

Abstract
A new family of methionine-sulfoxide reductase (Msr) was recently described. The enzyme, named fRMsr, selectively reduces the R isomer at the sulfoxide function of free methionine sulfoxide (Met-R-O). The fRMsrs belong to the GAF fold family. They represent the first GAF domain to show enzymatic activity. Two other Msr families, MsrA and MsrB, were already known. MsrA and MsrB reduce free Met-S-O and Met-R-O, respectively, but exhibit higher catalytic efficiency toward Met-O within a peptide or a protein context. The fold of the three families differs. In the present work, the crystal structure of the fRMsr from Neisseria meningitidis has been determined in complex with S-Met-R-O. Based on biochemical and kinetic data as well as genomic analyses, Cys118 is demonstrated to be the catalytic Cys on which a sulfenic acid is formed. All of the structural factors involved in the stereoselectivity of the l-Met-R-O binding were identified and account for why Met-S-O, DMSO, and a Met-O within a peptide are not substrates. Taking into account the structural, enzymatic, and biochemical information, a scenario of the catalysis for the reductase step is proposed. Based on the thiol content before and after Met-O reduction and the stoichiometry of Met formed per subunit of wild type and Cys-to-Ala mutants, a scenario of the recycling process of the N. meningitidis fRMsr is proposed. All of the biochemical, enzymatic, and structural properties of the N. meningitidis fRMsr are compared with those of MsrA and MsrB and are discussed in terms of the evolution of function of the GAF domain.
Keywords: Crystal Structure, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Mutation, Enzyme Structure, Met-O Reduction, Disulfide Bond, Sulfenic Acid
Introduction
Methionine (Met)4 is one of the two amino acids in proteins that are the most susceptible to oxidation by reactive oxygen species, forming Met-O (1). Prior to 2007, two families of methionine-sulfoxide reductase (Msr) enzymes, called MsrA and -B were known to reduce Met-O back into Met (2, 3). The MsrA family reduces the S isomer at the sulfoxide function, whereas MsrB is specific for the R isomer. Both Msrs, which reveal distinct unrelated folds, were shown to reduce more efficiently a Met-O within a polypeptide chain. Therefore, Msrs are repair enzymes that play important roles in the protection of cells against oxidative stress (3–5).
The catalytic mechanism of MsrA and MsrB is now well documented. For most MsrAs and MsrBs (i.e. those possessing a recycling Cys), the mechanism comprises three steps: a reductase step that leads to formation of a sulfenic acid intermediate on a catalytic Cys (6), a second step in which a disulfide bond is formed between the catalytic Cys and a recycling Cys, and finally, a step in which the intradisulfide bond is reduced by thioredoxin (Trx) or a Trx-like protein (7). A catalytic residue (i.e. Glu94 in MsrA and His103 in MsrB) was characterized, one of whose major roles is to protonate the oxygen of the sulfoxide function (8, 9). In all of the MsrAs and MsrBs studied to date, the reduction of the disulfide bond is rate-limiting, whereas the formation of the sulfenic acid intermediate is rate-determining in the process leading to intradisulfide bond formation. In other words, in the absence of Trx, only the intradisulfide intermediate accumulates.
Recently, a third family of Msr, named fRMsr, was discovered (10). The fRMsrs exhibit a GAF-type fold. GAF domains are one of the largest and most widespread domains found in all kingdoms of life. They are dimeric and are generally arranged in tandem in modular proteins to provide a large variety of regulation functions. However, most of the functions of GAF domains remain to be studied in detail. The fRMsr is the first case of a GAF domain that bears a catalytic activity. The fRMsrs are present in eubacteria and unicellular eukaryotes (11). The family displays a methionine-sulfoxide reductase activity, reducing selectively free Met-O with an R configuration at the sulfoxide (10). Such a function led Lowther and co-workers (10) to propose that Met-R-O can represent a signaling molecule in response to oxidative stress.
Only the crystal structures of fRMsr from Escherichia coli and Saccharomyces cerevisiae have been solved to date, without substrate (12, 13). Both structures are described as being composed of six β-strands, four α-helices, and two prominent loops, loop 1 and loop 2, located on the surface of the protein between β2 and β3 and between β4 and β5, respectively. Formation of a disulfide bond between Cys84 of loop 1 and Cys118 of loop 2 is assumed to close off the cavity in which a molecule of MES, which derives from the crystallization buffer, is bound in the E. coli enzyme (Protein Data Bank entry 1vhm). The sulfonic acid moiety is closed to another Cys94, which is located at the N terminus of an α-helix. The rest of the cavity is lined by several invariant residues. From the inspection of the crystal structure of E. coli fRMr in complex with a MES buffer molecule and by carrying out computational docking of Met-R-O into the cavity of the S. cerevisiae enzyme, a reductase mechanism was proposed in which the Cys94 located at one of the sides of the cavity is assumed to be the catalytic Cys (10). The sulfenic acid postulated to be formed on Cys94 is then attacked by Cys118, forming a disulfide bond. A disulfide bond exchange then occurs with Cys84, which finally leads to the Cys118–Cys84 disulfide intermediate.
In the present study, we report the crystal structure of the fRMsr from Neisseria meningitidis in complex with l-Met-R-O, solved at 1.25 Å resolution. By combining genomic analyses, structural information, and biochemical and kinetic data from the wild type and mutants in which Cys was substituted by Ala, Cys118 is shown to be the catalytic residue on which the sulfenic acid is formed.5 All of the molecular and structural factors involved in the stereoselective binding of the l-Met-R-O stereoisomer are identified from the crystal structure and explain why neither a Met-S-O, a Met-R-O in a peptide or a protein context, nor a DMSO molecule are substrates. Scenarios for both substrate binding and catalysis are proposed for the reductase step.
A disulfide bond between Cys84 and Cys118, present in all of the fRMsr crystal structures described so far, including the N. meningitidis fRMsr, stabilized the loop 2 as a flap that is an integral part of the active site. Formation of this disulfide bond seems to be a prerequisite to obtaining crystals. However, it is the free reduced form that is catalytically relevant to the reductase step. Based on the content of 1) the Cys in wild type and Cys-to-Ala mutants before and after Met-O reduction and 2) the Cys titrated under native conditions, a scenario for the Trx-recycling process is proposed in which a disulfide Cys118–Cys84 bond is formed first. All of the results are compared with those obtained for MsrA and MsrB and discussed in terms of the evolution of function of the GAF domain.
EXPERIMENTAL PROCEDURES
Plasmid Constructions, Site-directed Mutageneses, Production, and Purification of Wild Type and Cys-to-Ala Mutants of fRMsr from N. meningitidis
Expression plasmid pSKfRMsr for fRMsr was obtained by cloning the corresponding open reading frame by PCR from N. meningitidis 8013 genomic DNA, into the plasmid pSKMsr (6) between the NdeI and SacI sites. Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene).
