

Abstract
Genomewide analyses of the mammalian transcriptome have revealed that large tracts of sequence previously annotated as noncoding are frequently transcribed and give rise to stable RNA. Although the transcription of individual genes of the Kaposi's sarcoma-associated herpesvirus (KSHV) has been well studied, little is known of the architecture of the viral transcriptome on a genomewide scale. Here we have employed a genomewide tiling array to examine the lytic transcriptome of the Kaposi's sarcoma-associated herpesvirus, KSHV. Our results reveal that during lytic growth (but not during latency), there is extensive transcription from noncoding regions, including both intergenic regions and, especially, noncoding regions antisense to known open reading frames (ORFs). Several of these transcripts have been characterized in more detail, including (i) a 10-kb RNA antisense to the major latency locus, including many of its microRNAs as well as its ORFs; (ii) a 17-kb RNA antisense to numerous ORFs at the left-hand end of the genome; and (iii) a 0.7-kb RNA antisense to the viral homolog of interleukin-6 (vIL-6). These studies indicate that the lytic herpesviral transcriptome resembles a microcosm of the host transcriptome and provides a useful system for the study of noncoding RNAs.
Few segments of eukaryotic DNA have been as closely studied at the transcriptional level as those comprising the genomes of animal viruses. To date, the study of viral gene expression has largely been driven by interest in viral coding regions. In these regions, evolution has clearly selected for efficiencies in gene expression. Examples include the use of alternative splicing to derive multiple transcripts from a given locus (1), the translation of multiple overlapping open reading frames (ORFs) in a given mRNA (12), and the use of multiple initiation codons within a given ORF (4). Translational frameshifting (22) and RNA editing (8) are additional mechanisms that some viruses have deployed to further enhance the diversity of protein products engendered by a genomic region. In contrast, relatively little attention has been paid in virology to noncoding regions of viral genomes (other than those encoding microRNAs [miRNAs]) (40). For small viruses, like parvoviruses, polyomaviruses, and hepadnaviruses, such regions are few in number and limited in extent. But for larger DNA viruses, including members of the herpesvirus and poxvirus families, more extensive tracts of the genome are thought to lack coding potential. This includes not only intergenic regions (which are typically short even in these large viruses) but also long stretches of DNA complementary to known ORFs. Little attention has been given to transcription emanating from such regions, most likely reflecting the tacit assumption that such regions are not stably expressed.
However, recent genomewide analyses of the mammalian transcriptome have suggested that this assumption may be in error, and that disinterest in such regions may be a mistake. Use of both comprehensive cDNA identification (6) and genomic tiling arrays (3, 13) reveals that large regions of the genome previously thought to be noncoding are in fact represented in stable RNAs (reviewed in references 7 and 44). For example, in one analysis, 56% of mouse cDNAs were found to be noncoding (6). Moreover, a significant fraction of the genome can produce transcripts from both strands (9, 23). In the FANTOM 3 data set, at least 25% of confirmed murine coding RNAs have well-characterized overlapping antisense transcripts: when fragmentary cDNAs are included, this percentage can rise as high as 72% (23). The functions of these apparently noncoding RNAs have been the subject of much speculation and conjecture but few systematic experimental tests.
The genomes of herpesviruses may provide a useful system in which to examine this phenomenon. Because the functions of many herpesviral genes are known, and good assays exist for each important stage of viral replication, these viruses may provide an accessible system for probing the biochemical properties and biological roles of noncoding RNAs, if such exist. An early study of cytomegalovirus (CMV) cDNA clones (45) strongly suggests that they do: in that analysis, 45% of all viral cDNAs examined were predicted to be noncoding. Here, we have carried out a genomewide analysis of the lytic transcriptome of the Kaposi's sarcoma-associated herpesvirus (KSHV) using a different approach: hybridizing labeled RNA from infected cells to a custom KSHV genomic tiling array. Our results show that during lytic KSHV infection, large segments of the genome are represented in stable RNAs, including many tracts antisense to known coding regions. We present more detailed characterization of several RNAs whose transcripts are of particular regulatory interest, including (i) a 10-kb RNA antisense to the entire major latency locus (including key latent ORFs and many viral miRNAs); (ii) a huge (17- to 18-kb) RNA antisense to the leftmost 12% of the viral genome, including the genes encoding the viral SSB and polymerase proteins involved in DNA replication and the glycoprotein B required for viral entry; and (iii) a 0.7-kb RNA antisense to the viral homolog of interleukin-6 (vIL-6). Our results affirm that the overall architecture of herpesviral transcriptomes bears striking resemblance to that of their mammalian hosts and raise the possibility of previously unsuspected forms of viral gene regulation.
MATERIALS AND METHODS
Tissue culture, virus preparation, and infection.
Human umbilical vein endothelial cells (HUVECs) were cultured in EGM-2 supplemented with a bullet kit (CC-4176; Clonetics). TIME (telomerase-immortalized microvascular endothelial) cells were cultured as previously described (41). BJAB and BCBL-1 cells were cultured in RPMI supplemented with 10% fetal bovine serum (FBS), 55 μM β-mercaptoethanol, and 2 mM glutamine. KSHV stocks were prepared from BCBL-1 cells as previously described (2). HUVECs and TIME cells were infected with BCBL-1-derived KSHV as previously described for TIME cells (11). Infection of TIME cells resulted in >90% latent nuclear antigen-positive (LANA+) cells and <1% Orf59+ cells as judged by immunostaining. Infection of HUVECs resulted in ∼75% LANA+ cells and <3% Orf59+ cells. TIME cells were induced to enter the lytic phase by superinfection by adenovirus encoding replication and transcription activator (AdRTA) under conditions that resulted in >90% of KSHV-infected cells undergoing lytic replication as judged by Orf59 immunostaining 54 h postinfection.
