

SUMMARY
Atrial natriuretic factor receptor guanylate cyclase (ANF-RGC) is the receptor and the signal transducer of two natriuretic peptide hormones, ANF and BNP. It is a single transmembrane-spanning protein. It binds these hormones at its extracellular domain and activates its intracellular catalytic domain. This results in the accelerated production of cyclic GMP, a second messenger in controlling blood pressure, cardiac vasculature and fluid secretion. ATP is obligatory for the transduction of this hormonal signal. Two models of ATP action have been proposed. In Model 1, it is a direct allosteric transducer. It binds to the defined regulatory domain (ARM) juxtaposed to the C-terminal side of the transmembrane domain of ANF-RGC, induces a cascade of temporal and spatial changes and activates the catalytic module residing at the C-terminus of the cyclase. In Model 2, before ATP can exhibit its allosteric effect, ANF-RGC must first be phosphorylated by, yet unidentified, protein kinase. This initial step is obligatory in ANF signaling of ANF-RGC. Until now, none of these models has been directly validated because it has not been possible to segregate the allosteric and the phosphorylation effects of ATP in ANF-RGC activation. The present study accomplishes this aim through a novel probe, staurosporine. This unequivocally validates Model 1 and settles the over two-decade long debate on the role of ATP in ANF-RGC signaling. In addition, this study demonstrates that the mechanisms of allosteric modification of ANF-RGC by staurosporine and AMP-PNP, a non-hydrolyzable analogue of ATP are nearly (or totally) identical.
Keywords: ANF receptor guanylate cyclase, membrane guanylate cyclase, allosteric regulation, signal transduction, ATP, staurosporine
INTRODUCTION
ANF-RGC is the prototype mammalian membrane guanylate cyclase [1] whose discovery demonstrated that the membrane guanylate cyclases belong to the surface receptor family, ANF-RGC being the receptor of ANF and BNP [reviewed in 2–4]. With the discovery of two other—CNP-RGC and STa-RGC, the guanylate cyclase surface receptor family, composed of these three members was recognized [reviewed in: 2–5]. CNP-RGC is the receptor of C-type natriuretic peptide (CNP) and STa-RGC, the receptor of enterotoxin, guanylin and uroguanylin. These three guanylate cyclases have also been respectively termed as GC-A GC-B and GC-C [reviewed in: 2–5].
With the discovery of the Ca2+-modulated membrane guanylate cyclase, ROS-GC, that was solely modulated by the intracellular levels of Ca2+ within the photoreceptors outer segments, the membrane guanylate cyclase family branched into two subfamilies, peptide hormone receptor and Ca2+-modulated ROS-GC. The family became the transducer of both types of signals, generated outside and inside the cells. The ROS-GC subfamily consists of three members—ROS-GC1, ROS-GC2, and ONE-GC, they have alternately been termed as GC-E, GC-F, and GC-D, respectively [reviewed in: 2, 3, 6–9]. The recent discovery demonstrates that one member of this subfamily, ONE-GC, is also a receptor of an extracellular ligand, the odorant uroguanylin [10–12]. Also, recent studies show that prior to Ca2+ signal, illuminated rhodopsin recognized by the ROS-GC1 extracellular domain is required for the physiological level of ROS-GC1 activation during the recovery phase of phototransduction [13]. Therefore, as of today, there is evidence that two members of the Ca2+ modulated membrane guanylate cyclase subfamily are also regulated by signals directed toward their extracellular domains.
All members of the membrane guanylate cyclase family are single transmembrane-spanning proteins, composed of modular blocks [reviewed in: 2, 3]. In their functional forms, they are all homo-dimeric. In each mono-meric subunit, the transmembrane module divides the protein into two roughly equal portions, extracellular and intracellular. The individual modules within each portion provide functional uniqueness to each member of the guanylate cyclase family. Each modular block within the extracellular region of the receptor guanylate cyclases uniquely senses its peptide hormone signal and within the intracellular block of a ROS-GC its Ca2+ signal. The catalytic domain in each membrane guanylate cyclase resides in its intracellular region. However, topographical arrangement of this domain differs in the two sub-families. In the peptide hormone receptor, it is at the C-terminal end and in the ROS-GC it is followed by a C-terminal extension [reviewed in: 3, 4]. Similar topology holds for the third subfamily member, ONE-GC.
As ANF-RGC is a prototype of membrane guanylate cyclases, ANF is a prototype of the natriuretic peptide family [reviewed in: 14–16]. Gene-knock out studies link ANF and ANF-RGC with salt-sensitive [17] and salt-in-sensitive hypertension [18]. Thus, ANF and ANF-RGC are critical components of the renal and cardiovascular physiology.
Initial studies with the crude enzyme indicated that ATP facilitates and subsequently, reconstitution studies with the isolated ANF-RGC demonstrated that ATP is an obligatory transduction factor in ANF dependent ANF-RGC signaling [19–22; reviewed in: 5, 23, 24]. These studies resulted in the formulation of a two-step model (Model 1) for ANF signal transduction [23, 24]. In step 1, ANF binds to its extracellular receptor domain and exposes the intracellular ARM domain of the guanylate cyclase; in step 2, ATP binds to the exposed ARM domain, causes a cascade of structural changes and activates its catalytic domain located at its C-terminus. The final result is the transduction of the ANF signal into the production of its second messenger, cyclic GMP. This signal transduction model recognizes that one of the events involved is phosphorylation of ANF-RGC [23, 24]. But this event follows and is subordinate to the direct ATP-binding allosteric effect [23, 24].
In the subsequently proposed model (Model 2), ATP initiates the ANF signal by phosphorylating ANF-RGC through a hypothetical protein kinase [25, 26]. Important distinctive feature of this model is that only after this phosphorylation ANF-RGC is able to bind ANF [25]. In a successive step, ATP allosterically modifies and activates ANF-RGC [25].
The basis of the original proposal for the direct ATP modulation model for ANF-RGC signaling (Model 1) was that the non-hydrolyzable analogue of ATP, AMP-PNP, mimicked from 60 to 70% the ATP effect in ANF activation of ANF-RGC activity [22, 25]. The remaining 40 to 30 % of ATP activity in ANF-RGC activation was predicted to be due to phosphorylation of ANF-RGC which follows the allosteric step, a prediction in accord with the results obtained with another non-hydrolyzable analogue of ATP, ATPγS [22, 25]. ATPγS was the most potent effector in the ANF signaling of ANF-RGC, it caused approximately 20% higher activation of ANF-RGC than ATP [22, 25].
