


Abstract
Endonucleases in DNA repair must be able to recognize damaged DNA as well as cleave the phosphodiester backbone. These functional prerequisites are manifested in very short patch repair (Vsr) endonuclease through a common endonuclease topology that has been tailored for recognition of TG mismatches. Structural and biochemical comparison with type II restriction enzymes illustrates how Vsr resembles these endonucleases in overall topology but also how Vsr diverges in terms of the detailed catalytic mechanism. A histidine and two metal–water clusters catalyze the phosphodiester cleavage. The mode of DNA damage recognition is also unique to Vsr. All other structurally characterized DNA damage-binding enzymes employ a nucleotide flipping mechanism for substrate recognition and for catalysis. Vsr, on the other hand, recognizes the TG mismatch as a wobble base pair and penetrates the DNA with three aromatic residues on one side of the mismatch. Thus, Vsr endonuclease provides important counterpoints in our understanding of endonucleolytic mechanisms and of damaged DNA recognition.
INTRODUCTION
The genetic integrity of DNA is constantly being challenged by an array of DNA damaging agents, which can be either endogeneous or exogeneous in origin. Repair systems are necessary to counteract potentially mutagenic or cytotoxic consequences from the DNA damage. Base damage is repaired either directly, for example through simple dealkylation, or via complex and coordinated pathways involving multiple proteins. These latter systems include mismatch repair (MMR), base excision repair (BER) and nucleotide excision repair. Identification of the damage, removal of the damaged nucleotide and neighboring nucleotides and resynthesis of the strand are common to each of these pathways.
This review focuses on an endonuclease in one of the MMR pathways, the very short patch repair (VSR) system. The crystal structure of the Escherichia coli Vsr endonuclease alone and in a ternary complex with duplex DNA and magnesium has added to our knowledge of mechanisms for endonucleolytic cleavage, how MutL may differentially stimulate Vsr and NutH endonucleases, and how DNA repair proteins detect damaged DNA and, more specifically, mismatched DNA. Comparison of Vsr with type II restriction enzymes and two BER endonucleases highlight a distinguishing functional requirement for DNA damage proteins, that of pathway coordination.
VSR ENDONUCLEASE
Although Vsr shares no sequence homology with other endonucleases, it has endonuclease activity and cleaves on the 5′ side of a thymine opposite guanine, within the sequence 5′C↓T(A/T)GG3′/5′CC(T/A)GG3′ (1). Cleavage by Vsr endonuclease is a major component of the VSR pathway, which is responsible for the repair of damaged methylated cytosines (2). Methylated cytosines are vulnerable to spontaneous deamination and hydrolysis, which result in the formation of a mismatched thymine (Fig. 1). The general MMR process in bacteria, the MutHLS system, uses the inherent delay in methylation of newly synthesized strands and initiates repair by cleavage on the strand opposite a methylated adenine to preferentially repair daughter strands. Thus, in the case of damage to methylated cytosines, the MutHLS system may repair the newer non-methylated strand, resulting in a GC to AT transition and the loss of a methylation site. Incision on the thymine strand by Vsr thus is important for selecting the strand for repair and initiates repair by DNA PolI and a DNA ligase. The exonuclease of DNA PolI excises only a short stretch, approximately five residues (3), before resynthesis and ligation of the corrected strand. MutL and MutS are also required for the VSR pathway in vivo, and MutL has been shown in vitro to enhance Vsr binding to DNA substrate (4–7).
Figure 1.

Schematic of Vsr endonuclease’s role in the repair of TG mismatches. TG mismatches can result from spontaneous deamination and hydrolysis of methylated cytosines in dsDNA. Vsr recognizes the mismatch within this recognition sequence and cleaves on the 5′ side of the mismatched thymidine, indicated by the arrow.
Vsr has a three-layered α/β/α fold, which is stabilized by a structural zinc site (Fig. 2) (8). The coordination of the zinc, with three of the protein residues on one loop, is distinct from those of the majority of structural zinc sites, where the four coordinating residues are divided equally on two strands. However, it is similar to one of the sites in I-PpoI homing endonuclease, which is reviewed in this issue by Chevalier and Stoddard.
Figure 2.
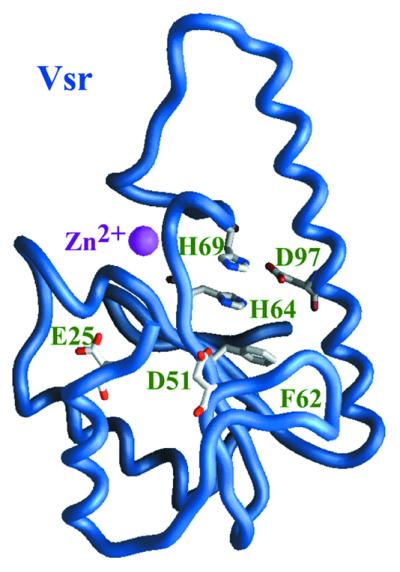
The structure of truncated Vsr endonuclease. Crystal studies of a truncated Vsr endonuclease, with the first 20 residues removed by limited proteolysis, revealed a central β-sheet with α-helices on either side. The main chain of Vsr [1vsr.pdb (8)] is depicted here as a worm representation. The core β-sheet is formed by two short and three long β-strands. Scanning alanine mutagenesis of conserved acidic residues has identified Asp51 as a critical active site residue and Glu25 and Asp97 as important for catalysis. Subsequent site-directed mutagenesis studies have found that His69 is also absolutely required for activity, while His64 is important for activity. These residues are depicted with carbon, nitrogen and oxygen atoms colored in white, blue and red, respectively. The zinc atom, displayed as a sphere, plays purely a structural role. The structure was determined to 1.8 Å. All structural figures have been drawn with the program GRASP (42).
