

Abstract
Patterns of genetic differentiation among taxa at early stages of divergence provide an opportunity to make inferences about the history of speciation. Here, we conduct a survey of DNA-sequence polymorphism and divergence at loci on the autosomes, X chromosome, Y chromosome and mitochondrial DNA in samples of Mus domesticus, M. musculus and M. castaneus. We analyzed our data under a divergence with gene flow model and estimate that the effective population size of M. castaneus is 200 000–400 000, of M. domesticus is 100 000–200 000 and of M. musculus is 60 000–120 000. These data also suggest that these species started to diverge approximately 500 000 years ago. Consistent with this recent divergence, we observed considerable variation in the genealogical patterns among loci. For some loci, all alleles within each species formed a monophyletic group, while at other loci, species were intermingled on the phylogeny of alleles. This intermingling probably reflects both incomplete lineage sorting and gene flow after divergence. Likelihood ratio tests rejected a strict allopatric model with no gene flow in comparisons between each pair of species. Gene flow was asymmetric: no gene flow was detected into M. domesticus, while significant gene flow was detected into both M. castaneus and M. musculus. Finally, most of the gene flow occurred at autosomal loci, resulting in a significantly higher ratio of fixed differences to polymorphisms at the X and Y chromosomes relative to autosomes in some comparisons, or just the X chromosome in others, emphasizing the important role of the sex chromosomes in general and the X chromosome in particular in speciation.
Keywords: ancestral polymorphism, effective population size, introgression, speciation
Introduction
Multilocus datasets of DNA sequence variation within and between closely related species can provide important insights into the history of speciation. A number of analytical approaches have been developed recently that take into account such data to estimate parameters in a coalescent framework and thereby evaluate different speciation models (e.g. Wakeley & Hey 1997; Nielsen & Wakeley 2001; Hey & Nielsen 2004, 2007). This analytical framework has become known as ‘divergence population genetics’. This general approach is growing in popularity and has been applied to closely related species or subspecies in a number of groups of both plants and animals (e.g. Machado et al. 2002; Won & Hey 2005; Kronforst et al. 2006; Lawton-Rauh et al. 2007; Stadler et al. 2008).
House mice have served as an important model for genetic studies of speciation, both in a well-studied hybrid zone (e.g. Teeter et al. 2008) and through crosses in the laboratory (e.g. Britton-Davidian et al. 2005), but relatively little is known about overall patterns of genetic differentiation. House mice include three main species (also referred to as subspecies): Mus domesticus in Western Europe, the Middle East and North Africa (and recently introduced worldwide), M. musculus in Eastern Europe and Northern Asia, and M. castaneus in Southeast Asia. Following previous authors (e.g. Sage et al. 1993), we refer to these taxa as species rather than subspecies because they are genetically distinct and exhibit partial reproductive isolation despite the presence of some gene flow, much like Drosophila pseudoobscura and D. persimilis (Hey & Nielsen 2004) or D. yakuba and D. santomea (Llopart et al. 2005). These lineages are thought to have diverged from an ancestral population in the Indian subcontinent (Boursot et al. 1996; Din et al. 1996). The timing of divergence is uncertain, with estimates ranging from 350 000 to 900 000 years ago (She et al. 1990; Boursot et al. 1996; Suzuki et al. 2004). M. domesticus is believed to have spread westward, and fossils dating to 12 000 bp are known from Israel (Auffray et al. 1990). From the Middle East, M. domesticus migrated into Western Europe during the Iron Age around 3000 years ago, after the spread of agriculture (Cucchi et al. 2005). The dispersal routes of M. musculus and M. castaneus are less well documented, but it is likely that M. musculus reached Eastern Europe via a northern Asian route, and that M. castaneus migrated eastwards (Boursot et al. 1993). M. domesticus and M. musculus meet in a hybrid zone that runs from Denmark to Bulgaria, and M. musculus and M. castaneus meet in a poorly studied hybrid region in northern China and have hybridized to form M. molossinus in Japan (Boursot et al. 1993). There is also evidence of hybridization between M. domesticus and M. castaneus in California (Orth et al. 1998). Mice from the Indian region have been referred to as bactrianus by some authors (reviewed in Boursot et al. 1993) and have been included within castaneus by others (e.g. Baines & Harr 2007). Here, we refer to mice from India as M. castaneus.
Studies of the hybrid zone between M. domesticus and M. musculus have documented extensive variation in patterns of introgression among loci (e.g. Macholan et al. 2007; Teeter et al. 2008). The X chromosome generally shows reduced introgression (Tucker et al. 1992; Dod et al. 1993; Munclinger et al. 2002; Macholan et al. 2007), while the Y chromosome shows reduced introgression in some transects of the hybrid zone (Vanlerberghe et al. 1986; Tucker et al. 1992; Dod et al. 1993), but not in others (Munclinger et al. 2002; Macholan et al. 2007). Laboratory crosses between M. domesticus (or B6, a strain largely derived from domesticus) and M. musculus, M. castaneus and M. molossinus reveal reduced fecundity or hybrid male sterility caused by loci on both the X chromosome and the autosomes (e.g. Forejt 1996; Oka et al. 2004, 2007; Storchova et al. 2004; Britton-Davidian et al. 2005; Davis et al. 2007; Good et al. 2008; Gregorova et al. 2008; Takada et al. 2008).
Important questions remain about the timing of divergence among the major lineages, the extent of historical gene flow, the effective population sizes for each lineage, and the consequences of population splitting and reproductive isolation for patterns of genetic differentiation. To begin to address these issues, we compared patterns of differentiation among loci residing on chromosomes with different modes of inheritance and different effective population sizes: mitochondrial DNA (mtDNA), the Y chromosome, the X chromosome and the autosomes. These differences lead to simple predictions for rates of differentiation under a neutral model with no gene flow following population splitting: mtDNA and Y-linked loci are expected to differentiate more quickly than X-linked loci which in turn will be more differentiated than autosomal loci.
We sequenced eight effectively unlinked loci, including one mitochondrial, one Y-linked, two X-linked and four autosomal regions, in population samples of M. domesticus, M. musculus and M. castaneus to address four main issues: (i) What is the level and pattern of genetic variation and effective population size of each species? (ii) When did these species start to diverge? (iii) Are patterns of genetic variation consistent with a simple allopatric model with no gene flow? If not, what is the extent and pattern of gene flow? (iv) are genomic regions with lower effective population sizes more differentiated, as predicted by theory?
Materials and methods
Samples
For nuclear loci, we sampled 60 Mus domesticus, 59 M. musculus and 59 M. castaneus from their native ranges (Fig. 1 and Table S1, Supporting information). For each species, at least two populations were included, one closer to the presumed ancestral range, the other derived. For M. domesticus, Israel (Is) is more ancestral and Western Europe (WE) is derived. For M. musculus, Kazakhstan (Kz) is ancestral and Russia (Ru) and Eastern Europe (EE) are derived. For M. castaneus, India (In) is ancestral and Taiwan (Tw) and China (Ch) are derived. All mice were collected at least 300 m apart to avoid sampling related individuals. DNA from one individual each of M. caroli, M. spicilegus and M. spretus was purchased from the Jackson laboratory, and these taxa were used as outgroups.
Fig. 1.
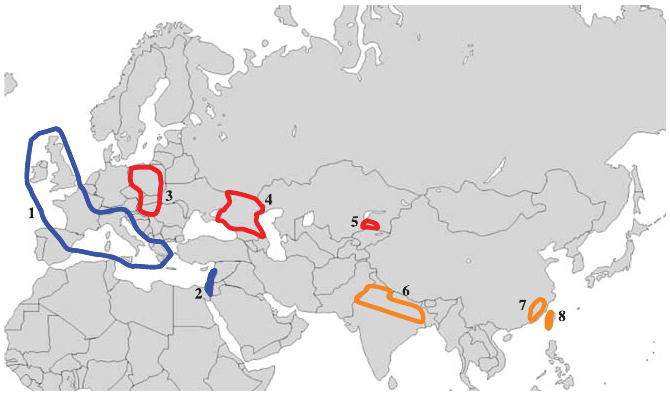
Approximate location of populations sampled in this study. Blue indicates Mus domesticus, red indicates M. musculus and orange indicates M. castaneus. 1, Western Europe (WE); 2, Israel (Is); 3, Eastern Europe (EE); 4, Russia (Ru); 5, Kazakhstan (Kz); 6, India (In); 7, China (Ch); 8, Taiwan (Tw). Sample sizes, sampling localities names and geographic coordinates are given in Table S1 (Supporting information).