The E. coli strain HB101 used for production of fRMsr was transformed with a pSKfRMsr plasmid. All wild type and mutants were produced in soluble form. For enzyme purification, cells were harvested by centrifugation, resuspended in buffer A (50 mm Tris-HCl, 2 mm EDTA, pH 8) containing 50 mm dithiothreitol (DTT) and sonicated. The enzyme was then precipitated at 50% (NH4)2SO4 saturation. The contaminating proteins were removed by applying the enzymatic solutions, after extensive dialysis against buffer A, onto a phenyl-Sepharose column (Amersham Biosciences) equilibrated with buffer A containing 1 m (NH4)2SO4, using a fast protein liquid chromatography system (Amersham Biosciences). Wild type and all mutants eluted at 150 mm (NH4)2SO4 from a linear gradient from 1 to 0 m (NH4)2SO4 in buffer A. Purified fractions were then pooled and applied to a Q-Sepharose column equilibrated with buffer A, followed by a linear gradient of KCl (0–0.4 m). All fRMsrs eluted at 250 mm KCl and were pure at this stage. The yield was ∼80 mg of fRMsr/liter of culture. An additional step was added to prepare samples for crystallization. Before (NH4)2SO4 precipitation, a treatment using 0.05% polyethyleneimine was carried out to remove most of the nucleic acids. The purity of the wild type and all mutants was checked by electrophoresis on a 12.5% SDS-polyacrylamide gel followed by Coomassie Brilliant Blue R-250 staining and by electrospray ionization mass spectrometry analyses. Purified enzymes were stored at −20 °C in the presence of 50 mm DTT and 70% (NH4)2SO4. Under these conditions, the wild type and all mutants were stable for several weeks. Their molar concentrations were determined spectrophotometrically, using extinction coefficients at 280 nm of 26,720 M−1 cm−1 for wild type and the mutants.
Mass Spectrometry Analyses
Samples were desalted by applying the enzymatic solutions onto an exclusion size column (HIPREP 26/10 Desalting, Amersham Biosciences) equilibrated in 50 mm ammonium bicarbonate buffer, pH 7.6. Electrospray ionization mass spectrometry measurements were performed on an electrospray ionization time-of-flight mass spectrometer (LCT, Micromass, Altrincham, UK) fitted with a standard Z-spray source. Measurements were performed in nondenaturing conditions by diluting enzymes to 10 μm in 100 mm ammonium acetate buffer, pH 6.8.
Quantification of the Free Cys Content of Wild Type and Mutants with 5,5′-Dithiobis(2-nitro)benzoate (DTNB) and Determination of the pKapp of Cys118 and Cys84 in the Wild Type with 2,2′-Dipyridyl Disulfide (2-PDS)
The Cys content of the wild type and mutants was determined using DTNB under native and denaturing conditions (final concentration of 9% SDS in buffer A) in the absence and presence of incubation with dl-Met-R,S-O at a concentration of 100 mm, as described previously (6).
Due to the high chemical reactivity of Cys84 and Cys118 and of the thionitrobenzoate (TNB−; pKapp of 7), fast kinetic measurements for determining the Cys pKapp were carried out on an Applied Photophysics SX18MV-R stopped flow apparatus with 2-PDS instead of DTNB, because the pKapp of pyridine-2-thione is 4.3. The experimental conditions were as previously described (14), with wild type and 2-PDS concentrations after mixing of 12.5 μm/subunit and 200 μm, respectively. The pseudo-first-order rate constant was determined at each pH by fitting the absorbance (A) at 343 nm versus time (t) to monoexponential Equation 1, where a is the burst amplitude and c is the end point. The second-order rate constants k2 were calculated by dividing kobs by 2-PDS concentration and then fitted to Equation 2, in which k2max represents the second rate constant for the thiolate form.
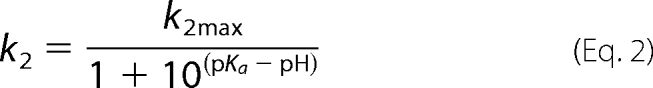 |
Quantification of Met Formed in the Absence of Trx
The reaction mixture containing dl-Met-R,S-O (100 mm) and the wild type or mutants (50–100 μm) was incubated at 25 °C for 10 min in buffer A, and then 250 μl of each assay was injected onto a Sephasil C18 column for Met quantification, as described earlier (15).
Steady-state Kinetics of Wild Type and Mutants with the Trx-regenerating System and with dl-Met-R,S-O as a Substrate
Steady-state kinetic parameters were determined with dl-Met-R,S-O as a substrate and E. coli Trx1 as a reductant in the presence of a Trx-regenerating system (1.25 μm E. coli Trx reductase and 0.3 mm NADPH), as previously described (15). Initial rate measurements were carried out at 25 °C in buffer A on a SAFAS UVmc2 spectrophotometer by following the decrease in the absorbance at 340 nm due to the oxidation of NADPH. Initial rate data were fitted to the Michaelis-Menten Equation 3 or to Equation 4, which fits the model of competitive inhibition by excess substrate. Using least squares analysis, kcat, Km, and KI values were determined using the program Sigmaplot (Jandel Scientific Software).
 |
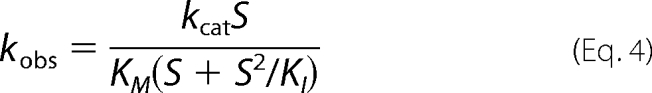 |
E. coli Trx1 and Trx reductase were prepared following published experimental procedures (15).
Steady-state Kinetics of the C84A/C136A fRMsr with the Trx-regenerating System and with l-Met-R,S-O or d-Met-R,S-O as Substrate
The kinetic experimental conditions were the same as those used with dl-Met-R,S-O. The experimental protocol used to prepare d-Met-R,S-O was adapted from Sharov et al. (16). Briefly, 0.3 g of d-Met was dissolved in 2 ml of H2O in a boiling water bath. After slight cooling, oxidation was carried out at 60 °C overnight with a 1.1-fold molar excess of H2O2. The d-Met-O was then precipitated by the addition of 200 ml of acetone and collected by centrifugation. The pellet was washed twice with acetone and dried under vacuum. Therefore, the d-Met-O was a mixture of R and S isomers at the sulfoxide function.
Crystallization and X-ray Diffraction Data Collection
The fRMsr used for crystallization was concentrated to 20 mg/ml in buffer (20 mm Tris-HCl, pH 8). Initial crystallization conditions were determined in 96-well Greiner plates using a nanoliter drop Mosquito robot (TTP LabTech) and the commercial Structure Screens I and II (Molecular Dimension Ltd.) and Wizard Screens 1 and 2 (Emerald Biostructure). Protein solution droplets of 200 nl were mixed with precipitant droplets of 100, 200, and 300 nl and then equilibrated against 80 μl of well buffer. Small crystal plates appeared after 2 days of sitting drop vapor diffusion using 30% MPD, 200 mm Mg(CH3COO)2, and 100 mm cacodylate, pH 6.5, at 20 °C. Diffraction quality crystals (0.3 × 0.4 × 0.5 mm) were obtained at 20 °C by the hanging drop vapor diffusion by mixing 4 μl of protein solution and 2 μl of 23% MPD, 120 mm Mg(CH3COO)2, and 100 mm cacodylate, pH 6.1–6.4, in 24-well plates filled with 1 ml of well buffer. Crystals were soaked for 30 s in a cryoprotectant solution containing 40% MPD, 120 mm Mg(CH3COO)2, 100 mm cacodylate, pH 6.5, and 10 mm dl-Met-R,S-O. The crystals were then flash-frozen under a gaseous nitrogen stream at 100 K. A data set was collected on the Proxima-1 beamline at SOLEIL (SOLEIL Synchrotron, Gif-sur-Yvette, France) using an ADSC Q315r detector. Diffraction data were indexed, integrated, and merged using XDS (17). The crystal diffracts at 1.25 Å resolution and belongs to the P1 space group with two molecules per unit cell. Data collection statistics are summarized in Table 1.
TABLE 1.