SIMILARITY OF VSR TO TYPE II RESTRICTION ENZYMES
The overall topology of Vsr endonuclease resembles type II restriction enzymes, which is comprehensively reviewed in this issue by Pingoud and Jeltsch (Fig. 3). Members of this family include a growing list of restriction enzymes as well as some of the DNA repair nucleases: MutH (9), λ exonuclease (10) and archeal Holliday junction resolvase (11). The central β-sheet, braced on either side by α-helices, is composed of two short and three to five long β-strands. The active site of this family falls into a niche formed in the β-sheet and is built on the catalytic motif PDX(6–30)D/EXK (12,13). These conserved residues are superimposable from enzyme to enzyme and are absolutely essential for activity, with the first two conserved residues binding an essential divalent cation and the last residue, which is the most variant between restriction enzymes, proposed either to stabilize the transition state (14,15) or to orient the attacking water (16,17). In the case of Vsr, superimposition of the β-sheet onto members of the type II restriction enzyme family shows that Asp51, a catalytically essential residue, superimposes directly onto the first conserved aspartate of the catalytic motif, further evidence of the similarity of Vsr to this family. However, the remaining part of the catalytic motif is not conserved in Vsr.
Figure 3.

Vsr is similar in topology to type II restriction endonucleases. Stereodiagram of truncated Vsr [green ribbon, 1vsr.pdb (8)] superimposed onto BamHI [magenta ribbon, 1bhm.pdb (43)] reveals the strong similarity of Vsr to type II restriction enzymes. Pairwise superimposition of only the Cα atoms of residues corresponding the type II restriction enzyme catalytic motif (residues 51, 62 and 64 in Vsr and, respectively, residues 94, 111 and 113 in BamHI) overlays the central β-sheets. Only one subunit of BamHI is displayed for clarity, and the main chain from residues 21–24 were removed from the truncated Vsr structure for clarity. Residues discussed in the text are depicted, and only Vsr residues are labeled.
The second conserved catalytic residue superimposes onto Phe62, which, while absolutely conserved among Vsr family members, cannot be involved in metal coordination. The third conserved residue superimposes onto a histidine. His64 is partially conserved as histidine or aspartate and is important but not critical for activity, the latter disqualifying it from strict conservation with the motif. To add to the conundrum, His69, which is close in vicinity to His64 but is positionally distinct from catalytic residues of the type II restriction enzymes, was found to be absolutely required for activity.
CATALYTIC MECHANISMS OF VSR
The crystal structure of Vsr in a ternary complex with magnesium and a cleaved DNA molecule (18), clarified the discrepancy with the catalytic motif (Fig. 4). Instead of the highly coordinated catalytic metal ion(s) that are typically found in the type II restriction enzyme family, Vsr uses two magnesium–water clusters. These metal–water clusters, characterized by little direct coordination with the protein, were first proposed for Serratia endonuclease (19) and are also found in the DNA complexes of I-PpoI and I-CreI homing endonucleases (20,21), BglII (17) and the Tn5 transposase (22). In Vsr, the essential residue Asp51 directly coordinates the two magnesium ions. The mainchain carbonyl of Thr63, which is one residue down from the predicted Phe62, coordinates to one of the metal ions and is the only other direct protein–metal ion interaction. His64 and Glu25 coordinate water molecules in the water–metal clusters, explaining the partial losses in activity after single alanine substitutions (8).
Figure 4.

The active site of Vsr endonuclease contains two magnesium–water clusters and a cleaved DNA intermediate. Stereoview of the active site from the Vsr–DNA complex [1cw0.pdb (18)] shows the interactions between the cleaved DNA termini and the surrounding protein residues. The scissile phosphate is coordinated by the two magnesium ions (magenta spheres) and is involved in a H-bond interaction with the Asp–His pair, Asp97–His69. The cleaved bond is represented by an orange line. The dark dashed lines represent interactions postulated to be absolutely essential for catalysis. The phosphate oxygen H-bonded to His69 is almost directly in line with the deoxyribose oxygen and is likely to have been the attacking water. His64 and Glu25, which are important but not absolutely required for catalysis, are involved in H-bond interactions with the waters (blue spheres) in the magnesium–water clusters. Atoms are displayed as in Figure 2, and phosphate is colored in yellow. The Vsr–DNA complex structure was determined to 2.3 Å resolution.