Molecular methods
We sequenced mostly intronic portions of Chrng, Med19, Prpf3 and Clcn6 on Chromosomes 1, 2, 3 and 4, respectively, G6pdx and Ocrl on the X chromosome, Jarid1d (Smcy) on the Y chromosome, and the mtDNA control region (Table 1). For nuclear loci, we selected genes that were widely expressed, defined as genes where the maximum expression in any tissue was 10% or less of the total expression (Su et al. 2004). For each locus, we amplified two overlapping fragments using polymerase chain reaction (PCR), and we sequenced both fragments. This allowed us to identify cases of allele-specific PCR. Both DNA strands were sequenced. The mitochondrial control region was chosen because it is variable and has been widely studied in these taxa (Prager et al. 1998). Fifty-six control region sequences of M. domesticus from WE were taken from Nachman et al. (1994), and 229 new sequences were generated from populations of M. domesticus, M. musculus and M. castaneus (Table S1, Supporting information). Outgroup sequences for this locus were retrieved from public databases. PCR and sequencing primers and amplicon details are provided in Table S2 (Supporting information).
Table 1.
Loci surveyed
| Gene | Chromosome | Region sequenced | Recombination rate (cM/Mb)* | Position in NCBI build 36 (bp) |
|---|---|---|---|---|
| Chrng | 1 | 5′UTR-Intron 6 | 0.36 | 89 036 568–89 040 081 |
| Med19 | 2 | Intron 1 | 0.22 | 84 483 105–84 485 675 |
| Prpf3 | 3 | Intron 3 | 0.71 | 95 934 441–95 937 152 |
| Clcn6 | 4 | Intron 8–11 | 0.77 | 146 861 451–146 864 028 |
| G6pdx | X | Intron 2 | 0.25 | 70 675 567–70 678 566 |
| Ocrl | X | Intron 1–4 | 0.51 | 44 205 361–44 208 191 |
| Jarid1d | Y | Intron 10 | 0.00 | 254 115–256 663 |
| Control region | Mitochondria | 0.00 | 15 373–16 299 |
The local recombination rate was calculated for a 10-Mb window centered on the sequenced region by regressing the genetic position of markers against their physical position on mouse NCBI build 36.
Data analyses
Sequences were trimmed to exclude short exonic regions. Assembly and editing were performed using phred/phrap/consed/polyphred (Nickerson et al. 1997; Ewing & Green 1998; Ewing et al. 1998; Gordon et al. 1998) coupled with automated shell scripts and Perl programs kindly provided by August Woerner (University of Arizona, USA). The resulting contigs were deposited in GenBank under Accession nos EU932966–EU933930 and EU938914–EU939142. Alignments generated with ClustalW (Thompson et al. 1994) were checked and manually edited with BioEdit (Hall 1999). All insertion/deletion polymorphisms were excluded from subsequent analyses. We excluded individuals with more than 10% missing data. We also excluded sites with more than 10% of the total individuals missing. This was done separately for each locus. Haplotypes were inferred with Phase 2.1.1 (Stephens et al. 2001; Stephens & Donnelly 2003) after checking for convergence of three independent runs for each data set.
The program sites (Wakeley & Hey 1997) was used to calculate a number of summary statistics, including π (Nei & Li 1979) and θ (Watterson 1975), two estimators of the population mutation parameter 4Neμ (where μ is the neutral mutation rate and Ne is the effective population size), and Dxy, the average pairwise divergence between populations or between species (Nei 1987). Due to the high mutation rate of the mtDNA control region the occurrence of multiple substitutions at single sites is likely. We estimated the appropriate model of nucleotide substitution using modeltest 3.06 (Posada & Crandall 1998) with the Akaike Information Criterion (Posada & Buckley 2004) and we then corrected for multiple substitutions. The ratio of the male to female mutation rate (α) was estimated with average Dxy at autosomes, X and Y chromosomes between the three species of house mice and Mus caroli, using the formulae in Miyata et al. (1987).
We tested for departures from a neutral model of molecular evolution using two tests based on the frequency spectrum of polymorphisms, Tajima's D (Tajima 1989) and Fu and Li's D (Fu & Li 1993). These tests were calculated for each population and also for each of the three species using sites (Wakeley & Hey 1997). The Hudson–Kreitman–Aguade (HKA) test (Hudson et al. 1987) was used to compare the ratio of polymorphism to divergence among loci. Multilocus HKA tests were performed using polymorphism in each species and also polymorphism in the three species together (i.e. a total of four tests) and uncorrected average pairwise divergence (Dxy) to M. caroli. Statistical significance for all neutrality tests was obtained by performing 1000 coalescent simulations conditioned on the parameters estimated from our data using the program hka (http://lifesci.rutgers.edu/∼heylab/HeylabSoftware.htm#HKA). FST between populations of a given species, and between species, was calculated using sites (Wakeley & Hey 1997). Evolutionary relationships among alleles were inferred using the neighbour-joining method (Saitou & Nei 1987) in mega 4 (Tamura et al. 2007). Trees were rooted with the M. caroli sequence and bootstrap values for each node were calculated after 1000 replicates (Felsenstein 1985).
To obtain maximum-likelihood (ML) estimates of population sizes, divergence times, and migration rates we used the computer program im which is an implementation of the Markov chain Monte Carlo (MCMC) method for analysis of genetic data under an isolation with migration model (Hey & Nielsen 2004). im assumes that there is no recombination within loci and free recombination between loci. We used the program imgc (Woerner et al. 2007) to obtain the longest region within each locus without four gametic types. Using this non-recombining dataset (Table S3, Supporting information), we performed three different pairwise analyses (M. domesticus and M. musculus, M. domesticus and M. castaneus, and M. musculus and M. castaneus) with three replicates for each. For each analysis, we ran the program under Metropolis Coupled MCMC, using 12 chains with a two-step heating scheme and parameters that allowed for proper chain swapping. We ran the program for at least 10 million steps. For each analysis we checked for convergence between the three replicates, and we present results from just one replicate of each analysis. We used im to estimate the effective population size of each species and the effective population size of the ancestral population that gave rise to the contemporary species. We also estimated the time since the ancestral population split, and the rate at which species exchange genes (2Nm) per generation. We recorded the distribution of the number of migration events for each locus over the course of the analyses. Output from im is expressed in units of 4Neμ, tμ, and m/μ, where μ is the neutral mutation rate per generation, t is the divergence time in generations and m is the migration rate per generation. To convert these parameters into Ne, t and m, we estimated μ for each locus assuming the divergence to M. caroli represents 4.3 million years (Suzuki et al. 2004) and a generation time of 0.5 or 1.0 years (see below). Likelihood ratio tests comparing models with and without gene flow were conducted with ima (Hey & Nielsen 2007).
There are several sources of error in these analyses. The im model includes gene flow between two populations which derive from a single ancestral population. The ancestral population and each of the derived populations may have different population sizes, but more complex demographic scenarios are not incorporated. The exact history of mouse populations is not known but is probably more complex. Our data include three species, each with two or three populations. This has several implications. First, our sample may contain structure that is not modelled appropriately by im. To address this, we redid all analyses using only the largest population from each species. Similar results were obtained and thus only the more complete analyses are reported. Second, since im compares only two populations at a time, it does not account for gene flow between those populations and any unsampled populations. We conducted analyses in all pairwise combinations for the three species and obtained similar estimates of parameters in different comparisons. For example, the estimate of Ne for M. domesticus is very similar in comparison to M. castaneus and in comparison to M. musculus (see Results). This suggests that gene flow with unsampled populations is not leading to substantial bias in the estimation of some parameters. Nonetheless, we also compared estimates of Ne obtained from im with estimates based on the neutral prediction that Ne = π/4μ for a single population at mutation–drift equilibrium without gene flow, and we obtained similar results.
Another potential source of error in these analyses comes from the estimate of mutation rate per generation, which requires assumptions about generation time and molecular clock calibrations from comparisons to other species. Our estimates of mutation rate per year (see Results) are in good agreement with previous estimates (e.g. Li et al. 1996; Waterston et al. 2002). However, estimates of Ne depend on estimates of mutation rate per generation. To convert mutation rates per year into rates per generation, we need to know the number of generations per year. Gestation in mice lasts three weeks, and mice are reproductively mature at about two months. In the lab, mice may have up to four generations per year. In the wild, commensal mice can breed year-round if food is available, but feral populations of mice typically breed seasonally (Bronson 1979). House mice have only recently evolved to be commensal, and abundant food for commensal mice has likely only occurred since the development of agriculture (i.e. within the last 8000 years). Thus, for the vast majority of their roughly 500 000-year evolutionary history, house mice have probably bred seasonally and had only one or two generations per year. To account for the uncertainty in generation time, we provide estimates of population parameters from im using generation times of 0.5 and 1.0 years. While our estimates of t depend on generation length, our estimates of divergence time in years do not.
