

Abstract
A very short patch repair system prevents mutations resulting from deamination of 5-methylcytosine to thymine. The Vsr endonuclease is the key enzyme of this system, providing sequence specificity. We identified two genes encoding Vsr endonucleases V.NgoAXIII and V.NgoAXIV from Neisseria gonorrhoeae FA1090 based on DNA sequence similarity to genes encoding Vsr endonucleases from other bacteria. After expression of the gonococcal genes in Escherichia coli, the proteins were biochemically characterized and the endonucleolytic activities and specificities of V.NgoAXIII and V.NgoAXIV were determined. V.NgoAXIII was found to be multispecific and to recognize T:G mismatches in every nucleotide context tested, whereas V.NgoAXIV recognized T:G mismatches in the following sequences: GTGG, CTGG, GTGC, ATGC, and CTGC. Alanine mutagenesis of conserved residues showed that Asp50 and His68 of V.NgoAXIII and Asp51 and His69 of V.NgoAXIV are essential for hydrolytic activity. Glu25, His64, and Asp97 of V.NgoAXIV and Glu24, Asp63, and Asp97 of V.NgoAXIII are important but not crucial for the activity of V.NgoAXIII and V.NgoAXIV. However, Glu24 and Asp63 are also important for the specificity of V.NgoAXIII. On the basis of our results concerning features of Vsr endonucleases expressed by N. gonorrhoeae FA1090, we postulate that at least two types of Vsr endonucleases can be distinguished.
The existence of methylated DNA in procaryotes and eucaryotes has been well documented, with 5-methylcytosine (m5C) being the most commonly modified base (1). Organisms use m5C as an epigenetic tag, but this modified base is very unstable and can undergo spontaneous deamination (15), resulting in a T:G mismatch. In the absence of an appropriate repair mechanism, cytosine deamination is highly mutagenic. Since the deamination usually occurs in a nonreplicating background, the lesion is refractory to methyl-directed mismatch repair. If the T:G mismatch is repaired by a general repair mechanism, the creation of an A·T substitution is as likely as the restoration of the original G·C base pair. In DNA, thymine resulting from deamination of m5C cannot be removed by general repair mechanisms because they do not recognize this thymine as erroneous. As a result, in the absence of a specific repair mechanism, deamination of m5C is highly mutagenic.
In Escherichia coli, a repair pathway counteracting the mutagenic effects of hydrolytic deamination of m5C is based on the action of a very short patch (VSP) repair system (2, 5, 8, 18, 23). The central enzyme of this pathway is Vsr, an endonuclease whose coding sequence overlaps the gene for M.EcoKDcm, an m5C methyltransferase (m5C-MTase) (19, 23). In genomes of other bacteria, the vsr genes are invariably associated with genes coding for m5C-MTases (3, 16, 20). The Vsr endonucleases that accompany m5C-MTases are believed to exhibit sequence specificity based on the recognition sequence of the accompanying MTase. However, only a few MTases have been studied in detail and the data indicate that methylation at sites other than that ascribed to the corresponding restriction endonuclease can occur with significant frequency (4), indicating that the recognition sequence of an MTase is somewhat arbitrarily assigned. The best-characterized Vsr endonuclease, V.EcoKDcm (9, 10, 29), is a gene product of E. coli K-12. This endonuclease recognizes the sequence CTWGG (W is A or T), where the underlined thymine is mispaired with guanine. The enzyme nicks the DNA backbone on the 5′ side of the mispaired thymine (12). The crystal structure of V.EcoKDcm shows that its catalytic center consists of two conserved aspartic acid residues (D51 and D97), glutamic acid (E25), threonine (T63), and two histidines (H64 and H69). Alanine-scanning mutagenesis of these conserved residues revealed that E25A, H64A, and D97A mutants have reduced activity, while D51A and H69A mutants have no detectable activity (28-30).
An individual strain of Neisseria gonorrhoeae may produce up to 16 different DNA MTases, with the bulk of these enzymes adding m5C to one of the cytosines in the recognition sequence (20, 25). Due to the high degree of potential cytosine methylation in the gonococcus, one might predict that genes containing any of these recognition sequences would represent hot spots for mutation. However, to date, no hot spots have been identified. Furthermore, we were only able to identify two potential Vsr endonucleases. While the genes encoding both of these proteins appear to be linked to restriction-modification system genes in a variety of gonococcal strains, these systems appear to be inactive (16). To understand the biochemical basis of VSP repair in the Neisseriaceae, we studied the properties of Vsr endonucleases from N. gonorrhoeae FA1090. Given the large number of m5C-MTases found in the gonococcus and the paucity of vsr genes identified using bioinformatic analysis based on amino acid sequence similarity with known Vsr proteins, it is possible that the Vsr endonucleases expressed by N. gonorrhoeae could have more general sequence recognition properties than those found in E. coli or Bacillus stearothermophilus. Alternatively, this species could have genes encoding more Vsr endonucleases which are too divergent structurally to be identified by bioinformatic methods. Our results indicate that N. gonorrhoeae FA1090 expresses two Vsr endonucleases. The first, V.NgoAXIII, recognizes T:G mismatches in all nucleotide contexts of known gonococcal MTases tested, and the second, V.NgoAXIV, recognizes only a subset. Moreover, comparison of their amino acid sequences has shown that these Vsr endonucleases differ in a region responsible for the recognition and cleavage of T:G mismatches, suggesting the existence of two different families of enzymes.
MATERIALS AND METHODS
Vsr endonuclease cloning, expression, and purification.
E. coli ER1821 [(McrA−) Δ(mcrC-mrr)114::IS10 (r− m−)] (New England Biolabs), M15 [K-12 Kmr (pREP4) Nals Strs Rifs Thi− Lac− Ara+ Gal+ Mtl− F− RecA+ Uvr+ Lon+] (Qiagen), and BL21(DE3) [F− ompT gal dcm lon hsdSB (rB− mB−) λ(DE3)] (Novagen) were used for cloning and expression experiments. These strains and their derivatives were grown in Luria-Bertani (LB) broth (21) with the addition of appropriate antibiotics when needed (ampicillin [100 μg/ml] or kanamycin [25 or 30 μg/ml]). The genes ngoAXIIIV (spanning nucleotides [nt] 301,365 to 301,778 in the N. gonorrhoeae FA1090 genome) and ngoAXIVV (spanning nt 1,110,897 to 1,111,319 in the gonococcal genome) were amplified by PCR from chromosomal DNA of N. gonorrhoeae FA1090. The DNA encoding V.NgoAXIII was cloned into the NcoI-to-XhoI sites of pET28a (Novagen), resulting in the formation of plasmid pAK15. The DNA encoding V.NgoAXIV was cloned into the BamHI site of pQE-30 (Qiagen), resulting in the formation of plasmid pAK16. The DNAs encoding the Vsr proteins were cloned such that they contained a His tag on the carboxy terminus (V.NgoAXIII) or on the amino terminus (V.NgoAXIV). The primers for DNA amplification were obtained from the Institute of Biochemistry and Biophysics (IBB), Warsaw, Poland. The V.NgoAXIII-coding sequence was amplified using primer 1 (5′ GCAGCCATGGATGGATAAATTAACC 3′) and primer 2 (5′ GATGCTCGAGTCTTGTTTGGATGA 3′). The V.NgoAXIV-coding sequence was amplified using primer 3 (5′ ATCGGGATCCACCGATATTTTCACTCCATC 3′) and primer 4 (5′ ATAGCATGGATCCTCAGACGGCATCTTTTATTTCCTC 3′). The PCRs were carried out using Pfu DNA polymerase (Fermentas) according to the manufacturer's recommendations.
