 0, 0.23 and 0.46 nM Ag NPs for 72 h using a comet assay (single cell gel electrophoresis assay) and the results are shown in Figure 4C: a-c, respectively. As a positive control experiment, we also studied the comet assay of the cells treated with H2O2, which led to the fragmentation of intracellular DNA (Figure 4C: d). The results indicate that the intracellular DNA of single cells was intact and no tail was observed in the gel electrophoresis of the cells cultured in the medium with 0, 0.23 and 0.46 nM Ag NPs for 72 (Figure 4C: a-c), suggesting that the DNA was not damaged by Ag NPs. On the contrary, a tail (comet) of DNA was observed for the cells treated by H2O2, illustrating the fragmentation of intracellular DNA by H2O2, as shown in Figure 4C: d.
The comet assay is a sensitive technique for quantifying DNA damage in single cells and has been widely used for the study of genotoxicity and effectiveness of chemotherapy.33-35 Comet assay is conducted by electrophoresis of single suspended lysed cells and fluorescence imaging of stained DNA of the cells. Their fluorescence images resemble comets with a distinct head and tail, which is composed of intact DNA, and damaged (single-strand or double-strand breaks) and fragmented DNA, respectively. The ratio of tail length to diameter of the head of EtBr-stained DNA of single cells is used to measure the degree of damaged DNA in this study.
Effects of Ag NPs on Nuclear Division and Viability of Single Tumor Cells
To further investigate the effects of Ag NPs on nuclear division and viability of cells with abnormal (large or multiple) nuclei, we cultured the cells on the coverslips in the petri-dishes, instead of flasks, which allowed us to image the cells with 100x objective and characterize the nuclei of single cells with higher resolution. Note that thickness of the flasks prohibits the use of 100x objective. The optical images of cells cultured on the coverslips in the medium with 0 and 0.46 nM Ag NPs for 72 h in Figures 5A and B show that the cells cultured in the absence of Ag NPs exhibit normal morphologies (spindle shape) with single nuclei in individual cells. On the contrary, the cells cultured in 0.46 nM Ag NPs illustrate abnormal morphologies (spherical shape) and multiple nuclei in single cells, suggesting that Ag NPs inhibit the cell division and serve as inhibitors of cytokinesis of tumor cells (L929).
Figure 5.

Imaging and characterization of effects of Ag NPs on morphology, viability, and membrane integrity of the tumor cells. Optical images of cells cultured on the coverslips in the medium with (A) 0 and (B) 0.46 nM Ag NPs for 72 h, show drastic effects of Ag NPs on the cellular morphologies.
(C) Selected images of single cells from (A) and (B) show (a) normal and (b) abnormal cells with undivided giant nuclei and (c-f) multiple nuclei.
(D) Fluorescence images of cells in (C), which are characterized using mitotracker-orange assay, show that the cells are viable.
(E) Absorbance of reduced MTS by intracellular dehydrogenase at λmax of 490 nm, for the cells cultured in the flasks containing the medium with 0, 0.23 and 0.46 nM Ag NPs for 72 h and assayed by MTS assay, shows that the cells are alive
(F) Plot of the percentage of viable cells, cultured in the flasks containing the medium with 0, 0.23 and 0.46 nM Ag NPs, and characterized using trypan blue assay, show that the majority of cells (> 94%) incubated with Ag NPs are alive and their membranes are intact.
The scale bar in (A) and (B) = 50 μm, (C) and (D) = 20 μm
The representative cells selected from Figures 5A and B are shown in Figure 5C, further illustrating the normal cells with single nucleus in (a), in contrast to single abnormal cells with a giant nucleus in (b) and multiple nuclei in (c-f). Mitotracker assay was used to characterize the viability of single cells (Figure 5D), showing that the normal cells with single nuclei in (a) and abnormal cells with a giant nucleus in (b) and multiple nuclei in (c-f) were alive. Mitochondria, cellular powerhouses, produce ATP (i.e., phosphorylation of ADP) via respiration and regulate cellular metabolism. Thus, active mitochondria can serve as a primary indicator of cellular viability. The mitotracker orange in its reduced form does not fluoresce until it diffuses into an actively respiring cell, where it is oxidized to a fluorescent mitochondrion-selective probe and then accumulated in the mitochondria.36 Only active mitochondria can oxidize the mitotracker orange (non-fluorescent form) to its fluorescent form, which allows us to use the fluorescence of mitotracker orange to detect the viability of cells. Fluorescence images of cells characterized using mitotrack orange in Figure 5D show that the normal and abnormal cells are alive and mitochondria are located in cytoplasm of the cells.