RNA preparation.
HUVEC and TIME cell samples were harvested in RLT buffer, and RNA was isolated using the RNeasy minikit according to the manufacturer's protocol (Qiagen). BJAB and BCBL-1 samples were harvested in RNA-Bee solution, and RNA was isolated according to the manufacturer's protocol (Tel-Test, Inc.). BJAB and BCBL-1 RNAs that were subjected to microarray analysis were then repurified using the RNeasy minikit according to the manufacturer's protocol (Qiagen); RNAs that were subjected to Northern analysis were used without further purification. Poly(A)-enriched RNA's were made using the Oligotex mRNA Midi kit according to the manufacturer's protocol (Qiagen).
KSHV tiling microarray construction, sample labeling, and microarray hybridization.
The KSHV genome sequence (GenBank accession no. U75698.1) was used for the microarray design. Using Agilent technology, 13,746 60-mer KSHV-specific oligonucleotide probes were printed in duplicate. Each probe overlapped a neighboring probe by 40 nucleotides (nt), resulting in a tiling design with probes offset by 20 nucleotides. Probes were designed from sequences of both strands of the KSHV genome; in addition, the microarray design contained control probes (approximately 1,500) and probes detecting host transcripts (16,311). This custom KSHV tiling microarray can be obtained from Agilent (AMADID: 017577).
Total RNA from the experimental samples was purified as indicated above. Reference RNA was a mixture of RNAs of both infected and uninfected cells, including HUVECs and TIME, BJAB, BCBL-1, and SLK cells. The integrity of the RNAs was analyzed using the 2100 Bioanalyzer (Agilent), or the RNAs were analyzed using conventional formaldehyde-agarose gel electrophoresis and staining. RNAs were quantified using the ND1000 spectrophotometer (Nanodrop). The Quick Amp labeling kit (Agilent) was used according to the manufacturer's protocol to generate labeled cRNA from 450 ng of total RNA. Experimental samples were labeled with Cy5, and reference samples were labeled with Cy3, which were then competitively hybridized to the custom KSHV tiling microarray according to the manufacturer's protocol for the Whole Human Genome Oligo Microarrays (Agilent). Hybridized microarrays were washed according to the manufacturer's protocol (Agilent) and scanned on the GenePix 4000B scanner (Axon instruments), and all feature intensities were collected using the GenePix Pro 6.0 software. TIFF images of scanned slides were analyzed using Feature Extraction Software version 9.5.3 (Agilent) with a custom grid file (017577_D_F_20070822). All data are reported as log2 values of the dye-normalized Cy5/Cy3 ratios. The log2 ratios of all samples were normalized to the average log2 ratios of the corresponding mock-infected (or control) samples.
Northern blotting.
Total RNA (10 μg) or poly(A)-enriched RNA (200 ng) was resolved by formaldehyde-agarose gel electrophoresis in MOPS (morpholinepropanesulfonic acid) buffer. Resolved RNAs were transferred to Nytran SuPerCharge membrane according the Turboblotter manufacturer's protocol (Whatman, Schleicher & Schuell). Membranes were UV cross-linked using the UV Stratalinker 2400 (Stragene). Membranes were prehybridized using Ultrahyb buffer (Ambion) at 65°C for more than 1 h. UTP-32P-labeled riboprobes were generated by performing T3 or T7 in vitro transcription reactions (Ambion) using PCR templates. PCR products, obtained using T3- or T7-linked primers spanning specific regions of the KSHV genome (see supplemental material file S1), served as templates for these transcription reactions. Labeled riboprobes were hybridized to the membranes in Ultrahyb buffer at 65°C for approximately 16 h. Membranes were first washed two times with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 0.1% SDS at 65°C for 5 min. Then the membranes were washed in a high-stringency buffer (0.2× SSC, 0.1% SDS) for various amounts of time at 65°C. The length of time was empirically determined for each probe such that the wash conditions resulted in minimal nonspecific hybridization. Blots were exposed to a phosphor storage screens (GE Healthcare). Screens were analyzed using the Storm 860 scanner (Molecular Dynamics).
RPA.
Total RNA was subject to RNase protection mapping using the RNase protection assay (RPA) III kit according to the manufacturer's protocol (Ambion). The undigested probe that was used in this study spanned positions 120748 to 12283 and contained non-KSHV sequence on both sides of the KSHV sequence, resulting in a probe length of 532 nt. The UTP-32P-labeled RPA probe was generated by performing T3 transcription from a linearized plasmid template bearing the indicated sequences downstream of a T3 promoter. The RPA reactions were resolved by 6% acrylamide denaturing urea gel electrophoresis. Gels were dried and exposed to phosphor storage screens (GE Healthcare). Screens were analyzed using the Storm 860 Scanner (Molecular Dynamics).
Microarray data accession number.
All reported raw microarray data are MIAME compliant and are stored in NCBI's Gene Expression Omnibus (GEO) (19), accessible through GEO accession no. GSE21972 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE21972).
RESULTS
Microarray analysis of viral RNAs.
To examine the transcription of KSHV on a genomewide scale, we designed a custom DNA microarray that tiles across the KSHV genome using synthetic 60-mer oligonucleotides, offset by 20-bp intervals. The arrayed oligonucleotides span the entire 137.5 kb of unique genomic DNA but do not include the terminal repeats, which are 85% G+C and are therefore likely to hybridize nonspecifically to many sequences. Each strand of KSHV DNA was represented in its entirety.