In the crystal structure of the complex, the DNA duplex was also found in direct coordination with the magnesium ions. As determined in biochemical studies, the DNA was cleaved leaving 5′ phosphate and 3′ hydroxyl-termini. The phosphate at the cleavage termini is coordinated with both metal ions, and His69, a catalytically essential residue, coordinates one phosphate oxygen, providing the foundation for a potential catalytic mechanism. In the model, the scissile phosphate is coordinated and stabilized by both metal–water clusters. His69 abstracts a proton from one of the waters in the magnesium–water clusters, and the activated water attacks the phosphate. As described in the classical two-metal based mechanism (23), the pentacoordinate intermediate is stabilized between the two magnesium ions, leading to cleavage of the phosphodiester backbone. In the crystal structure, the geometry of the deoxyribose oxygen relative to the proposed attacking water, the phosphate oxygen coordinated by His69 and by one of the magnesium–water clusters, is in agreement with an in-line attack. As the cleaved deoxyribose oxygen was within 2.3 Å of one of the magnesiums, the final step of protonation could not yet have occurred. The lack of significant side chain conservation in the vicinity of the deoxyribose oxygen suggests that the protein does not donate the proton but instead, one of the waters in the magnesium–water cluster could easily play that role. As a caveat in the Pingoud and Jeltsch review, this mechanism is based on only the structures of the apoenzyme and of the protein in complex with a cleaved DNA and requires confirmation by further biochemical and structural investigation.
The sugar conformations on either side of the cleavage site for Vsr were C3′-endo, and these conformations, characteristic of A-type DNA, were also found in the DNA complexes of I-PpoI homing and BglII endonucleases, where metal–water clusters were also identified. It is possible that for these enzymes, unwinding to an A-type DNA, and thereby repositioning the scissile phosphate, is critical for catalysis.
RECOGNITION OF THE DNA
One of the more striking features from the Vsr–DNA complex is the mode of DNA recognition (Figs 5 and 6). Although the DNA duplexes in most type II restriction enzymes follow along the same path, the DNA bound by Vsr diverges almost perpendicularly on the 5′ side of the cleavage (Fig. 7). This is due to a remarkable intercalation of three aromatic residues into the major groove of the DNA on the side of the TG mismatch opposite to the cleavage site. Trp68 penetrates the deepest and stacks with the thymine base (Figs 5 and 6). Phe67 stacks midway between the AT base pair, and Trp86 is embedded between the sugars. Met14 and Ile17 come in from the minor groove and completely close off any interaction between the two base pairs and leave only enough room for the phosphate backbone to pass on either side. Although intercalation has been observed in other DNA-binding proteins, these proteins bend the DNA by inserting a small wedge into the minor groove and compressing the major groove (24). Vsr, on the other hand, penetrates from both sides of the DNA and predominantly from the major groove side. Base stacking is completely disrupted by a 6 Å increase in rise, the base pair to base pair distance, with both the major and minor grooves extensively widened. The finding that only a small percentage of Vsr endonuclease is active at one time (25) may be due to the kinetic difficulty in intercalating residues into the DNA.
Figure 5.

Intercalation of Vsr into the DNA in this stereoview of the Vsr–DNA complex. Full-length Vsr [1cw0.pdb (18)], the main chain depicted here as a worm model, clamps the DNA onto the main core of the protein with an N-terminal helical arm. Three aromatic residues, Phe67, Trp68 and Trp86 (green) intercalate into the DNA duplex on one side of the TG mismatch. The catalytically essential residues, Asp51 and His69, (magenta) are proximal to the cleaved DNA termini. The major groove has been flattened out, while the minor groove has been expanded to the point where the N-terminal helix can fit deep into the minor groove. Blue and green spheres show the location of the zinc and magnesium atoms, respectively. Atoms of the DNA are colored as in Figure 4.
Figure 6.

Recognition of the mismatch is through H-bond interactions, intimate surface complementarity with the shifted wobble conformation and intercalation with aromatic residues. Stereoview of the TG mismatch and neighboring region from the Vsr–DNA complex [1cw0.pdb (18)] reveals the multiple mechanisms for specific recognition of a TG mismatch. The mismatch forms a wobble base pair with the thymine base moved into the major groove; the cytosine in a GC Watson–Crick base pair would be significantly higher in the figure. The neighboring GC base pair shown in the figure can be used for comparison. The guanine base is recognized from both sides by Lys89 and the carbonyl of Met14, while Asn93 recognizes the lowered thymine base. The main chain of N-terminal residues Ile17 to Thr19 push down onto the mismatched thymidine, thus further sterically encoding an interface allowing only the TG wobble base pair. Trp68 stacks directly onto the mismatched thymine base, while Trp86 wedges between the sugars on the other side of the DNA duplex. The side chains of Ile17 and Met14 meet those of Trp68 and Trp86 to completely prevent base pair stacking on one side of the TG mismatch. Atoms are colored as in Figure 4, with sulfur colored in green.