To express and purify the Vsr endonucleases, a single colony of E. coli BL21(DE3)(pAK15) or M15(pAK16) generated by fresh transformation was used to inoculate 100 ml of LB broth containing appropriate antibiotics, kanamycin for pAK15 and ampicillin and kanamycin for pAK16. Cultures were incubated at 37°C, and when the optical density of the culture at 600 nm reached 0.6, isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. Incubation was continued at 30°C for an additional 12 h. The cells were collected by centrifugation, and the bacterial pellet was resuspended in 10 ml of buffer containing 50 mM NaHPO4, 300 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol, 0.1% Tween 20, 55 μM phenylmethylsulfonyl fluoride (PMSF), and 1 tablet of EDTA-free complete Mini (protease inhibitor cocktail) (Roche Diagnostics). After sonication, the cellular debris was removed by centrifugation at 40,000 × g for 1 h and the supernatant applied to a 3-ml Ni-nitrilotriacetic acid (NTA) agarose column previously equilibrated with 100 ml of the above-described buffer. The column was washed with 250 ml of buffer containing 50 mM NaHPO4, 300 mM NaCl, 35 mM imidazole, and 10% glycerol. The proteins were eluted with a gradient of imidazole (50 mM to 0.5 M) in the same buffer. V.NgoAXIII and V.NgoAXIV eluted at 0.2 to 0.25 M imidazole. The homogeneity of the purified enzymes was determined by electrophoresis on a 15% SDS-PAGE gel. The amount of purified protein was determined by using Bradford Reagent (Sigma-Aldrich), with bovine serum albumin (BSA) as the protein standard. The yields of purified protein were ∼3 mg/liter for V.NgoAXIII and ∼29 mg/liter for V.NgoAXIV. PageRuler prestained protein ladder (170, 130, 100, 70, 55, 40, 35, 25, 15, and 10 kDa) (Fermentas) was used as the protein molecular mass marker.
Enzymes and chemicals.
Restriction enzymes, T4 DNA ligase, Pfu DNA polymerase, IPTG, polymerase I Klenow fragment, and DNA and protein markers were purchased from Fermentas and used under conditions recommended by the manufacturer. Kits for DNA purification and plasmid DNA preparation were purchased from A&A Biotechnology, Gdansk, Poland. Ni-NTA agarose was purchased from Qiagen. All other chemicals were purchased from Sigma-Aldrich, unless otherwise noted.
Preparation of substrate DNA.
Two oligonucleotides (obtained from Sigma-Aldrich) were used to generate substrate DNA. The base sequence for all oligonucleotides is given below. Oligonucleotides 5′ ATATTCAAACTGGCGCCGAGCGTATGCCGCATGACCTTTCCCATCTTGGCTTCCTTGCTGGTCAGATTGGTCGTCTTATTACCATTTCAACTACTNNNNNNGATATCNNNNNNCGACTCC 3′ (120-mer) and 5′ AGCAAGGCCACGACGCAATGGAGAAAGACGGAGAGCGCCAACGGCGTCCATCTCGAAGGAGTCGNNNNNNGATATCNNNNNNAGTAGTTG 3′ (90-mer) were used to create substrate DNA. Each oligonucleotide contained an ∼33-nt complementary region (underlined) (see Table 1 for the composition of each primer set). Primer pairs were generated for derivatives of all MTase recognition sequences, where the only difference in each substrate DNA was the composition of the NNNNNN sequence (in boldface). The NNNNNN sequence in each primer pair corresponds to the sequence derived from known specific gonococcal m5C-MTase recognition sequences. The italics in the sequence indicate the region that varies from the MTase recognition sequence that includes thymine mismatched to guanine. See Table 1 for the DNA sequence of the conserved substrate DNA region of each primer set. To generate substrate DNA, each oligonucleotide pair was mixed, heated to 95°C for 5 min in 1× SSC (0.15 M NaCl, pH 7.2, 0.015 M sodium citrate), and slowly cooled to room temperature. Single-stranded ends were filled in by polymerase I Klenow fragment (Fermentas) in the presence of deoxynucleoside triphosphates (dNTPs) under conditions recommended by the manufacturer. Each substrate DNA contained two sequences derived from the sequence modified by the individual m5C-MTases (in substrate DNA, the modified cytosine was replaced by thymine). On the opposite strand in the sequence modified by the m5C-MTase, in the site of the modified cytosine, there was thymine mismatched to guanine. In this way, each substrate DNA contained two T:G mismatches. If the Vsr endonuclease incised the 5′ side of mismatched thymine, two products would be created (∼110 bp and ∼80 bp). The control substrate DNAs did not contain a T:G mismatch in the NNNNNN region.
TABLE 1.
Substrate DNAs used in this study
| Substrate | Sequencea |
|---|---|
| Control (6N) | 5′CAACTACTAGCGCCGATATCAGCGCCCGACTCC3′ |
| 3′GTTGATGATCGCGGCTATAGTCGCGGGCTGAGG5′ | |
| Control (4N) | 5′CAACTACTCCGGGATATCCCGGCGACTCC3′ |
| 3′GTTGATGAGGCCCTATAGGGCCGCTGAGG5′ | |
| M.NgoAI-sub | 5′CAACTACTAGCGCTGATATCAGTGCTCGACTCC3′ |
| 3′GTTGATGATCGTGACTATAGTCGCGAGCTGAGG5′ | |
| M.NgoAII-sub | 5′CAACTACTGGCCGATATCGGTCCGACTCC3′ |
| 3′GTTGATGACTGGCTATAGCCGGGCTGAGG5′ | |
| M.NgoAIII-first-sub | 5′CAACTACTCCGCGGGATATCTCGCGGCGACTCC3′ |
| 3′GTTGATGAGGCGCTCTATAGGGCGCCGCTGAGG5′ | |
| M.NgoAIII-second-sub | 5′CAACTACTCCGCGGGATATCCTGCGGCGACTCC3′ |
| 3′GTTGATGAGGCGTCCTATAGGGCGCCGCTGAGG5′ | |
| M.NgoAIII-third-sub | 5′CAACTACTCCGCGGGATATCCCGTGGCGACTCC3′ |
| 3′GTTGATGAGGTGCCCTATAGGGCGCCGCTGAGG5′ | |
| M.NgoAIV-sub | 5′CAACTACTGCCGGCGATATCGTCGGCCGACTCC3′ |
| 3′GTTGATGACGGCTGCTATAGCGGCCGGCTGAGG5′ | |
| M.NgoAVII-sub | 5′CAACTACTGCGGCGATATCGTGGCCGACTCC3′ |
| 3′GTTGATGACGCTGCTATAGCGCCGGCTGAGG5′ | |
| M.NgoAORF675P-sub | 5′CAACTACTGGCTCCGATATCGGCTTCCGACTCC3′ |
| 3′GTTGATGACTGAGGCTATAGCCGAGGGCTGAGG5′ | |
| M.NgoAORF302P-sub | 5′CAACTACTGCCGGTGATATCGTCGGTCGACTCC3′ |
| 3′GTTGATGACGGCTACTATAGCGGCCAGCTGAGG5′ | |
| M.NgoAORF1175P-sub | 5′CAACTACTCCGGGATATCCTGGCGACTCC3′ |
| 3′GTTGATGAGGTCCTATAGGGCCGCTGAGG5′ | |
| M.HhaI-sub | 5′CAACTACTGCGCGATATCGTGCCGACTCC3′ |
| 3′GTTGATGACGTGCTATAGCGCGGCTGAGG5′ | |
| M.EcoKDcm-sub | 5′CAACTACTCCAGGGATATCCTAGGCGACTCC3′ |
| 3′GTTGATGAGGTTCCTATAGGGTCCGCTGAGG5′ |
The bases in boldface are the recognition sequence for the various MTases, and an underlined T indicates the thymine that would arise from spontaneous deamination of m5C.