KSHV infection can produce one of two outcomes: (i) latency, in which all but a handful of viral genes are silenced, and the viral genome persists in the nucleus as a plasmid; and (ii) lytic replication, in which all viral genes are expressed in a temporally ordered cascade, with the production and release of progeny virions and death of the cell. In cell culture, de novo infection generally results in latency, although a small subpopulation of latently infected cells do undergo spontaneous lytic reactivation. From latency, lytic replication can be induced by chemicals (phorbol esters, histone deacetylase [HDAC] inhibitors) or by ectopic expression of the KSHV lytic switch protein RTA (replication and transcription activator) (27, 31, 37). We infected primary human umbilical vein endothelial cells (HUVECs) or telomerase-immortalized microvascular endothelial (TIME) cells (25) acutely with wild-type KSHV at a high multiplicity of infection (MOI). From each cell line, total RNA was prepared at different time points following de novo KSHV infection (in which most cells establish a latent infection) or after induction of lytic replication by superinfection with an adenoviral vector expressing RTA (AdRTA). We also examined BCBL-1 cells, a B-cell line derived from KSHV-related primary effusion lymphoma (31); this line bears stably latent KSHV genomes, but it can be induced to lytic reactivation with valproic acid (VA) (35).
From each sample, total RNA was labeled using the Eberwine protocol, resulting in Cy5 (or Cy3)-labeled cRNA (17). This protocol employs a T7-based transcription step that resulted in a 20- to 40-fold amplification. Figure 1 shows the results of this experiment. De novo infection of HUVECs and TIME cells showed low levels of gene expression initially, but by 2 days postinfection, substantial levels of expression were observed across large regions of the viral genome (Fig. 1, left and center panels). This pattern is not expected of the restricted gene expression in latency (46); rather, it is more in keeping with lytic reactivation. Since HUVECs and TIME cells are known to harbor substantial subpopulations of lytically infected cells following de novo infection (14, 25), we speculated that this pattern was derived from that subpopulation. This was affirmed by our finding that the pattern of hybridization seen in HUVECs and TIME cells was closely mimicked by that observed in a reconstruction experiment in which RNA from a population of lytically infected TIME cells was admixed with RNA isolated from a latently infected cell population in various proportions, such that the RNA mixture modeled RNA harvested from a population of cells experiencing a 0.5% to 9% spontaneous reactivation rate (see Fig. S1 in the supplemental material). Similarly, the broad pattern of hybridization observed with uninduced BCBL-1 RNA (Fig. 1, right panel) is likely emanating from the 2 to 5% of these primary effusion lymphoma (PEL) cells that are known to spontaneously enter the lytic cycle (31).
FIG. 1.

KSHV transcriptome analysis. KSHV tiling microarray data (ordered by genome position) are displayed for 13,746 unique probes. Expression analysis was performed on indicated cells (top line). In each group of microarray data, the color bar describes the fold changes relatively to the appropriate uninfected or infected cells. HUVECs (left panel) and TIME cells (center panel) were infected de novo with BCBL-1-derived KSHV, and RNAs were prepared at the indicated times postinfection. KSHV-infected TIME cells were treated with AdRTA to induce entry into lytic cycle, and RNA harvested at 54 h postinfection (hpi) was similarly examined (center panel, rightmost lanes). (Right panel) RNAs were prepared and analyzed from BJAB and BCBL-1 cells and BCBL-1 cells treated with 600 μM valproic acid (VA) for 48 h.
When the lytic cycle in BCBL-1 was deliberately induced with valproic acid (VA), intense hybridization to nearly all probes on the array was observed (Fig. 1, right panel). The same was true of RNA extracted from KSHV-infected TIME cells induced to lytic replication by superinfection with an adenovirus encoding RTA (AdRTA) (Fig. 1, center panel); this indicates that the pattern was due to bona fide reactivation, not the result of aberrant chromatin remodeling caused by the HDAC-inhibitory activity of valproate. Figure 2 displays the locations of the hybridizing regions on the genome of KSHV, with reference to the known viral open reading frames (ORFs). As can be seen, extensive hybridization was detected to both strands of the viral genome, including many regions that lacked annotated coding regions. Most of the latter corresponded to the noncoding strands that are antisense to known ORFs.
FIG. 2.

Genomic position of microarray hybridization signal from induced BCBL-1 cells. Approximate genomic positions of microarray hybridization signal are transposed on a KSHV genomic map. Genes in blue are carried by the top strand of the genome and are transcribed left to right. Genes in yellow are carried by the bottom strand and are transcribed right to left. The red bars indicate the genomic location of increased microarray hybridization signal in lytically reactivated BCBL-1 cells.
Large noncoding RNAs of KSHV.
As an example of this class of noncoding RNAs, we first focused on the signals derived from the left-hand end of the viral genome. As shown in Fig. 2, viral RNA showed prominent annealing to numerous oligonucleotides derived from the bottom (noncoding) strand of KSHV DNA, spanning ca. 17 to 18 kb—the largest contiguous noncoding region in the viral genome. To explore transcription of this area further, we examined total RNA from lytically infected cells by Northern blotting, using a series of strand-specific RNA probes from this region that were complementary to the antisense strand of KSHV (Fig. 3). The locations of the probes are shown as red bars; the associated Northern blot is displayed directly beneath each probe. As shown in Fig. 3, a single large (17 to 18 kb) and abundant transcript can be detected with every probe from this region. This enormous RNA is antisense to eight open reading frames for known viral proteins, including several involved in viral DNA replication (ORF6 [SSB] and ORF9 [DNA polymerase]), immune evasion (ORFs 4, 10, and 11), viral entry (ORF8 [gB]), and signal transduction (ORF K1). Most probes also detect a slightly smaller band (15 to 16 kb), and some probes detected additional species of RNAs, most likely representing multiple smaller transcripts that overlap portions of the larger transcript. The analysis of Fig. 3 thus indicates that the array hybridization pattern can underestimate the complexity of RNAs produced in such a region, since the array signatures of smaller transcripts can be masked by those derived from overlapping larger RNAs. Clearly, the actual KSHV transcriptome structure cannot be accurately defined solely by array hybridization. Nonetheless, because the array allows scanning of the entire length of the genome, it is useful for highlighting transcripts from regions of potential interest; these can then be characterized using methods (Northern blotting, nuclease mapping and reverse transcription [RT]-PCR) that can resolve overlapping RNAs and map their structures.