Figure 7.

The path of the DNA on the 5′ of the scissile phosphate is different for Vsr and for type II restriction enzymes. The DNA taken from Vsr/DNA [green, 1vsr.pdb (8)], BamHI/DNA (magenta, 1bhm) and EcoRV/DNA [blue, 2rve.pdb (44)] complexes are depicted after pairwise superimposition of type II restriction enzyme catalytic motif ‘equivalent’ residues as described in Figure 3. The perspective is that from the top of Figure 5 and shows that the Vsr-bound DNA diverges almost 90° from the DNA bound by restriction enzymes. The proteins were removed for clarity, but the normal path of DNA bound by restriction enzymes superimposes directly into the upper region of Vsr protein, shown in Figure 5.
BINDING OF THE TG MISMATCH
Unlike most DNA repair enzymes, which use nucleotide flipping (reviewed in 26–29), Vsr endonuclease recognizes the damaged TG mismatch in the context of a base pair (Figs 5 and 6). TG forms a classical wobble base pair, with the thymine off-set toward the major groove. The only direct H-bond interaction, which would differentiate thymine from the original cytosine, is from Asn93 to O4 of the thymine. The 60% decrease in single turnover catalytic rate with uracil as the substrate demonstrated that the 5-methyl group is important for activity (30), probably due to the sensitivity of catalysis to the exact positioning of the scissile phosphate relative to His69. Recognition of the guanine base is through Met14 and Lys89 to N2 and O6, respectively. Inosine, which is missing the N2 substituent, shows an order of magnitude decrease in activity (30).
However, simple recognition of either base is not sufficient to explain substrate specificity. Vsr does not cleave substrates containing Watson–Crick base pairs, CG or TA (1), but actually retains some activity against substrates where the TG positions are singly or doubly replaced by 1′,2′-dideoxyribose to mimic abasic site(s) (30). The complementarity of the active site surface to the TG base pair would select for the significantly shifted wobble base pairs. An abasic site, which would not have the steric constraints placed on it by Watson–Crick base pairing, would be able to flexibly place its phosphodiester backbone in a catalytically active position. Consistent with this idea, DNA containing the abasic sites, are slightly better substrates than TG analogs which form Watson–Crick base pairs (30).
Another factor to select for the TG wobble base pair may be the ability to intercalate into the DNA. The shift of the wobble base pair into the major groove would disrupt base pair to base pair stacking on only one side of the TG mismatch. In crystallographic studies of B-type DNA, there was a small but significant increase in the base pair to base pair distance on one side of the TG mismatch (31). This side of the TG mismatch corresponds to the side where Vsr intercalates the three aromatic residues. In support of this theory, the crystal structure of MutS bound to a TG mismatch revealed that MutS stacks a phenylalanine on this same side of the thymine base, despite the fact that there is no other similarity in the recognition of the TG mismatch between Vsr and MutS (32). The findings that MutS binds from the mismatch only from the minor groove of the DNA and Vsr primarily from the major groove and given that MutL and MutS are required for the VSR pathway in vivo, suggest that MutL and MutS either present to Vsr the major groove side of the TG mismatch as a loop (33) or somehow directly pass the TG mismatch to Vsr. The former model of MutL presenting the TG mismatch in a DNA-binding competent conformation to Vsr would agree with the results that MutL can catalytically enhance Vsr binding to DNA and that there is no stable complex of Vsr–MutL–DNA detected (7).
As a further note, the importance of this intercalation for catalytic activity is partially encoded by the primary sequence: two of the intercalating residues, Phe67 and Trp68, and one of the essential catalytic residues, His69, are successive in the sequence and represents one of the most invariant regions in the Vsr family. These residues are also probably conformationally constrained by being on the same loop that contains three of the four residues that coordinate the zinc: Cys66, His71 and Cys73. The zinc loop may also play a similar role of positioning catalytic residues in I-PpoI homing endonuclease. The effect of the uracil substitution of the mismatched thymine to Vsr activity (30) indicates the sensitivity of this region to subtle changes in structure. It is also notable that the indole ring of Trp68 has rotated 110° upon intercalating into the DNA and is likely to be involved in the first steps of substrate recognition.
That specificity in the exact cleavage site is retained in substrates containing abasic sites (30) illustrates the importance of the four other base pairs in the recognition site, 5′C*T(A/T)GG3′/5′CC(T/A)GG3′. In the crystal structure, the DNA is approached from all directions through a network of direct and water-mediated protein interactions. There are as many H-bond interactions to the phosphate backbone as to the bases, and many of the residues are interacting with more than one DNA base. Many of the interactions are made by peptide backbone atoms, further contributing to formation of a more rigid DNA-binding surface.