Since its original proposition, the ATP-dependent two-step model of ANF-RGC signaling has been comprehensively tested and mechanistically advanced. Most significantly, the ATP signaling of ANF-RGC has been demonstrated through direct binding of 8-azido-ATP [27, 28]. In addition, the systematic analysis through the techniques of steady state, time resolved tryptophan fluorescence and Förster Resonance Energy transfer (FRET), site-directed and deletion mutagenesis, reconstitution and molecular modeling studies have validated the basic operational principles of the model and revealed many of its structural elements [29]. The ATP-binding ARM domain of ANF-RGC has two distinct structural elements. One is the ATP-binding pocket and second, the transduction region. The binding pocket resides in the smaller, N-terminal lobe of the ARM domain and the transduction region in the larger, predominantly helical, C-terminal lobe [29, 30], reviewed in [5, 23, 24]. The ATP binding to the pocket causes a cascade of sequential stereo-specific changes, which lead to exposure of the 669WTAPELL675 transduction motif; the exposure, in turn, facilitates activation of the catalytic module [29].
Despite the overwhelming evidence in support of the direct ATP-modulated two-step model, the direct biochemical segregation of the ATP allosteric activation versus indirect ATP phosphorylation, have not been accomplished. The present investigation addresses this issue. Through a novel probe, staurosporine, the study shows that ATP stimulates ANF signaling of ANF-RGC in a direct allosteric fashion. And this stimulation is independent of its phosphorylation activity.
RESULTS
Rationale for development of the staurosporine probe
To segregate the ATP allosteric step from the phoshorylation step in the ANF signaling of ANF-RGC, the alkaloid, staurosporine, was used in place of ATP. There were four reasons for this choice: 1) ANF-RGC ARM domain, the site of ATP binding, exhibits sequence homology with the catalytic domains of tyrosine kinases [30]; 2) staurosporine binds to the same as ATP site of various protein kinases’ including tyrosine kinases [31–34]; 3) staurosporine uses the same as ATP hydrogen bond interactions in its binding to protein kinases [31–34]; and 4) staurosporine exhibits higher than ATP affinity for the specific binding site [31–34]. It was, therefore, hypothesized that staurosporine, like ATP, should also bind to the ARM domain of ANF-RGC and mimic the allosteric aspect of ATP action in ANF signaling of ANF-RGC. But this process will not involve phosphorylation. This hypothesis was tested and found to be correct as described below.
Staurosporine binds to the ATP-binding pocket
To determine if staurosporine binds to the ATP site of the ARM domain, the approach of competitive displacement was used. It was reasoned that if staurosporine binds to the ATP-binding pocket, it should competitively displace ATP from this site. This proposition was tested using the isolated ARM domain residues 486–692 [29] and [γ32P]-8-azido-ATP. Specificity of [γ32P]-8-azido-ATP binding to the ARM domain has been shown previously [27].
The ARM domain was incubated with 1 μCi (100 pmoles) of [γ32P]-8-azido-ATP in the presence or absence of staurosporine and UV irradiated (cross-linked). In side-by-side performed control experiment, unlabelled ATP was added instead of staurosporine and the reaction mixture was UV irradiated. Each reaction mixture was resolved by SDS-PAGE and the radiolabeled protein was visualized by autoradiography (Fig. 1A). As anticipated, addition of nonradioactive ATP to the reaction mixture prevented binding of [γ32P]-8-azido-ATP to the ARM domain [Fig. 1A: compare lane “0” (only [γ32P]-8-azido-ATP present) with lane “1 mM ATP”]. These results were identical to those previously reported [27, 29] and confirmed that the ARM domain used for the study was functional.
Figure 1. Displacement of [γ32P]-8-azido-ATP by staurosporine.

(A) Qalitative analysis. The ANF-RGC ARM domain protein (1 μg of protein for one reaction) was UV cross-linked with [γ32P]-8-azido-ATP in the absence or presence of 100 μM staurosporine or 1 mM ATP. Lane “0” - only [γ32P]-8-azido-ATP present; lane “1 mM ATP” – 1 mM ATP was present in addition to [γ32P]-8-azido-ATP; lane “100 μM S” – 100 μM staurosporine was present in addition to [γ32P]-8-azido-ATP. The reaction mixtures were analyzed through SDS-15%PAGE and autoradiographed. The radioactive band corresponding to the [γ32P]-8-azido-ATP cross-linked ARM domain is indicated on both sides of the autoradiogram by arrows. (B) Quantitative analysis. The ANF-RGC ARM domain protein (1 μg of protein for one reaction) was UV cross-linked with [γ32P]-8-azido- ATP in the presence of indicated concentrations of staurosporine (closed circles) or ATP (open circles). Radiolabeled bands were cut out from the gel, counted for radioactivity and the percentage of radioactivity retained was calculated. The experiment was repeated three times and the results presented are mean ± SD of these experiments.
When the cross-linking of the ARM domain with [γ32P]-8-azido-ATP was performed in the presence of 100μM staurosporine, the intensity of the band corresponding to the radiolabeled protein on the autoradiogram was significantly lower than the intensity of the band corresponding to the cross-linked protein in the absence of staurosporine (Fig. 1A: compare lane “0” with lane “100 μM S”). These results show that staurosporine, displaces [γ32P]-azido-ATP from the ARM domain thus, it binds to the same as ATP binding site.
To determine the kinetics of the ATP displacement by staurosporine, [γ32P]-azido-ATP was UV cross-linked with the ARM domain in the presence of increasing concentrations of staurosporine. After visualization of the cross-linked protein by autoradiography, the original gel was aligned with the autoradiogram, the bands corresponding to the radiolabeled proteins were excised from the gel and counted for radioactivity. The results are presented in figure 1B. They show that staurosporine displaces [γ32P]-azido-ATP in a dose-dependent fashion. Fifty percent of the bound [γ32P]-8-azido-ATP was displaced by 70 μM staurosporine and 250 μM staurosporine displaced almost 90% of the bound [γ32P]-8-azido-ATP. When, in parallel experiments, ATP was used, 0.45 mM ATP was needed to displace 50% and 4 mM ATP was needed to displace about 85% of [γ32P]-8-azido-ATP (Fig. 1C and ref [27]). The fact that almost one-order of magnitude lower concentration of staurosporine than of ATP is needed to displace half of the [γ32P]-8-azido-ATP bound shows that staurosporine has higher than ATP affinity for the binding site in the ARM domain.