FIG. 3.

Tiling northern analysis to detect transcripts that are antisense to genes at the left end of the genome. Total RNA from BJAB cells (A) or BCBL-1 cells treated with valproic acid (VA) for 24 h (B) was prepared and resolved by formaldehyde-agarose gel electrophoresis, transferred to nylon membranes, and hybridized to radioactively labeled riboprobes, which detect transcripts that span the regions indicated by the red bars. The Northern data are presented directly below the indicated riboprobe used in each blot (please see supporting information file S1 in the supplemental material for information regarding riboprobe design).
As an example of how the tiling array can focus attention on interesting regulatory regions, we note that many probes from the region antisense to the major latency locus (encoding LANA [ORF73], v-cyclin [ORF72], and v-FLIP [ORF71]) also hybridized to lytic viral RNA, albeit in a somewhat discontinuous fashion (summarized in Fig. 2). To clarify the nature of the antisense transcripts in this region, we carried out a Northern blotting analysis, again using labeled probes from across the locus to anneal to RNA from induced BCBL-1 cells. Figure 4 shows the result, indicating the presence of an abundant RNA of ca 10 kb that anneals to all probes detecting transcripts antisense to ORFs 71, 72, and 73. This RNA extends several kb 5′ to the region antisense to ORF71—although it does not extend to the adjacent kaposin ORF. This means that this region of the RNA is also antisense to many of the KSHV miRNAs (see Fig. 6B), as well as to portions of the latent kaposin pre-mRNA. At its 3′ end, the RNA spans the coding regions for ORF K14 and ORF74 (virus-encoded G protein-coupled receptor [vGPCR]). (Because of its extremely long 5′ untranscribed region [UTR], we would not expect this RNA to encode significant amounts of either K14 or vGPCR proteins.) As expected, probes for the latter two ORFs also detect the known 2.8-kb K14-vGPCR bicistronic RNA (Fig. 4) (24). A downstream probe from the ORF75 region did not anneal to this transcript, suggesting it may be coterminal with the known K14-GPCR RNA. Fine-mapping studies validated this prediction (see below and Fig. 6B). We provisionally designate this 10-kb transcript as ALT (antisense to latency transcripts) RNA.
FIG. 4.

Tiling Northern analysis to detect transcripts that are antisense to the latency locus. Total RNA from BJAB cells (A) or BCBL-1 cells (B) treated with valproic acid (VA) for 24 h was subject to Northern blotting analysis using radioactively labeled riboprobes, which detect transcripts that span the regions indicated by the orange bars. The Northern data are presented directly above the indicated riboprobe used in each blot.
FIG. 6.

Fine-structure mapping of ALT RNA. (A) The RNase protection assay (RPA) was performed on total RNA from yeast cells (lane 1), BJAB cells (lane 2), or VA-induced BCBL-1 cells at 24 h postinduction (+RNase; lane 3) using an undigested riboprobe that spans nucleotide positions 120748 to 120283 (−RNase; lane 1). After hybridization of the radiolabeled riboprobe to the indicated RNAs, the probes were subject to a treatment with RNase A or T1. The protected fragments were analyzed by acrylamide gel electrophoresis. Lane M, molecular size markers. (B) Schematic of ALT RNA transcript structure, based on 5′- and 3′-RACE analyses, RT-PCR analysis, and RPA. The indicated splice site is the same splice site (129219 to 129367) found in the bistronic K14 or vGPCR message. The 3′ end of ALT RNA is coterminal (130546) with the bistronic K14 or vGPCR message. The 5′-RACE clone and the RPA support the conclusion that nt 120297 is the 5′ nucleotide, but other downstream start sites may also exist. Assuming this start site, KSHV pre-miRNAs that are spanned by ALT RNA are indicated by the red symbols, while the pre-miRNAs that are not spanned by the ALT RNA are indicated by the green symbols.
Because it is antisense to so many key latent gene products, it was important to determine whether ALT RNA is unique to BCBL-1 cells or can be detected in other KSHV-infected cells. Figure 5A and B show that a similar RNA can be detected in a second B-cell line (BC-3), as well as in iSLK.219 cells. These are SLK endothelial cells that harbor (i) a doxycycline-inducible RTA transgene and (ii) rKSHV.219, a recombinant, replication-competent KSHV genome that constitutively expresses green fluorescent protein (GFP) (42). As such, iSLK.219 cells were induced by exposure to doxycycline; BCBL-1 and BC-3 were induced with VA. In all three cases, ALT RNA is only observed following lytic induction. The transcript is found in oligo(dT)-selected RNA populations (Fig. 5A), indicating it is likely polyadenylated.
FIG. 5.

Northern analysis of ALT RNA in poly(A)-enriched RNA and in multiple cell types. Total or poly(A)-enriched RNA from the indicated cells was subjected to Northern blotting analysis using a radioactively labeled riboprobe that detects ALT RNA. Total RNA or poly(A)-enriched RNA was prepared from BJAB cells, untreated BCBL-1 cells, or BCBL-1 cells treated with valproic acid (VA; 600 μM) for 24 h; untreated BC-3 cells or BC-3 cells treated with valproic acid (600 μM) for 24 h; or SLK cells, untreated iSLK.219 cells, or iSLK.219 cells treated with doxycycline (Dox; 1 μg/ml) for 48 h. Arrows indicate ALT transcript.