Summary of data collection and refinement statistics
| Parameters | Values |
|---|---|
| Crystal | |
| Space group | P1 |
| Unit cell | a = 39.32 Å, b = 39.48 Å, c = 52.25 Å, α = 104.17°, β = 105.16°, γ = 100.50° |
| Data collection | |
| Beamline | Proxima I |
| Wavelength (Å) | 0.886 |
| Resolution (Å) | 20-1.25 (1.30-1.25)a |
| Reflections observed | 624,099 (30,697) |
| Unique reflections | 73,065 (7048) |
| Redundancy | 8.54 (4.35) |
| Completeness (%) | 92.8 (80.3) |
| I/σI | 22.2 (3.97) |
| Rmerge (%)b | 5.3 (47.0) |
| Refinement | |
| Resolution (Å) | 20-1.25 |
| No. of atoms | 5809 |
| Rwork/Rfree (%)c | 11.78/15.68 |
| r.m.s. deviation bonds (Å) | 0.022 |
| r.m.s. deviation angles (degrees) | 2.286 |
| Mean B values (Å2) | |
| Wilson B overall | 19.3 |
| Overall | 18.68 |
| Protein | 17.08 |
| Water molecules | 33.68 |
a Highest resolution shell data are shown in parenthesis.
b Rmerge = Σhkl Σi|Ihkli − 〈Ihkli〉|/Σhkl Σi 〈Ihkli〉.
c Rcryst = Σhkl‖Fo| − |Fc‖/Σ|Fo|.
Structure Determination and Refinement
Initial phases were obtained by the molecular replacement method using the crystal structure of the dimeric protein YebR from E. coli (Protein Data Bank entry 1vhm) (12) as a search model and the program MOLREP (18). The structure refinement was initiated by a rigid body refinement of both subunits between 20 and 3 Å resolution. Refinements using REFMAC5 (19) were then carried out between 20 and 1.25 Å resolution. After each cycle of refinement, the structure was manually built using the graphics program COOT (20). The quality of the 2Fo − Fc maps and the Fo − Fc residual maps during the refinement was excellent and allowed the construction of a protein model between the ninth residue and the C-terminal residue, as well as localization of two substrate molecules (i.e. l-Met-R-O). Water molecules as well as MPD and acetate molecules were added to the model using the available options in COOT. Hydrogen atoms were then automatically generated, and temperature factors were refined anisotropically using REFMAC. During the last steps of refinement, weak 2Fo − Fc and Fo − Fc residual maps appeared at the N-terminal parts and allowed the first six amino acids of both subunits to be constructed with low occupancy. On the basis of the Fo − Fc residual maps and their temperature factors, 11 atoms were refined as magnesium ions. Atomic ligands located within the active site were modeled and refined as water molecules but not as magnesium ions. The final model includes residues 1–167 of chains A and B, two l-Met-R-O molecules, four MPD molecules, two acetates, 11 magnesium ions, and 318 water molecules. The quality of the refined structure, which was checked using MolProbity, indicates that 98.1% of the residues lie in the favored regions of the Ramachandran plot, and 99.5% of all of the residues are in the allowed region. 0.61% are present in the disallowed region and concern the Ala76 residue of the A and B subunits that are well defined in the electron 2Fo − Fc map. Within the dimer, both Ala76 residues that precede in sequence Pro77 have a constraint geometry that leads to ϕ, ψ angles of (−123.4°,−160.2°) and (−125.3°,−162.6°), respectively.
RESULTS
Biochemical and Kinetic Properties of Wild Type and fRMsr Mutants
The fRMsrs were overexpressed in E. coli. About 10% of the soluble proteins were fRMsrs. The molecular mass determined by mass spectrometry under non-denaturing conditions (36,333 ± 2 Da) is in accord with the dimeric state (theoretical value: 36,330 Da) of a GAF domain. Size exclusion chromatography coupled to static light scattering also showed that the N. meningitidis fRMsr is in a dimeric form (data not shown). The use of DTNB reagent revealed four Cys residues under denaturating conditions (Table 2). This result was expected from the fRMsr sequence of N. meningitidis, which contains four Cys at positions 84, 94, 118, and 136 (Fig. 1).
TABLE 2.
Free sulfhydryl content and stoichiometry of Met formed in the absence of Trx-regenerating system of wild type and Cys-to-Ala fRMsrs
The values indicated represent the average of two independent measurements of at least two enzyme concentrations (S.D. range 10%). Cys content was determined spectrophotometrically using DTNB in the presence or absence of 9% SDS, in the absence or in the presence of 100 mm dl-Met-R,S-O without any regenerating system (see “Experimental Procedures”).
| fRMsr | No. of Cys | No. of Cys measured under native conditions | No. of Cys measured under denaturated conditions |
Stoichiometry of Met formed | ||
|---|---|---|---|---|---|---|
| Without dl-Met-R,S-O | With dl-Met-R,S-O | Decrease in free thiola | ||||
| mol Cys/mol subunit | mol Cys/mol subunit | mol Met/mol subunit | ||||
| Wild type | 4 | 2.2 | 3.9 | 2.1 | 1.8 | 1.0 |
| C84A | 3 | 1.9 | 2.9 | 1.0 | 1.9 | 1.0 |
| C94A | 3 | 2.1 | 3.0 | 1.0 | 2.0 | 1.1 |
| C118A | 3 | 2.8 | 3.1 | 3.0 | 0.1 | 0.0 |
| C136A | 3 | 2.1 | 3.1 | 1.1 | 2.0 | 1.1 |
| C84A/C136A | 2 | 1.8 | 2.1 | 0.1 | 2.0 | 0.9 |
| C94A/C136A | 2 | 1.9 | 1.9 | 0.0 | 1.9 | 0.8 |
| C84A/C94A/C136A | 1 | 1 | 1.0 | 0.0 | 1.0 | 0.9 |
a The difference in the number of free Cys thiols upon treatment with dl-Met-R,S-O versus no treatment.
FIGURE 1.

Alignment of sequences of fRMsrs from E. coli (E c), N. meningitidis (N m), and S. cerevisiae (S c). The sequences were aligned with Bioedit software. The numbering of the amino acid residues indicated is based on the numbering of the E. coli fRMsr sequence. The conserved residues in the 358 putative fRMsr sequences are indicated in white on black boxes, whereas residues Cys84, Cys94, and Phe78 are in white on gray boxes. Cys84 and Cys94 are present in 94.5 and 98.6%, respectively, of the sequences of all putative fRMsrs. In terms of signatures, Cys118/Cys84/Cys94, Cys118/Cys94, and Cys118/Cys84 are present in 93, 5.6, and 1.4% of the putative fRMsr sequences. Phe78 is present in 92.7% of the sequences but is replaced by Tyr in the other sequences. The secondary structures depicted above the alignment are those from the fRMsr of E. coli (12).
To evaluate the role of these Cys in the catalytic mechanism, they were substituted, individually, in tandem, and three at once (Cys84, Cys94, and Cys136), by Ala. The C118A mutant was inactive, whereas all of the other mutants were active under steady-state conditions, including the triple mutant C84A/C94A/C136A. In all of the mutants except for the C118A mutant, 1 mol of Met/subunit was formed in the absence of Trx.
To investigate how the recycling process occurs, the free thiol content of the wild type and the different mutants was determined under denaturating conditions by DTNB, before and after incubation with Met-O and in the absence of reductant. As shown in Table 2, 2 Cys residues/subunit were no longer titrated with DTNB in all of the fRMsrs except for the C118A mutant, in which no difference in Cys titration was observed, and the C84A/C94/C136 mutant, in which only one Cys was lost.