Molecular modeling: Staurosporine and ATP bind to the same site - ARM domain model explains the competition results
To explain the ATP/staurosporine competitive binding results in 3D-terms, the ARM domain model [30] was analyzed. The original 3-D model of the ARM domain was built using crystal structures of insulin receptor kinase (IRK) and haematopoietic cell kinase (Hck) as the templates [30, PDB 1T53]. To analyze the binding of staurosporine to the ARM domain through molecular modeling it was necessary to align the domain’s structure with a structure of a protein kinase complexed with staurosporine. For this purpose the structure of the spleen protein tyrosine kinase (SYK) catalytic domain co-crystallized with staurosporine (PDB file 1XBC) was used. The structure of the SYK catalytic domain was superimposed on the structure of the ARM domain along the C-αs of all the residues and that are conserved in the protein kinase family. These residues, G503, G509, L511, K535, E551, T580, N633, V635, D646 of the ARM domain and G378, G383, V385, K402, E420, M448, N499, L501 and D512 of the SYK catalytic domain (Table 1), are indicated in figure 2 showing the superimposed structures of the ARM domain and SYK catalytic domains.
Table 1. Conserved amino acid residues in the ANF-RGC ARM domain and the catalytic domain of spleen tyrosine kinase (SYK).
The C-αs of the conserved in the ARM domain and SYK catalytic domain amino acid residues were used to align 3-D structures of these domains. These residues are listed as corresponding pairs.
| ARM domain | SYK |
|---|---|
| G503 | G378 |
| G509 | G383 |
| L511 | V385 |
| K535 | K402 |
| E551 | E420 |
| T580 | M448 |
| N663 | N499 |
| V635 | L501 |
| D646 | D512 |
Figure 2. Structure comparison of the SYK catalytic domain and the ARM domain.
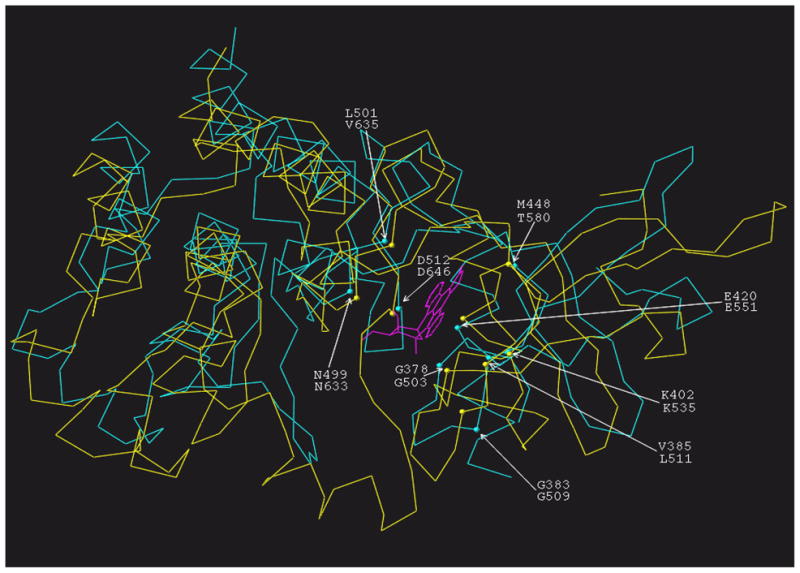
3D modeled structure of the ANF-RGC ARM domain (PDB file 1T53, shown in yellow) and the crystal structure of the spleen tyrosine kinase (SYK) catalytic domain co-crystallized with staurosporine (PDB file 1XBC; shown in cyan) were superimposed along the amino acid residues present in the ATP binding pocket and conserved in protein kinases. The C-alpha atoms of amino acid residues of the ARM and SYK domains used for the alignment are indicated by yellow and cyan balls, respectively and identified for their positions. For clarity the amino acid residues are denoted in one-letter code; the upper letter indicates the residue in the SYK catalytic domain and the lower, the residue in the ARM domain. The staurosporine molecule is shown in magenta. As can be seen in the figure, both structures have similar structural arrangement with the RMS deviation of 1.9 Å.
This figure shows that the structural features of both proteins are almost identical. In the quantitative terms, the high degree of overlap of the two structures is reflected in the value of the root mean square deviation (RMSD). It is the measure of the average distance between the backbones of the superimposed proteins. For the SYK catalytic domain and the ARM domain the RMSD value is 1.9 Å.
Docking of staurosporine to the ARM domain
It has been shown through co-crystallography studies that staurosporine binds to the ATP binding pocket of protein kinases and the binding induces conformational changes that mimic ATP binding [31–34]. Most of the residues in and around the ATP binding pocket of the ARM domain are homologous to the corresponding residues in protein kinases (see Table 1). This fact made it possible to use the molecular replacement logic [35] for docking the staurosporine molecule to the ARM domain. In this approach the information about the position and relative orientation of a known structure is used to map the position of non-crystallographic operators with respect to the crystallographic symmetry elements.
The staurosporine molecule extracted from the complex SYK-staurosporine was merged into the ARM domain, the torsion angles of the side chains of the amino acid residues surrounding staurosporine were optimized for generating possible hydrogen bonds and the bad steric contacts that are invariably introduced during the merging procedure were removed. Finally, the staurosporine-ARM domain complex was energy optimized. The complex is shown in figure 3A and its features are described below.
Figure 3.