Detailed mapping of the termini of ALT RNA is shown in Fig. 6. To determine the 5′ end of the RNA, we carried out RNase protection analysis (RPA) with a riboprobe spanning nt 120748 to 120283. As shown in Fig. 6A, the predominant protected band was ca. 450 nt long, although several less-abundant shorter products were also detected. We next carried out 5′ rapid amplification of cDNA ends (RACE) analysis, which again yielded multiple products, but the product most consistent with the RPA mapped to nucleotide position 120297. (A second product with the 5′ end mapping to ca. 120570 could account for the lower band seen in the RPA of Fig. 6A.) We think it likely that additional minor start sites exist, but further work will be required to rigorously assign their termini.
To determine whether there may also be small splices in the body of ALT RNA, we carried out RT-PCR studies across the body of the transcript and used agarose gel electrophoresis to compare the size of the products to those derived from PCR of genomic DNA with the same primers. In all cases, these products were of identical mobility (data not shown), save for the region between ORF K14 and ORF74—a region known to undergo splicing to remove a small (149-nt) intron (24). The same intron that is processed in the smaller (2.8-kb) K14 or vGPCR mRNA is also excised from ALT RNA as well. Splicing is incomplete, and both unspliced and spliced versions exist in the ALT RNA population (Fig. 6B). However, the remainder of the ALT transcript appears to be unspliced.
The Northern blotting of Fig. 4 suggested that the 3′ end of ALT RNA might be coterminal with the previously mapped 2.8-kb K14 or GPCR mRNA. To map the 3′ end, we carried out an RT-PCR with an anchored oligo(dT) primer and an upstream primer (nt 127799 to 127818) derived from sequences 5′ to ORF K14 that are unique to ALT RNA. Sequencing of this product revealed that the poly(A) addition site was at nt 130546, exactly as previously mapped for the 2.8-kb K14 or vGPCR mRNA (24); this site is 29 nt 3′ to a canonical AAUAAA polyadenylation signal.
The kinetics of expression of ALT are examined in Fig. S2 in the supplemental material. In both BCBL-1 cells and infected SLK cells, the transcript accumulates in parallel with the mRNA for K14 or GPCR, a known delayed early transcript (24). Consistent with this, its accumulation is not blocked by phosphonoformic acid (PFA), an inhibitor of viral DNA synthesis.
An RNA antisense to vIL-6.
Another striking feature of the array hybridization pattern of lytic viral RNA was the presence of hybridization to probes antisense to ORF K2, the gene encoding vIL-6, the viral homolog of IL-6 (Fig. 2). To characterize this transcript further, Northern blotting was performed on oligo(dT)-selected RNA from iSLK.219 cells before and after induction with doxycycline, using probes from the K2 or vIL-6 ORF. Figure 7 shows that these probes detected a discrete ∼0.7-kb polyadenylated RNA complementary to vIL-6; this RNA was strongly induced during lytic growth. Since vIL-6 mRNA is also upregulated during lytic growth (28), these two RNAs are coexpressed during the lytic cycle. This behavior mimics that of the RTA sense-antisense pair we previously identified (26, 43); coexpression is also a feature of the majority of sense-antisense pairs found in the mammalian genome (29).
FIG. 7.
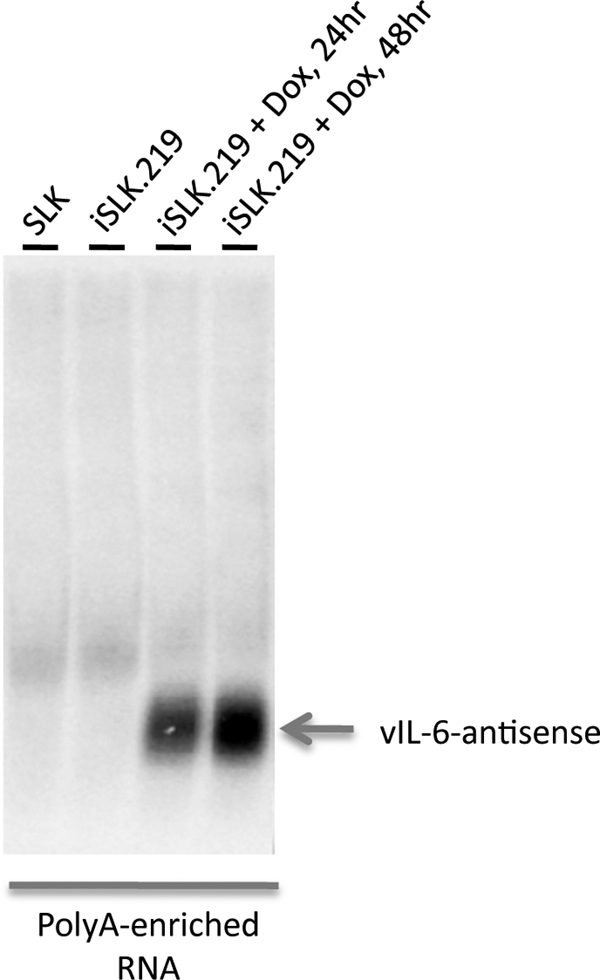
Expression of RNA antisense to vIL-6 in lytic infection. RNA extracted from the indicated cell lines was oligo(dT) selected and then analyzed by electrophoresis through a 1% agarose gel, transfer to a solid support, and hybridization with a probe complementary to the antisense region of the vIL-6 gene.