We also studied the viability of bulk cells cultured in the flasks using MTS and trypan blue assays, which determined the cellular viability by detecting active dehydrogenase enzymes and integrity of cellular membrane (Figures 5E and F), respectively. The reduction of MTS occurs only when dehydrogenase enzymes are active. In the presence of phenazine methosulfate (PMS), the reduced MTS generates a water-soluble formazan product with an absorbance maximum at 490-500 nm in PBS buffer, serving as a colorimetric assay to measure viable (living) cells. Absorbance of 105 cells in each well assayed using MTS in Figure 5E shows that the cells cultured in the medium with 0, 0.23 and 0.46 nM Ag NPs for 72 h are alive. Interestingly, the highest absorbance was observed for the cells cultured in 0.46 nM Ag NPs, and the least absorbance for the cells cultured in the absence of Ag NPs, suggesting the highest concentration of dehydrogenase enzymes for the cells cultured in the presence of Ag NPs. This interesting phenomenon is attributed to more undivided cells in the presence of Ag NPs. Single undivided cells with multiple nuclei were counted as single cells, instead of multiple cells, for MTS assay, which led to the higher amount of dehydrogenases and the higher absorbance. Notably, control experiments show that Ag NPs alone do not contribute to the absorbance of the cells assayed using MTS. The results from trypan blue assay in Figure 5F show that membranes of the cells cultured with 0, 0.23 and 0.46 nM Ag NPs for 72 h are intact and the cells are alive.
We further characterized nuclei and viability of single living cells over time using DRAQ5 and calcein AM assay, respectively. DRAQ5 is highly permeable to nuclei of living cells and has a high affinity with DNA.37 Its signature fluorescence emission at 665 nm allows us to image the intra-nuclear DNA of single living cells. Calcein AM is a non-fluorescence compound and can passively diffuse into living cells, where calcein AM is hydrolyzed by intracellular esterases, leading to intensive green fluorescence at 516 nm.38 This colorimetric assay allows us to determine viable cells, based upon the activity of esterases.
Optical images of the cells cultured with 0.46 nM Ag NPs for 72 h in Figure 6A show the normal morphology of cells in (a) and single abnormal cells with a giant nucleus in (b) and multiple nuclei in (c-f). Their fluorescence images in Figure 6B clearly illustrate their nuclei lighted up by red fluorescence of DRAQ5. Their intensive green fluorescence images in Figure 6C, resulted from calcein AM assay, show that these cells are viable.
Figure 6.
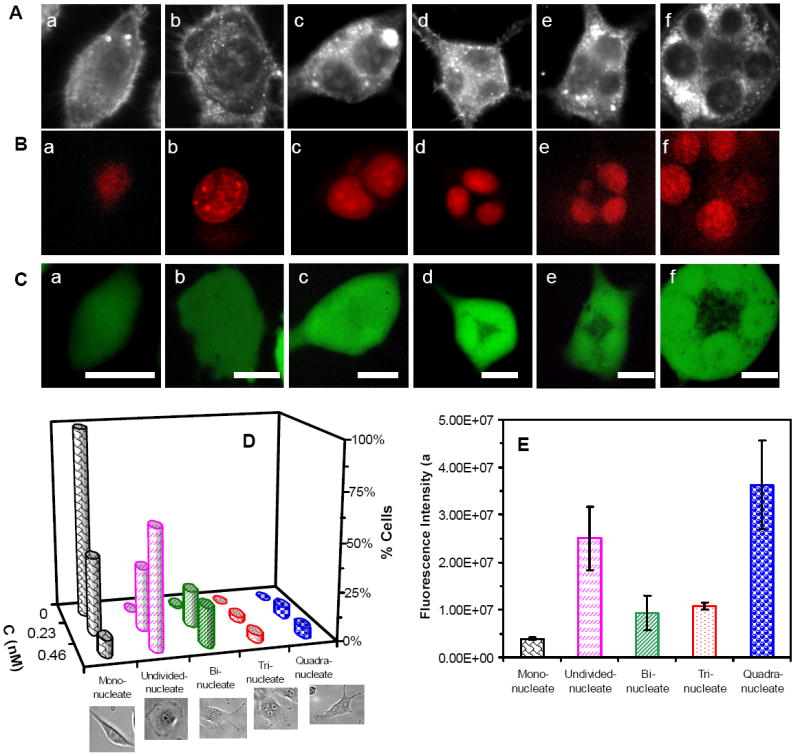
Imaging and study of effects of Ag NPs on nuclear division and viability of single tumor cells.
(A) Optical images of cells cultured on the coverslips in the medium containing 0.46 nM Ag NPs for 72 h, show (a) normal, (b) abnormal and (c-f) undivided cells.