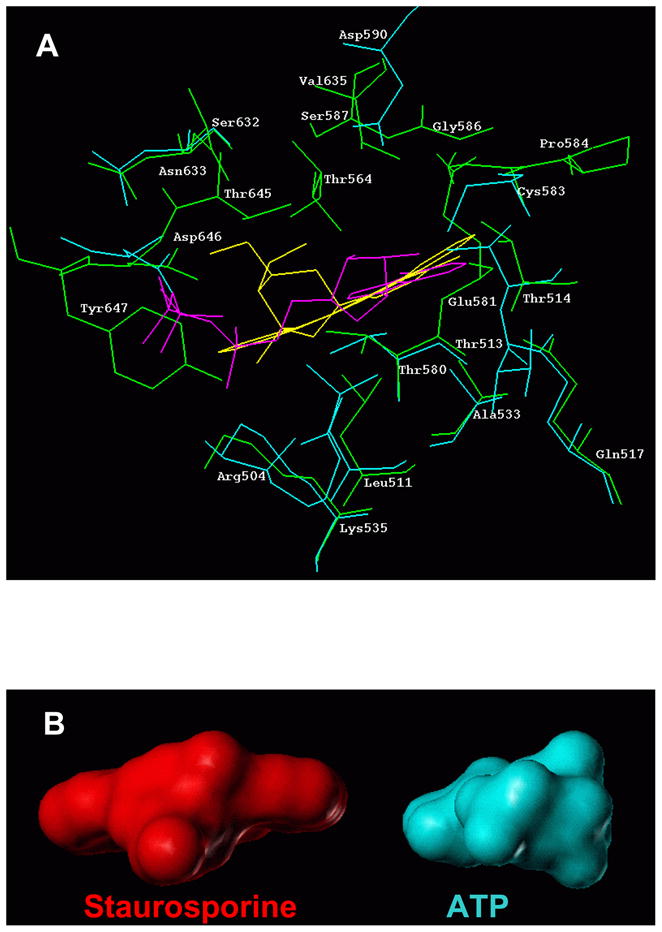
(A) Docking of staurosporine into the ARM domain of ANF-RGC - Comparison with ATP docking. Staurosporine molecule (shown in yellow) extracted from its complex with SYK catalytic domain (PDB file 1XBC) was docked into the ARM domain. For comparison, docking of ATP (shown in magenta) is also provided. Amino acid residues constituting the staurosporine binding pocket are shown in green while those constituting the ATP binding pocket are in cyan. For clarity, only residues within 4 Å radius around the respective molecule are shown. (B) Models of staurosporine and ATP. The space filling models of staurosporine and ATP are shown side-by-side to illustrate the difference if sizes of the two molecules.
Staurosporine is a relatively rigid molecule with tetrahydropyran ring adopting a boat conformation. Its total surface area of 411Å2 is nearly 3 times larger than the surface of the purine ring of the ATP molecule (to illustrate the difference in sizes between staurosporine and ATP, the space filling models of these molecules are presented in figure 3B). In complex with the ARM domain, staurosporine occupies the ATP binding site (Fig. 3A: the ATP molecule is shown in magenta and staurosporine in yellow). Because of its size, staurosporine induces conformational changes to fit perfectly (induced fit) in the cleft between the N- and C-terminal lobes of the ARM domain.
When binding to the tyrosine protein kinases, staurosporine exploits similar as ATP hydrogen bond interactions. Therefore the model was analyzed to determine whether the same is true for staurosporine interaction with the ARM domain. 18 amino acid residues of the ARM domain fall within ~4 Å sphere around the staurosporine molecule; they are: L511, T514, Q517, A533, K535, T564, T580, E581, C583, P584, Gly586, S632, N633, V635, T645, Y647, D646 and Y657. Out of these, L511, T514, Q517, A533, K535, T580, C583, N33, V635, T645 and D646 are part of the original ATP binding pocket. There are at least two hydrogen bonds in the complex; one is formed between the carbonyl oxygen of the staurosporine and C583 and another, between T645 and the glycosyl portion of the staurosporine molecule (Fig. 4A). There is also a strong possibility that K535 forms a hydrogen bond with staurosporine’s endo-cyclic oxygen when present in solution. In addition to the hydrogen bonds, staurosporine interacts with L511, T514, T580 and Y647 of the ARM domain through non-binding/van der Waals’s force. To illustrate the position of staurosporine within its binding pocket the space filling model of staurosporinew and the pocket is shown in figure 4B whereas figure 4C shows the staurosporine binding pocket as a part of the entire ARM domain. These results confirm the biochemical findings that: 1) staurosporine binds to the ARM domain; and 2) the staurosporine binding site is the same as the ATP binding site.
Figure 4. Staurosporine binding pocket in the ARM domain of ANF-RGC.

(A) The ARM domain residues forming the staurosporine (shown in yellow) binding pocket are shown. Two hydrogen bonds (shown by dotted lines), anchor staurosporine while non-bonding van der Waals’ interactions, provide a stable complex. Although Lys535 does not show direct hydrogen bonding in the existing conformation it may form a hydrogen bond with staurosporine’s endo-cyclic oxygen when present in solution. (B) The ARM domain residues (yellow) located within the 4-Å sphere from the interacting staurosporine (red) are depicted in a space filling model. (C) The localization of the staurosporine binding pocket within the ARM domain. Red-staurosporine; yellow – amino acid residues forming the staurosporine binding pocket; green – ARM domain residues outside the staurosporine binding pocket.
Docking of staurosporine to the ARM domain also explains staurosporine’s higher than ATP affinity for the binding pocket. It can be attributed to the fact that staurosporine molecule is bigger than ATP. There are 18 amino acid residues of the ARM domain surrounding staurosporine but only 12 surrounding ATP adenine ring. Therefore, staurosporine has higher possibility than ATP to interact with the surrounding residues through van der Waals’ forces providing for more stable complex.
Staurosporine allosterically modulates ANF-dependent activation of ANF-RGC
Having determined that staurosporine binds to the same as ATP site in the ARM domain, the next question was: On a functional level, can staursporine mediate the ANF-dependent ANF-RGC activation thus, mimic ATP activity?
To address this issue, membranes of COS cells expressing ANF-RGC were exposed to 10−7 M ANF in the presence of increasing (10 nM -100 μM) concentrations of staurosporine. In control experiments the membranes were exposed to 10−7 M ANF only. ANF alone stimulated ANF-RGC activity minimally, from 7 to 11 pmol cyclic GMP formed/min/mg protein. In the presence of ANF and staurosporine the ANF-RGC activity was stimulated in a dose-dependent fashion (Fig. 5A). The half-maximal stimulation was observed at 50 nM staurosporine and the maximal stimulation, of 4.5 ± 0.5 fold above the basal activity, at approx. 500 nM (Fig. 5A). The concentration of staurosporine resulting in the half-maximal stimulation of ANF-RGC was ~4 orders of magnitude lower than the 0.3 mM concentration of ATP necessary for half-maximal stimulation of ANF-dependent ANF-RGC activity [22, 36] and is comparable with staurosporine concentration exhibiting half-maximal effect on protein kinases activities [31]. These results show that staurosporine is an efficient modulator of ANF-dependent signaling of ANF-RGC. And, importantly, because staurosporine cannot act as a substrate of any protein kinase they show, that phosphorylation is not indispensable for the activation of ANF-RGC.