Results
Intraspecific polymorphism and effective population size
We observed considerable variation in levels of polymorphism among loci and among species (Table 2). Averaged over all nuclear loci, Mus castaneus was the most variable (π = 0.43%, SE = 0.11%), followed by M. domesticus (π = 0.14%, SE = 0.03%) and M. musculus (π = 0.13%, SE = 0.07%). In these comparisons, π for X-linked loci was multiplied by 4/3, and π for Jarid1d was multiplied by 4 to account for differences in effective population size. Nucleotide diversity for mtDNA showed the same trend among species (Table 2). In general, the proportion of segregating sites (θ) was higher than the average number of pairwise differences (π), and thus Tajima's D was negative for many locus/population combinations (values for each species are given in Table 2, and values for each population are given in Table S4, Supporting information). A smaller number of locus/population combinations had positive Tajima's D-values. The same was observed for Fu and Li's D. Of the 35 significant tests of Tajima's D and Fu and Li's D at nuclear genes, 29 tests were associated with significantly negative values (including all genes except Clcn6) while six tests were associated with significantly positive values, and these all involved Clcn6 (Table S4, Supporting information). The observation of widespread rare polymorphisms (i.e. negative Tajima's D) is consistent with population expansions, although Clcn6 may be subject to different evolutionary forces (see below).
Table 2.
Levels of polymorphism within species of house mice, and divergence between these species and Mus caroli
| Locus (chromosome) | Species | N† | L (bp)‡ | S§ | π (%)¶ | Θ (%)¶ | Tajima's D†† | Fu and Li's D†† | Dxy‡‡ |
|---|---|---|---|---|---|---|---|---|---|
| Chrng (1) | M. domesticus | 92 | 2218 | 30 | 0.284 | 0.266 | 0.211 | −0.038 | 3.382 |
| M. musculus | 108 | 2214 | 19 | 0.046 | 0.163 | −2.050** | −2.157* | 3.526 | |
| M. castaneus | 62 | 2124 | 62 | 0.671 | 0.622 | 0.270 | 0.391 | 3.471 | |
| Med19 (2) | M. domesticus | 102 | 1699 | 13 | 0.048 | 0.147 | −1.808* | −2.792** | 5.489 |
| M. musculus | 84 | 1658 | 7 | 0.135 | 0.084 | 1.443 | −0.562 | 5.592 | |
| M. castaneus | 76 | 1679 | 15 | 0.056 | 0.182 | −1.990** | −1.047 | 5.494 | |
| Prpf3 (3) | M. domesticus | 108 | 2423 | 21 | 0.071 | 0.165 | −1.641* | −3.226** | 2.380 |
| M. musculus | 108 | 2399 | 29 | 0.062 | 0.230 | −2.194 | −2.395 | 2.441 | |
| M. castaneus | 100 | 2430 | 34 | 0.126 | 0.270 | −1.640* | −3.384** | 2.250 | |
| Clcn6 (4) | M. domesticus | 104 | 2028 | 29 | 0.216 | 0.274 | −0.645 | 1.305 | 3.745 |
| M. musculus | 106 | 2012 | 46 | 0.547 | 0.437 | 0.791 | 0.731 | 3.833 | |
| M. castaneus | 92 | 1986 | 62 | 0.763 | 0.613 | 0.794 | 0.634 | 3.747 | |
| Average of autosomal loci | M. domesticus | 102 | 2092 | 23 | 0.155 | 0.213 | 3.749 | ||
| M. musculus | 102 | 2071 | 25 | 0.198 | 0.229 | 3.848 | |||
| M. castaneus | 83 | 2055 | 43 | 0.404 | 0.422 | 3.741 | |||
| G6pdx (X) | M. domesticus | 56 | 2386 | 5 | 0.060 | 0.046 | 0.769 | −0.923 | 2.591 |
| M. musculus | 59 | 2386 | 5 | 0.026 | 0.045 | −1.012 | −2.981** | 2.617 | |
| M. castaneus | 43 | 2354 | 23 | 0.174 | 0.226 | −0.755 | −0.242 | 2.679 | |
| Ocrl (X) | M. domesticus | 55 | 2123 | 17 | 0.122 | 0.175 | −0.933 | −1.997* | 3.477 |
| M. musculus | 55 | 2100 | 8 | 0.017 | 0.083 | −2.128** | −3.245** | 3.341 | |
| M. castaneus | 30 | 1983 | 32 | 0.336 | 0.407 | −0.634 | −0.227 | 3.538 | |
| Average of X-linked loci | M. domesticus | 56 | 2255 | 11 | 0.091 | 0.111 | 3.034 | ||
| M. musculus | 57 | 2243 | 7 | 0.022 | 0.064 | 2.979 | |||
| M. castaneus | 37 | 2169 | 28 | 0.255 | 0.317 | 3.109 | |||
| Jarid1d (Y) | M. domesticus | 52 | 2329 | 4 | 0.034 | 0.038 | −0.247 | −0.131 | 4.740 |
| M. musculus | 36 | 2335 | 3 | 0.023 | 0.031 | −0.544 | −1.644 | 4.882 | |
| M. castaneus | 28 | 2315 | 13 | 0.185 | 0.144 | 0.948 | −0.320 | 4.904 | |
| control region (mtDNA) | M. domesticus | 67 | 889 | 37 | 0.563 | 0.872 | −1.154 | 0.483 | 12.631 |
| M. musculus | 138 | 889 | 26 | 0.378 | 0.532 | −0.836 | −0.949 | 11.735 | |
| M. castaneus | 80 | 889 | 44 | 0.712 | 0.999 | −0.928 | 0.476 | 12.134 |
Number of chromosomes;
Average sequence length;
Number of polymorphic nucleotide sites;
π and θ are estimators of the population mutation parameter; see Materials and methods;
P < 0.05,
P < 0.01;
Dxy is the average pairwise divergence per site compared to M. caroli (Nei 1987).
We compared ancestral and derived populations to see if derived populations were associated with population bottlenecks and consequent lower levels of diversity and higher average values of Tajima's D, as seen in humans (e.g. Akey et al. 2004). For M. castaneus, we focused on the population from Taiwan since it has a larger sample size. Average nucleotide diversity was similar in ancestral and derived populations of M. domesticus (πanc = 0.13%, SE = 0.03%; πder = 0.13%, SE = 0.03%) and M. musculus (πanc = 0.12%, SE = 0.07%; πder = 0.12%, SE = 0.08%), while in M. castaneus, the ancestral population harboured more variation than the derived population (πanc = 0.36%, SE = 0.11%; πder = 0.18%, SE = 0.12%). Similar levels of polymorphism in ancestral and derived populations of M. domesticus and M. musculus could be due in part to the fact that the samples for the derived populations span a larger geographic range than the ancestral populations (Fig. 1). For Tajima's D, we observed no consistent differences between ancestral and derived populations of M. musculus and M. domesticus, but for M. castaneus, Tajima's D was often higher in the derived population than in the ancestral population (Table S4, Supporting information). These results suggest that the derived population of M. castaneus from Taiwan may have been associated with a bottleneck.
We tested the neutral prediction of equal ratios of polymorphism to divergence among loci in an HKA framework (Hudson et al. 1987) using polymorphism from each species separately as well as all three species together. Divergence was calculated in comparison to M. caroli. Each of these four tests rejected a neutral model (P < 0.001 for each). The largest deviations in these tests were caused by a lack of divergence (or excess of polymorphism) at mtDNA. We then corrected for multiple substitutions at mtDNA using modeltest 3.06 (Posada & Crandall 1998) and performed HKA tests with corrected values. Only the test involving M. musculus polymorphism remained significant (P = 0.003). In this test, the greatest deviation from neutral expectations was due to an excess of polymorphism at Clcn6 relative to divergence (46 observed polymorphisms when only 24 were expected). When this locus was removed, the resulting test was not significant. These results suggest that with the exception of Clcn6 in M. musculus, patterns of polymorphism and divergence in this multilocus dataset are consistent with neutral predictions.
We used im to estimate Ne of each species under a model of divergence with gene flow. The ML estimates and 90% highest posterior density (HPD90) intervals are shown in Fig. 2 and Table 3. Assuming one generation per year, average Ne for M. castaneus was 203 626, average Ne for M. domesticus was 100 923, and average Ne for M. musculus was 60 450; estimates were twice as large assuming two generations per year. Notably, the estimates for each species were in reasonable agreement with each other, regardless of which species was used in comparison, and the likelihood surfaces in all cases had single clear sharp peaks. For example, Ne for M. domesticus was 101 400 when compared to M. musculus and 100 446 when compared to M. castaneus with one generation per year. In contrast to the sharp likelihood surfaces for current Ne, the likelihood surfaces for ancestral Ne were relatively flat (Fig. 2). We also estimated population size from the expectation Ne = π/4μ following a simple model of mutation–drift equilibrium, and obtained similar results. For example, for M. domesticus autosomes, π = 0.155% (Table 2) and μ = 4.1 × 10−9 (see below), resulting in Ne = 95 000 assuming one generation per year.
Fig. 2.