(B-C) Fluorescence images of cells in (A), assayed by (B) Draq 5 and (C) calcein AM, show that (B) giant and multiple nuclei in single cells, and (C) these cells are alive.
(D) Plots of histograms of cells with normal and abnormal nuclei, for the cells that are cultured in the medium containing 0, 0.23, and 0.46 nM Ag NPs, show normal cells in the absence of NPs, and abnormal cells with giant nuclei and multiple nuclei in the presence of NPs. The percentage of abnormal cells increases with Ag NP concentration.
(E) Plot of fluorescence intensity of single cells with normal mono nucleus and abnormal nuclei, assayed by DRAQ5, as those in (B), shows that single abnormal cells with single undivided giant nuclei or multiple nuclei (two, three and four nuclei) contain much higher amounts of nuclear DNA than single normal cells with mono nucleus. The cells are cultured as described in (D). The scale bar in (A-C) = 20 μm
Plots of percentage of cells with normal mono nucleus, a giant (undivided) nucleus, bi-nuclei, tri-nuclei, and quadri-nuclei for the cells cultured with 0, 0.23 and 0.46 nM Ag NPs for 72 h are illustrated in Figure 6D. The results show that in the absence of Ag NPs (control experiment), 99% of cells exhibit normal morphology (spindle shape) and single nuclei, and 1% of cells show bi-nuclei, that are attributed to on-going cell division. In the presence of 0.23 nM Ag NPs, 40% of cells show normal morphology with single normal nuclei, while 32% of cells exhibit abnormal morphology (spherical shape) with single giant undivided nuclei, and 19%, 3% and 6% of cells show abnormal morphologies with bi-nuclei, tri-nuclei and quadri-nuceli in individual cells, respectively. In 0.46 nM Ag NPs, only 9% of cells exhibit normal morphology with single normal nuclei, while 61% of cells show abnormal morphology (spherical shape) with single giant undivided nuclei, and 20%, 4% and 5% of cells display abnormal morphologies with bi-nuclei, tri-nuclei and quadri-nuceli in individual cells, respectively. The ratios of cells to their nuclei in Figure 6D agree with those observed in Figures 4A and B. The nuclear diameters for cells cultured in the medium with 0, 0.23 and 0.46 nM Ag NPs for 72 h are 7.6-13.8, 5.4-21.0 and 7.6-23.4 μm, respectively. Take together, the results show the dose-dependent effects of Ag NPs on the nuclear and cellular division, and Ag NPs can inhibit cytokinesis and karyokinesis of the tumor cells.
We further characterized the amounts of DNA in individual cells with single normal nuclei, single abnormal giant nuclei and multiple nuclei by measuring the integrated fluorescence intensity of DRAQ5 stained-DNA in the nuclei of single cells. The results in Figure 6E show that the undivided giant nuclei contain higher amount of DNA than single normal nuclei; the bi-nuclei, tri-nuclei and quadri-nuclei have roughly twice, three and four times more DNA than single normal nuclei, showing that single undivided giant nuclei and multiple nuclei in single cells are indeed attributed to the undivided nuclei and cells, respectively. The results suggest that Ag NPs did not inhibit the replication of DNA in the nuclei. Ag NPs (zeta potential -34 mV) and DNA are both negatively charged which prevents them from interactions and avoids the effect of Ag NPs on the replication of DNA.
Imaging and Characterization of Intracellular Single Ag NPs
We characterized intracellular Ag NPs by imaging the optical cross section of living cells using DFOMS. Representative optical images of the cells in Figure 7A show that multiple colors of Ag NPs are presence in both nucleus and cytoplasm of the cell in (a) and in cytoplasm alone in (b). We studied total number of intracellular Ag NPs with different colors (sizes) in 25 representative cells over 72 h of cell culture time. The results in Figure 7B show that the number of intracellular Ag NPs increases with time and most blue NPs (5-14 nm), less green NPs (14-20 nm) and least red NPs (20-32 nm) are found inside the cells. Intracellular single Ag NPs are characterized using their LSPR spectra, which are similar to those observed in the medium in Figure 1E. Color distributions of intracellular Ag NPs in Figure 7B are similar to those Ag NPs in medium (Figure 1F) prior to 24 h of incubation while the percentage of red NPs increases slightly over time after 24 h of incubation. The red shift of LSPR spectra (colors) of single NPs is likely attributed to the adsorption of biomolecules (e.g., proteins) on the surface of NPs.10, 11 Notably, only small portion (< 5%) of NPs show the red-shift of their LSPR spectra. The results show that transport of single Ag NPs into the cells are independent upon their sizes (5-32 nm) and the majority of Ag NPs remain non-aggregated in the medium and inside the cells over time.
Figure 7.