Searching for potential polysome associations of these transcripts.
Recently, we characterized a pair of seemingly noncoding RNAs that are antisense to the major mRNA encoding RTA and found that they are likely mRNAs for small peptides (43). One the early pieces of evidence pointing to this finding was their localization on polysomes. Accordingly, we have conducted a preliminary screen for polysome localization of KSHV transcripts genomewide, by preparing polysomal RNA as previously described (43), labeling it, and hybridizing the labeled product to the KSHV tiling array. The results are displayed in Fig. S3 in the supplemental material. As shown there, ALT sequences are not represented in the polysome fraction, while sequences from the region antisense to the vIL-6 gene and from the region antisense to ORFs K1 and 4 to 11 are so represented. It is therefore possible that the latter two transcripts may also encode previously unsuspected peptides. However, we note that considerable direct experimentation will be required to validate this suggestion. This is particularly true for the 17- to 18-kb RNA mapped in Fig. 3; since this RNA is 10 times the size of most other host of viral mRNAs, RNPs bearing such a transcript may well have cosedimented with polysomes in this experiment.
DISCUSSION
Genomewide analyses of the transcription of higher eukaryotic genomes have led to a startling change in our perception of the architecture of the mammalian transcriptome. These studies (reviewed in reference 44) reveal that, contrary to expectation, large tracts of seemingly noncoding DNA nonetheless can template the production of stable RNAs. These transcripts emanate from both intergenic regions and from the antisense strand of known coding regions. Our findings, together with those recently reported for CMV (45), indicate that these features of the host transcriptome are also preserved in the lytic transcriptomes of herpesviruses that infect these hosts. Interestingly, they are not preserved in the latent transcriptomes of KSHV—the complex transcripts documented here in uninduced cultures of KSHV-infected cells can be accounted for entirely by contamination with cells that have spontaneously entered the lytic state (Fig. 1). Indeed, we have identified certain cell lines (e.g., HFF and SLK) with extremely low backgrounds of spontaneous lytic reactivation, and array profiling in such cells reveals the expected restriction of transcription to a handful of RNAs (10). These emanate principally from coding regions, although low levels of the vIL-6 antisense transcript may also be expressed in some latent lines (10). Presumably, the dramatic repression that accompanies latent infection can override the signals that enable the more promiscuous transcription observed during the lytic cycle.
Because of the imperfect correlation between array hybridization pattern and transcript structure, tiling array data alone do not suffice to allow a detailed map of the KSHV lytic transcriptome. This issue is well illustrated by our findings with ALT RNA. The array hybridization pattern (Fig. 2) suggested discontinuities in hybridization, but detailed transcript mapping revealed that ALT transcripts span a contiguous block of the genome in and around the major latency locus (Fig. 6B). Clearly, not all oligonucleotides on the array hybridize efficiently to stable transcripts that span their length; this produces apparent discontinuities in the transcript map whose origins cannot be understood without independent studies of transcript structure. (This feature of tiling arrays has also been noted in studies of other cellular transcriptomes [32].) In addition to this problem, we note that hybridization signals from longer RNAs will obscure the identification of shorter RNAs that overlap them—a phenomenon that was encountered in both ALT RNA (Fig. 4) and the large 17-kb antisense transcript from the left-hand end of the genome (Fig. 3). Nevertheless, the tiling array allows a genomewide perspective on the transcriptome that allows the investigator to focus in on interesting genomic regions with unusual or provocative transcript patterns.
Our array analyses, in addition to revealing the similarity of the lytic transcriptome's architecture to that of its host, also allow us to identify previously unsuspected RNAs of potential regulatory significance. Perhaps the most dramatic example of this is ALT RNA, which is antisense to the major latency locus, including ORFs 71 to 73 and many of the virus-encoded miRNAs (Fig. 4). This RNA is strongly induced during lytic growth. Could it have a regulatory role in attenuating the expression of latent proteins or miRNAs during the lytic cycle? Most studies show that LANA (15) and most of the miRNAs (5, 34) do not increase in abundance during the lytic cycle, despite the substantial increase in viral copy number. Although many possible mechanisms can be involved to explain these facts, our studies raise additional possibilities: e.g., (i) that accumulation of an antisense transcript interferes with stability or translation of mRNA for ORFs 71 to 73 and (ii) that the antisense RNA can act as an “miRNA sponge” (16) to inhibit miRNA accumulation or function during lytic growth. All of these possibilities can now be subjected to direct experimental tests.
Our finding of a small transcript directly antisense to the vIL-6 transcript provides another example of a sense-antisense pair of RNAs in the KSHV genome. Such pairs are frequent in the mammalian transcriptome, and their functions have been extensively debated (although little investigated) over the years. Most discussions begin with the idea that these RNAs serve as regulators that target the cognate coding transcript for inhibition (reviewed in reference 29). Proposed inhibitory mechanisms include impaired transcriptional elongation in the overlap region (30), altered regional chromatin remodeling (33, 36, 39), altered RNA processing (20, 21), or impaired translation (18, 38). Consistent with such inhibitory models, a reciprocal relationship is sometimes seen between expression of the antisense RNA and the accumulation of its cognate RNA (23). More often, though, the two RNAs are concordantly regulated, accumulating in tandem (as is observed with the vIL-6 sense-antisense pair). Very few examples of such coordinately regulated sense-antisense pairs have been rigorously studied, and proposals about their function have been largely conjectural. We believe that the KSHV genome, whose genes are much more better understood than those of its host, will provide an excellent experimental system for parsing the function(s) of such antisense RNAs.
Supplementary Material
Acknowledgments
This work was funded by the Howard Hughes Medical Institute.