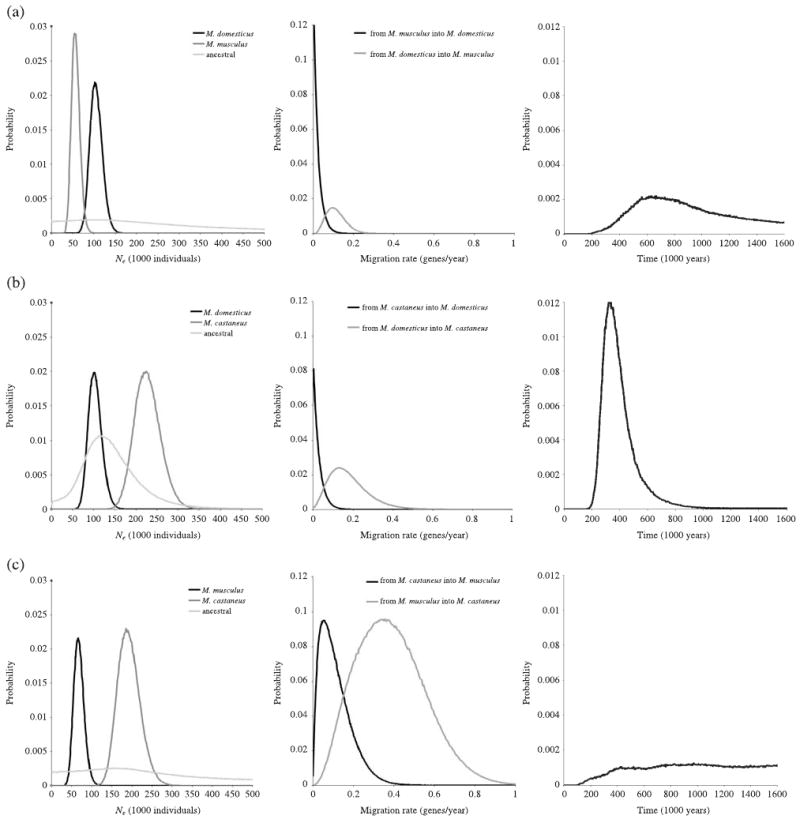
Marginal posterior probability distributions for im model parameters. Curves are shown for the analysis with (a) Mus domesticus and M. musculus, (b) M. domesticus and M. castaneus and (c) M. musculus and M. castaneus.
Table 3.
Maximum-likelihood (ML) estimates and 90% posterior density intervals (in parentheses) of demographic parameters obtained with im between species of house mice for generation length of 1 and 0.5 years
| Generation Length | Species 1 | Species 2 | NeSpecies 1 | NeSpecies 2 | Neancestral | t† | 2Nm1‡ | 2Nm2§ |
|---|---|---|---|---|---|---|---|---|
| 1 year | M. musculus | M. castaneus | 65 833 (46 928–88 788) |
184 148*** (145 301–236 985) |
149 961 | 0.054 (0.005–0.219) |
0.342 (0.104–0.644) |
|
| M. musculus | M. domesticus | 55 067 (39 632–72 601) |
101 400*** (80 258–128 805) |
98 266 | 627 876 | 0.094* (0.034–0.186) |
0.002¶ (0.002¶–0.053) |
|
| M. domesticus | M. castaneus | 100 446 (76 398–129 145) |
222 765*** (174 516–276 633) |
116 597 (26 674–250 721) |
329 586 (220 897–579 617) |
0.001¶ (0.001¶–0.063) |
0.129* (0.024–0.319) |
|
| 0.5 years | M. musculus | M. castaneus | 131 666 (93 856–177 576) |
368 296*** (290 602–473 970) |
299 922 | 0.054 (0.005–0.219) |
0.342 (0.104–0.644) |
|
| M. musculus | M. domesticus | 110 134 (79 264 – 145 202) |
202 800*** (160 516 – 257 610) |
196 532 | 125 5752 | 0.094* (0.034–0.186) |
0.002¶ (0.002¶–0.053) |
|
| M. domesticus | M. castaneus | 200 892 (152 796–158 290) |
445 530*** (349 032–553 266) |
233 194 (53 348–501 442) |
659 172 (441 794–1 159 234) |
0.001¶ (0.001¶–0.063) |
0.129* (0.024–0.319) |
Missing values are where parameters could not be reliably estimated;
P < 0.05;
P < 0.01;
P < 0.005 in comparisons between species;
The time since Species 1 and 2 split in numbers of generations;
The population migration rate into Species 1 from Species 2 per generation;
The population migration rate into Species 2 from Species 1 per generation;
Corresponds to the first bin of the parameter space, and therefore represents zero.
Interspecific divergence, mutation rates and age of species
Comparisons between species allowed us to estimate mutation rates and divergence times. We also took advantage of comparisons between genes with different modes of inheritance to estimate mutation rates separately for males and females. Average divergence (D) between M. caroli and M. domesticus, M. musculus or M. castaneus was on the order of 2–5% for introns of nuclear genes (Table 2). We used these data to estimate mutation rates (μ) per generation per site assuming a divergence time between M. caroli and the three species of 4.3 million years (Suzuki et al. 2004) and a generation time of one year. Under a neutral model, D = 2μt + 4Nancμ, where Nanc is the ancestral population size and t is the divergence time measured in generations. If we assume that the ancestral population size is similar to current population sizes (Table 3), then 4Nancμ is small relative to D (Table 2) and D = 2μt approximately. Using this approximation, average mutation rates were 4.1 × 10−9 for the autosomes, 3.3 × 10−9 for the X chromosome and 5.4 × 10−9 for the Y chromosome. The mutation rate for the mitochondrial control region was roughly one order of magnitude higher (μ = 4.1 × 10−8). These estimates should be viewed as approximations owing to the uncertainty in generation length and divergence time (She et al. 1990; Chevret et al. 2005). However, we note that our estimates per year are in good agreement with previous estimates (e.g. Li et al. 1996). If mice have two generations per year rather than one, all estimates of μ per generation are half as large.
By comparing divergence among X, Y and autosomal loci we estimated α, the ratio of the male to female mutation rates as in Miyata et al. (1987). Each of these comparisons yielded slightly different estimates of α (X vs. autosomes, α = 3.9; X vs. Y, α = 2.3; autosomes vs. Y, α = 1.8). These estimates suggest that 2–4 times as many mutations come from males compared to females, in general agreement with previous estimates for rodents (Chang et al. 1994; Chang & Li 1995; Sandstedt & Tucker 2005).
Levels of divergence among M. domesticus, and M. musculus, and M. castaneus are shown in Table 4, and neighbour-joining trees showing relationships of haplotypes for each locus are shown in Fig. 3 and Fig. S1 (Supporting information). For some loci, each species formed a monophyletic group (e.g. Ocrl), while at other loci species were intermingled on the phylogeny (e.g. Clcn6). These differences among loci are consistent with a recent origin for these species and may reflect unsorted ancestral polymorphism as well as gene flow (discussed below). Divergence among these species was less than 1% in all comparisons (Table 4). The average interspecific divergence in pairwise comparisons was nearly identical for each of the three possible comparisons (domesticus-musculus Dxy = 0.54%; domesticus-castaneus Dxy = 0.51%; musculus-castaneus Dxy = 0.51%), presumably reflecting separation from an ancestral population at roughly the same time. Using the mutation rates calculated above and an ancestral population size of 120 000, we estimate that musculus and domesticus began to diverge approximately 495 000 years ago {for autosomes, t = (D − 4Nancμ)/(2μ) = [(6.02 × 10−3) − (4.8 × 105)(4.1 × 10−9)]/(8.2 × 10−9) = 495 000 years}. Roughly similar estimates are obtained for the other species pairs and for comparisons involving the X chromosome.
Table 4.