Imaging and characterization of intracellular single Ag NPs in single living tumor cells:
(A) Optical images of single cells show NPs accumulated in (a) both the cytoplasm and nuclei of the cells, and (b) in the cytoplasm only.
(B) Total number of intracellular (a) blue (5-14 nm), (b) green (14-20 nm) and (c) red (20-32 nm) NPs in 25 cells increases over time.
(C) Total number of intracellular NPs in the (a) nuclei and (b) cytoplasm of 25 cells shows more NPs in the cytoplasm.
(D) Percentage of the cells with NPs versus time shows that it increases with time and the majority of cells, 69%, 81%, 94% and 100% of cells, have NPs at 6, 12, 24, and 48-72 h, respectively.
The cells in (A-D) are cultured on the coverslips in the medium containing 0.46 nM Ag NPs, and imaged at 6, 12, 24, 48, and 72 h, respectively. Total 25 cells at each given time in (B-D) are analyzed. The scale bar in (A) = 20 μm
We determined distribution of the amounts of Ag NPs in the nuclei and cytoplasm of single cells (Figure 7C), showing that 2-7 times more Ag NPs are located in cytoplasm than in nuclei and the amounts of Ag NPs in both the cytoplasm and nuclei increase with time. The number of cells with intracellular Ag NPs increases with incubation time (Figure 7D), showing that 69%, 81%, 94%, and 100% of the cells have intracellular Ag NPs at 6, 12, 24, and 48-72 h, respectively. Taken together, these results show that Ag NPs are well distributed in the medium and in the cells, and the majority of cells have intracellular NPs, which effectively inhibits the division and growth of tumor cells and offers the possibility of using Ag NPs for effective therapy.
Summary
In summary, we have successfully synthesized and prepared purified and stable Ag NPs (11.3 ± 2.3 nm) and developed new imaging tools to characterize their stability in cell culture medium and in single cells at single NP resolution using DFOMS. The imaging approaches overcome the uncertainty of using an ensemble method (UV-vis spectroscopy) to determine the concentration of NPs over time. We found that the number of Ag NPs in the medium remained constant for 72 h, showing that NPs were stable in the medium over time. The stability of NPs in the medium allowed us to culture the tumor cells (L929) in the presence of 0.23 and 0.46 nM Ag NPs, and characterize their effects on the intracellular DNA, nuclei and cells over time. Our results show that Ag NPs effectively inhibited the cell growth by disrupting cytokinesis and karyokinesis of cells (division of cells and nuclei), leading to undivided giant nuclei and multiple nuclei in single cells. The results suggest that Ag NPs affected the segregation of DNA (chromosomes), leading to single giant undivided nuclei in single cells. Interestingly, Ag NPs neither damaged DNA nor affected their replication. Ag NPs were well distributed in the entire cell population and the majority of intracellular NPs remained non-aggregated in the cells. The number of intracellular Ag NPs increased with time and they were located in the nuclei and cytoplasm with higher number in the cytoplasm. Work is in progress to identify the mechanisms of inhibitory effects of Ag NPs on the tumor cells and to explore the possibility of using Ag NPs as effective therapeutic agents to treat the tumor growth.
Experimental Section
Reagents and Supplies
Silver perchlorate monohydrate (≥ 99%, Alfa Aesar), sodium citrate dihydrate (≥ 99%, Sigma), and sodium borohydride (≥ 98%, Sigma) were used, as received, for synthesis of Ag NPs. The mouse fibroblast cells (L929, ATCC) at 2-4 passages were used for the study. Modified eagle powdered medium without phenol red (MEM) (sigma), sodium bicarbonate (sigma), fetal bovine serum (ATCC), 100X penicillin-streptomycin-glutamine (ATCC), and sodium pyruvate (Bio Whittaker), were used to prepare cell culture medium (MEM 1X with 1% PS-G and 10% FBS). Hanks’ Balanced Salt Solution (HBSS, without Ca2+, Mg2+ and phenol red; pH = 7.4) (ATCC) was used to rinse and maintain the cells. Trypan blue (0.4%, Signa) and ethidium bromide (Sigma) were used to study cellular viability and DNA damage in single cells using trypan blue assay and comet assay, respectively. Calcein-AM (Invitrogen), mitotracker-Orange (Invitrogen), MTS/PMS (Promega), and DRAQ5 (Axxora) were used as received, and as intracellular fluorescence probes to further characterize cellular viability. Calcein-AM, MTS, PMS, and DRAQ5 represent calcein acetoxymethyl ester, (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium), phenazine methosulfate, and 1,5-bis{[2-(di-methylamino) ethyl]amino}-4, 8-dihydroxyanthracene-9,10-dione, respectively. We used nanopure deionized (DI) water (18 MΩ, Barnstead) to prepare solutions and rinse glassware.