Endonuclease activity assay.
Endonuclease activity was assayed by incubating 0.15 μM substrate DNA with 1.5 μM purified Vsr endonuclease in a final volume of 20 μl containing 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 0.1 mg/ml BSA (for V.NgoAXIII) or 66 mM Tris-acetate (pH 7.9 at 37°C), 20 mM magnesium acetate, 132 mM potassium acetate, and 0.2 mg/ml BSA (for V.NgoAXIV) for 60 min at 37°C. Occasionally other metals, pHs, concentrations of NaCl, or concentrations of MgCl2 were tested. The cleavage products were analyzed using 10% polyacrylamide gels.
Site-directed mutagenesis of Vsr endonuclease genes.
Site-directed mutagenesis of Vsr endonuclease genes was performed using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. The DNA sequence of the mutated DNA was determined to verify that the constructs contained only the desired mutation.
Kinetic experiments.
Hydrolysis of substrate DNA was performed in 200 μl of 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 0.1 mg/ml BSA (for V.NgoAXIII) or 66 mM Tris-acetate (pH 7.9 at 37°C), 20 mM magnesium acetate, 132 mM potassium acetate, 0.2 mg/ml BSA (for V.NgoAXIV). Vsr (2.4 μM) was incubated with 0.15 μM DNA at 37°C. Samples (20 μl) were taken periodically and added to 1 μl phenol/chloroform solution. Substrate and products were separated on a 10% polyacrylamide gel. The amount of DNA contained in each band visualized on the gel was quantified using Quantity One (Bio-Rad). The amounts present in individual bands were expressed as a fraction of the total intensity of DNA as visualized by ethidium bromide staining. Rate constants were determined by fitting the data to the first-order rate equation (7, 13) % productt = 100[1 − exp(−kstt)] (where t is time and kst is the first-order rate constant) using Origin 6.1 (OriginLab, Northampton, MA).
Other methods.
All other methods were carried out in accordance with protocols described by Sambrook and Russell (21). For gel electrophoresis, reaction products were analyzed on a 10% polyacrylamide gel using a Tris-borate buffer system. DNA was visualized by ethidium bromide staining. The GeneRuler 50-bp DNA ladder (band sizes of 1,000, 900, 800, 700, 600, 500, 400, 300, 250, 200, 150, 100, and 50 bp) (Fermentas) was used as a standard.
Computer analysis.
DNA and protein sequences were compared with GenBank and SWISS-PROT databases on the BLAST server hosted by the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/blast). We used the genomic sequence of N. gonorrhoeae strain FA1090 (GenBank accession no. NC_002946). Phylogenetic and molecular evolutionary analyses were conducted using MEGA version 4 (27).
RESULTS
Identification and analysis of Vsr-encoding genes in N. gonorrhoeae FA1090.
Analysis of the published DNA sequence of N. gonorrhoeae FA1090 allowed us to identify genes encoding potential Vsr endonucleases. Two open reading frames (ORFs) were identified where the predicted protein products showed significant homology to V.EcoKDcm and other putative Vsr endonucleases. A phylogenetic tree was constructed, and the data indicate that while Vsr nucleases have significant homology, the putative gene products cluster as distinct families of proteins (Fig. 1). These data indicate that V.NgoAXIII and V.NgoAXIV represent two different groups of Vsr enzymes. Vsr endonucleases have conserved amino acid sequences and motifs. The data in Fig. 2 demonstrate the presence of key conserved residues necessary for catalysis in V.EcoKDcm (28-30). While the enzymes form distinct phylogenic clusters, all key residues are retained in all of the enzymes identified. It would appear that there are two distinct classes of enzymes that can be distinguished by the nature of the amino acid in position 63 or 64, with one class containing an aspartic acid and other class containing a histidine.
FIG. 1.

Phylogenetic tree of Vsr endonucleases. Sequences used for phylogram are from N. gonorrhoeae FA1090 (V.NgoAXIII, GenBank accession no. AAW89048.1, and V.NgoAXIV, GenBank accession no. AAW89837.1), Haemophilus parainfluenzae (V.HpaIIP, GenBank accession no. AAA20480.1), N. lactamica (V.NlaL17ORFAP, GenBank accession no. AAO17335.1), Flavobacterium johnsoniae UW101 (V.FjoUWORF652P, GenBank accession no. YP_001193008.1), Heliobacterium modesticaldum Ice1 (V.HmoORF2858P, GenBank accession no. YP_001681379.1), Clostridium thermocellum ATCC 27405 (V.CthORF1749P, GenBank accession no. YP_001038161.1), Geobacillus stearothermophilus (V.BssHIII, GenBank accession no. CAC50878.1), N. meningitidis FAM18 (V.Nme18ORF1992P, GenBank accession no. YP_975913.1), Actinobacillus pleuropneumoniae L20 (V.AplORF1202P, GenBank accession no. YP_001053893.1), Xanthomonas oryzae pv. oryzae KACC10331 (V.XorKI, GenBank accession no. YP_199245.6), Maricaulis maris MCS10 (V.Mma10ORF3057P, GenBank accession no. YP_758275.1), E. coli strain K-12 substrain MG1655 (V.EcoKDcm, GenBank accession no. NP_416469.1), Methylobacterium sp. 4-46 (V.Msp446ORF1487P, GenBank accession no. YP_001768429.1), Laribacter hongkongensis HLHK9 (V.LhoHORF3244P, GenBank accession no. YP_002797229.1), Sinorhizobium meliloti 1021 (V.SmaORF3763P, GenBank accession no. CAC47728.1), Shigella sonnei Ss046 (V.Sso46DcmP, GenBank accession no. YP_310917.1), Salmonella enterica serovar Typhimurium LT2 (V.StyLT2DcmP, GenBank accession no. NP_460942.1), Klebsiella pneumoniae 342 (V.Kpn342ORF1851P, GenBank accession no. YP_002237694.1), Serratia proteamaculans 568 (V.Spr568ORF573P, GenBank accession no. YP_001476806.1), Bacillus subtilis strain R (V.BsuRI, GenBank accession no. X02988.1), Lactobacillus delbrueckii subsp. bulgaricus (V.LdeBBORF1147P, GenBank accession no. YP_813120.1), and an unidentified thermophile (V.TspMI, GenBank accession no. ABO38151.1). MEGA version 4 software was used as a drawing tool for creating the phylogenetic tree. V.NgoAXIII and V.NgoAXIV are underlined. The size bar indicates estimated evolutionary distance.
FIG. 2.

Sequence alignment of Vsr endonucleases. Sequences used for alignment are the same as listed in Fig. 1. The residues important for endonucleolytic activity are indicated by a closed circle. Amino acid sequence numbers listed are from V.NgoAXIII.
Properties of V.NgoAXIII and V.NgoAXIV.