Under native conditions, two Cys reacted with DTNB in the wild type and C94A/C136A mutant (Table 2). This observation indicates that either these two Cys reacted individually and formed an interdisulfide bond with the TNB− moiety or one of the two Cys reacted more rapidly and the second formed a disulfide bond with the first via an intramolecular attack. To differentiate between these two possibilities, the two oxidized fRMsrs were isolated, and DTT was added. No absorbance at 412 nm corresponding to release of TNB− was observed (data not shown). In mutants in which either Cys84 and Cys136 or Cys118 and Cys136 were substituted by Ala, two Cys residues were also titrated under native conditions, therefore corresponding to either Cys118 and Cys94 or Cys94 and Cys84, respectively. In contrast to the wild type, an absorbance at 412 nm corresponding to release of 2 mol of TNB− was observed when DTT was added. Altogether, these results indicate that only Cys118 and Cys84 are accessible in the wild type and form a disulfide bond under DTNB titration.
Reaction of 2-PDS with the wild type obeyed pseudo-first-order kinetics, with formation of 2 mol of pyridine-2-thione per subunit of fRMsr, as shown from the absorbance increase at 343 nm. This result is similar to that found with DTNB. Moreover, no absorbance at 343 nm corresponding to release of pyridine-2-thione was observed when DTT was added to the oxidized fRMsr (data not shown). Again, similar to DTNB, a disulfide bond is thus formed, probably between Cys118 and Cys84. For all pH values used, stopped-flow traces fitted to monoexponential Equation 1, with amplitude corresponding to the release of 2 mol of pyridine-2-thione/subunit. The pH-k2 profile fitted to monosigmoidal Equation 2 with a pKapp value of 8.5 and k2max value of 1.5 × 105 m−1 s−1 (see Fig. 2). This finding supports a pKapp of 8.5 for both Cys118 and Cys84. The fact that the k2 value is high also indicates that both Cys residues are highly accessible.
FIGURE 2.

pH dependence of the second-order rate constant k2 for the reaction of the thiol group of wild type fRMsr with 2-PDS. Reaction kinetics were performed at 25 °C in 120 mm Tris-HCl, 30 mm acetic acid, 30 mm imidazole buffer at a constant ionic strength of 0.15 m over a pH range of 5–9.5. The concentrations of wild type fRMsr and 2-PDS were 12.5 and 200 μm, respectively. The values represent the average of three independent measurements (S.D. range <2%). Values of second-order rate constants k2 (●) were fitted to Equation 2 (solid line), leading to k2max and pKapp values of (1.50 ± 0.04) × 105 m−1 s−1 and 8.5 ± 0.1, respectively.
As shown in Table 3, all of the mutants (except the C118A mutant), including the triple mutant C84A/C94A/C136A, are active under steady-state conditions using Trx as the reductant and using dl-Met-R,S-O as substrate. The kcat values vary from 8.5 to 18 s−1 for wild type and mutants in which Cys84 is present and from 1 to 2.6 s−1 when Cys84 is absent. The Km values for Met-O vary from 2 for the wild type up to 32 mm for the C84A mutant. Because the l-Met-O is preferentially reduced (see below), the S isomer at the sulfoxide function is not active, and the Met-O used is a mixture of four stereoisomers, the Km value must be divided by 4. The catalytic efficiency (kcat/Km) of the wild type relative to l-Met-R-O is better by a factor of at least 1000 when compared with MsrB from N. meningitidis (15), due to both kcat and Km effects. Inhibition by excess substrate is observed but only with the wild type and the C136A mutant. This observation remains to be explained but seems to be correlated with the presence of both Cys84 and Cys94. To determine whether the d-Met-O was a substrate, the kinetic parameters for both l-Met-O and d-Met-O were determined in the presence of the Trx-regenerating system not with the wild type but with the double mutant C94A/C136A because this mutant does not show substrate inhibition. The d-Met-O is not efficient as substrate. No saturating effect was observed up to 500 mm, and the k2 constant, which represents the kcat/Km value, is ∼0.4 m−1 s−1. Therefore, the Km value is at least 0.5 m, and thus the kcat value is lower than 0.8 s−1. In contrast, kcat and Km values for l-Met-O are in the range of those found with dl-Met-O (i.e. 18 s−1 and 10 mm, respectively), with a Km value decreased by a factor of ∼2, as expected. The catalytic efficiency, which is better by a factor of 4.104 compared with d-Met-O, is the consequence of both kcat and Km effects.
TABLE 3.
Kinetic parameters of wild type and Cys-to-Ala fRMsrs under steady-state conditions
Reactions were carried out at 25 °C in 50 mm Tris-HCl, 2 mm EDTA buffer at pH 8.0, as described under “Experimental Procedures.” Enzyme concentrations were 0.1, 0.5, or 1 μm. The substrate used was dl-Met-R,S-O. For determining the Km(Met-O), Trx concentration was 100 μm, and Met-O concentration varied from 0.1 to 500 mm. For Km(Trx) determination, Trx concentration varied from 1 to 200 μm, and Met-O concentration was 100 mm, except for wild type and C136A fRMsrs (20 mm). Data presented were obtained by fitting the experimental data to the Michaelis-Menten equation, except data of wild-type and C136A fRMsrs which were obtained by using the equation of competitive inhibition by excess substrate. At least two independent determinations were performed to determine each constant. The Km values must be divided by 4, considering the fact that the S isomer at the sulfoxide function is not a substrate, the d-Met-O is not efficient as a substrate, and the Met-O used is a mixture of the four stereoisomers. ND, activity not detectable.
| fRMsr | kcat | Km(Met-O) | KI | Km(Trx) |
|---|---|---|---|---|
| s−1 | mm | mm | μm | |
| Wild type | 14 ± 2 | 2 ± 1 | 34 ± 19 | 40 ± 7 |
| C118A | ND | |||
| C136A | 8.5 ± 0.6 | 2.2 ± 0.5 | 56 ± 16 | 24 ± 3 |
| C84A | 1.0 ± 0.1 | 32 ± 9 | 50 ± 4 | |
| C94A | 18 ± 1 | 30 ± 6 | 27 ± 4 | |
| C84A/C136A | 2.6 ± 0.8 | 12 ± 4 | 40 ± 6 | |
| C94A/C136A | 13 ± 1 | 18 ± 1 | 15 ± 2 | |
| C84A/C94A/C136A | 1.0 ± 0.1 | 30 ± 8 | 16 ± 4 |
Overall Structure
The crystal structure of fRMsr of N. meningitidis in complex with l-Met-R-O was solved at 1.25 Å resolution. As expected, the fRMsr structure shares the overall topology of the GAF domains (Fig. 3). Structural superimposition of the dimeric fRMsr of N. meningitidis with two known structures of fRMsrs from S. cerevisiae (13) and E. coli (12) (Protein Data Bank entries 1f5m and 1vhm, respectively), reveals that most of the differences are located within the protein loops (r.m.s. deviations of 1.218 and 1.151 Å, respectively, calculated with 266 Cα and 245 atoms). In the following, the residue numbering is that from the sequence of the E. coli enzyme according to the sequence alignment (Fig. 1).
FIGURE 3.