Synthesis and Characterization of Ag Nanoparticles
We synthesized Ag NPs (11.3 ± 2.3 nm), as we described previously.6, 18 Briefly, sodium borohydride (10 mM) and sodium citrate (3 mM) were well mixed in ice-cold nanopure DI water (247.5 mL). Silver perchlorate (2.5 mL, 10 mM) was added into the mixture, and stirred overnight at room temperature. The solution was then filtered using 0.2 μm sterilized membrane filters (Whatman). Ag NPs were immediately washed twice with DI water using centrifugation at 15,000 rcf (relative centrifugal force) to prepare purified and stable Ag NPs. The washed Ag NPs were resuspended in DI water, and stored in dark at 4° C until use. As we reported previously,6, 18 the washed NPs dispersed in DI water are stable (non-aggregation) for months, because the washing steps help to reduce ionic strength of solution and thereby enhance zeta potentials of NPs and higher stability. The NP concentrations were calculated as described in our previous studies.6, 7, 39
We suspended the washed Ag NPs (0.46 nM) in cell culture medium, and incubated them in a CO2 incubator (37 °C, 5% CO2, 100% humidity) for 72 h. We characterized the stability of Ag NPs in the medium over time, at single NP resolution using our dark-field optical microscopy and spectroscopy (DFOMS) and high-resolution transmission electron microscopy (HRTEM) (FEI Tecnai G2 F30 FEG, at 300 kV), and with ensemble average of bulk NPs using dynamic light scattering (DLS, Nicomp 380ZLS particle sizing system) and UV-Vis spectroscopy (Hitachi U-2010).
We have described DFOMS (also named as SNOMS by us) in our previous studies for real-time imaging and spectroscopic characterization of single NPs in solutions, in single living cells and in single zebrafish embryos, and for single molecule detection.6, 7, 14, 40-44 In this study, we used EMCCD (PhotonMAX) equipped with a spectrograph (SpectraPro-150) (Roper Scientific) to characterize LSPR spectra of single Ag NPs. We also used EMCCD, a high-resolution CCD camera (Micromax, 5 MHz Interlin) (Roper Scientific), and color digital camera to image and characterize single Ag NPs in solution, in cell culture media and in single cells. The DFOMS is equipped with a dark-field condenser (Oil 1.43-1.20, Nikon) and a 100x objective (Nikon Plan fluor 100x oil, iris, SL. N.A. 0.5-1.3, W.D. 0.20 mm).
Study of Effects of Ag NPs on Single Tumor Cells: Cell Culture, Imaging and Assays
In Flasks
We cultured the cells in the flasks as described previously.10 Briefly, we first grew the cells (L929) in a culture flask using the medium (MEM 1X with 1% PS-G and 10% FBS). As the monolayer cells reached 80% confluence (106 cells/mL) on the surface of the flask, we transferred 5×105 cells to each of three flasks (T-25) containing the medium (5 mL) with 0, 0.23 and 0.46 nM (or 11 μg/mL or 22 μg/mL) Ag NPs, respectively, and placed the flasks in a CO2 incubator (Forma; 37 °C, 5% CO2, 100% humidity). We followed the cell growth for 72 h by imaging individual cells in ten desired surface areas (each with 0.15 mm2) of the flasks using a bright field optical microscopy (Nikon, 20x objective) at each given time. Note that each experiment was repeated at least 3 times.
At 72 h, we determined cell viability using MTS assay 45, 46 and integrity of cellular membrane (cell viability) using trypan blue assay. The assays were carried out as described in the following. We removed the medium from each flask that contained cultured cells with 0. 0.23 and 0.46 nM Ag NPs. Note that the cells (L929) are adherent cells and they remained on the surface of the flasks. We thoroughly rinsed the cells using HBSS buffer. We then removed the cells from the surface of the flasks by incubating trypsin-EDTA with the cells in the incubator for 1 min. The cell suspension was then centrifuged to remove the buffer. The cells in the pellet were re-suspended in the HBSS buffer and washed three times with HBSS using centrifugation. We determined the concentration of the cells (the number of cells in a given volume) from each flask using a hemocytometer.
For MTS assay, based upon the cell concentration determined using the hemocytometer, we pipetted the volume of each cell suspension from each flask to have 105 cells into each well of three wells in a 96-well plate. The volume of cell solution in each well was adjusted to 100 μL using HBSS buffer. Three wells in the plate for each experiment were filled out with HBSS buffer (100 μL) as a blank control experiment. We added freshly prepared MTS/PMS solution (20 μL) into each well. The plate was then covered with the Saran wrap and placed in the CO2 incubator for 2 h. The reaction was stopped by adding 1% SDS in HBSS buffer (25 μL) into each well. The absorbance at 490 nm of each well in the plate was measured using an ELISA plate reader.