Footnotes
Published ahead of print on 9 June 2010.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Akusjarvi, G. 2008. Temporal regulation of adenovirus major late alternative RNA splicing. Front. Biosci. 13:5006-5015. [DOI] [PubMed] [Google Scholar]
- 2.Bechtel, J. T., Y. Liang, J. Hvidding, and D. Ganem. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertone, P., V. Stolc, T. E. Royce, J. S. Rozowsky, A. E. Urban, X. Zhu, J. L. Rinn, W. Tongprasit, M. Samanta, S. Weissman, M. Gerstein, and M. Snyder. 2004. Global identification of human transcribed sequences with genome tiling arrays. Science 306:2242-2246. [DOI] [PubMed] [Google Scholar]
- 4.Bruss, V. 2007. Hepatitis B virus morphogenesis. World J. Gastroenterol. 13:65-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai, X., S. Lu, Z. Zhang, C. M. Gonzalez, B. Damania, and B. R. Cullen. 2005. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:5570-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carninci, P., T. Kasukawa, S. Katayama, J. Gough, M. C. Frith, N. Maeda, R. Oyama, T. Ravasi, B. Lenhard, C. Wells, R. Kodzius, K. Shimokawa, V. B. Bajic, S. E. Brenner, S. Batalov, A. R. Forrest, M. Zavolan, M. J. Davis, L. G. Wilming, V. Aidinis, J. E. Allen, A. Ambesi-Impiombato, R. Apweiler, R. N. Aturaliya, T. L. Bailey, M. Bansal, L. Baxter, K. W. Beisel, T. Bersano, H. Bono, A. M. Chalk, K. P. Chiu, V. Choudhary, A. Christoffels, D. R. Clutterbuck, M. L. Crowe, E. Dalla, B. P. Dalrymple, B. de Bono, G. Della Gatta, D. di Bernardo, T. Down, P. Engstrom, M. Fagiolini, G. Faulkner, C. F. Fletcher, T. Fukushima, M. Furuno, S. Futaki, M. Gariboldi, P. Georgii-Hemming, T. R. Gingeras, T. Gojobori, R. E. Green, S. Gustincich, M. Harbers, Y. Hayashi, T. K. Hensch, N. Hirokawa, D. Hill, L. Huminiecki, M. Iacono, K. Ikeo, A. Iwama, T. Ishikawa, M. Jakt, A. Kanapin, M. Katoh, Y. Kawasawa, J. Kelso, H. Kitamura, H. Kitano, G. Kollias, S. P. Krishnan, A. Kruger, S. K. Kummerfeld, I. V. Kurochkin, L. F. Lareau, D. Lazarevic, L. Lipovich, J. Liu, S. Liuni, S. McWilliam, M. Madan Babu, M. Madera, L. Marchionni, H. Matsuda, S. Matsuzawa, H. Miki, F. Mignone, S. Miyake, K. Morris, S. Mottagui-Tabar, N. Mulder, N. Nakano, H. Nakauchi, P. Ng, R. Nilsson, S. Nishiguchi, S. Nishikawa, et al. 2005. The transcriptional landscape of the mammalian genome. Science 309:1559-1563. [DOI] [PubMed] [Google Scholar]
- 7.Carninci, P., J. Yasuda, and Y. Hayashizaki. 2008. Multifaceted mammalian transcriptome. Curr. Opin. Cell Biol. 20:274-280. [DOI] [PubMed] [Google Scholar]
- 8.Casey, J. L. 2006. RNA editing in hepatitis delta virus. Curr. Top. Microbiol. Immunol. 307:67-89. [DOI] [PubMed] [Google Scholar]
- 9.Cawley, S., S. Bekiranov, H. H. Ng, P. Kapranov, E. A. Sekinger, D. Kampa, A. Piccolboni, V. Sementchenko, J. Cheng, A. J. Williams, R. Wheeler, B. Wong, J. Drenkow, M. Yamanaka, S. Patel, S. Brubaker, H. Tammana, G. Helt, K. Struhl, and T. R. Gingeras. 2004. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116:499-509. [DOI] [PubMed] [Google Scholar]
- 10.Chandriani, S., and D. Ganem. 2010. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 84:5565-5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandriani, S., and D. Ganem. 2007. Host transcript accumulation during lytic KSHV infection reveals several classes of host responses. PLoS One 2:e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, L. J., P. Pryciak, D. Ganem, and H. E. Varmus. 1989. Biosynthesis of the reverse transcriptase of hepatitis B viruses involves de novo translational initiation not ribosomal frameshifting. Nature 337:364-368. [DOI] [PubMed] [Google Scholar]
- 13.Cheng, J., P. Kapranov, J. Drenkow, S. Dike, S. Brubaker, S. Patel, J. Long, D. Stern, H. Tammana, G. Helt, V. Sementchenko, A. Piccolboni, S. Bekiranov, D. K. Bailey, M. Ganesh, S. Ghosh, I. Bell, D. S. Gerhard, and T. R. Gingeras. 2005. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308:1149-1154. [DOI] [PubMed] [Google Scholar]
- 14.Ciufo, D. M., J. S. Cannon, L. J. Poole, F. Y. Wu, P. Murray, R. F. Ambinder, and G. S. Hayward. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebert, M. S., J. R. Neilson, and P. A. Sharp. 2007. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 4:721-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eberwine, J. 1996. Amplification of mRNA populations using aRNA generated from immobilized oligo(dT)-T7 primed cDNA. Biotechniques 20:584-591. [DOI] [PubMed] [Google Scholar]
- 18.Ebralidze, A. K., F. C. Guibal, U. Steidl, P. Zhang, S. Lee, B. Bartholdy, M. A. Jorda, V. Petkova, F. Rosenbauer, G. Huang, T. Dayaram, J. Klupp, K. B. O'Brien, B. Will, M. Hoogenkamp, K. L. Borden, C. Bonifer, and D. G. Tenen. 2008. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev. 22:2085-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar, R., M. Domrachev, and A. E. Lash. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30:207-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastings, M. L., H. A. Ingle, M. A. Lazar, and S. H. Munroe. 2000. Post-transcriptional regulation of thyroid hormone receptor expression by cis-acting sequences and a naturally occurring antisense RNA. J. Biol. Chem. 275:11507-11513. [DOI] [PubMed] [Google Scholar]