The coding sequence for V.NgoAXIII consists of 414 bp and would encode a protein of 137 amino acids (aa) with a calculated molecular mass of 16 kDa. The coding sequence for V.NgoAXIV is 423 bp and would encode a protein containing 140 aa with a calculated molecular mass of 16.7 kDa. V.NgoAXIII and V.NgoAXIV were purified to apparent homogeneity from E. coli BL21(DE3)(pAK15) or M15(pAK16) cells by metal affinity chromatography on a Ni-NTA agarose column (Fig. 3). The mobilities of purified V.NgoAXIII and V.NgoAXIV were consistent with those predicted on the basis of their amino acid sequences plus the additional amino acids incorporated into these proteins as a result of the cloning methods. The identity and mass of purified V.NgoAXIII and V.NgoAXIV were confirmed by mass spectrometry (data not shown).
FIG. 3.

Purification of Vsr endonucleases. After purification by metal affinity chromatography, V.NgoAXIII and V.NgoAXIV were separated on a 15% SDS-PAGE gel and stained with Coomassie brilliant blue R250. Lane M, PageRuler prestained protein ladder; lane 1, V.NgoAXIV; lane 2, V.NgoAXIII.
Determination of the optimal conditions for Vsr endonuclease activity.
To determine the optimal conditions for enzymatic activity of the Vsr endonucleases, different buffers were tested, varying the ionic strength, pH, and Mg2+ ion concentrations. The maximum activity of V.NgoAXIII was observed at 37°C in the presence of 10 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 0.1 mg/ml BSA in the absence of any salt. The maximal activity of V.NgoAXIV was seen at 37°C in the presence of 66 mM Tris-acetate (pH 7.9 at 37°C), 20 mM magnesium acetate, 132 mM potassium acetate, and 0.2 mg/ml BSA (data not shown). Both enzymes required Mg2+ for optimal enzyme activity. We observed about 10% of the maximum activity with Mn2+. No activity was observed when Ca2+, Co2+, Zn2+, or Ni2+ was used as the divalent cation source. The enzymes were active over a pH range from 7.2 to 8.0, with the optimum activity at 7.5, and were most active at 37°C (data not shown).
Determination of the specificity of V.NgoAXIII and V.NgoAXIV.
N. gonorrhoeae FA1090 carries genes encoding eight well-characterized m5C-MTases (rebase.neb.com): M.NgoAI (RGCGCY), M.NgoAII (GGCC), M.NgoAIII (CCGCGG), M.NgoAIV (GCCGGC), M.NgoAVII (GCSGC), M.NgoAORF675P (GGNNCC), M.NgoAORF302P (RCCGGY), and M.NgoAORF1175P (CCGG). Recognition sequences, where R is A or G, Y is C or T, S is C or G, and N is A, T, C, or G, are given in parentheses. The bolded C is the cytosine that is modified by the m5C-MTase and that can deaminate to thymine. The specificities of V.NgoAXIII and V.NgoAXIV were tested using 0.15 μM substrate DNA and 1.5 μM purified enzymes. Each substrate DNA contained a T:G mismatch within a sequence derived from the specific recognition sequences for individual gonococcal m5C-MTases (in substrate DNAs, the methylated cytosine was replaced by a thymine). For sequences derived from M.NgoAI, M.NgoAII, M.NgoAIV, M.NgoAVII, M.NgoAORF675P, M.NgoAORF302P, and M.NgoAORF1175P, single substrate DNAs were used. For sequences derived from M.NgoAIII (CCGCGG), three different substrate DNAs were used because the modified cytosine is undetermined. The substrate M.NgoAIII-first-sub contains thymine in the place of the first cytosine in the recognition sequence (CCGCGG), the substrate M.NgoAIII-second-sub in the place of the second (CCGCGG), and the M.NgoAIII-third-sub in the place of the third (CCGCGG). The hydrolytic activity of Vsr endonucleases is demonstrated by the appearance of two bands on an acrylamide gel (∼110 and ∼80 bp) after endonuclease treatment. The results presented in Fig. 4 A indicate that V.NgoAXIII is able to introduce a nick in substrate DNA containing the T:G mismatch within the sequences specific for all m5C-MTases expressed by N. gonorrhoeae FA1090. No cleavage products were observed when substrate DNA without a T:G mismatch was used.
FIG. 4.
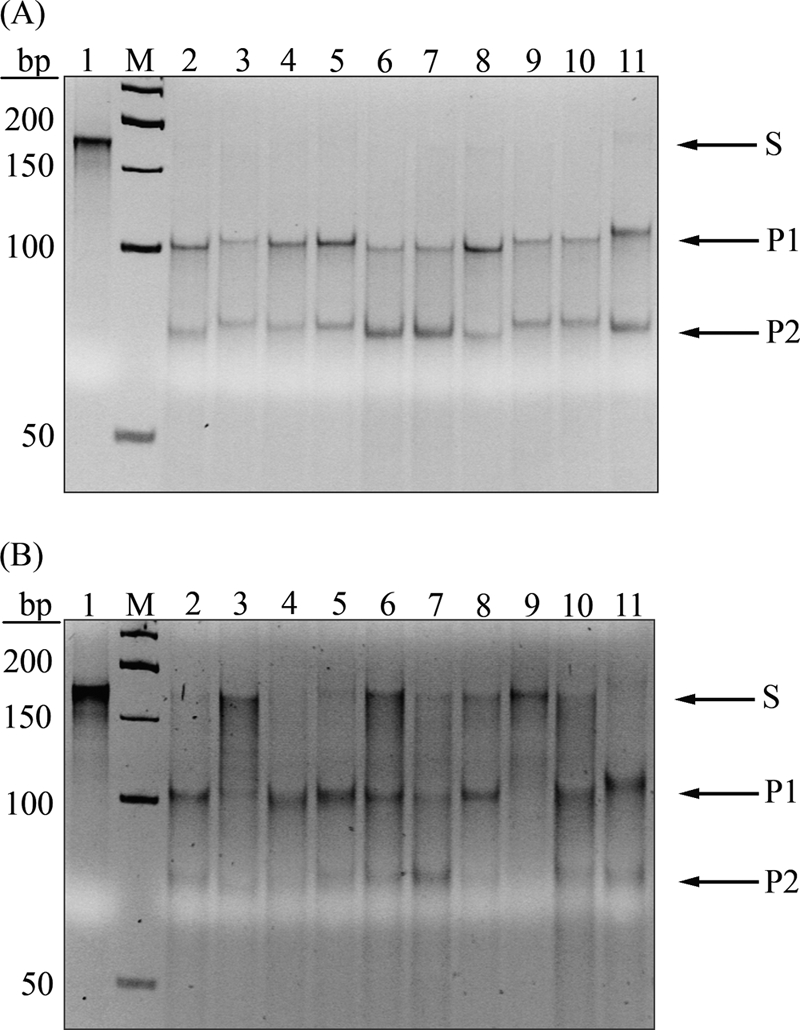
Determination of the specificity of Vsr endonucleases expressed by N. gonorrhoeae FA1090. (A) Specificity of V.NgoAXIII. (B) Specificity of V.NgoAXIV. Lane 1, control substrate DNA without T:G mismatch; lane M, GeneRuler 50-bp DNA ladder; lane 2, substrate DNA M.NgoAI-sub containing the sequence AGTGCT/AGCGCT; lane 3, substrate DNA M.NgoAII-sub containing the sequence GGTC/GGCC; lane 4, substrate DNA M.NgoAIII-first-sub containing the sequence TCGCGG/CCGCGG; lane 5, substrate DNA M.NgoAIII-second-sub containing the sequence CTGCGG/CCGCGG; lane 6, substrate DNA M.NgoAIII-third-sub containing the sequence CCGTGG/CCGCGG; lane 7, substrate DNA M.NgoAIV-sub containing the sequence GTCGGC/GCCGGC; lane 8, substrate DNA M.NgoAVII-sub containing the sequence GTGGC/GCCGC; lane 9, substrate DNA M.NgoAORF675P-sub containing the sequence GGCTTC/GGAGCC; lane 10, substrate DNA M.NgoAORF302P-sub containing the sequence ATCGGC/GCCGGT; lane 11, substrate DNA M.NgoAORF1175P-sub containing the sequence CTGG/CCGG. The T:G mismatch resulting from deamination of m5C to thymine is in boldface. Arrows indicate substrate (S) and reaction products (P1 and P2) obtained after cleavage by purified Vsr endonucleases.