For trypan blue assay, we incubated the cells with trypan blue (0.4%) for 5 min and imaged the cells using the bright field microscopy. The stained (dead) and unstained (viable) cells were counted, and viability of cells was calculated by dividing the viable (unstained) cells with total number of the cells. More than 300 cells in total were investigated at each time point for each experiment
At 72 h, we also investigated the possible fragmentation of intracellular DNA and potential apoptotic cells using a comet assay (single cell gel electrophoresis assay). The cells (1 mL of 106 cells/mL) were characterized in a low melting point agarose gel using electrophoresis at 25 V for 30 min, then stained with EtBr and imaged using the epi-fluorescence microscopy. We measured the length of fluorescent intracellular EtBr-DNA of single cells to determine the possible fragmentation of intracellular DNA. As a positive control, we incubated 880 μM of H2O2 with the cells, cultured in the absence of NPs, for 15 min, to fragment intracellular DNA, and determined the length of fluorescent intracellular EtBr-DNA using the comet assay. We analyzed at least 100 cells for each experiment.
On Coverslips
We also cultured the cells on the coverslips by transferring the cells to the sterile coverslips (0.08 mm thickness), immersing them in petri dishes that contained the medium with 0 and 0.46 nM Ag NPs, and placing petri dishes in the CO2 incubator (37 °C, 5% CO2, 100% humidity). We took the coverslips with the cells from the medium at each given time, well rinsed them with HBSS buffer, and imaged morphologies and number of the cells, and characterized intracellular DNA and Ag NPs at the single-cell and single-NP resolution using DFOMS.
At 72 h, we characterized the viability of cells on the coverslips using calcein AM and mitotracker-orange (mitochondrion-select) assays. Furthermore, we used DRAQ5 to identify nuclei of single living cells by imaging fluorescence of intracellular DNA-DRAQ5 of single cells. We performed the assays as follows: (i) we well rinsed the cells on the coverslips with HBSS buffer, and incubated them with 0.5 μM calcein AM, 2 μM mitotracker-orange, or 20 μM DRAQ5 for 20 min, respectively. (ii) We then imaged the cells using epi-fluorescence microcopy, equipped with CCD camera (Micromax, Roper Scientific) and fluorescence filter cubes (Chroma Tech) containing a band-pass excitation filter (460 ± 80 nm; 488 ± 75 nm; 530 ± 30 nm), bandpass emission filter (530 ± 30 nm; 605 ± 30 nm; 650 ± 30 nm), and a dichroic mirror (475 nm; 565 nm; 565 nm), respectively.
Imaging and Characterization of Single Ag Nanoparticles in Single Living Cells
To determine the sizes of intracellular single Ag NPs and their locations (e.g., cytoplasm or nucleus) inside the cells, we imaged and characterized the NPs in the cells cultured on the coverslips using our DFOMS. We took the cells cultured on the coverslips out of the medium at each given time, well rinsed the cells using HBSS, and imaged the cells and intracellular NPs using DFOMS. We imaged the surface of the cells by focusing on the cell surface that is near to the objective, then focusing on the center of the cells and finally on the surface (other side) of the cells that is away from the objective by gradually moving the fine focus knob of the microscope. This approach ensured that we imaged intracellular NPs, but not NPs on the cell surface. We characterized single Ag NPs inside the cells using their unique LSPR spectra by DFOMS, similar to those shown in Figure 1E. At each given time, we imaged and characterized single Ag NPs in 25 selected cells.
Data Analysis and Statistics
For characterization of single NPs, a minimal of 100 individual NPs similar to those shown in Figure 1A, were imaged using HRTEM, to determine the sizes and shapes of single Ag NPs. A minimal of 360 NPs were imaged using DFOMS to determine the number of NPs dispersed in cell culture medium at each given time (Figure 2A), and distribution of LSPR spectra (colors) of single NPs in the medium for correlation of the colors with the sizes of single NPs (Figure 1F). These approaches allow us to determine the sizes, number and LSPR properties of bulk NPs at single NP resolution. The sizes of NPs in the medium were also determined using DLS. Each experiment was repeated at least three times.
At each given time, the cells at ten desired surface locations of the flasks and the coverslips were imaged using the bright-field (20x objective) and dark-field (100x objective) optical microscopy, respectively. At least 100 cells in each flask and 25 cells on each coverslip were studied for each measurement. Each experiment was repeated at least three times. Therefore, at least 300 cells in the flasks and 75 cells on the coverslips were imaged and characterized at each given time to gain sufficient statistics, which allowed us to study the effects of Ag NPs on bulk cells at single cell resolution.