- 21.Hastings, M. L., C. Milcarek, K. Martincic, M. L. Peterson, and S. H. Munroe. 1997. Expression of the thyroid hormone receptor gene, erbAalpha, in B lymphocytes: alternative mRNA processing is independent of differentiation but correlates with antisense RNA levels. Nucleic Acids Res. 25:4296-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacks, T., M. D. Power, F. R. Masiarz, P. A. Luciw, P. J. Barr, and H. E. Varmus. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280-283. [DOI] [PubMed] [Google Scholar]
- 23.Katayama, S., Y. Tomaru, T. Kasukawa, K. Waki, M. Nakanishi, M. Nakamura, H. Nishida, C. C. Yap, M. Suzuki, J. Kawai, H. Suzuki, P. Carninci, Y. Hayashizaki, C. Wells, M. Frith, T. Ravasi, K. C. Pang, J. Hallinan, J. Mattick, D. A. Hume, L. Lipovich, S. Batalov, P. G. Engstrom, Y. Mizuno, M. A. Faghihi, A. Sandelin, A. M. Chalk, S. Mottagui-Tabar, Z. Liang, B. Lenhard, and C. Wahlestedt. 2005. Antisense transcription in the mammalian transcriptome. Science 309:1564-1566. [DOI] [PubMed] [Google Scholar]
- 24.Kirshner, J. R., K. Staskus, A. Haase, M. Lagunoff, and D. Ganem. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006-6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagunoff, M., J. Bechtel, E. Venetsanakos, A. M. Roy, N. Abbey, B. Herndier, M. McMahon, and D. Ganem. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J. Virol. 76:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 28.Nicholas, J., V. R. Ruvolo, W. H. Burns, G. Sandford, X. Wan, D. Ciufo, S. B. Hendrickson, H. G. Guo, G. S. Hayward, and M. S. Reitz. 1997. Kaposi's sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nat. Med. 3:287-292. [DOI] [PubMed] [Google Scholar]
- 29.Prasanth, K. V., and D. L. Spector. 2007. Eukaryotic regulatory RNAs: an answer to the ‘genome complexity’ conundrum. Genes Dev. 21:11-42. [DOI] [PubMed] [Google Scholar]
- 30.Prescott, E. M., and N. J. Proudfoot. 2002. Transcriptional collision between convergent genes in budding yeast. Proc. Natl. Acad. Sci. U. S. A. 99:8796-8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 32.Royce, T. E., J. S. Rozowsky, P. Bertone, M. Samanta, V. Stolc, S. Weissman, M. Snyder, and M. Gerstein. 2005. Issues in the analysis of oligonucleotide tiling microarrays for transcript mapping. Trends Genet. 21:466-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sado, T., Y. Hoki, and H. Sasaki. 2005. Tsix silences Xist through modification of chromatin structure. Dev. Cell 9:159-165. [DOI] [PubMed] [Google Scholar]
- 34.Samols, M. A., J. Hu, R. L. Skalsky, and R. Renne. 2005. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:9301-9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw, R. N., J. L. Arbiser, and M. K. Offermann. 2000. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 14:899-902. [DOI] [PubMed] [Google Scholar]
- 36.Sun, B. K., A. M. Deaton, and J. T. Lee. 2006. A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol. Cell 21:617-628. [DOI] [PubMed] [Google Scholar]
- 37.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thenie, A. C., I. M. Gicquel, S. Hardy, H. Ferran, P. Fergelot, J. Y. Le Gall, and J. Mosser. 2001. Identification of an endogenous RNA transcribed from the antisense strand of the HFE gene. Hum. Mol. Genet. 10:1859-1866. [DOI] [PubMed] [Google Scholar]
- 39.Tufarelli, C., J. A. Stanley, D. Garrick, J. A. Sharpe, H. Ayyub, W. G. Wood, and D. R. Higgs. 2003. Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat. Genet. 34:157-165. [DOI] [PubMed] [Google Scholar]
- 40.Umbach, J. L., and B. R. Cullen. 2009. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 23:1151-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venetsanakos, E., A. Mirza, C. Fanton, S. R. Romanov, T. Tlsty, and M. McMahon. 2002. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp. Cell Res. 273:21-33. [DOI] [PubMed] [Google Scholar]
- 42.Vieira, J., and P. M. O'Hearn. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225-240. [DOI] [PubMed] [Google Scholar]
- 43.Xu, Y., and D. Ganem. 2010. Making sense of antisense: seemingly noncoding RNAs antisense to the master regulator of Kaposi's sarcoma-associated herpesvirus lytic replication do not regulate that transcript but serve as mRNAs encoding small peptides. J. Virol. 84:5465-5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasuda, J., and Y. Hayashizaki. 2008. The RNA continent. Adv. Cancer Res. 99:77-112. [DOI] [PubMed] [Google Scholar]
- 45.Zhang, G., B. Raghavan, M. Kotur, J. Cheatham, D. Sedmak, C. Cook, J. Waldman, and J. Trgovcich. 2007. Antisense transcription in the human cytomegalovirus transcriptome. J. Virol. 81:11267-11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. U. S. A. 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.