Ribbon representation of fRMsr from N. meningitidis in complex with l-Met-R-O. The two subunits are in white and yellow. The flap of the subunit A is in red. The side chains of Cys118 and Cys84 are shown in stick representations and colored in green. The l-Met-R-O is represented in a stick representation with the carbon atoms in purple, the oxygen atoms in red, the nitrogen in blue, and sulfur atoms in yellow. The side chains of Arg91, Asp114, Phe61, and Trp62 are represented in sticks colored yellow for the carbon atoms, red for oxygen atoms, and blue for nitrogen atoms.
The interface between the two subunits involves the β-strand β2, the α-helix α2, the loop (Gly80–Arg86), and the first 6 amino acids that are not well defined in the 2Fo − Fc electron density maps. The 2125-Å2 area between the two interacting subunits, which is similar to that found for the E. coli enzyme, represents 25% of the overall area of each subunit and provides a strong dimer association. Molecular contacts between both subunits mainly involve direct van der Waals interactions, hydrogen bonds, and two salt bridges between Arg86 and Glu39 of each subunits. In addition, indirect hydrogen bonds and salt bridges are mediated via water molecules and magnesium ions.
The structure of a subunit is organized around a central twisted antiparallel β-sheet consisting of six strands (β3-β2-β1-β6-β5-β4) flanked on one side by a three-α-helix bundle that encompasses the helices α1, α2, and α4. On the other side, the “loop-β3-α3-loop” motif encompassing residues Gly80–Thr103 links the strands β2 to β4 and girdles the overall β-sheet and sits on the short loop located between β1 and β2 strands. In addition, the loop between β4 and β5 strands (residues Gly107–Ser124) acts as a capping flap that shields the active site.
A disulfide bond is observed between Cys84 and Cys118. The disulfide bond is formed during the crystallization process similarly to the two other crystal structures of fRMsr described so far. Indeed, the addition of Met-O, which is substrate, was done after obtaining the crystals and just before flash-freezing.
The Flap
Like the other fRMsr structures, the flap is locked within the crystal structure by a disulfide bridge between Cys84 and Cys118 (Fig. 3). The side chain of Cys84 adopts a double conformation. The conformation of the flap is stabilized by van der Waals interactions with main and side chain atoms of residues located in the loop between the strand β6 and the helix α4. A loop located between the helix α2 and the strand β1 contains 3 residues that also participate in the stabilization of the loop β6-α4 via van der Waals interactions with the side chain atom of Trp62. Indeed, at one side of the active site, Phe61 directly interacts with 3 residues of the flap (Ser119, Leu121, and Ser122). At the other side of the active site, the Arg91 that precedes the helix α3 is involved in a salt bridge with Asp114 of the flap. As a consequence, the disulfide bridge (Cys84–Cys118), the van der Waals interactions of Phe61 with residues 119–122, and the salt bridge (Arg86–Asp114) maintain the conformation of the flap. As shown below, residues of the flap also interact with the substrate.
The mean isotropic thermal values were calculated with the atoms of the flap of both subunits A and B (20.27 and 24.37 Å2, respectively), which are significantly higher than the overall B factors of the subunits A and B (15.54 and 18.62 Å2, respectively). The temperature factors of the atoms of the flaps of each subunit reveal and assess the dynamic properties as well as the flexibility of the flap. Finally, the superimposition of the overall dimer with the structure of the fRMsr from E. coli shows r.m.s. deviations of 1.84 and 2.07 Å between residues of the flap (residues 99–113), as calculated with 14 Cα of the subunits A and B, respectively. This structural difference could be related to the flexibility of the flap.
By contrast, the r.m.s. deviations with the S. cerevisiae enzyme reveal that the flap adopts the same conformation (r.m.s. deviations of 1.16 and 1.00 Å calculated on 14 Cα atoms for subunits A and B, respectively).
Active Site
The two active sites within the dimer are separated from each other by 23.4 Å. Both bind an l-Met-R-O substrate and water molecules. The l-Met-R-O is bound in a small cavity that excludes solvent accessibility. The substrate lies down full-length on the central antiparallel β-sheet and interacts with the β1-, β6-, and β5-strands. The “loop-β3-α3-loop” motif surrounds the substrate, forming a cavity that ensures tight binding, whereas the N-terminal part of α3 helix plays a major role in substrate recognition and positioning via its carboxylic group. Finally, the flap that caps the active site shields the substrate molecule within the cavity and assumes the active site architecture.
Substrate Binding
The l-Met-R-O is tightly bound within the active site (Fig. 4A). The partial positive charge of the helix α3 dipole points toward the carboxylate of the l-Met-R-O, which makes strong interactions with invariant Val93 and Cys94 as well as an additional residue, invariant Ile116, all via their main chain peptidic nitrogens. Indeed, the oxygen O1 of the carboxylate is located 2.84 Å from the NH of Cys94, whereas the oxygen O2 atom points toward the NH of Val93 and Ile116 (2.84 and 2.90 Å, respectively). Val93 and Cys94, which are both situated at the N terminus of helix α3, as well as Ile116 are positioned on one side of the active site and form an oxyanion hole. In addition, the NH3+ is sandwiched between the oxygen of the carbonyl of Ile116 (2.79 Å) and the Oϵ1 and Oϵ2 atoms of Glu125 (2.88 and 3.01 Å, respectively), to which it forms salt bridges. In addition, the imidazole ring of His115 which is located at the protein surface, acts like a wall for both the NH3+ group and the O2 atom of the carboxylate of the substrate, which both point perpendicularly toward the side chain.
FIGURE 4.

Stick representation of the active site of fRMsr from N. meningitidis with l-Met-R-O bound. A, the l-Met-R-O is colored according to atom type with carbon atoms in yellow, whereas the surrounding amino acids are colored according to atom type with carbon atoms in white. Dashed lines, polar interactions. Water molecules are represented in a ball representation. The Pro82 and Cys84 are in double conformation. The side chain of Ser122 was refined in three conformations. The side chain of Asp143 was refined in double conformation. The minor conformation of Asp143 side chain (occupancy 0.3) interacts with Wat125 that could interact with the side chain of Cys118 by rotation of its side chain. B, wide eye stereo view of the active site, showing the selected residues (labeled) and omit map. The omit map was calculated around l-Met-R-O. The electron density map was contoured at 4σ and calculated with all of the reflections between 20 and 1.25 Å. The colors of the atoms are the same as were used in A.