Supplementary Material
Acknowledgments
This work is supported in part by NSF (NIRT: BES 0507036) and NIH (R01 GM076440). Nallathamby are grateful for the support of Dominion Scholar Fellowship. We thank CharFac of U. of Minnesota (a NNIN site funded by NSF) for their assistance to characterize Ag nanoparticles using HRTEM.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Figure 1S: Study of Ag NPs dispersed in nanopure DI water and cell culture medium using an ensemble method, UV-vis spectroscopy.
References
- 1.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Acc Chem Res. 2008;41:1721. doi: 10.1021/ar800035u. and references therein. [DOI] [PubMed] [Google Scholar]
- 2.Sun RW, Chen R, Chung NP, Ho CM, Lin CL, Che CM. Chem Comm. 2005:5059. doi: 10.1039/b510984a. and references therein. [DOI] [PubMed] [Google Scholar]
- 3.Tiwari SB, Amiji MM. Curr Drug Delivery. 2006;3:219. doi: 10.2174/156720106776359230. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.Xu XHN, Patel RP. In: Imaging and Assembly of Nanoparticles in Biological Systems. Nalwa HS, editor. 2005. and references therein. [Google Scholar]
- 5.Agrawal A, Sathe T, Nie S. In: Nanoparticle Probes for Ultrasensitive Biological Detection and Imaging. Xu X-HN, editor. New Jersey: 2007. and references therein. [Google Scholar]
- 6.Lee KJ, Nallathamby PD, Browning LM, Osgood CJ, Xu X-HN. ACS Nano. 2007;1:133. doi: 10.1021/nn700048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu X-HN, Brownlow WJ, Kyriacou SV, Wan Q, Viola JJ. Biochemistry. 2004;43:10400. doi: 10.1021/bi036231a. [DOI] [PubMed] [Google Scholar]
- 8.Xu X-HN, Song Y, Nallathamby PD. In: Probing Membrane Transport of Single Live Cells Using Single Molecule Detection and Single Nanoparticle Assay. Xu X-HN, editor. New Jersey: 2007. [Google Scholar]
- 9.Haes AJ, Zou SL, Schatz GC, Van Duyne RP. J Phys Chem B. 2004;108:6961. [Google Scholar]
- 10.Huang T, Nallathamby PD, Gillet D, Xu XHN. Anal Chem. 2007;79:7708. doi: 10.1021/ac0709706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang T, Nallathamby PD, Xu X-HN. J Am Chem Soc. 2008;130:17095. doi: 10.1021/ja8068853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaidyanathan R, Kalishwaralal K, Gopalram S, Gurunathan S. Biotechnol Adv. 2009;27:924. doi: 10.1016/j.biotechadv.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Rahman MF, Duhart HM, Newport GD, Patterson TA, Murdock RC, Hussain SM, Schlager JJ, Ali SF. Neurotoxicology. 2009;30:926. doi: 10.1016/j.neuro.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Xu X-HN, Chen J, Jeffers RB, Kyriacou SV. Nano Letters. 2002;2:175. [Google Scholar]
- 15.Fabrega J, Fawcett SR, Renshaw JC, Lead JR. Environ Sci Technol. 2009;43:7285. doi: 10.1021/es803259g. [DOI] [PubMed] [Google Scholar]
- 16.Miura N, Shinohara Y. Biochem Biophys Res Commun. 2009;390:733. doi: 10.1016/j.bbrc.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 17.Browning LM, Lee KJ, Huang T, Nallathamby PD, Lowman J, Xu X-HN. Nanoscale. 2009;1:138. doi: 10.1039/b9nr00053d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nallathamby PD, Lee KJ, Xu X-HN. ACS Nano. 2008;2:1371. doi: 10.1021/nn800048x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia Y, Xiong Y, Lim B, Skrabalak SE. Angew Chem Int Ed Engl. 2009;48:60. doi: 10.1002/anie.200802248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan X, Liu T, Zhong J, Liu X, Li G. Chem Biochem. 2004;5:1686. doi: 10.1002/cbic.200400080. [DOI] [PubMed] [Google Scholar]