To test the specificity of V.NgoAXIV, the same substrate DNAs were used. The results presented in Fig. 4B indicate that V.NgoAXIV is able to introduce a nick in substrate DNA containing the T:G mismatch (in boldface) within the following sequences: AGTGCT/AGCGCT, TCGCGG/CCGCGG, CTGCGG/CCGCGG, CCGTGG/CCGCGG, GTCGGC/GCCGGC, GTGGC/GCCGC, ATCGGC/GCCGGT, and CTGG/CCGG. No cleavage products were observed when substrate DNA without a T:G mismatch was used. To determine if V.NgoAXIII and V.NgoAXIV recognize T:G mismatches within sequences other than those modified by the gonococcal m5C-MTases, substrate DNAs containing NGTGCN and NCT(A/T)GG were used. The sequence GCGC is modified by M.HhaI, and CCA/TGG by M.EcoKDcm. The results presented in Fig. 5 A indicate that V.NgoAXIII recognizes T:G mismatches within both GTGC/GCGC and CTTGG/CCAGG sequences. The results presented in Fig. 5B indicate that V.NgoAXIV recognizes a T:G mismatch only within the GTGC/GCGC sequence.
FIG. 5.

Recognition of alternative DNA sequences by V.NgoAXIII and V.NgoAXIV. (A) Results of reaction catalyzed by V.NgoAXIII. (B) Results of reaction catalyzed by V.NgoAXIV. Lane 1, substrate DNA M.HhaI-sub containing the sequence GTGC/GCGC; lane 2, substrate DNA M.EcoKDcm-sub containing the sequence CTAGG/CCTGG; lane M, GeneRuler 50-bp DNA ladder. T:G mismatch is in boldface. Arrows indicate substrate (S) and reaction products (P1 and P2) obtained after cleavage by purified Vsr endonucleases.
Single-turnover experiments.
Because V.NgoAXIII and V.NgoAXIV recognize more than one sequence, the sequence preferences of V.NgoAXIII and V.NgoAXIV toward different substrates containing T:G mismatches were tested. Single-turnover experiments were carried out using 0.15 μM substrate DNA and 2.4 μM purified enzymes (sixteen times excess enzyme over DNA). This enabled the direct observation of the conversion of substrates into intermediates and products in a single pass of the reactants through the enzymatic pathway.
The percentage of created product was plotted versus reaction time, and the first-order rate constants (kst) were calculated (Fig. 6). The rate constants were normalized to the results for the most efficiently processed substrates (Table 2). The relative rate constants for V.NgoAXIII for the best substrate (GTCGGC/GCCGGC; derived from the specific recognition sequence for M.NgoAIV) and for the slowest (GGCTTC/GGAGCC; derived from the specific recognition sequence for M.NgoAORF675P) differ by a factor of ∼4. Since all tested sequences are not closely related, this suggests that V.NgoAXIII does not exhibit a pronounced preference for a particular T:G mismatch-containing sequence. An analysis of the sequences flanking the T:G mismatch in the substrate DNAs cleaved by V.NgoAXIII shows that this enzyme was able to cleave the substrate when the T:G mismatch occurred in the sequences G(T/G)G, G(T/G)C, C(T/G)G, T(T/G)C, A(T/G)C, C(T/G)T, and C(T/G)C, where the T/G in parentheses indicates the location of the mismatch.
FIG. 6.

Hydrolysis of substrate DNAs containing T:G mismatches by Vsr endonucleases using single-turnover conditions. (A) Results of reaction catalyzed by V.NgoAXIII. (B) Results of reaction catalyzed by V.NgoAXIV. In the top part of each panel is an example of the electrophoresis profile of reaction products in a 10% polyacrylamide gel that was used to quantify the digestions. The gel for V.NgoAXIII endonuclease shows the results of the reaction with substrate DNA M.NgoAI-sub, and the gel for V.NgoAXIV endonuclease shows the results of the reaction with substrate M.NgoAORF1175P-sub. The bottom part of each panel shows the analysis of the data obtained by electrophoresis of the reaction products and fitted to a first-order rate equation using Origin 6.1. The graphs show results with substrates M.NgoAII-sub, M.NgoAIV-sub, M.NgoAORF302P-sub, and M.EcoKDcm-sub for V.NgoAXIII endonuclease and substrates M.NgoAI-sub, M.NgoAIII-third-sub, M.NgoAORF1175P-sub, and M.HhaI-sub for V.NgoAXIV endonuclease. Lane M is the GeneRuler 50-bp DNA ladder. Arrows indicate substrate (S) and reaction products (P1 and P2) obtained after cleavage by Vsr endonucleases.
TABLE 2.
Effect of nucleotide context on the efficiency of hydrolytic reactions catalyzed by the N. gonorrhoeae endonucleases
| Endonuclease and substrate | Sequence contexta | First-order rate constant (kst) (min−1)c | Factor of decrease in efficiencyb |
|---|---|---|---|
| V.NgoAXIII | |||
| M.NgoAI-sub | AGTGCT | 0.124 | 3.35 |
| M.NgoAII-sub | GGTC | 0.119 | 3.49 |
| M.NgoAIII-first-sub | TCGCGG | 0.276 | 1.50 |
| M.NgoAIII-second-sub | CTGCGG | 0.226 | 1.84 |
| M.NgoAIII-third-sub | CCGTGG | 0.389 | 1.06 |
| M.NgoAIV-sub | GTCGGC | 0.416 | 1 |
| M.NgoAVII-sub | GTGGC | 0.102 | 4.07 |
| M.NgoAORF675P-sub | GGCTTC | 0.101 | 4.11 |
| M.NgoAORF302P-sub | ATCGGC | 0.328 | 1.26 |
| M.NgoAORF1175P-sub | CTGG | 0.160 | 2.60 |
| M.HhaI-sub | GTGC | 0.122 | 3.40 |
| M.EcoKDcm-sub | CTAGG | 0.280 | 1.48 |
| V.NgoAXIV | |||
| M.NgoAI-sub | AGTGCT | 0.104 | 1 |
| M.NgoAIII-first-sub | TCGCGG | 0.074 | 1.40 |
| M.NgoAIII-second-sub | CTGCGG | 0.093 | 1.11 |
| M.NgoAIII-third-sub | CCGTGG | 0.033 | 3.15 |
| M.NgoAIV-sub | GTCGGC | 0.043 | 2.41 |
| M.NgoAVII-sub | GTGGC | 0.041 | 2.53 |
| M.NgoAORF302P-sub | ATCGGC | 0.044 | 2.36 |
| M.NgoAORF1175P-sub | CTGG | 0.102 | 1.01 |
| M.HhaI-sub | GTGC | 0.094 | 1.10 |
A boldface T shows the thymine mispaired with guanine. Italics show the immediate surroundings of the T:G mismatch.