The Cβ and the Cγ of the substrate are shape-stabilized within the active site. In particular, the side chains of Ile87 that were refined in a double conformation, of Tyr66, and of Ile116 participate in van der Waals interactions with these two CH2 groups, introducing steric constraints that prevent any tipping of the SOCH3 around the χ angle. The side chains of Ile87 and Leu75 are located on either side of the aromatic side chain of Tyr66. Facing the side chain of Leu75, the indole ring of the invariant Trp62 ensures the stabilization of the ϵ-methyl of the sulfoxide that points perpendicularly toward the indole moiety (Fig. 4B). The aromatic side chain of Phe78 (or a Tyr in ∼7% of the fRMsr sequences; see the alignment shown in supplemental Fig. S1) acts like a platform upon which both Leu75 and Trp62 are stacked, strictly determining their side chain conformations. Finally, the side chains of Ile87, Tyr66, and Trp62 create a hydrophobic step shape arrangement within the active site that is necessary for substrate binding. The oxygen atom of the sulfoxide is hydrogen-bonded to a water molecule (Wat99) (i.e. 2.67 Å), which is itself hydrogen-bonded to the main chain NH of Ser122 (i.e. 2.98 Å). Therefore, the oxygen atom of the sulfoxide is located at a distance of 2.88 Å from the Oδ1 atom of Asp143. In the later stages of the refinement, the quality of the 2Fo − Fc maps as well as the Fo − Fc residual maps around the side chain of Asp143 allowed us to refine the side chain to two conformations. Whereas the minor conformation (occupancy 0.3) of the side chain of Asp143 is able to interact directly with the oxygen atom of the sulfoxide, this interaction does not occur in the major conformation (occupancy 0.7), and instead a contact is formed with Asp141. The major conformation of Asp143 allows a network of hydrogen bonds to form between two other important and conserved acidic residues, Asp141 and Glu125. The distance between the Oδ2 of both aspartates 143 and 141 is 2.76 Å, whereas the oxygen Oδ1 of Asp141 strongly interacts with the Oϵ2 atom of Glu125 (2.50 Å), which, as mentioned previously, is involved in a salt bridge with NH3+. The carboxylates of these residues that belong to the central β sheet give rise to a relatively flat acidic surface. In addition, another water molecule (Wat67) is located in a distal position with respect to the oxygen atom of the sulfoxide (3.22 Å). The side chains of Asp143, His115, Ser122, and another water molecule, Wat99, are closely hydrogen-bonded to the water molecule Wat67 (2.75, 2.64, 2.70, and 3.03 Å, respectively). Finally, all of the residues of the active site impose geometrical constraints on the substrate, illustrated by the proximity of the amino group to the sulfoxide (distance N–O, 2.79 Å). All of the enzyme-substrate interactions raise the question of how these geometrical constraints affect the electronic state of the sulfoxide that could be catalytically relevant. The crystal structure reveals that the sulfur atom of the sulfoxide is placed almost linearly between the Oδ2 atom of Asp141 and the thiol of Cys118 (3.77 and 3.64 Å, respectively). In addition, the final residual Fo − Fc maps reveal a small negative peak at 5.5σ on the thiol of Cys118. In that context, according to the 1.25 Å resolution, it is difficult to deduce whether the crystal suffered from radiation damages during the data collection or whether this peak revealed a lack of electron density on the thiol of Cys118 with respect to electronic effects. In addition, a small positive peak at 4σ very close to the sulfur atom of the substrate (1.5 Å) but distinct from that in the 2Fo − Fc map is located between the sulfur atom of the substrate and Asp141.
The Cys84 adopts two conformations and makes a disulfide bridge with Cys118 in the crystal, as observed in the two other crystal structures solved so far. Although the thiols of Cys94 and Cys84 are distant from the sulfur atom of the l-Met-R-O (5.73 and 4.84 Å, respectively), the thiol of Cys118 is located only 3.64 Å away. In contrast to the other Cys within the active site, the positioning of the side chain of Cys118 is appropriate to allow a nucleophilic attack on the sulfur atom of the sulfoxide by side chain rotation, when the disulfide bond between Cys118 and Cys84 is reduced. In addition, the angle of attack is consistent with a catalytic trigonal bipyramidal geometry of the transition state, with the oxygen atom of the sulfoxide positioned in a distal location. A search of other rotamers of the Cys118 side chain reveals that one rotamer allows for the thiol to be brought close to the sulfur atom of the sulfoxide (2.51 Å).
DISCUSSION
A scenario of the catalytic mechanism, similar to that of MsrA and MsrB, can be proposed for fRMsr based on the genomic, biochemical, kinetic, and structural analyses carried out in the present study. First, a sulfenic acid intermediate is formed on the catalytic Cys118. This conclusion is based on four types of data. First, only the substitution of Cys118 by Ala leads to a complete loss of activity, whereas the mutants in which the other three Cys are individually substituted with Ala, in particular the C84A and C94A mutants, remain active under steady-state conditions. These latter results contradict those described for fRMsr from S. cerevisiae, for which substitution of Cys94 and Cys84 by Ser led to a complete loss of activity (11). Second, the C84A/C94A/C136A mutant is active when tested under steady-state conditions with the Trx-regenerating system. Third, in the absence of Trx, several species with the C84A/C94A/C136A mutant are efficiently formed, including a covalent intermediate, which migrates as a dimer of fRMsr on denaturating SDS-polyacrylamide gel. Its mass corresponds to a dimer linked by a thiosulfinate bond (data not shown). Because formation of a thiosulfinate bond can only proceed via condensation of two sulfenic acids, this result demonstrates that reduction of Met-O passes through the sulfenic acid chemistry. Fourth, inspection of the alignment of the 358 protein sequences of putative fRMsrs deduced from the DNA sequences available to date shows that Cys118 is invariant, whereas Cys84 and Cys94 are 94.5 and 98.6% conserved (see the alignment shown in supplemental Fig. S1). Taken together, these data definitively prove that 1) Cys118 is the catalytic Cys and 2) the catalytic mechanism involves the formation of a sulfenic acid intermediate on Cys118. Therefore, these results exclude Cys94 as the catalytic Cys as postulated from molecular modeling on the S. cerevisiae fRMsr (11) and from inspection of the E. coli fRMsr in complex with a MES molecule trapped from the crystallization buffer (10).
The crystal structure of the N. meningitidis fRMsr in complex with l-Met-R-O illuminates not only the catalysis of the reductase step but also how fRMsr binds Met-O with a strong preference for the l-Met-R-O isomer. As already indicated, the catalysis implies a proton transfer from the catalytic Cys to the oxygen of the sulfoxide. Such a function in fRMsr is probably played by the invariant Asp143, which is located on strand β6, possibly via Wat125. In accord with this assumption, a D143A fRMsr shows a 350-fold decrease in activity (data not shown), similar to that observed for E94A MsrA and H103A MsrB, when determined under similar steady-state conditions (8, 9). The crystal structure also shows that the oxygen of the sulfoxide is located in a hydrophilic cavity, where it strongly interacts not only with Asp143 but also with Wat99, which is itself hydrogen-bonded with the main chain nitrogen of Ser119. Wat99 recalls the water molecule observed in MsrB that is stabilized through a hydrogen-bonding network with the side chains of Thr26, His100, and Asn119 (21). In the scenario, a sulfurane intermediate/transition state is formed that rearranges into a sulfenic acid intermediate (see Scheme 1A). Therefore, Asp143 and Wat99 should interact not only with the oxygen of the sulfoxide and its polarized form but probably also with the OH of the sulfurane transition state and directly or indirectly with the OH of the sulfenic acid, as observed in the crystal structure of the MsrA sulfenic acid intermediate (22). To form a sulfurane, the Cys118 has to be under the thiolate form to attack the sulfur of the sulfoxide, the geometry of which is tetrahedral. Clearly, because the pKapp of Cys118 in the free enzyme is 8.5, binding of the substrate should contribute to the decrease of the pKapp of Cys118. This situation is similar to that observed in MsrA and MsrB. As shown by the crystal structure, the sulfur is closer to Cys118 than to Cys84, and consequently it is in a better position to form a covalent bond with Cys118. Moreover, if Cys118 is reduced, the thiolate can adopt a favorable orientation that permits formation of the sulfurane with the SH and the OH groups in apical positions.
SCHEME 1.