- 21.Braydich-Stolle L, Hussain S, Schlager JJ, Hofmann MC. Toxicol Sci. 2005;88:412. doi: 10.1093/toxsci/kfi256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hussain SM, Hess KL, Gearhart JM, Geiss KT, Schlager JJ. Toxicol in Vitro. 2005;19:975. doi: 10.1016/j.tiv.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 23.AshaRani PV, Mun GL, Hande MP, Valiyaveettil S. ACS Nano. 2009;3:279. doi: 10.1021/nn800596w. [DOI] [PubMed] [Google Scholar]
- 24.Abken H, Bützler C, Willecke K. Proc Natl Acad Sci U S A. 1988;85:468. doi: 10.1073/pnas.85.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blackburn RV, Galoforo SS, Berns CM, Armour EP, McEachern D, Corry PM, Lee YJ. Int J Cancer. 1997;72:871. doi: 10.1002/(sici)1097-0215(19970904)72:5<871::aid-ijc26>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 26.Chang NS. Int J Mol, Med. 1998;2:653. [PubMed] [Google Scholar]
- 27.Eldeniz AU, Mustafa K, Orstavik D, Dahl JE. Int Endod J. 2007;40:329. doi: 10.1111/j.1365-2591.2007.01211.x. [DOI] [PubMed] [Google Scholar]
- 28.Maksimovic-Ivanic D, Mijatovic S, Harhaji L, Miljkovic D, Dabideen D, Fan CK, Mangano K, Malaponte G, Al-Abed Y, Libra M, Garotta G, Nicoletti F, Stosic-Grujicic S. Mol Cancer Ther. 2008;7:510. doi: 10.1158/1535-7163.MCT-07-2037. [DOI] [PubMed] [Google Scholar]
- 29.Musiol IM, Feldman D. Endocrinology. 1997;138:12. doi: 10.1210/endo.138.1.4858. [DOI] [PubMed] [Google Scholar]
- 30.Tonetti M, Millo E, Sturla L, Bisso A, De Flora A. Biochem Biophys Res Commun. 1997;230:636. doi: 10.1006/bbrc.1996.6022. [DOI] [PubMed] [Google Scholar]
- 31.Vercammen D, Vandenabeele P, Declercq W, Van de Craen M, Grooten J, Fiers W. Cytokine. 1995;7:463. doi: 10.1006/cyto.1995.0063. [DOI] [PubMed] [Google Scholar]
- 32.Coimbra VC, Yamamoto D, Khusal KG, Atayde VD, Fernandes MC, Mortara RA, Yoshida N, Alves MJ, Rabinovitch M. Infect Immun. 2007;75:3700. doi: 10.1128/IAI.00194-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McArt DG, McKerr G, Howard CV, Saetzler K, Wasson GR. Biochem Soc Trans. 2009;37:914. doi: 10.1042/BST0370914. [DOI] [PubMed] [Google Scholar]
- 34.Liao W, McNutt MA, Zhu WG. Methods. 2009:46. doi: 10.1016/j.ymeth.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 35.Ostling O, Johanson KJ. Biochem Biophys Res Commun. 1984;123:291–298. doi: 10.1016/0006-291x(84)90411-x. [DOI] [PubMed] [Google Scholar]
- 36.Haugland RP. Probes for Organelles. Eugene, OR: 2002. [Google Scholar]
- 37.Edward R. Mol Cells. 2009;27:391. doi: 10.1007/s10059-009-0066-3. [DOI] [PubMed] [Google Scholar]
- 38.Haugland RP. Assays for Cell Viability, Proliferation and Function. Eugene, OR: 2002. [Google Scholar]
- 39.Xu X-HN, Huang S, Brownlow W, Salatia K, Jeffers R. J Phys Chem B. 2004;108:15543. [Google Scholar]
- 40.Kyriacou SV, Brownlow WJ, Xu XH. Biochemistry. 2004;43:140. doi: 10.1021/bi0351110. [DOI] [PubMed] [Google Scholar]
- 41.Kyriacou SV, Nowak ME, Brownlow WJ, Xu X-HN. J Biomed Opt. 2002;7:576. doi: 10.1117/1.1506707. [DOI] [PubMed] [Google Scholar]
- 42.Song Y, Nallathamby PD, Huang T, Elsayled-Ali H, Xu X-HN. J Phys Chem C. 2010;114:74. doi: 10.1021/jp9083019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu X-HN, Brownlow WJ, Huang S, Chen J. Biochem Biophys Res Commun. 2003;305:79. doi: 10.1016/s0006-291x(03)00692-2. [DOI] [PubMed] [Google Scholar]
- 44.Xu X-HN, Jeffers RB, Gao J, Logan B. Analyst. 2001;126:1285. doi: 10.1039/b104180k. [DOI] [PubMed] [Google Scholar]
- 45.Cory AH, Owen TC, Barltrop JA, Cory JG. Cancer Commun. 1991;3:207. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 46.O’Toole SA, Sheppard BL, McGuinness EP, Gleeson NC, Yoneda M, Bonnar J. Cancer Detect Prev. 2003;27:45. doi: 10.1016/s0361-090x(02)00171-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.