A value of 1 indicates substrate DNA that is the most efficiently digested by Vsr endonucleases. The efficiency of reactions catalyzed by the tested enzymes decreases with an increase of this factor.
All values are the average of three measurements and have a maximum error of ∼20%.
An analogous analysis of the results received for V.NgoAXIV indicated that the enzyme has some sequence preferences toward different substrate DNAs containing T:G mismatches. The relative rate constants of 9 tested sequences differ by a factor of ∼3.15. The sequences recognized by V.NgoAXIV seem to be related to derivatives of CTGG or GTGC. This indicates that V.NgoAXIV can have some sequence preference toward a T:G mismatch within CTGG/CCGG or GTGC/GCGC sequences. Moreover, analysis of the direct surroundings of the T:G mismatch in substrates digested by V.NgoAXIV demonstrated that the enzyme V.NgoAXIV cleaves only when the T:G mismatch is surrounded by GG, CG, GC, AC, and CC. The sequences recognized by V.NgoAXIV can be divided into two groups. The sequences AGTGCT/AGCGCT, TCGCGG/CCGCGG, CTGCGG/CCGCGG, CTGG/CCGG, and GTGC/GCGC are more efficiently cleaved by the V.NgoAXIV enzyme, and GTCGGC/GCCGGC, GTGGC/GCGGC, ATCGGC/GCCGGT, and CCGTGG/CCGCGG are cleaved less efficiently. From these data, we concluded that V.NgoAXIII is a multispecific Vsr endonuclease that cleaves all substrate DNAs containing T:G mismatches independent of the nucleotide context. V.NgoAXIV recognizes a variety of sequences, but its ability to nick DNA is limited to a smaller number of nucleotide contexts.
Cleavage of mismatches other than T:G.
To determine if V.NgoAXIII and V.NgoAXIV recognize mismatches other than T:G, substrate DNA containing an A:C mismatch was used in our nicking assay. The A:C mismatch was in the sequence RCCAGY/RCCCGY (R is A or G and Y is C or T; the A:C mismatch is in boldface). In the reaction catalyzed by V.NgoAXIII, as well as that catalyzed by V.NgoAXIV, no cleavage products were observed (data not shown). The presence of undigested substrate and the absence of hydrolysis bands indicate that V.NgoAXIII and V.NgoAXIV do not hydrolyze DNA containing A:C mismatches.
Determination of the molar concentration of Vsr endonucleases required to cleave substrate.
Due to the fact that single-turnover experiments were carried out with the enzyme in excess over the substrate, the amounts of V.NgoAXIII and V.NgoAXIV required to generate cleavage products were determined. In reaction mixtures with the substrate DNA containing a T:G mismatch in the sequence GTCGGC/GCCGGC (substrate M.NgoAIV-sub) in an equimolar ratio of DNA to V.NgoAXIII enzyme, ∼40% of the substrate was cleaved; at a 1.5-fold molar excess of V.NgoAXIII over DNA, ∼69% of the substrate was cleaved; and at a 10-fold excess of V.NgoAXIII, 100% of the substrate was digested. The results obtained for V.NgoAXIII in reactions with all substrate DNAs are shown in Table 3. A parallel analysis was performed for V.NgoAXIV. The reactions were carried out with substrate DNA containing a T:G mismatch in the sequence CTGG/CCGG. In reaction mixtures where the ratio of DNA to protein was 1:1, ∼20% of the substrate DNA was cleaved; at 1:1.5, 40%; and at 1:16, 90%.
TABLE 3.
Effects of variation in the molar amount of V.NgoAXIII on DNA cleavage
| Substrate | % of substrate digested at the indicated molar excess of enzyme over substrate |
||||
|---|---|---|---|---|---|
| 1.5:1 | 3:1 | 5:1 | 10:1 | 16:1 | |
| M.NgoAI-sub | 48.7 | 72.5 | 89.9 | 100 | 100 |
| M.NgoAII-sub | 44.7 | 69.3 | 85.2 | 100 | 100 |
| M.NgoAIII-first-sub | 34.8 | 87.6 | 88.2 | 100 | 100 |
| M.NgoAIII-second-sub | 25.5 | 48.6 | 68.4 | 100 | 100 |
| M.NgoAIII-third-sub | 52.1 | 78 | 82.1 | 100 | 100 |
| M.NgoAIV-sub | 69.2 | 89.3 | 100 | 100 | 100 |
| M.NgoAVII-sub | 42.5 | 74.7 | 83.8 | 100 | 100 |
| M.NgoAORF675P-sub | 5.2 | 14.7 | 30.4 | 84.8 | 92 |
| M.NgoAORF302P-sub | 26.8 | 46.2 | 64.1 | 100 | 100 |
| M.NgoAORF1175P-sub | 25.6 | 42.2 | 55.3 | 100 | 100 |
| M.HhaI-sub | 40.1 | 70.7 | 87 | 100 | 100 |
| M.EcoKDcm-sub | 42.9 | 67.8 | 90.2 | 100 | 100 |
Identification of amino acids important for specificity.
The comparison of the amino acid sequences of V.NgoAXIII and V.NgoAXIV to that of V.EcoKDcm enabled us to predict amino acids important for the specificity and the hydrolytic activity of V.NgoAXIII and V.NgoAXIV (Fig. 2). Alanine mutagenesis of Glu24, Asp50, His68, or Asp97 of V.NgoAXIII and Glu25, Asp51, His64, His69, or Asp97 of V.NgoAXIV was performed. A mutant of V.NgoAXIII that had Asp63 converted to His63 was also created. The specificities and activities of all mutants created were tested. Reactions were carried out with 0.15 μM substrate DNA containing T:G mismatches specific for Vsr sequences and 1.5 μM purified enzymes. Reaction products were analyzed by 10% polyacrylamide gel electrophoresis. Analysis of the activities of the mutants V.NgoAXIIID50A and V.NgoAXIIIH68A showed that these proteins were unable to introduce a nick in substrate DNA containing the T:G mismatch within the sequences derived from sequences modified by any m5C-MTase expressed by N. gonorrhoeae FA1090 (data not shown). The mutants V.NgoAXIVD51A (Fig. 7 B) and V.NgoAXIVH69A (data not shown) also were unable to incise mismatched thymine within sequences specific to V.NgoAXIV. The replacement of Glu25, His64, or Asp97 by Ala in the V.NgoAXIV endonuclease resulted in reduced activity toward all DNAs for which this Vsr endonuclease is specific. The reductions of the activities of these proteins were proportional to the efficiency of cleavage characteristic to each sequence in reactions catalyzed by wild-type V.NgoAXIV. V.NgoAXIVE25A had ∼22% activity in comparison to that of wild-type protein, the H64A mutant had ∼45%, and the D97A mutant had ∼44% (data not shown). The replacement of Glu24 or Asp97 by Ala and of Asp63 by His in V.NgoAXIII also resulted in reduced activity of these proteins (data not shown). Moreover, V.NgoAXIIIE24A and V.NgoAXIIID63H had not only reduced activity but also narrowed specificity, recognizing TCGCGG/CCGCGG, CTGCGG/CCGCGG, and GTCGGC/GCCGGC but with various reductions in efficiency. The analysis of the specificity of V.NgoAXIIIE24A is shown in Fig. 7A. V.NgoAXIIIE24A has 75% of the activity of the wild-type protein toward the TCGCGG/CCGCGG sequence, 18% toward CTGCGG/CCGCGG, and 20% toward GTCGGC/GCCGGC. A parallel analysis of the results obtained for V.NgoAXIIID63H revealed that this protein has 30% of the activity of the wild-type protein toward the TCGCGG/CCGCGG sequence, 17% toward CTGCGG/CCGCGG, and 23% toward GTCGGC/GCCGGC, suggesting that the presence of aspartate in position 63 provides the enzyme with not only broader specificity but also increased enzymatic function.