Patterns of differentiation between Mus domesticus (dom), M. musculus (mus) and M. castaneus (cast), and patterns of differentiation between populations within each of these species (NA, not available)
| Locus (chromosome) | Interspecific comparisons | Intraspecific comparisons | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dom-mus | dom-cast | mus-cast | dom WE/Is | mus EE/Kz | mus EE/Ru | mus Kz/Ru | cast In/Tw | cast In/Ch | cast Tw/Ch | ||
| Chrng (1) | FST | 0.637 | 0.311 | 0.455 | 0.197 | 0.107 | 0.007 | 0.152 | 0.176 | 0.536 | 0.200 |
| Dxy(%) | 0.454 | 0.688 | 0.646 | 0.309 | 0.057 | 0.012 | 0.057 | 0.734 | 0.717 | 0.326 | |
| Med19 (2) | FST | 0.721 | 0.801 | 0.689 | 0.149 | 0.688 | 0.680 | −0.109 | 0.110 | 0.096 | NA |
| Dxy(%) | 0.327 | 0.262 | 0.307 | 0.072 | 0.206 | 0.204 | 0.055 | 0.041 | 0.041 | 0.000 | |
| Prpf3 (3) | FST | 0.930 | 0.767 | 0.883 | 0.093 | 0.186 | 0.221 | 0.012 | 0.305 | 0.160 | 0.017 |
| Dxy(%) | 0.956 | 0.424 | 0.811 | 0.070 | 0.075 | 0.047 | 0.080 | 0.143 | 0.139 | 0.083 | |
| Clcn6 (4) | FST | 0.434 | 0.366 | 0.128 | 0.097 | 0.243 | 0.142 | −0.028 | 0.075 | 0.080 | 0.048 |
| Dxy(%) | 0.672 | 0.767 | 0.750 | 0.243 | 0.628 | 0.573 | 0.496 | 0.838 | 0.746 | 0.733 | |
| G6pdx (X) | FST | 0.904 | 0.763 | 0.485 | 0.355 | −0.007 | −0.017 | 0.050 | 0.340 | 0.510 | 0.224 |
| Dxy(%) | 0.447 | 0.494 | 0.193 | 0.078 | 0.022 | 0.032 | 0.033 | 0.212 | 0.167 | 0.073 | |
| Ocrl (X) | FST | 0.867 | 0.640 | 0.774 | 0.478 | 0.000 | 0.074 | 0.067 | 0.372 | 0.372 | NA |
| Dxy(%) | 0.524 | 0.630 | 0.677 | 0.173 | 0.009 | 0.033 | 0.039 | 0.273 | 0.273 | 0.000 | |
| Jarid1d (Y) | FST | 0.921 | 0.664 | 0.502 | 0.528 | 0.889 | 0.889 | NA | 0.939 | 0.909 | 0.000 |
| Dxy(%) | 0.364 | 0.325 | 0.209 | 0.051 | 0.044 | 0.044 | 0.000 | 0.327 | 0.335 | 0.011 | |
| control region (mtDNA) | FST | 0.766 | 0.669 | 0.650 | 0.266 | 0.437 | 0.257 | 0.236 | 0.137 | 0.167 | 0.102 |
| Dxy(%) | 2.012 | 1.924 | 1.558 | 0.564 | 0.475 | 0.375 | 0.279 | 0.782 | 0.738 | 0.661 | |
Fig. 3.
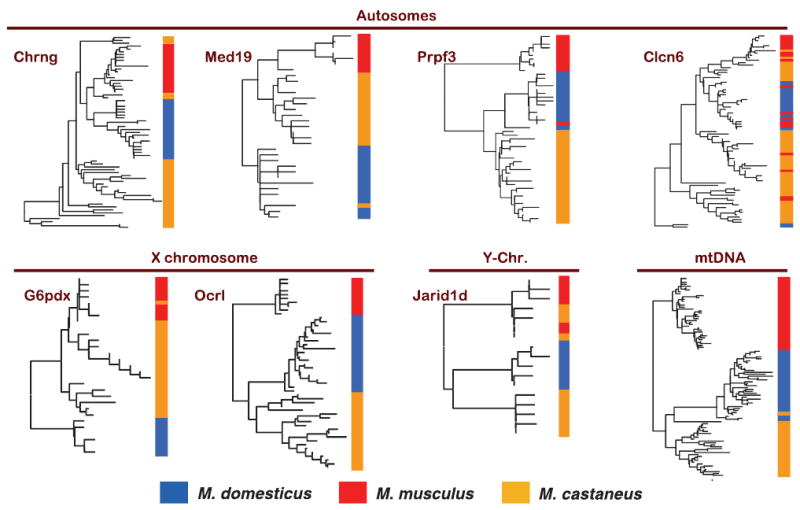
Neighbour-joining trees of haplotypes for four autosomal, two X-linked, one Y-linked and one mitochondrial locus in samples of Mus domesticus, M. musculus, and M. castaneus. Neighbour-joining trees were rooted with the M. caroli sequence.
The phylogeny of these species has been debated, although current phylogenetic evidence supports a musculus + castaneus clade, with domesticus as basal (Tucker et al. 2005). The difficulty of inferring the correct population history can be seen from the trees in Fig. 3. For some loci, such as G6pdx, M. domesticus is the sister clade to a clade containing M. musculus and M. castaneus. For other loci, M. musculus is basal (e.g. Ocrl), and for yet other loci M. castaneus is basal (e.g. Chrng). Variation in phylogenetic patterns among loci is expected when the time between successive population splits is small. Of the eight trees in Fig. 3, four support a close relation between domesticus and castaneus with musculus branching off first (Prpf3, Ocrl, Jarid1d and mtDNA), two support a close relation between musculus and castaneus with domesticus branching off first (Med19, G6pdx), one supports a close relation between domesticus and musculus with castaneus in a basal position (Chrng), and one shows very little concordance between species and phylogeny (Clcn6). The discordance among these trees suggests that all three species split from an ancestral population at nearly the same time and that data from many loci may be needed to resolve the correct bifurcating topology, if one exists.
ML estimates for divergence time between musculus and castaneus using im were generally unreliable, with different runs converging on different values. In some cases, the likelihood surfaces were quite flat (Fig. 2). The estimated divergence time for musculus and domesticus was 628 000 years, and the estimated divergence time for domesticus and castaneus was 330 000 years. The HPD90 intervals, which were quite broad (Table 3), include the estimate of ∼500 000 years from the calculation above assuming a simple molecular clock.
Levels and patterns of gene flow
We studied patterns of differentiation both within and between species (Table 4). As expected, FST was generally higher between species than within species, although there was also considerable differentiation between ancestral and derived populations within species. Average FST between species was 0.66 (range 0.13–0.93), while average FST within species was 0.25 (range 0.00–0.94). The highest levels of differentiation within species were between Indian and Chinese or Indian and Taiwanese populations of M. castaneus (Table 4).
We used these data to test the hypothesis that M. musculus, M. domesticus and M. castaneus diverged in allopatry with no subsequent gene flow. ML estimates of gene flow revealed asymmetric patterns (Table 3 and Fig. 2). While no gene flow was detected into M. domesticus, significant gene flow was detected into both M. castaneus and M. musculus. We compared nested models with a likelihood ratio test using ima (Hey & Nielsen 2007). In all three pairwise comparisons, a model allowing gene flow was a significantly better fit to the data than a model with no gene flow (P < 0.01 for each).
Genetic differentiation is higher at sex chromosomes
Patterns of differentiation and gene flow differed among loci with different modes of inheritance. The average FST between species for autosomal loci (0.59) was lower than for X-linked loci (0.74), the Y-linked Jarid1d (0.70) or the mitochondrial control region (0.70). The greater differentiation for loci on the X chromosome compared to those on autosomes can also be seen in the relative numbers of polymorphisms within species and fixed differences between species (Table 5 and Table S5, Supporting information). The ratio of total polymorphisms to fixed differences was significantly greater on the autosomes compared to the X chromosome in 2 × 2 contingency tables for each of the three pairwise species comparisons (Fisher's Exact Test, FET, P < 0.05 for each). The ratio of polymorphisms to fixed differences was also significantly greater on the autosomes compared to the Y chromosome in domesticus-musculus and domesticus-castaneus comparisons (FET, P < 0.01 for each) but not in the castaneus-musculus comparison (FET, P > 0.05). The ratio of polymorphisms to fixed differences was significantly greater on the autosomes than in the mtDNA control region in the musculus-domesticus comparison (FET, P < 0.01), but not in the other comparisons (FET, P > 0.05 for both).
Table 5.
Numbers of polymorphic nucleotide sites within species and fixed differences between species for different regions of the genome
| Species pair | Genome region | Polymorphism | Fixed differences | P-value* |
|---|---|---|---|---|
| Mus domesticus — M. musculus | Autosomes | 171 | 6 | |
| X-Chromosome | 35 | 18 | < 10−6 | |
| Y-Chromosome | 5 | 7 | < 10−6 | |
| mtDNA | 56 | 9 | 0.01 | |
| M. domesticus — M. castaneus | Autosomes | 253 | 4 | |
| X-Chromosome | 77 | 9 | 0.0008 | |
| Y-Chromosome | 16 | 3 | 0.008 | |
| mtDNA | 70 | 0 | 0.41 | |
| M. musculus — M. castaneus | Autosomes | 241 | 3 | |
| X-Chromosome | 66 | 4 | 0.046 | |
| Y-Chromosome | 16 | 0 | 1.00 | |
| mtDNA | 60 | 3 | 0.35 |
P-values are for Fisher's Exact Tests in comparison to autosomal values.