In terms of specific recognition of the substrate sequence outside of the TG mismatch, there is a surprising singularity to the direct protein–base interactions in the Vsr/DNA structure. In biochemical studies, Fritz and coworkers (1,34) found that the fourth base pair, GC, is absolutely required for substrate recognition, with partial requirements for the remaining sequence. Of the two direct side chain interactions, only Asn13 would select for a guanosine at that position since the other direct interaction, Arg10 to the O2 position of cytidine, would not be able to differentiate it from the O2 of thymine, The first CG base pair of the recognition sequence is also important for Vsr activity (34) and is similarly selected by a single residue to one of the bases, Lys89 to the O6 position of the guanosine. It should be noted that Lys89 is also involved in specific recognition of the guanosine in the TG base pair. Biochemical analysis of mutants in these residues will be necessary to determine whether these direct but sparse interactions are required for recognition or whether the water-mediated interactions also contribute significantly to specific recognition. Recently, it has been shown that there is a significant increase in Vsr activity with hemimethylated substrate (25). A methyl group on the cytidine of the fourth base pair may lead to better packing against the aliphatic region of the Lys77 side chain and thus enhance the protein/DNA interface. Sequence comparison with other Vsr-like endonucleases shows that only the residues interacting with the TG base pair and residues coming from the minor groove are invariantly conserved, consistent with the fact that the sequence specificity of the methylases and presumably the Vsr-like endonucleases are dissimilar from one species to another.
MutH, another DNA repair endonuclease
Structural comparison of Vsr and MutH endonucleases, which use, respectively, cytosine and adenine methylation sites to recognize their DNA substrates, underscores small but interesting differences in their regulation and function. In the major MMR system, MutH is responsible for generating a nick in hemimethylated DNA in cis with the mismatch and is stimulated by MutL and MutS to cleave the strand opposite to the adenine methylation site (35–38). Thus, MutH and Vsr share similar properties: both are methylation-directed endonucleases and both are stimulated by MutL in vitro.
Like Vsr, the structure of MutH is similar in topological fold to type II restriction enzymes, with the central β-sheet core and active site residues which localize in the notch formed by short and long β-strands (9,39). These active site residues (QD70 ... E77LK) resemble the catalytic motif of type II restriction enzymes. However, the active site of MutH is disordered and the catalytic residues (Asp70, Glu77, Lys79) from this motif do not superimpose well onto the type II restriction enzyme catalytic motif, represented by Vsr in Figure 8. In particular, Asp70 is close to a disordered loop and the average B value for Asp70 is remarkably ∼20 Å2 higher than that for its neighboring residue, Phe71. Furthermore, most members of the type II restriction endonucleases, including Vsr, place a proline N-terminal to the first Asp of the catalytic motif, and its absence in MutH may further destabilize this region. This inherent disorder in the active site is a likely explanation for the relatively low activity of MutH compared with other endonucleases and lends itself to a simple model where MutL, which has been shown to physically interact with MutH (38), stabilizes the active site and thereby stimulates catalytic activity. There is an additional level of flexibility encoded in subdomain motion, postulated from three independently determined crystal structures of MutH (9,39). A cleft divides key catalytic residues with the three type II restriction-like catalytic residues on the N-terminal subdomain and another catalytically required residue, Lys116, on the C-terminal subdomain (39). This cleft varies in size from one crystal form to another, leading to a model where MutL may control MutH by shifting the cleft from catalytically important open and closed conformations. The role of the fourth residue, Lys116, in catalysis is not yet understood. Because of specific details in the active site conformation, Vsr is different from MutH. First, the Vsr active site already appears structurally optimized for catalytic activity as there is little difference between the position of active site residues in the apo-enzyme and the DNA product-bound form. Secondly, there has been no evidence for a stable complex between MutL and Vsr (7) and there is no region of conserved residues outside of the active site that would be suggestive of a MutL-binding site. A prediction would thus be that MutL will primarily enhance catalytic activity of MutH but will increase substrate binding and not kcat in the VSR system. In addition, there is potentially a third difference between MutH and Vsr. As described above, the path of the bound DNA 5′ to the cleavage in Vsr is distinctly different from type II restriction enzymes (Fig. 7). The superimposition of MutH and Vsr suggests that the path for MutH-bound DNA would be similar to that of type II restriction enzymes and thus very different from that of Vsr.
Figure 8.

Active site residues in MutH are more spread apart than those in Vsr. Pairwise superimposition of only the Cα atoms of type II restriction enzyme catalytic motif-equivalent residues [residues 51, 62 and 64 in the truncated Vsr [green ribbon, 1vsr.pdb (8)] and, respectively, residues 70, 77 and 79 in MutH [blue ribbon, 1azo.pdb (9)] highlights the non-canonical location of these type II restriction enzyme catalytic motif residues in MutH. Important catalytic residues for both proteins, with the exception of Lys116 in MutH, are depicted and labeled. The right portion of MutH, which is not topologically conserved with type II restriction enzymes and residues 22–24 in the truncated Vsr structure were removed for clarity. The perspective is similar to that of Figure 3.