FIG. 7.

Analysis of the specificity of V.NgoAXIIIE24A and V.NgoAXIVD51A endonucleases. (A) Results generated using V.NgoAXIIIE24A. (B) Results generated using V.NgoAXIVD51A. Lane 1, substrate M.NgoAI-sub; lane 2, substrate DNA M.NgoAII-sub; lane 3, substrate DNA M.NgoAIII-first-sub; lane 4, substrate DNA M.NgoAIII-second-sub; lane 5, substrate DNA M.NgoAIII-third-sub; lane 6, substrate DNA M.NgoAIV-sub; lane 7, substrate DNA M.NgoAVII-sub; lane 8, substrate DNA M.NgoAORF675P-sub; lane 9, substrate DNA M.NgoAORF302P-sub; lane 10, substrate DNA M.NgoAORF1175P-sub; lane 11, substrate DNA M.HhaI-sub; lane 12, substrate DNA M.EcoKDcm-sub; lane M, GeneRuler 50-bp DNA ladder. Arrows indicate substrate (S) and reaction products (P1 and P2) obtained after cleavage by purified Vsr endonucleases.
DISCUSSION
Most bacterial strains possess a small number of m5C-MTases and a single Vsr endonuclease. As a species, the gonococcus contains numerous m5C-MTases, with most strains tested expressing at least 8 different m5C-MTases. Here, we show that the two Vsr endonucleases from N. gonorrhoeae FA1090 are functional and have different but overlapping activities. This makes N. gonorrhoeae the first bacterium which has been experimentally shown to have genes encoding two different active Vsr endonucleases. The V.NgoAXIII enzyme was capable of recognizing all possible T:G mismatches that might arise from spontaneous deamination of m5C that were produced by the known m5C-MTases of the gonococcus, whereas V.NgoAXIV was able to cleave only a subset of these recognition sequences. These enzymes appear to be specific for T:G mismatches, as they were incapable of digesting A:C mismatches. The fact that N. gonorrhoeae expresses two different Vsr endonucleases, with one of them being multispecific and recognizing T:G mismatches in every nucleotide context found in the gonococcus, raises questions about the origin and the biological implications of the retention of the genes encoding both enzymes. There are genomic sequences available for 15 different gonococcal strains in various forms in nucleotide databases, and both V.NgoAXIII and V.NgoAXIV are present in all strains (D.C.S., unpublished observations). This is in contrast to the other Neisseria spp., where V.NgoAXIII is rarely found. V.NgoAXIV is found in almost all neisserial isolates (D.C.S., unpublished observations).
It is very interesting that genes of active gonococcal Vsr endonucleases are associated with genes of inactive restriction-modification systems. One possible explanation is connected to bacterial evolution and acquisition of new genes. Probably genes of these restriction-modification systems were acquired together with Vsr endonuclease genes. Then, during evolution, genes of m5C-MTases M.NgoAORF302P and M.NgoAORF1175P were inactivated. The acquisition of new genes would occur naturally by DNA-mediated transformation of N. gonorrhoeae. This is the main mechanism by which the Neisseria species have acquired their genetic diversity (24). The acquisition of new genes by horizontal gene transfer is connected to the evolution of bacteria and is particularly related to their environmental adaptation and virulence.
We have previously shown that at least one of the restriction modification systems found in most gonococcal strains was most likely acquired from a Haemophilus sp. (11). Saunders and Snyder (22) suggested the existence of a minimal mobile element that utilizes natural transformation and homologous recombination, independent of transposases and other mobilization mechanisms. Such a minimal mobile element could be flanked by the pheS and pheT genes (22). In the gonococcal genome between the pheS and pheT genes is a region which includes ngoAXIIIV. Between the pheS and pheT genes in other Neisseria species are genes encoding other m5C-MTases, which may be solitary MTases or components of a restriction-modification system (17, 20, 22). The acquisition of the ngoAXIIIV gene by such a minimal mobile element is consistent with Kobayashi's hypothesis (14) that restriction-modification systems are selfish mobile genetic elements.
While gonorrhea is an ancient disease, meningococcal disease seems to have appeared in the last 200 years (26). This implies that the gonococcus has had a much longer period of time to acquire new genetic material. Analysis of gonococcal genomes finds a wide diversity in the number of functional restriction-modification systems compared to their numbers in other neisserial genomes. From an evolutionary standpoint, V.NgoAXIII would have had a significant time to evolve into an enzyme with broad specificity that could repair mismatches resulting from deamination of any of the gonococcal m5C-MTases. The ngoAXIVV gene is found close to the 10-nt Neisseria DNA uptake sequence (GCCGTCTGAA), whose presence would enhance its ability to be taken up by the gonococcus (6). The existence of a multispecific Vsr protein would allow the gonococcus to acquire new genes encoding MTases without increasing the mutagenic potential of this acquisition. On the other hand, the possession of two Vsr endonucleases by the gonococcus could be required because the m5C-MTases could methylate noncanonical DNA sites (4). If an MTase methylated DNA at noncanonical sites, then the number of potential hot spots would increase. The presence of V.NgoAXIII or V.NgoAXIV could prevent mutations resulting from the increased frequency of deamination of m5C to thymine due to the increased presence of m5C in the genome.
Site-directed mutagenesis of V.NgoAXIII and V.NgoAXIV enabled us to identify amino acids important for the specificity and hydrolytic activity of the Vsr endonucleases. Our results show that Asp51 in V.NgoAXIV and Asp50 in V.NgoAXIII are the critical active-site residues and that Glu25 and Asp97 in V.NgoAXIV and Glu24 and Asp97 in V.NgoAXIII are important for catalysis. His69 in V.NgoAXIV and His68 in V.NgoAXIII are also absolutely required for the activity of neisserial Vsr endonucleases. Furthermore, His64 in V.NgoAXIV and Asp63 in V.NgoAXIII are important for hydrolytic reactions catalyzed by these proteins. Based on comparison of our results to published data demonstrating the significance of Glu25, Asp51, His64, His69, and Asp97 for reactions catalyzed by V.EcoKDcm (28-30), we can predict the roles of analogous amino acids in catalytic reactions carried out by the V.NgoAXIII and V.NgoAXIV endonucleases. V.EcoKDcm uses two magnesium-water clusters (28). V.NgoAXIII and V.NgoAXIV require Mg2+ ions for catalysis, and by similarity to V.EcoKDcm and high conservation among Vsr endonucleases, we postulate that neisserial Vsr endonucleases also use metal-water clusters. Probably Asp51 in V.NgoAXIV and Asp50 in V.NgoAXIII also directly coordinate the two magnesium ions. His64 in V.NgoAXIV and Asp63 in V.NgoAXIII and Glu25 in V.NgoAXIV and Glu24 in V.NgoAXIII could be involved in H-bond interactions with the water molecules in the magnesium-water clusters. In the complex of DNA and V.EcoKDcm, the scissile phosphate is coordinated by the two magnesium ions and is involved in an H-bond interaction with the pair Asp97-His69. His69 abstracts a proton from one of the water molecules in the metal-water cluster, and the activated water molecule attacks the phosphate (29). It is possible that Asp97 and His69 in V.NgoAXIV and His68 in V.NgoAXIII have the analogous functions. The features of the mutant proteins V.NgoAXIIIE24A and V.NgoAXIIID63H suggest that Glu24 and Asp63 in V.NgoAXIII are important not only for its activity but also for its specificity. Asp63 could be one of the amino acids that is particularly significant for the multispecificity of V.NgoAXIII. This hypothesis is supported by fact that the multispecific V.BssHIII also has an Asp at aa 64, whereas the monospecific V.EcoKDcm and V.NgoAXIV that recognizes T:G mismatches only in particular nucleotide contexts have a His in the analogous position. Moreover, analysis of amino acid sequences of Vsr endonucleases indicates that a putative target recognition domain of Vsr endonucleases includes residues 70 to 84 (residue positions for V.NgoAXIII). Our results support the conclusion concerning the specificity of V.NgoAXIIID63H and imply that this domain includes amino acids at least from aa 63.