The im analysis and the neighbour-joining trees in Fig. 3 reveal that shared polymorphisms between species result from gene flow in some cases and unsorted ancestral polymorphism in others. For example, the tree for Jarid1d reveals three deep lineages corresponding to castaneus, domesticus, and a group containing both musculus and castaneus together. The im analysis shows that the clade containing both musculus and castaneus is a result of migration of the musculus Y chromosome into castaneus (Table 6). The castaneus containing the musculus Y included all of the castaneus individuals from China and Taiwan but none of the individuals from India (Fig. S1 and Table S1, Supporting information). This suggests that the castaneus Y has been replaced by the musculus Y over a large geographic region. In contrast, im identified no gene flow into domesticus at Clcn6 (Table 6), yet some domesticus individuals were widely dispersed on the tree in Fig. 3, suggesting that domesticus contains unsorted ancestral variation.
Table 6.
The modal number of migration events between species of house mice inferred using the im analysis
| Species 1 | Species 2 | Locus | Migration events into species 1 | Migration events into species 2 |
|---|---|---|---|---|
| Mus domesticus | M. musculus | Chrng | 0 | 1 |
| Med19 | 0 | 1 | ||
| Prpf3 | 0 | 1 | ||
| Clcn6 | 0 | 3 | ||
| G6pdx | 0 | 0 | ||
| Ocrl | 0 | 0 | ||
| Jarid1d | 0 | 0 | ||
| control region | 0 | 1 | ||
| M. domesticus | M. castaneus | Chrng | 0 | 0 |
| Med19 | 0 | 1 | ||
| Prpf3 | 0 | 0 | ||
| Clcn6 | 0 | 3 | ||
| G6pdx | 0 | 0 | ||
| Ocrl | 0 | 0 | ||
| Jarid1d | 0 | 0 | ||
| control region | 0 | 2 | ||
| M. musculus | M. castaneus | Chrng | 0 | 5 |
| Med19 | 0 | 0 | ||
| Prpf3 | 0 | 0 | ||
| Clcn6 | 3 | 6 | ||
| G6pdx | 0 | 3 | ||
| Ocrl | 0 | 0 | ||
| Jarid1d | 0 | 2 | ||
| control region | 0 | 0 |
The greater differentiation of the X chromosome compared to the autosomes appears to be due at least partly to differences in the level of gene flow for X-linked compared to autosomal loci. For example, the im analyses identified gene flow from domesticus into musculus for the autosomes but not for the X chromosome. The different levels of gene flow between the X chromosome and the autosomes, as well as the asymmetry of gene flow, are consistent with clinal patterns over a much smaller geographic scale in the musculus-domesticus hybrid zone (e.g. Tucker et al. 1992; Teeter et al. 2008).
Discussion
We conducted a survey of nucleotide variation at eight loci in populations of Mus domesticus, M. musculus and M. castaneus to make inferences about the history of speciation in this group. We discovered that: (i) M. castaneus harboured the most genetic variation, followed by M. domesticus and then M. musculus, with inferred effective population sizes of approximately 200 000–400 000, 100 000–200 000 and 60 000–120 000, respectively; (ii) these species began to diverge about 500 000 years ago, with all three species diverging within a short time interval; (iii) patterns of genetic variation are inconsistent with a simple allopatric model of speciation with no gene flow; instead, gene flow occurred and was asymmetric between the species; and (iv) the X chromosome was more differentiated between species than the autosomes, due to both more gene flow and the presence of ancestral polymorphism on the autosomes compared to the X chromosome.
Levels of polymorphism and effective population sizes
These data add to a growing literature documenting the amount and structure of DNA sequence variation in wild house mice (Nachman 1997; Harr 2006; Baines & Harr 2007; Laurie et al. 2007; Salcedo et al. 2007). Our results are consistent with other studies in suggesting that M. castaneus harbours more variation than M. domesticus or M. musculus (Baines & Harr 2007). Much of that variation is found within India, as shown earlier for allozymes (Din et al. 1996) and mtDNA (Boursot et al. 1996), consistent with the suggestion that this region represents the ancestral range for the species complex (Boursot et al. 1993).
Our data indicate that the species-wide effective population size for M. castaneus is about 200 000–400 000, while it is about 100 000–200 000 for M. domesticus and 60 000–120 000 for M. musculus. While the absolute populations sizes are subject to uncertainty in generation length, the relative sizes are not (assuming the three species have the same generation length). The current and historical range of M. castaneus was probably less affected by Pleistocene climate changes than the ranges of M. domesticus or M. musculus, both of which have more northern distributions. M. domesticus and M. musculus have colonized regions that were extensively glaciated as recently as 10 000 years ago. The smaller effective population sizes of these species may reflect contractions during periods when their ranges were more restricted.
We found similar levels of variability in ancestral and derived populations of both M. domesticus and M. musculus for both the autosomes and the X chromosome. These observations argue against a strong bottleneck during the colonization of Western Europe by mice from the Middle East or the colonization of Eastern Europe by mice from central Asia. Baines & Harr (2007) reported reduced variation on the X chromosome relative to the autosomes in derived populations of both M. domesticus and M. musculus, and they attributed this pattern to hitchhiking effects associated with adaptation to novel environments. We found no evidence for such a reduction on the X in our data (Table S4, Supporting information). This difference between our results and theirs may be due to the different genes that were sampled or to different geographic sampling. For example, Baines & Harr (2007) sampled Iran rather than Israel for their ancestral population of M. domesticus, and Iran is likely to be closer to the ancestral range of the species. Moreover, the derived populations of domesticus and musculus in the present study were sampled over a larger geographic region.
Estimates of nucleotide variability in mice allow us to make comparisons with similar data from humans, the mammalian species for which the best data are available. While the average level of nucleotide diversity at non-coding sites in humans is low (π = 0.11%, e.g. Li & Sadler 1991), in mice, values range from 0.13% in M. musculus and 0.14% in M. domesticus to 0.43% in M. castaneus. House-mouse populations therefore have up to four times as much variation as human populations. Differences in estimates of Ne between humans and house mice are even greater. Ne for humans is in the order of 10 000, while for M. castaneus Ne is about 200 000. This 20-fold difference in estimates of Ne between humans and mice is due to a roughly five-fold lower mutation rate per generation in mice (∼4 × 10−9, see Results) compared to humans (2 × 10−8, Nachman & Crowell 2000). Although mice have higher substitution rates per year than humans (e.g. Li et al. 1996), they have lower rates per generation.
Humans and house mice both expanded their ranges fairly recently and on similar timescales when expressed in generations. Humans moved out of Africa roughly 60 000 years ago (or about 3000 generations), while mice colonized northern Europe and Asia about 3000 years ago (or about 3000–6000 generations). Despite these similarities, patterns of nucleotide variability in ancestral and derived regions are different in humans and mice. In humans, non-African populations have reduced variation and fewer rare variants than in African populations (e.g. Akey et al. 2004). Derived populations of M. domesticus and M. musculus show neither of these characteristics compared to ancestral populations.
Age of the species
Our data indicate that M. domesticus, M. musculus and M. castaneus diverged recently from each other and did so within a short period of time. The average divergence among each of the three pairs of species suggests a divergence time of about 500 000 years ago, and this is roughly consistent with the ML estimates of divergence time obtained using im. On average, alleles within a species are expected to coalesce within 4Ne generations, although the variance is very large. If our estimates of Ne and divergence time are approximately correct, then we would expect to see some ancestral variation segregating among these species. For example, we estimated that Ne for M. castaneus is 200 000 and that it therefore diverged less than 4Ne generations ago. Patterns of variation at some genes, such as Clcn6, appeared to be consistent with this expectation. We also note that this expectation is independent of assumptions about generation time, since different generations times would affect our estimates of both population size and divergence expressed in numbers of generations.
A key unresolved issue concerning speciation in this group is the order in which the species separated. Current evidence supports a phylogeny in which M. domesticus diverged first, with M. castaneus and M. musculus as sister species (Tucker et al. 2005). Two of the loci in our study support this phylogeny with M. domesticus in a basal position (G6pdx and Med19, Fig. 3). However, the most notable aspect of our data with regard to the relationship among species is the absence of a consistent pattern among loci. Some loci support a phylogeny in which M. musculus is basal (Prpf3, Ocrl, mtDNA) while other loci support a phylogeny in which M. castaneus is basal (Chrng). This discordance among loci is similar to the discordance among loci in resolving the human, chimp and gorilla trichotomy (e.g. Ruvolo 1997) and is expected in situations where the time between successive speciation events is small or the ancestral population size is large (Hudson 1983). In such cases, a large number of loci may be required to resolve the true bifurcating phylogeny, if one exists. An alternative hypothesis is that all three species diverged at roughly the same time from an ancestral population. Resolving this issue will require sampling not only more loci but also sufficient geographic sampling to capture populations that may contain ancestral variation. For example, the phylogenetic analysis in Tucker et al. (2005) was based on a single M. castaneus from Thailand (CAST/Ei) and may not reflect the topology that would be obtained using M. castaneus from India.