Other DNA damage-recognizing endonucleases
Only two other protein–DNA complex structures of endonucleases that, like Vsr, bind to damaged DNA and cleave the phosphodiester backbone, have been determined (40,41). Ape1 and EndoIV are endonucleases that cleave the phosphodiester backbone on the 5′ side of an apurinic/apyrimidinic site (AP) site. Given the similarity of the three endonucleases’ function, it has been surprising to find that they are completely distinct from each other in terms of overall topology, DNA damage recognition and catalytic mechanism. Vsr is the only one that is similar to type II restriction enzymes in topology and catalytic mechanism, as described above.
Unlike Vsr, which recognizes the TG mismatch as a base pair within the context of dsDNA, Ape1 stabilizes the flipped out base and kinks the DNA 35° using a pre-formed DNA-binding face on one end of a double β-sheet (40). Met270 packs against the orphan base, while Arg177 inserts through the major groove and interacts with the phosphate 3′ to the AP site (Fig. 9). The attacking water is activated by Asp210, and the pentacoordinate phosphate intermediate is stabilized through a single magnesium atom. Like Vsr, Ape1 has an Asp–His pair (Asp287–His309) in the active site. In the substrate complex, His309 makes a H-bond to one of the phosphate oxygen atoms, and the authors proposed that His309 uses this H-bonding to orient and polarize the scissile P–O3′ bond (40). However, it is also possible that His309 plays a similar role to His69 in Vsr and has a more direct role in activation of the water in conjunction with Asp210. Further experimental work will be required to clarify the role of the Asp–His pair in Ape1 catalysis.
Figure 9.

The DNA-binding mode of Vsr is distinct from the other DNA repair endonucleases, Ape1 [1de8.pdb (40)] and EndoIV [1qum.pdb (41)]. Vsr has intercalated three aromatic residues into the dsDNA with all base pairing maintained, while Ape1 and EndoIV insert residues into the former positions of the flipped out AP sites and/or nucleotide. All three proteins clamp down on the bound DNA. The proteins are represented as surface representations, although the N-terminus of Vsr and Arg177 of Ape1 are depicted, respectively, as worm and liquorice models for clarity. Residues that insert into the DNA are labeled. The abasic sites in the DNA of Ape1 and EndoIV are demarked with an arrow. The magnesium atoms in Vsr and one of the three zinc atoms in EndoIV are depicted as green spheres and are visible close to the respective scissile phosphates. Atoms of the DNA are colored as in Figure 4.
The EndoIV active site, on the other hand, is at one end of a TIM barrel-like structure with three zinc atoms (41). Tyr72 and Arg37 insert through the DNA minor groove, displacing, respectively, the AP site and the base opposite (Fig. 9). The AP site is flipped out into a pocket deep in the DNA-binding cleft, and the DNA is bent ∼90°. In a three metal-based mechanism, the proposed attacking water is coordinated by two of the zinc atoms, Zn1 and Zn2, while the pentacoordinate intermediate is stabilized by Zn2 and Zn3.
Although Vsr, Ape1 and EndoIV each recognize damaged DNA and cleave the phosphodiester backbone on the 5′ side of the damage, their approach is very different. (i) Vsr has a single β-sheet core, Ape1 two β-sheets and EndoIV a β-barrel. (ii) Vsr recognizes the TG mismatch in the context of a wobble base pair, Ape1 flips out the AP site and EndoIV flips out both the AP site and the opposing base. (iii) Vsr intercalates three aromatic residues into the duplex DNA through the major groove, clamping the DNA from the other side with two hydrophobic residues; Ape1 inserts a methionine residue through the minor groove, clamping down with an arginine; and EndoIV inserts only from the minor groove side with an arginine and a tyrosine. (iv) Although the three endonucleases use metal ions(s) to stabilize the pentacoordinate intermediate, Vsr uses two magnesium–water clusters in catalysis, Ape1 a single magnesium ion and EndoIV three zinc ions. Thus, the structures and mechanisms of DNA repair endonucleases are clearly not defined by the presence of DNA damage, by even the same exact type of DNA damage as evidenced in the structures of the two 5′ AP endonucleases or by the fact that they are all have the same endonucleolytic activity which cleaves dsDNA on the 5′ side of the damage.
So what, if anything, do these structures have in common? All three of these endonucleases have a dramatic distortion of the DNA and penetration of residues deep into the helical core of the DNA duplex (Fig. 9). Although type II restriction endonucleases also bend the DNA by as much as 50°, base pairing and base stacking are maintained. This difference may pertain to the opposing function of restriction enzymes and DNA repair endonucleases. Restriction enzymes are used as a defense by generating deleterious double-strand breaks into invading phage DNA. Thus, their goal is to damage the DNA and leave. DNA repair enzymes, however, repair native DNA, and their actions must be beneficial to the cell. Thus, it is critical for them to coordinate with other enzymes along the pathway and to never leave the nicked DNA without one protein or another present at all times (27–29). This coordination may be mediated in part by slowing product release through insertion of side chains deep into the DNA duplex. In the case of Ape1, alanine substitution of the lysine clamp actually increased specific activity by 25% relative to wild-type enzyme (40). For Vsr, there is no direct experimental evidence for rate-limited product release. However, the first order rate constant for Vsr was three orders of magnitude lower than restriction enzymes (30); a Vsr/product–DNA complex was able to be observed in the gel mobility shift analysis (25; and unpublished data); and Vsr–DNA crystals that took 3 weeks to grow at 20°C contained a cleaved 3mer oligonucleotide product (18). Thus, DNA repair endonucleases may be distinguished from other endonucleases by this functional requirement to prevent premature release of product, where insertion of side chains into the DNA plays a role not only in DNA damage recognition but also slows product release.