Acknowledgments
This work was conducted with the support of the Ministry of Science and Higher Education in Poland (grants no. NN301165135-MNiSzW and 2 P04A 053 26) and a grant from the National Institutes of Health to D.C.S. (AI 24452).
We thank Anna Mac-Skrzeczkowska for technical assistance with site-directed mutagenesis of Vsr endonucleases.
The authors declare no conflict of interest.
Footnotes
Published ahead of print on 28 May 2010.
REFERENCES
- 1.Ahmad, I., and D. N. Rao. 1996. Chemistry and biology of DNA methyltransferases. Crit. Rev. Biochem. Mol. Biol. 31:361-380. [DOI] [PubMed] [Google Scholar]
- 2.Bhagwat, A. S., and M. Lieb. 2002. Cooperation and competition in mismatch repair: very short patch repair and methyl-directed mismatch repair in Escherichia coli. Mol. Microbiol. 44:1421-1426. [DOI] [PubMed] [Google Scholar]
- 3.Choi, S. H., and J. E. Leach. 1994. Identification of the XorII methyltransferase gene and a vsr homolog from Xanthomonas oryzae pv. oryzae. Mol. Gen. Genet. 244:383-390. [DOI] [PubMed] [Google Scholar]
- 4.Cohen, H. M., D. S. Tawfik, and A. D. Griffiths. 2002. Promiscuous methylation of non-canonical DNA sites by HaeIII methyltransferase. Nucleic Acids Res. 30:3850-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dar, M. E., and A. S. Bhagwat. 1993. Mechanism of expression of DNA repair gene vsr, an Escherichia coli gene that overlaps the DNA cytosine methylase gene, dcm. Mol. Microbiol. 9:823-833. [DOI] [PubMed] [Google Scholar]
- 6.Elkins, C., C. E. Thomas, H. S. Seifert, and P. F. Sparling. 1991. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J. Bacteriol. 173:3911-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott, S. L., J. Brazier, R. Cosstick, and B. A. Connolly. 2005. Mechanism of the Escherichia coli DNA T:G-mismatch endonuclease (Vsr protein) probed with thiophosphate-containing oligodeoxynucleotides. J. Mol. Biol. 353:692-703. [DOI] [PubMed] [Google Scholar]
- 8.Fox, K. R., S. L. Allinson, H. Sahagun-Krause, and T. Brown. 2000. Recognition of GT mismatches by Vsr mismatch endonuclease. Nucleic Acids Res. 28:2535-2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gläsner, W., R. Merkl, V. Schellenberger, and H.-J. Fritz. 1995. Substrate preferences of Vsr DNA mismatch endonuclease and their consequences for the evolution of the Escherichia coli K-12 genome. J. Mol. Biol. 245:1-7. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Nicieza, R., D. P. Turner, and B. A. Connolly. 2001. DNA binding and cleavage selectivity of the Escherichia coli DNA G:T-mismatch endonuclease (Vsr protein). J. Mol. Biol. 310:501-508. [DOI] [PubMed] [Google Scholar]
- 11.Gunn, J. S., and D. C. Stein. 1993. Natural variation of the NgoII restriction-modification system of Neisseria gonorrhoeae. Gene 132:15-20. [DOI] [PubMed] [Google Scholar]
- 12.Hennecke, F., H. Kolmar, K. Brundl, and H.-J. Fritz. 1991. The vsr gene product of E. coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature 353:776-778. [DOI] [PubMed] [Google Scholar]
- 13.Johnson, K. A. 1992. Transient-state kinetic analysis of enzyme reaction pathways. Academic Press, Inc., New York, NY.
- 14.Kobayashi, I. 2001. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29:3742-3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kow, Y. W. 2002. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 33:886-893. [DOI] [PubMed] [Google Scholar]
- 16.Kwiatek, A., M. Kobes, K. Olejnik, and A. Piekarowicz. 2004. DNA methyltransferases from Neisseria meningitidis and Neisseria gonorrhoeae FA1090 associated with mismatch nicking endonucleases. Microbiology 150:1713-1722. [DOI] [PubMed] [Google Scholar]
- 17.Kwiatek, A., and A. Piekarowicz. 2007. The restriction endonuclease R.NmeDI from Neisseria meningitidis that recognizes a palindromic sequence and cuts the DNA on both sides of the recognition sequence. Nucleic Acids Res. 35:6539-6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lieb, M., and A. S. Bhagwat. 1996. Very short patch repair: reducing the cost of cytosine methylation. Mol. Microbiol. 20:467-473. [DOI] [PubMed] [Google Scholar]
- 19.Lieb, M., and S. Rehmat. 1995. Very short patch repair of T:G mismatches in vivo: importance of context and accessory proteins. J. Bacteriol. 177:660-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts, R. J., T. Vincze, J. Posfai, and D. Macelis. 2007. REBASE—enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 35:D269-D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual (3rd ed.). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 22.Saunders, N. J., and L. A. Snyder. 2002. The minimal mobile element. Microbiology 148:3756-3760. [DOI] [PubMed] [Google Scholar]
- 23.Sohail, A., M. Lieb, M. Dar, and A. S. Bhagwat. 1990. A gene required for very short patch repair in Escherichia coli is adjacent to the DNA cytosine methylase gene. J. Bacteriol. 172:4214-4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stein, D. C. 1991. Transformation of Neisseria gonorrhoeae: physical requirements of the transforming DNA. Can. J. Microbiol. 37:345-349. [DOI] [PubMed] [Google Scholar]
- 25.Stein, D. C., J. S. Gunn, M. Radlinska, and A. Piekarowicz. 1995. Restriction and modification systems of Neisseria gonorrhoeae. Gene 157:19-22. [DOI] [PubMed] [Google Scholar]
- 26.Stephens, D. S. 2009. Biology and pathogenesis of the evolutionarily successful, obligate human bacterium Neisseria meningitidis. Vaccine 27:B71-B77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. [DOI] [PubMed] [Google Scholar]
- 28.Tsutakawa, S. E., H. Jingami, and K. Morikawa. 1999. Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell 99:615-623. [DOI] [PubMed] [Google Scholar]
- 29.Tsutakawa, S. E., and K. Morikawa. 2001. The structural basis of damaged DNA recognition and endonucleolytic cleavage for very short patch repair endonuclease. Nucleic Acids Res. 29:3775-3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsutakawa, S. E., T. Muto, T. Kawate, H. Jingami, N. Kunishima, M. Ariyoshi, D. Kohda, M. Nakagawa, and K. Morikawa. 1999. Crystallographic and functional studies of very short patch repair endonuclease. Mol. Cell 3:621-628. [DOI] [PubMed] [Google Scholar]