Gene flow
The data presented here allow us to reject a model of allopatric speciation with no gene flow. The highest posterior density intervals on ML estimates of migration using im did not include zero for at least one member of each species pair. Likewise, models with gene flow revealed a significantly better fit to the data compared to models without gene flow in likelihood ratio tests implemented in ima. The inferred gene flow can also be seen in the topologies of some of the loci in Fig. 3. For example, at both Med19 and Prpf3, there are three lineages corresponding nearly perfectly to the three species. In each case, there is a single mismatched haplotype on an otherwise sorted genealogy. Similarly, the genealogy for the mtDNA control region is generally well sorted, with the exception of a few domesticus haplotypes in castaneus mice from Taiwan and China. These mice contain castaneus alleles at other loci. The observation of introgression of domesticus mtDNA into castaneus has been confirmed in other samples from these same localities (H. T. Yu, unpublished results). Despite the evidence for gene flow, the actual amount appears to be low, with estimates of Nm well below one (Table 3).
Notably, the analyses provide no evidence of gene flow into M. domesticus but suggest that gene flow has occurred into both M. castaneus and M. musculus. This asymmetry between M. domesticus and M. musculus is also seen in the hybrid zone formed between these two species. Considerable variation in cline width is observed for different markers, but when introgression occurs, it is almost always due to M. domesticus alleles moving into M. musculus (e.g. Teeter et al. 2008). This agreement between hybrid zone studies of cline width (sampled over tens of km) and gene genealogies from animals across the range of the species (sampled over thousands of km) further strengthens the inference of gene flow.
It is important to point out that our analyses do not directly address the timescale over which gene flow has occurred. The current hybrid zone between M. domesticus and M. musculus is believed to be quite young, but it is unknown whether these species have had multiple periods of isolation and contact, or if they evolved primarily in isolation until recently. It is noteworthy that the mismatched alleles in the trees in Fig. 3 come from individuals in both ancestral and derived populations (Fig. S1, Supporting information). This suggests that not all of the gene flow is recent.
Sex chromosomes and speciation
The X chromosome is significantly more differentiated than the autosomes in comparisons between species (Table 5). In principle, this could be due to either faster lineage sorting on the X, reduced gene flow on the X or some combination of both. Faster sorting is expected as a simple consequence of the effective population size of the X chromosome, which is three-quarter that of the autosomes. Faster lineage sorting could also be driven by a greater incidence of positive selection on the X chromosome and associated genetic hitchhiking (e.g. Begun & Whitley 2000).
Patterns of gene flow in the hybrid zone between M. domesticus and M. musculus indicate reduced gene flow on the X chromosome (e.g. Tucker et al. 1992). Laboratory crosses also consistently reveal a role for the X chromosome in hybrid male sterility (e.g. Oka et al. 2004; Storchova et al. 2004; Good et al. 2008). Our im analysis is consistent with these observations in revealing little evidence for gene flow on the X chromosome compared to the autosomes (Table 6). However, we cannot rule out the possibility that the greater differentiation seen on the X chromosome is also partly a consequence of faster lineage sorting due to either positive selection or a simple consequence of smaller effective population size. For example, the pattern seen at Clcn6 on Chromosome 4, in which all three species are intermingled on the genealogy, is probably most consistent with unsorted ancestral variation. This pattern is not seen for either of the two X-linked loci sampled here (Fig. 3) or any of the 11 X-linked loci studied by Salcedo et al. (2007) in smaller samples of M. musculus and M. domesticus.
Patterns of differentiation on the Y chromosome are slightly more complicated. The neighbour-joining tree for Jarid1d reveals three deep lineages, probably reflecting complete lineage sorting. One of these lineages includes all of the M. musculus as well as the M. castaneus from Taiwan and China. This pattern is most easily explained by introgression of the M. musculus Y into M. castaneus in this geographic region. Introgression of the M. musculus Y chromosome into some populations of M. castaneus has previously been reported (Boissinot & Boursot 1997), as well as the introgression of the Y chromosome in some areas of the European hybrid zone between M. musculus and M. domesticus (Munclinger et al. 2002). These observations suggest that the Y chromosome may be less important in reproductive isolation between species of house mice than the X chromosome.
Supplementary Material
Acknowledgments
We thank Diethard Tautz and members of the Max Plank Institute for Evolutionary Biology in Ploen, Germany for providing a stimulating environment for MWN while on sabbatical. We also thank the members of the Nachman lab for discussion, and Ms Yulia Koval'skaya who collected Russian mice. We thank J. Pialek and the members of his lab who helped BG with field work in Poland, Hungary and Slovakia. We acknowledge the Fundacao para a Ciencia e a Tecnologia for a Post-Doctoral fellowship (SFRH/BPD/24743/2005) to Armando Geraldes, the Swiss National Science Foundation for a Post-Doctoral fellowship (PBLAA-111572) to Patrick Basset, and NSF and NIH grants to MWN for financial support.
Footnotes
Armando Geraldes and Patrick Basset are postdoctoral fellows working on the genetics of speciation in house mice in Michael Nachman's lab. All authors share an interest in evolutionary genetics broadly and the biology of house mice in particular.
Supporting information: Additional supporting information may be found in the online version of this article:
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Akey JM, Eberle MA, Rieder MJ, et al. Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biology. 2004;2:e286. doi: 10.1371/journal.pbio.0020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray JC, Vanlerberghe F, Brittondavidian J. The house mouse progression in Eurasia — a paleontological and archaeozoological approach. Biological Journal of the Linnean Society. 1990;41:13–25. [Google Scholar]
- Baines JF, Harr B. Reduced X-linked diversity in derived populations of house mice. Genetics. 2007;175:1911–1921. doi: 10.1534/genetics.106.069419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Whitley P. Reduced X-linked nucleotide polymorphism in Drosophila simulans. Proceedings of the National Academy of Sciences, USA. 2000;97:5960–5965. doi: 10.1073/pnas.97.11.5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissinot S, Boursot P. Discordant phylogeographic patterns between the Y chromosome and mitochondrial DNA in the house mouse: selection on the Y chromosome? Genetics. 1997;146:1019–1034. doi: 10.1093/genetics/146.3.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursot P, Auffray JC, Brittondavidian J, Bonhomme F. The evolution of house mice. Annual Review of Ecology and Systematics. 1993;24:119–152. [Google Scholar]
- Boursot P, Din W, Anand R, et al. Origin and radiation of the house mouse: mitochondrial DNA phylogeny. Journal of Evolutionary Biology. 1996;9:391–415. [Google Scholar]
- Britton-Davidian J, Fel-Clair F, Lopez J, et al. Postzygotic isolation between the two European subspecies of the house mouse: estimates from fertility patterns in wild and laboratory-bred hybrids. Biological Journal of the Linnean Society. 2005;84:379–393. [Google Scholar]
- Bronson FH. The reproductive ecology of the house mouse. The Quarterly Review of Biology. 1979;54:265–299. doi: 10.1086/411295. [DOI] [PubMed] [Google Scholar]
- Chang BH, Li WH. Estimating the intensity of male-driven evolution in rodents by using X-linked and Y-linked Ube 1 genes and pseudogenes. Journal of Molecular Evolution. 1995;40:70–77. doi: 10.1007/BF00166597. [DOI] [PubMed] [Google Scholar]
- Chang BH, Shimmin LC, Shyue SK, et al. Weak male-driven molecular evolution in rodents. Proceedings of the National Academy of Sciences, USA. 1994;91:827–831. doi: 10.1073/pnas.91.2.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevret P, Veyrunes F, Britton-Davidian J. Molecular phylogeny of the genus Mus (Rodentia: Murinae) based on mitochondrial and nuclear data. Biological Journal of the Linnean Society. 2005;84:417–427. [Google Scholar]
- Cucchi T, Vigne JD, Auffray JC. First occurrence of the house mouse (Mus musculus domesticus Schwarz & Schwarz, 1943) in the Western Mediterranean: a zooarchaeological revision of subfossil occurrences. Biological Journal of the Linnean Society. 2005;84:429–445. [Google Scholar]
- Davis RC, Jin A, Rosales M, et al. Genome-wide set of congenic mouse strains derived from CAST/Ei on a C57BL/6 background. Genomics. 2007;90:306–313. doi: 10.1016/j.ygeno.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Din W, Anand R, Boursot P, et al. Origin and radiation of the house mouse: clues from nuclear genes. Journal of Evolutionary Biology. 1996;9:519–539. [Google Scholar]
- Dod B, Jermiin LS, Boursot P, et al. Counterselection on sex-chromosomes in the mus-musculus European hybrid zone. Journal of Evolutionary Biology. 1993;6:529–546. [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Research. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Forejt J. Hybrid sterility in the mouse. Trends in Genetics. 1996;12:412–417. doi: 10.1016/0168-9525(96)10040-8. [DOI] [PubMed] [Google Scholar]