An old dog with a new trick
It seems as if every new structure solved, especially in DNA repair, has demonstrated the diverse repertoire of structures and mechanisms available to the cell. Vsr is a good example as it uses the type II restriction structural topology but with a modified active site and DNA-binding mode to reflect its role in DNA damage repair: (i) recognition of a TG mismatch, (ii) cleavage of the phosphodiester backbone on one side of the damaged thymine base and (iii) coordination with other enzymes in the VSR pathway.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by the Biomolecular Engineering Research Institute (Osaka, Japan) and at the Lawrence Berkeley National Laboratory by the US Department of Energy under contract no. DE-AC03-76F00098.
References
- 1.Hennecke F., Kolmar,H., Brundl,K. and Fritz,H.J. (1991) The vsr gene product of E.coli K-12 is a strand- and sequence-specific DNA mismatch endonuclease. Nature, 353, 776–778. [DOI] [PubMed] [Google Scholar]
- 2.Lieb M. (1985) Recombination in the lambda repressor gene: evidence that very short patch (VSP) mismatch correction restores a specific sequence. Mol. Gen. Genet., 199, 465–470. [DOI] [PubMed] [Google Scholar]
- 3.Dzidic S. and Radman,M. (1989) Genetic requirements for hyper-recombination by very short patch mismatch repair: involvement of Escherichia coli DNA polymerase I. Mol. Gen. Genet., 217, 254–256. [DOI] [PubMed] [Google Scholar]
- 4.Lieb M. (1987) Bacterial genes mutL, mutS and dcm participate in repair of mismatches at 5-methylcytosine sites. J. Bacteriol ., 169, 5241–5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zell R. and Fritz,H.J. (1987) DNA mismatch-repair in Escherichia coli counteracting the hydrolytic deamination of 5-methyl-cytosine residues. EMBO J., 6, 1809–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieb M. and Rehmat,S. (1995) Very short patch repair of T:G mismatches in vivo: importance of context and accessory proteins. J. Bacteriol ., 177, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drotschmann K., Aronshtam,A., Fritz,H.J. and Marinus,M.G. (1998) The Escherichia coli MutL protein stimulates binding of Vsr and MutS to heteroduplex DNA. Nucleic Acids Res., 26, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsutakawa S.E., Muto,T., Kawate,T., Jingami,H., Kunishima,N., Ariyoshi,M., Kohda,D., Nakagawa,M. and Morikawa,K. (1999) Crystallographic and functional studies of very short patch repair endonuclease. Mol. Cell, 3, 621–628. [DOI] [PubMed] [Google Scholar]
- 9.Ban C. and Yang,W. (1998) Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J., 17, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovall R. and Matthews,B.W. (1997) Toroidal structure of lambda-exonuclease. Science, 277, 1824–1827. [DOI] [PubMed] [Google Scholar]
- 11.Nishino T., Komori,K., Tsuchiya,D., Ishino,Y. and Morikawa,K. (2001) Crystal structure of the archaeal holliday junction resolvase Hjc and implications for DNA recognition. Structure, 9, 197–204. [DOI] [PubMed] [Google Scholar]
- 12.Aggarwal A.K. (1995) Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol., 5, 11–19. [DOI] [PubMed] [Google Scholar]
- 13.Pingoud A. and Jeltsch,A. (1997) Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem., 246, 1–22. [DOI] [PubMed] [Google Scholar]
- 14.Jeltsch A., Alves,J., Maass,G. and Pingoud,A. (1992) On the catalytic mechanism of EcoRI and EcoRV. A detailed proposal based on biochemical results, structural data and molecular modelling. FEBS Lett., 304, 4–8. [DOI] [PubMed] [Google Scholar]
- 15.Jeltsch A., Alves,J., Wolfes,H., Maass,G. and Pingoud,A. (1993) Substrate-assisted catalysis in the cleavage of DNA by the EcoRI and EcoRV restriction enzymes. Proc. Natl Acad. Sci. USA, 90, 8499–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horton N.C., Newberry,K.J. and Perona,J.J. (1998) Metal ion-mediated substrate-assisted catalysis in type II restriction endonucleases. Proc. Natl Acad. Sci. USA, 95, 13489–13494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukacs C.M., Kucera,R., Schildkraut,I. and Aggarwal,A.K. (2000) Understanding the immutability of restriction enzymes: crystal structure of BglII and its DNA substrate at 1.5 Å resolution. Nature Struct. Biol., 7, 134–140. [DOI] [PubMed] [Google Scholar]
- 18.Tsutakawa S.E., Jingami,H. and Morikawa,K. (1999) Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell, 99, 615–623. [DOI] [PubMed] [Google Scholar]