Figure 5. Staurosporine mimics the allosteric ATP effect on ANF-dependent ANF-RGC activity.

(A) Membranes of COS cells expressing ANF-RGC were incubated with 10−7 M ANF in the presence of indicated concentrations of staurosporine, AMP-PNP or ATP. (B) Membranes of COS cells expressing ANF-RGC were incubated with 10−7 M ANF and/or 0.5 mM AMP-PNP, 10 μM staurosporine or were incubated with 10−7 M ANF, 0.5 mM AMP-PNP and 10 μM staurosporine. The experiments were done in triplicate and repeated three times. The values presented are average (± SD) from these experiments.
This conclusion was further validated by comparing the effectiveness of staurosporine with those of ATP and its non-hydrolyzable analog AMP-PNP. AMP-PNP caused ANF signaling of ANF-RGC with almost identical as staurosporine Vmax value whereas with ATP the Vmax value was ~40% higher (Fig. 5). These results show that the phosphorylation independent allosteric step results in partial activation of ANF-RGC. When this step is followed by phosphorylation, the cyclase becomes fully active. Thus, phosphorylation is subordinate to the allosteric effect and it is not the primary requirement. This conclusion is in agreement with the two-step signal transduction model of ANF-RGC.
Finally, the ANF signaling of ANF-RGC was assessed by the simultaneous presence of both AMP-PNP and staurosporine in the reaction mixture (Fig. 5B). ANF, AMP-PNP or staurosporine alone did not stimulate significantly the activity of ANF-RGC. ANF (10−7 M) together with 0.5 mM AMP-PNP or 10 μM staurosporine stimulated the activity approx. 5 fold above the basal value (from 6.8 to 34 and 29 pmol cyclic GMP/min/mg prot, for AMP-PNP and staurosporine, respectively) (Fig. 5B). Similar stimulated activity, 30±2.5 pmol cyclic GMP/min/mg prot, was observed when 0.5 mM AMP-PNP and 10 μM staurosporine were present together (Fig. 5B: solid bar). Because the effects of AMP-PNP and staurosporine on ANF-dependent ANF-RGC activity are not additive, these results provide additional confirmation that staurosporine acts through the same as ATP signaling site of ANF-RGC.
The mechanism of staurosporine activation of ANF-RGC
Systematic analysis of the ARM domain has established that it contains a signature transduction domain motif that is critical for the ANF/ATP signaling of ANF-RGC. The structure of this motif is 669WTAPELL675 [23, 24, 29]. It resides in the larger lobe of the ARM domain [23, 24, 29]. The two-step model predicts that the ATP binding-dependent configurational changes in the smaller lobe are transmitted to the larger lobe and cause a movement of the EF helix by 2–5 Å [23, 24, 29]. The 669WTAPELL675 motif constitutes the EF helix. As the consequence of the movement this hydrophobic motif becomes exposed and able to directly or indirectly activate the ANF-RGC catalytic domain. Critical role of the 669WTAPELL675 motif and ANF/ATP-dependent activation of ANF-RGC was experimentally validated [29]. It was, therefore, predicted that because staurosporine mimics ATP, it should function through the 669WTAPELL675 motif of the ARM in the activation of ANF-RGC.
To assess this possibility, the 669WTAPELL675 deletion mutant of ANF-RGC was analyzed for ANF/staurosporine-dependent activation of the cyclase catalytic domain. It has been shown previously that deletion of the 669WTAPELL675 sequence form ANF-RGC does not affect expression of the mutant-protein in the membrane compartment of the cell, its basal cyclase activity and ability to bind ANF [29].
The truncated-669WTAPELL675- ANF-RGC mutant was expressed in COS cells and their membrane fraction was exposed to 10−7 M ANF and increasing concentrations of staurosporine. As positive control, membranes of COS cells expressing wild type ANF-RGC were treated identically. The results are shown in figure 6. As anticipated, deletion of the 669WTAPELL675 motif resulted in complete loss of the staurosporine-mediated ANF stimulation of the ANF-RGC. These results indicate that similar to ATP, 669WTAPELL675 is the signature transduction motif for the staurosporine-mediated ANF signaling of ANF-RGC and the motif is critical for activation of the catalytic domain.
Figure 6. Role of the 669WTAPELL675 motif in ANF-RGC signal transduction.

Wild type ANF-RGC, the 669WTAPELL675 deletion, the W669A substitution and W669F substitution mutants were individually expressed in COS cells and their membranes were analyzed for ANF/staurosporin-dependent cyclase activity. Cyclic GMP formed was measured by radioimmunoassay. The experiment was done in triplicate and repeated two times for reproducibility. The results presented (mean ± SD) are from these experiments.
Although all residues of the 669WTAPELL675 motif contribute to its functional significance the W669 residue is pivotal [29]. The ATP binding-dependent reorientation of its side chain pushes the remainder of the motif, 670TAPELL675, to the surface. This function of the W699 residue is due to its bulky, aromatic ring structure [29]. Therefore, the next question was: Does the same principle apply to the staurosporine action? The question was answered by comparing the responses to staurosporine and ANF of two ANF-RGC mutants: one, in which the W669 residue was mutated to small aliphatic amino acid alanine (W669A mutant) and the other, in which W669 was mutated to another aromatic amino acid phenylalanine (W669F mutant). The basal activities of these mutants were respectively 6.8 and 7.1 pmol cyclic GMP formed/min/mg protein, and were comparable to the 7 pmol cyclic GMP formed/min/mg protein activity of the wild-type ANF-RGC. The mutants, however, responded in different manners to the ANF/staurosporine stimulation (Fig. 6). The saturation activity of wild type ANF-RGC was 4.8-fold over its basal value, it was 2.3-fold for W669A mutant and 4.3-fold for the W669F mutant. These results are comparable to that obtained previously with ANF and ATP [29]. They validate the proposed mechanism of ANF-RGC catalytic domain activation through the 669WTAPELL675 transduction motif and validate the notion that an aromatic residue at the 669 position is necessary for the functionality of this motif.