Proposal for the catalysis of the fRMsr reductase step (A) and the mechanism up to the formation of the disulfide Cys118–Cys84 intermediate (B). A, the substrate binds to the active site with its sulfoxide function largely polarized, leading to a transition state compatible with a sulfurane of bipyramidal geometry. The rearrangement of the transition state leads to the formation of a sulfenic acid intermediate on the catalytic Cys118 residue and of Met. In this scenario, Asp143 and Wat99, which is itself hydrogen-bonding with the main chain nitrogen of Ser119, stabilize the sulfurane transition state via interaction with the OH group, a situation similar to that described for MsrB (7). B, the reductase step (the first one) leads to the formation of a sulfenic acid intermediate on the catalytic Cys118 and of Met. In the second step, attack of Cys84 on the sulfur atom of the sulfenic acid intermediate leads to the formation of a disulfide bond between Cys118 and Cys84 and the release of a water molecule. a, return of the active site to a fully reduced state can proceed via reduction of the Cys118–Cys84 disulfide bond formed (see “Discussion”) by Trx or if the Cys94–Cys84 disulfide bond forms (which is rather unlikely), via the reduction of the Cys94–Cys84 disulfide bond by Trx.
As shown previously (10), fRMsrs only reduce the R isomer at the sulfoxide function. This stereochemical preference is confirmed by the structure of N. meningitidis fRMsr in complex with l-Met-R-O. The methyl group of the sulfoxide points perpendicularly toward the indole ring of the invariant Trp62, whose conformation is strongly stabilized within the active site. Therefore, the indole ring is the major factor involved in the binding and stabilization of the ϵ-methyl group, as already observed in MsrA and MsrB, where at minimum a Trp residue is involved. The β- and δ-methylene groups are stabilized by van der Waals interactions with the side chains of the invariant Ile87, Ile116, and Tyr66. Together, these structural data explain why only the R isomer at the sulfoxide function is recognized by the fRMsrs.
As shown in the present study from kinetic studies, fRMsr is highly selective for the l-isomer at the Cα carbon. Moreover, as already described, it is the free form of Met-O that is active. Inspection of the crystal structure of fRMsr shows the following. 1) The negatively charge of the carboxylate function, which is completely delocalized, is buried and points toward the positive helix dipole at the N-terminal end of the helix α3. In addition, the backbone amides of Val93, Cys94, and Ile116 are within hydrogen distance and form an oxyanion hole. A similar situation is observed with the negative charge of the sulfonic acid function of the MES molecule in the E. coli fRMsr (10), the negatively charged 3′,5′-cyclic phosphate of cGMP and cAMP in the phosphodiesterase 5A (23), and adenylate cyclase from Anabaena (24), respectively. 2) The NH3+ group is strongly stabilized by a hydrogen-bonding network involving (i) the side chain of the invariant Glu125, which itself is stabilized by interactions with the invariant Asp141 and Asp143, and (ii) the main chain oxygen of Ile116. All of these stabilizing interactions 1) favor binding of the l-Met-O epimer, 2) prevent the binding of a Met-R-O within a peptide or protein context due to steric hindrance, and 3) are responsible for the absence of activity with DMSO because of the absence of COO− and NH3+ groups in DMSO.
As clearly revealed by the crystal structure, the l-Met-R-O is strongly bound in a small cavity that is capped by the flap (residues 107–124, including Cys118), which thus protects the active site from the solvent. In the structure, the Cys84–Cys118 disulfide bond should restrict the conformational freedom of the flap, which is situated between strands β4 and β5, respectively. Because Cys118 has to be free to play its catalytic role, the disulfide-bonded Cys118–Cys84 form is not catalytically relevant for the reductase step. Nevertheless, the positioning of Cys118 is compatible with a nucleophilic attack of the sulfoxide function of the substrate. The disulfide bond Cys118–Cys84 is, however, essential to obtaining crystals because the addition of DTT dissolves them (data not shown) and thus prevents obtention of crystals of the reduced form.
Formation of a sulfenic acid intermediate on Cys118 implies a recycling process in the wild type that can involve formation of an intradisulfide bond, which is then reduced by Trx. Two Cys, Cys94 or Cys84, could be implicated. All of the fRMsr mutants in which either Cys84 or Cys94 was substituted by Ala were active. In agreement with these results is the fact that 5.6 and 1.4% of the putative sequences of fRMsrs only contain the signatures Cys118/Cys94 and Cys118/Cys84, respectively (see the alignment shown in supplemental Fig. S1). When Cys118 and Cys94 were individually mutated to Ala in the wild type from N. meningitidis, a stoichiometry of 1 mol of Met/subunit was observed in the absence of Trx with formation of a disulfide bond between Cys118 and Cys94 or between Cys118 and Cys84, depending on the mutant considered. Such results are based on the difference in thiol content observed before and after the addition of Met-O. For the wild type and the C136A fRMsr, a stoichiometry of 1 mol of Met/subunit was also observed with formation of a disulfide bond, the nature of which remains to be determined. Because 1) the disulfide bond in the crystal is between Cys118 and Cys84 when Met-O is bound, 2) Cys94 is located at the N terminus of helix α3 on one of the two sides of the active site far away from Cys118 and Cys84 (i.e. ∼8 Å), and 3) Cys118 is located within the flap, it is probable that the first disulfide formed for recycling the activity of the fRMsr from N. meningitidis is Cys118–Cys84 (see Scheme 1B). This is in accord with the DTNB titration results obtained under native conditions, which showed that Cys118 and Cys84 are titrated by DTNB and formed a disulfide bond in the wild type and C136A/C94A mutant. In fact, one of the two Cys residues, probably Cys118 that is located on the flexible flap forms an adduct with the TNB− moiety, which, by an intramolecular attack of Cys84, leads to formation of the Cys84–Cys118 disulfide bond. When one of these two Cys residues, either Cys84 or Cys118, is substituted by Ala, Cys94 is now titrated together with either Cys118 or Cys84, respectively, and thus is accessible. But no disulfide bond between Cys94 and either Cys118 or Cys84 is formed, in contrast to that observed between Cys118 and Cys84. Clearly, formation of the Cys84–Cys118 disulfide bond in the wild type prevents accessibility of the DTNB to Cys94, whereas in the absence of the Cys84–Cys118 disulfide bond, as is the case for the C118A or C84A mutant, Cys94 becomes accessible to DTNB.
The fact that only one Met molecule was formed in the wild type and in the C136A mutant requires explanation. Either Cys118 remains engaged in a disulfide bond with Cys84, it participates in a new disulfide bond with Cys94, or it is free. The last hypothesis, although less probable, implies formation of a Cys84–Cys94 disulfide bond and an active site that is no longer catalytically competent to reduce another Met-O molecule. In other words, formation of the Cys84–Cys94 disulfide bond would perturb the conformation of the active site, which in return would prevent efficient binding of l-Met-R-O. In any case, the behavior of the wild-type fRMsr is clearly different from that of the MsrA from E. coli (6). Indeed, the E. coli MsrA, which also possesses three Cys residues, forms two successive disulfide bonds, first between the catalytic Cys51 and the recycling Cys198 and then between Cys198 and Cys206. In that case, formation of the second disulfide bond renders the catalytic Cys51 free and renders the active site competent to reduce a second molecule of Met-O.
The fact that oxidized disulfide forms of fRMsr are formed (i.e. containing a Cys118–Cys84 or Cys118–Cys94 bond) or would be formed in the absence of reductant (i.e. Cys84–Cys94) does not mean that all of these oxidized forms are catalytically competent under steady-state conditions (i.e. in the presence of Trx). For instance, in the 5.6% of putative fRMsrs where only Cys94 is present, a disulfide bond probably forms between Cys118 and Cys94 in the absence of reductant, as shown for the C84A/C136A fRMsr from N. meningitidis. However, inspection of the crystal structure shows, as already pointed out, that Cys94 is situated at one of the sides of the active site far away from Cys118. Thus, Cys94 is not in a good position to form a Cys118–Cys94 bond efficiently unless the enzyme is highly flexible. At present, we do not know the rate at which this disulfide bond is formed in the C84A mutant and whether it is this form that is recognized and reduced by Trx.