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good JM, Handel MA, Nachman MW. Asymmetry and polymorphism of hybrid male sterility during the early stages of speciation in house mice. Evolution. 2008;62:50–65. doi: 10.1111/j.1558-5646.2007.00257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Research. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Gregorova S, Divina P, Storchova R, et al. Mouse consomic strains: exploiting genetic divergence between Mus m. musculus and Mus m. domesticus subspecies. Genome Research. 2008;18:509–515. doi: 10.1101/gr.7160508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user friendly biological sequence alignment editor and analyses program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–98. [Google Scholar]
- Harr B. Genomic islands of differentiation between house mouse subspecies. Genome Research. 2006;16:730–737. doi: 10.1101/gr.5045006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics. 2004;167:747–760. doi: 10.1534/genetics.103.024182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Nielsen R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proceedings of the National Academy of Sciences, USA. 2007;104:2785–2790. doi: 10.1073/pnas.0611164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. Testing the constant-rate neutral allele model with protein sequence data. Evolution. 1983;37:203–217. doi: 10.1111/j.1558-5646.1983.tb05528.x. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR, Young LG, Blume LM, Gilbert LE. Multilocus analyses of admixture and introgression among hybridizing Heliconius butterflies. Evolution. 2006;60:1254–1268. [PubMed] [Google Scholar]
- Laurie CC, Nickerson DA, Anderson AD, et al. Linkage disequilibrium in wild mice. PLoS Genetics. 2007;3:e144. doi: 10.1371/journal.pgen.0030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawton-Rauh A, Robichaux RH, Purugganan MD. Diversity and divergence patterns in regulatory genes suggest differential gene flow in recently derived species of the Hawaiian silversword alliance adaptive radiation (Asteraceae) Molecular Ecology. 2007;16:3995–4013. doi: 10.1111/j.1365-294X.2007.03445.x. [DOI] [PubMed] [Google Scholar]
- Li WH, Sadler LA. Low nucleotide diversity in man. Genetics. 1991;129:513–523. doi: 10.1093/genetics/129.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Ellsworth DL, Krushkal J, et al. Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Molecular Phylogenetics and Evolution. 1996;5:182–187. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- Llopart A, Lachaise D, Coyne JA. Multilocus analysis of introgression between two sympatric sister species of Drosophila: Drosophila yakuba and D. santomea. Genetics. 2005;171:197–210. doi: 10.1534/genetics.104.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado CA, Kliman RM, Markert JA, Hey J. Inferring the history of speciation from multilocus DNA sequence data: the case of Drosophila pseudoobscura and close relatives. Molecular Biology and Evolution. 2002;19:472–488. doi: 10.1093/oxfordjournals.molbev.a004103. [DOI] [PubMed] [Google Scholar]
- Macholan M, Munclinger P, Sugerkova M, et al. Genetic analysis of autosomal and X-linked markers across a mouse hybrid zone. Evolution. 2007;61:746–771. doi: 10.1111/j.1558-5646.2007.00065.x. [DOI] [PubMed] [Google Scholar]
- Miyata T, Hayashida H, Kuma K, et al. Male-driven molecular evolution: a model and nucleotide substitution analysis. Cold Spring Harbour Symposia of Quantitative Biology. 1987;52:863–967. doi: 10.1101/sqb.1987.052.01.094. [DOI] [PubMed] [Google Scholar]
- Munclinger P, Bozikova E, Sugerkova M, et al. Genetic variation in house mice (Mus, muridae, rodentia) from the Czech and Slovak republics. Folia Zoologica. 2002;51:81–92. [Google Scholar]
- Nachman MW. Patterns of DNA variability at X-linked loci in Mus domesticus. Genetics. 1997;147:1303–1316. doi: 10.1093/genetics/147.3.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Boyer SN, Searle JB, Aquadro CF. Mitochondrial DNA variation and the evolution of Robertsonian chromosomal races of house mice, Mus domesticus. Genetics. 1994;136:1105–1120. doi: 10.1093/genetics/136.3.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York: 1987. [Google Scholar]
- Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences, USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson DA, Tobe VO, Taylor SL. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Research. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Wakeley J. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics. 2001;158:885–896. doi: 10.1093/genetics/158.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Mita A, Sakurai-Yamatani N, et al. Hybrid breakdown caused by substitution of the X chromosome between two mouse subspecies. Genetics. 2004;166:913–924. doi: 10.1534/genetics.166.2.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Aoto T, Totsuka Y, et al. Disruption of genetic interaction between two autosomal regions and the X chromosome causes reproductive isolation between mouse strains derived from different subspecies. Genetics. 2007;175:185–197. doi: 10.1534/genetics.106.062976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth A, Adama T, Din W, Bonhomme F. Hybridation naturelle entre deux sous-especes de souris domestique, Mus musculus domesticus et Mus musculus castaneus, pres du lac Casitas (Californie) Genome. 1998;41:104–110. doi: 10.1139/g97-109. [DOI] [PubMed] [Google Scholar]
- Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Systematic Biology. 2004;53:793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Prager EM, Orrego C, Sage RD. Genetic variation and phylogeography of central Asian and other house mice, including a major new mitochondrial lineage in Yemen. Genetics. 1998;150:835–861. doi: 10.1093/genetics/150.2.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo M. Molecular phylogeny of the hominoids: inferences from multiple independent DNA sequence data sets. Molecular Biology and Evolution. 1997;14:248–265. doi: 10.1093/oxfordjournals.molbev.a025761. [DOI] [PubMed] [Google Scholar]
- Sage RD, Atchley WR, Capanna E. House mice as models in systematic Biology. Systematic Biology. 1993;42:523–561. [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Salcedo T, Geraldes A, Nachman MW. Nucleotide variation in wild and inbred mice. Genetics. 2007;177:2277–2291. doi: 10.1534/genetics.107.079988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstedt SA, Tucker PK. Male-driven evolution in closely related species of the mouse genus Mus. Journal of Molecular Evolution. 2005;61:138–144. doi: 10.1007/s00239-004-0279-1. [DOI] [PubMed] [Google Scholar]
- She JX, Bonhomme F, Boursot P, et al. molecular phylogenies in the genus Mus — comparative-analysis of electrophoretic, scnDNA hybridization, and mtDNA RFLP data. Biological Journal of the Linnean Society. 1990;41:83–103. [Google Scholar]
- Stadler T, Arunyawat U, Stephan W. Population genetics of speciation in two closely related wild tomatoes (Solanum section lycopersicon) Genetics. 2008;178:339–350. doi: 10.1534/genetics.107.081810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. American Journal of Human Genetics. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova R, Gregorova S, Buckiova D, et al. Genetic analysis of X-linked hybrid sterility in the house mouse. Mammalian Genome. 2004;15:515–524. doi: 10.1007/s00335-004-2386-0. [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proceedings of the National Academy of Sciences, USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Shimada T, Terashima M, Tsuchiya K, Aplin K. Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Molecular Phylogenetics and Evolution. 2004;33:626–646. doi: 10.1016/j.ympev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada T, Mita A, Maeno A, et al. Mouse inter-subspecific consomic strains for genetic dissection of quantitative complex traits. Genome Research. 2008;18:500–508. doi: 10.1101/gr.7175308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. Mega 4: Molecular Evolutionary Genetics Analysis (MEGA) Software, Version 4.0. Molecular Biology and Evolution. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Teeter KC, Payseur BA, Harris LW, et al. Genome-wide patterns of gene flow across a house mouse hybrid zone. Genome Research. 2008;18:67–76. doi: 10.1101/gr.6757907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. Clustal-W — improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker PK, Sage RD, Warner J, et al. Abrupt cline for sex-chromosomes in a hybrid zone between 2 species of mice. Evolution. 1992;46:1146–1163. doi: 10.1111/j.1558-5646.1992.tb00625.x. [DOI] [PubMed] [Google Scholar]
- Tucker PK, Sandstedt SA, Lundrigan BL. Phylogenetic relationships in the subgenus Mus (genus Mus, family Muridae, subfamily Murinae): examining gene trees and species trees. Biological Journal of the Linnean Society. 2005;84:653–662. [Google Scholar]
- Vanlerberghe F, Dod B, Boursot P, et al. Absence of Y-chromosome introgression across the hybrid zone between Mus musculus domesticus and Mus musculus musculus. Genetics Research. 1986;48:191–197. doi: 10.1017/s0016672300025003. [DOI] [PubMed] [Google Scholar]
- Wakeley J, Hey J. Estimating ancestral population parameters. Genetics. 1997;145:847–855. doi: 10.1093/genetics/145.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Watterson GA. On the number of segregating sites in genetical models without recombination. Theoretical Population Biology. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- Woerner AE, Cox MP, Hammer MF. Recombination-filtered genomic datasets by information maximization. Bioinformatics. 2007;23:1851–1853. doi: 10.1093/bioinformatics/btm253. [DOI] [PubMed] [Google Scholar]
- Won YJ, Hey J. Divergence population genetics of chimpanzees. Molecular Biology and Evolution. 2005;22:297–307. doi: 10.1093/molbev/msi017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.