DISCUSSION
The objective of the study was to segregate the ATP allosteric modulation from ATP phosphorylation in the process of ANF-dependent ANF-RGC activation. To separate these two ATP roles, the availability of an ATP substitute that would mimic its allosteric but not phosphorylating function was necessary. In a search for such a substitute, advantage was taken from the knowledge that the ARM domain exhibits sequence homology with catalytic domain of protein tyrosine kinases (hence this region was originally termed “kinase homology domain” [37]) and that staurosporine has higher than ATP affinity for the catalytic domains of various protein kinases [31–34]. Based on these facts it was reasoned that staurosporine should bind to the ATP binding pocket of ANF-RGC ARM domain and mimic the ATP allosteric effect in ANF signaling of ANF-RGC activation. Validity of this reasoning was tested experimentally by answering the following questions:
Question 1
Does staurosporine bind to the ARM domain of ANF-RGC?
This issue was analyzed through binding competition and molecular modeling. Increasing concentrations of staurosporine displace [γ32P]-8-azido-ATP cross-linked with the ARM domain in a dose-dependent fashion. Kinetics of the displacement show that the affinity of staurosporine to the ARM domain is higher than that of ATP.
Molecular modeling, involving docking of staurosporine to the ARM domain shows that it binds to the same as ATP site. Out of the 18 amino acid residues of the ARM domain that surround staurosporine molecule 12 belong to the ATP binding pocket. The ability of staurosporine to interact through hydrogen or van der Waals’ bonds with higher number of amino acid residues of the ARM domain explains its higher affinity for the binding site. Thus, the two lines of experiments allow answering the posed question in affirmative.
Question 2
If staurosporine binds to the same as ATP site in the ARM domain, can it also modulate ANF-dependent activity of ANF-RGC?
This question was answered through assessing the ANF-RGC activity in the presence of physiological concentrations of ANF and varying concentrations of staurosporine, ATP or its nonhydrolyzable analog, AMP-PNP. Results of these experiments show that both staurosporine and AMP-PNP cause similar maximal stimulated activity. The stimulation was approx. 40% higher when ATP was present.
Question 3
How the staurosporine modulatory effect relates to the ATP signaling model?
The results presented show that staurosporine and AMP-PNP, two effectors that cannot act as substrates of any protein kinase are efficient modulators of the ANF-dependent ANF-RGC activity. Therefore, the allosteric modulation reflected in the effect of staurosporine and AMP-PNP is an independent step sufficient to activate, although not to the full extent, ANF-RGC. The ~40% increase in Vmax caused by ATP may be attributed to phosphorylation of ANF-RGC by ATP in a subsequent step, leading to full activation of the cyclase. These sequence of events was predicted in the original model [23] and has been now validated: Upon ATP binding to the ARM domain there is a movement and rotation (ATP allosteric effect) of the region (strands β1, β2 and the loop between them within the smaller lobe of the ARM domain [23]) where the residues determined to be phosphorylated [26] are located. The consequence of the movement is a change in the positions of their side chains from buried to exposed, thus they become available for phosphorylation (ATP phosphorylation effect).
Question 4
Is the modulatory effect of staurosporine transduced through the same mechanism as that of ATP?
Another aspect of the ATP allosteric regulation of ANF-RGC activity is centered on a conserved hydrophobic motif 669WTAPELL675. Based on initial modeling studies involving comparison of the ARM domain structure in its apo and ATP-bound states it was hypothesized that this motif, distal to the ATP binding pocket, is involved in ANF/ATP-dependent stimulation of ANF-RGC [23, 24] in a following manner: As a result of ATP binding, the entire ARM domain acquires a more compact structure, there is a reorientation of tryptophan-669 (W669) side chain and movement of the side chains of T670, E673, L674 and L675 toward the protein surface [29]. The movement of the 669WTAPELL675 motif towards the surface of the protein facilitates its interaction with subsequent transduction motif, possibly within the catalytic domain, propagation of the ANF/ATP binding signal and activation of the catalytic domain. This hypothesis was previously experimentally validated through tryptophan fluorescence and mutagenesis/expression experiments [29].
If the 669WTAPELL675 motif is indeed part of the ATP-allosteric mechanism then it should be also involved in the staurosporine-mediated activation of ANF-RGC? Using the ANF-RGC 669WTAPELL675 deletion mutant the present study shows that the 669WTAPELL675 motif is absolutely critical for ANF/staurosporine signaling of ANF-RGC (Fig. 6) indicating that both, ATP and staurosporine use the same transduction motif in their stimulatory modes.
Previous studies have also shown that W669 is the key residue for the functionality of the 669WTAPELL675 motif [29]. It was reasoned and experimentally validated that the special function of the W699 residue is due to its bulky and/or aromatic side chain which enables it to act as a lever which upon ATP binding pushes the 670–675 residues, TAPELL, to the surface. Bigger the side chain of the 669 amino acid residue, more efficient it is in pushing the other residues up. This study shows that the same logic applies to staurosporine as the allosteric regulator. Phenylalanine substitutes for tryptophan at the 669 position with over 90% efficiency but alanine substitutes in only 50%.
CONCLUSION
In conclusion, the present study segregates the ATP allosteric effect from the phosphorylation effect, and demonstrates that ATP stimulates ANF signaling of ANF-RGC in a direct allosteric fashion. And this stimulation is independent of its phosphorylation activity.
Together with the earlier massive evidence on the mechanistic steps involved in the ARM modification upon ATP binding, the basic principles of the two-step ANF signal transduction mechanism are depicted in figure 7 and summarized as follows: The dimer form of the extracellular receptor domain binds to one molecule of ANF [38–40]. The binding modifies the juxtamembrane region [41] where the disulfide 423Cys-Cys432 structural motif is a key element in this modification [42]. The signal twists the transmembrane domain and induces the structural change in ARM domain allowing it to bind ATP [40]. Upon interaction with its binding pocket, ATP induces a cascade of temporal and spatial changes in the entire ARM domain [23, 24, 29; reviewed: 4, 5]. One of these changes is the exposure of the 669WTAPELL675 motif, which facilitates its direct (or indirect) interaction with and activation of the catalytic module [29]. The catalytic domain is in a form of an antiparallel dimer [43, 44]. Another change is the exposure of the six phosphorylation sites within the ARM domain [26], which subsequently become phosphorylated. Collectively, these two ATP effects result in full activation of ANF-RGC.