- 19.Miller M.D., Cai,J. and Krause,K.L. (1999) The active site of Serratia endonuclease contains a conserved magnesium-water cluster. J. Mol. Biol., 288, 975–987. [DOI] [PubMed] [Google Scholar]
- 20.Flick K.E., Jurica,M.S., Monnat,R.J.,Jr and Stoddard,B.L. (1998) DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature, 394, 96–101. [DOI] [PubMed] [Google Scholar]
- 21.Chevalier B.S., Monnat,R.J.,Jr and Stoddard,B.L. (2001) The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nature Struct. Biol., 8, 312–316. [DOI] [PubMed] [Google Scholar]
- 22.Davies D.R., Goryshin,I.Y., Reznikoff,W.S. and Rayment,I. (2000) Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science, 289, 77–85. [DOI] [PubMed] [Google Scholar]
- 23.Beese L.S. and Steitz,T.A. (1991) Structural basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J., 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werner M.H., Gronenborn,A.M. and Clore,G.M. (1996) Intercalation, DNA kinking and the control of transcription. Science, 271, 778–784. [DOI] [PubMed] [Google Scholar]
- 25.Turner D.P. and Connolly,B.A. (2000) Interaction of the E.coli DNA G:T-mismatch endonuclease (vsr protein) with oligonucleotides containing its target sequence. J. Mol. Biol., 304, 765–778. [DOI] [PubMed] [Google Scholar]
- 26.Vassylyev D.G. and Morikawa,K. (1997) DNA-repair enzymes. Curr. Opin. Struct. Biol., 7, 103–109. [DOI] [PubMed] [Google Scholar]
- 27.Parikh S.S., Mol,C.D., Hosfield,D.J. and Tainer,J.A. (1999) Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol., 9, 37–47. [DOI] [PubMed] [Google Scholar]
- 28.Mol C.D., Parikh,S.S., Putnam,C.D., Lo,T.P. and Tainer,J.A. (1999) DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu. Rev. Biophys. Biomol. Struct., 28, 101–128. [DOI] [PubMed] [Google Scholar]
- 29.Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]
- 30.Fox K.R., Allinson,S.L., Sahagun-Krause,H. and Brown,T. (2000) Recognition of GT mismatches by Vsr mismatch endonuclease. Nucleic Acids Res., 28, 2535–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunter W.N., Brown,T., Kneale,G., Anand,N.N., Rabinovich,D. and Kennard,O. (1987) The structure of guanosine-thymidine mismatches in B-DNA at 2.5-Å resolution. J. Biol. Chem., 262, 9962–9970. [DOI] [PubMed] [Google Scholar]
- 32.Lamers M.H., Perrakis,A., Enzlin,J.H., Winterwerp,H.H., de Wind,N. and Sixma,T.K. (2000) The crystal structure of DNA mismatch repair protein MutS binding to a G × T mismatch. Nature, 407, 711–717. [DOI] [PubMed] [Google Scholar]
- 33.Allen D.J., Makhov,A., Grilley,M., Taylor,J., Thresher,R., Modrich,P. and Griffith,J.D. (1997) MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J., 16, 4467–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glasner W., Merkl,R., Schellenberger,V. and Fritz,H.J. (1995) Substrate preferences of Vsr DNA mismatch endonuclease and their consequences for the evolution of the Escherichia coli K-12 genome. J. Mol. Biol., 245, 1–7. [DOI] [PubMed] [Google Scholar]
- 35.Langle-Rouault F., Maenhaut-Michel,G. and Radman,M. (1987) GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO J., 6, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Welsh K.M., Lu,A.L., Clark,S. and Modrich,P. (1987) Isolation and characterization of the Escherichia coli mutH gene product. J. Biol. Chem., 262, 15624–15629. [PubMed] [Google Scholar]
- 37.Au K.G., Welsh,K. and Modrich,P. (1992) Initiation of methyl-directed mismatch repair. J. Biol. Chem., 267, 12142–12148. [PubMed] [Google Scholar]
- 38.Hall M.C. and Matson,S.W. (1999) The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J. Biol. Chem., 274, 1306–1312. [DOI] [PubMed] [Google Scholar]
- 39.Yang W. (2000) Structure and function of mismatch repair proteins. Mutat. Res., 460, 245–256. [DOI] [PubMed] [Google Scholar]
- 40.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 41.Hosfield D.J., Guan,Y., Haas,B.J., Cunningham,R.P. and Tainer,J.A. (1999) Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell, 98, 397–408. [DOI] [PubMed] [Google Scholar]
- 42.Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- 43.Newman M., Strzelecka,T., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1995) Structure of BamHI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science, 269, 656–663. [DOI] [PubMed] [Google Scholar]
- 44.Winkler F.K., Banner,D.W., Oefner,C., Tsernoglou,D., Brown,R.S., Heathman,S.P., Bryan,R.K., Martin,P.D., Petratos,K. and Wilson,K.S. (1993) The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J., 12, 1781–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]