Like all GAF domains, fRMsr is dimeric. An obvious question is whether not only both subunits of fRMsr are active as shown in the absence of Trx but also express their activity with the same catalytic efficiency. Another question concerns the nature of the rate-limiting step. This has to be determined in order to validate the amino acids that play a role in the catalysis and in the substrate specificity in the reductase step.
The fRMsr represents a unique case in which a GAF fold behaves as an enzyme and moreover as an independent folded unit not included in a modular larger protein. Like MsrA and MsrB, it catalyzes the reduction of Met-O and therefore constitutes the third protein fold to exhibit a Met-O reductase activity that involves the sulfenic acid chemistry. From a structural point of view, the active site of fRMsr is clearly different from those of MsrA and MsrB. All of the amino acids within the active site that underlie the l-Met-R-O reductase activity are not present in the other GAF domains. From an evolutionary point of view, this observation raises the question of how a GAF fold has evolved to acquire a broad range of different functions by binding various molecules, including cyclic nucleotides, heme, amino acids, or Met-O.
Supplementary Material
Acknowledgments
We thank Dr. A. Thompson, Dr. B. Guimaraes, and Dr. P. Legrand for help during data collection at beamline PROXIMA 1 at synchrotron Soleil (GIF-sur-YVETTE); Dr. G. Chevreux, Dr. S. Sanglier-Cianferani, and Dr. A. Van Dorsselear for mass spectrometry analyses; Sabrina Hecht for efficient technical help in fRMsr purification; and Dr. K. Weissman for careful reading of the manuscript. Access to the Bruker X8-Proteum and to the TDA 302 VISCOTEK (SEC-Light Scattering) (Service Commun de Biophysicochimie des Interactions Moléculaires, Nancy I) is deeply appreciated.
This work was supported by the CNRS, the University of Nancy I, and the IFR 111 Bioingénierie.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
The atomic coordinates and structure factors (code 3MMH) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The numbering of fRMsr amino acid residues is based on the numbering of the E. coli fRMsr sequence according to sequence alignment presented in Fig. 1.
- Met
- methionine
- Met-O
- methionine sulfoxide (dl-Met-R,S-O)
- MES
- 2-(N-morpholino)ethanesulfonic acid
- 2-PDS
- 2,2′-dithiodipyridine
- DTNB
- 5,5′-dithiobis(2-nitro)benzoate
- fRMsr
- free methionine-R-sulfoxide reductase
- DTT
- dithiothreitol
- MPD
- 2-methylpentane-2,4-diol
- r.m.s.
- root mean square.
REFERENCES
- 1.Schöneich C. (2005) Biochim. Biophys. Acta 1703, 111–119 [DOI] [PubMed] [Google Scholar]
- 2.Boschi-Muller S., Olry A., Antoine M., Branlant G. (2005) Biochim. Biophys. Acta 1703, 231–238 [DOI] [PubMed] [Google Scholar]
- 3.Moskovitz J. (2005) Biochim. Biophys. Acta 1703, 213–219 [DOI] [PubMed] [Google Scholar]
- 4.Stadtman E. R., Van Remmen H., Richardson A., Wehr N. B., Levine R. L. (2005) Biochim. Biophys. Acta 1703, 135–140 [DOI] [PubMed] [Google Scholar]
- 5.Weissbach H., Resnick L., Brot N. (2005) Biochim. Biophys. Acta 1703, 203–212 [DOI] [PubMed] [Google Scholar]
- 6.Boschi-Muller S., Azza S., Sanglier-Cianferani S., Talfournier F., Van Dorsselear A., Branlant G. (2000) J. Biol. Chem. 275, 35908–35913 [DOI] [PubMed] [Google Scholar]
- 7.Boschi-Muller S., Gand A., Branlant G. (2008) Arch. Biochem. Biophys. 474, 266–273 [DOI] [PubMed] [Google Scholar]
- 8.Antoine M., Gand A., Boschi-Muller S., Branlant G. (2006) J. Biol. Chem. 281, 39062–39070 [DOI] [PubMed] [Google Scholar]
- 9.Neiers F., Sonkaria S., Olry A., Boschi-Muller S., Branlant G. (2007) J. Biol. Chem. 282, 32397–32405 [DOI] [PubMed] [Google Scholar]
- 10.Lin Z., Johnson L. C., Weissbach H., Brot N., Lively M. O., Lowther W. T. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 9597–9602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le D. T., Lee B. C., Marino S. M., Zhang Y., Fomenko D. E., Kaya A., Hacioglu E., Kwak G. H., Koc A., Kim H. Y., Gladyshev V. N. (2009) J. Biol. Chem. 284, 4354–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Badger J., Sauder J. M., Adams J. M., Antonysamy S., Bain K., Bergseid M. G., Buchanan S. G., Buchanan M. D., Batiyenko Y., Christopher J. A., Emtage S., Eroshkina A., Feil I., Furlong E. B., Gajiwala K. S., Gao X., He D., Hendle J., Huber A., Hoda K., Kearins P., Kissinger C., Laubert B., Lewis H. A., Lin J., Loomis K., Lorimer D., Louie G., Maletic M., Marsh C. D., Miller I., Molinari J., Muller-Dieckmann H. J., Newman J. M., Noland B. W., Pagarigan B., Park F., Peat T. S., Post K. W., Radojicic S., Ramos A., Romero R., Rutter M. E., Sanderson W. E., Schwinn K. D., Tresser J., Winhoven J., Wright T. A., Wu L., Xu J., Harris T. J. (2005) Proteins 60, 787–796 [DOI] [PubMed] [Google Scholar]
- 13.Ho Y. S., Burden L. M., Hurley J. H. (2000) EMBO J. 19, 5288–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antoine M., Boschi-Muller S., Branlant G. (2003) J. Biol. Chem. 278, 45352–45357 [DOI] [PubMed] [Google Scholar]
- 15.Olry A., Boschi-Muller S., Marraud M., Sanglier-Cianferani S., Van Dorsselear A., Branlant G. (2002) J. Biol. Chem. 277, 12016–12022 [DOI] [PubMed] [Google Scholar]
- 16.Sharov V. S., Ferrington D. A., Squier T. C., Schöneich C. (1999) FEBS Lett. 455, 247–250 [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W. (1993) J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 18.Vagin A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 19.Murshudov G. N., Vagin A. A., Lebedev A., Wilson K. S., Dodson E. J. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 247–255 [DOI] [PubMed] [Google Scholar]
- 20.Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 21.Ranaivoson F. M., Neiers F., Kauffmann B., Boschi-Muller S., Branlant G., Favier F. (2009) J. Mol. Biol. 394, 83–93 [DOI] [PubMed] [Google Scholar]
- 22.Ranaivoson F. M., Antoine M., Kauffmann B., Boschi-Muller S., Aubry A., Branlant G., Favier F. (2008) J. Mol. Biol. 377, 268–280 [DOI] [PubMed] [Google Scholar]
- 23.Heikaus C. C., Stout J. R., Sekharan M. R., Eakin C. M., Rajagopal P., Brzovic P. S., Beavo J. A., Klevit R. E. (2008) J. Biol. Chem. 283, 22749–22759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez S. E., Bruder S., Schultz A., Zheng N., Schultz J. E., Beavo J. A., Linder J. U. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 3082–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.