Figure 7. Two-step activation of ANF-RGC – a model.
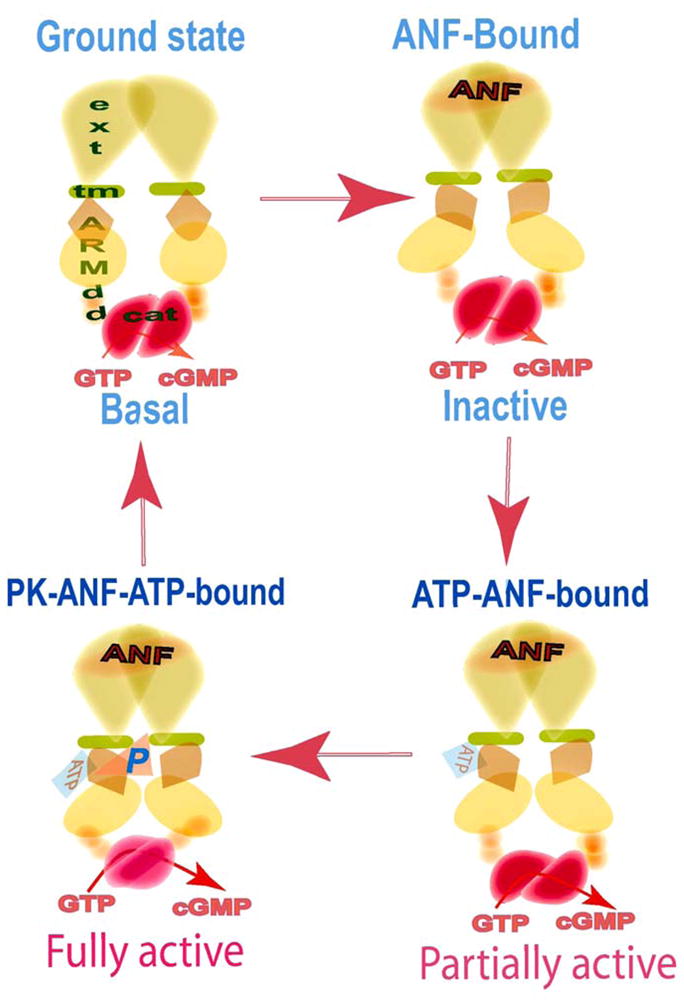
The functional domains are denoted as: ext, extracellular domain; tm, transmembrane domain; ARM, ATP regulatory domain; dd, dimerization domain; cat, catalytic domain. Ground state - In its basal state ANF-RGC exists as a dimer. ANF-Bound - The signal transduction process is initiated by binding of one molecule of ANF to the extracellular domain dimer. The binding modifies and twists the hinge juxtamembrane region and induces the structural change in ARM domain allowing it to bind ATP. The cyclase activity remains at its basal value. ATP-ANF-bound - Upon interaction with its binding pocket, ATP induces a cascade of temporal and spatial changes in the entire ARM domain. The cyclase is partially activated. PK-ANF-ATP-bound - The six serine/threonine residues within the ARM domain become phosphorylated and the 669WTAPELL675 interacts with and activates the dimeric catalytic domain. The cyclase is fully active. Note: the protein kinase (PK) involved in the phosphorylation of the ARM domain has not been identified yet.
EXPERIMENTAL PROCEURES
Materials
ATP and AMP-PNP were purchased from Roche, ANF from Bachem AG, 8-azido-ATP from Affinity Photoprobes, Inc., and staurosporine from Sigma.
Mutagenesis
Point- and deletion-mutants of ANF-RGC were constructed using Quick-change mutagenesis kit (Stratagene) and appropriate mutagenic primers as described in [29].
Expression in COS cells
COS-7 cells were transfected with ANF-RGC or its mutants’ cDNA using a calcium-phosphate coprecipitation technique [45]. Sixty hours after transfection, the cells were harvested and their membranes prepared [27, 29, 30].
Guanylate cyclase activity assay
Membranes of COS cells expressing ANF-RGC or its mutants were preincubated with or without 10−7 M ANF and varying concentrations of ATP, AMP-PNP or staurosporine for 10 min on ice. The assay system contained 10 mM theophylline (phosphodiesterase inhibitor), 15 mM phosphocreatine, 20 μg creatine kinase, 50 mM Tris-HCI, pH 7.5. The total assay volume was 25 μl. The reaction was initiated by the addition of the substrate solution containing 4 mM MgCl2 and 1 mM GTP, continued for 10 min at 37 °C, and terminated by the addition of 225 μl of 50 mM sodium acetate buffer (pH 6.25) followed by heating in a boiling water-bath for 3 min. The amount of cyclic GMP formed was quantified by radioimmunoassay [46]. All experiments were done in triplicate.
Expression and purification of ANF-RGC ARM domain
The ARM domain fragment aa 486–692, was amplified from ANF-RGC cDNA by PCR and directly cloned into the ligation independent site of pET-30aXa/LIC vector (Novagen). The protein was expressed and purified to homogeneity as described in [29].
UV cross-linking
According to the previous protocol [27, 29], 1 μg (50 pmol) of the purified protein (ARM domain) in 20 mM phosphate buffer pH 7.5 was incubated for 5 min with 100 pmol of 8-azido-ATP, 1 μCi [γ32P]-8-azido-ATP (specific activity 10–15 Ci/mmol), 1 mM MgCl2 and ATP or staurosporine in a total volume of 25μl. The reaction mixture was UV irradiated (254 nm) and analyzed by SDS-15%PAGE followed by autoradiography and liquid scintillation counting.
Molecular modeling
Molecular modeling was done on a Silicon Graphics workstation using Sybyl molecular modeling package [SYBYL modeling software (version 6.6) Tripos Associate Inc.] and the figures were generated using Molmol [47]. Crystal structure of the spleen protein tyrosine kinase (SYK) catalytic domain co-crystalized with staurosporine (PDB file 1XBC) and the previously modeled structure of the ANF-RGC ARM domain [23, 24, 30, PDB file 1T53] were used. The structure of the SYK kinase was superimposed on the structure of the ARM domain along C-α atoms of the amino acid residues that are conserved in both proteins, SYK catalytic domain and ARM domain. These residues are given in Table 1. Using this overlapping protocol the structure of the staurosporine molecule was extracted from the SYK and merged into the ARM domain. Torsion angles of the side chains of amino acid residues surrounding the staurosporine molecule were scanned and optimized for possible hydrogen bonds and the bad contacts were removed. Finally, the structure of the staurosporine-ARM domain complex was energy optimized using Tripos force field and molecular mechanics method [48].
Acknowledgments
The authors thank Mr. Dawid Wojtas for his technical assistance in purification of the ARM domain protein and cross-linking experiments.
FUNDING. This study was supported by the National Institutes of Health National Heart Lung and Blood Institute [grant HL084584].
Footnotes
FINANCIAL INTERESTS. None
References
- 1.Paul AK, Marala RB, Jaiswal RK, Sharma RK. Coexistence of guanylate cyclase and atrial natriuretic factor receptor in a 180-kD protein. Science. 1987;235:1224–1226. doi: 10.1126/science.2881352. [DOI] [PubMed] [Google Scholar]
- 2.Sharma RK. Evolution of the membrane guanylate cyclase transduction system. Mol Cell Biochem. 2002;230:3–30. [PubMed] [Google Scholar]
- 3.Sharma RK. Membrane guanylate cyclase is a beautiful signal transduction machine: overview. Mol Cell Biochem. 2010;334:3–36. doi: 10.1007/s11010-009-0336-6. [DOI] [PubMed] [Google Scholar]
- 4.Garbers DL, Lowe DG. Guanylyl cyclase receptors. J Biol Chem. 1994;269:30741–30744. [PubMed] [Google Scholar]
- 5.Duda T. Atrial natriuretic factor-receptor guanylate cyclase signal transduction mechanism. Mol Cell Biochem. 2010;334:37–48. doi: 10.1007/s11010-009-0335-7. [DOI] [PubMed] [Google Scholar]
- 6.Foster DC, Wedel BJ, Robinson SW, Garbers DL. Mechanisms of regulation and functions of guanylyl cyclases. Rev Physiol Biochem Pharmacol. 1999;135:1–39. doi: 10.1007/BFb0033668. [DOI] [PubMed] [Google Scholar]
- 7.Dizhoor AM, Hurley JB. Regulation of photoreceptor membrane guanylyl cyclases by guanylyl cyclase activator proteins. Methods. 1999;19:521–531. doi: 10.1006/meth.1999.0894. [DOI] [PubMed] [Google Scholar]
- 8.Gibson AD, Garbers DL. Guanylyl cyclases as a family of putative odorant receptors. Annu Rev Neurosci. 2000;23:417–439. doi: 10.1146/annurev.neuro.23.1.417. [DOI] [PubMed] [Google Scholar]
- 9.Sharma RK, Duda T, Venkataraman V, Koch K-W. Calcium-modulated mammalian membrane guanylate cyclase ROS-GC transduction machinery in sensory neurons: A universal concept. Research Trends, Current Topics in Biochemical Research. 2004;6:111–144. [Google Scholar]
- 10.Leinders-Zufall T, Cockerham RE, Michalakis S, Biel M, Garbers DL, Reed RR, Zufall F, Munger SD. Contribution of the receptor guanylyl cyclase GC-D to chemosensory function in the olfactory epithelium. Proc Natl Acad Sci USA. 2007;104:14507–14512. doi: 10.1073/pnas.0704965104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duda T, Sharma RK. ONE-GC membrane guanylate cyclase, a trimodal odorant signal transducer. Biochem Biophys Res Commun. 2008;367:440–445. doi: 10.1016/j.bbrc.2007.12.153. [DOI] [PubMed] [Google Scholar]
- 12.Duda T, Sharma RK. Ca2+-modulated ONE-GC odorant signal transduction. FEBS Lett. 2009;583:1327–1330. doi: 10.1016/j.febslet.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 13.Yamazaki A, Yamazaki M, Yamazaki RK, Usukura J. Illuminated rhodopsin is required for strong activation of retinal guanylate cyclase by guanylate cyclase-activating proteins. Biochemistry. 2006;45:1899–1909. doi: 10.1021/bi0519396. [DOI] [PubMed] [Google Scholar]
- 14.de Bold AJ. Atrial natriuretic factor: a hormone produced by the heart. Science. 1985;230:767–770. doi: 10.1126/science.2932797. [DOI] [PubMed] [Google Scholar]
- 15.Pandey KN. Biology of natriuretic peptides and their receptors. Peptides. 2005;26:901–932. doi: 10.1016/j.peptides.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 16.de Bold AJ, de Bold ML. Determinants of natriuretic peptide production by the heart: basic and clinical implications. J Investig Med. 2005;53:371–377. doi: 10.2310/6650.2005.53710. [DOI] [PubMed] [Google Scholar]
- 17.John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG, Smithies O. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995;267:679–681. doi: 10.1126/science.7839143. Erratum in: Science267, 1753. [DOI] [PubMed] [Google Scholar]
- 18.Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J, Garbers DL, Beuve A. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995;378:65–68. doi: 10.1038/378065a0. [DOI] [PubMed] [Google Scholar]
- 19.Kurose H, Inagami T, Ui M. Participation of adenosine 5′-triphosphate in the activation of membrane-bound guanylate cyclase by the atrial natriuretic factor. FEBS Lett. 1987;219:375–379. doi: 10.1016/0014-5793(87)80256-9. [DOI] [PubMed] [Google Scholar]
- 20.Chang CH, Kohse KP, Chang B, Hirata M, Jiang B, Douglas JE, Murad F. Characterization of ATP-stimulated guanylate cyclase activation in rat lung membranes. Biochim Biophys Acta. 1990;1052:159–160. doi: 10.1016/0167-4889(90)90071-k. [DOI] [PubMed] [Google Scholar]
- 21.Chinkers M, Singh S, Garbers DL. Adenine nucleotides are required for activation of rat atrial natriuretic peptide receptor/guanylyl cyclase expressed in a baculovirus system. J Biol Chem. 1991;266:4088–4093. [PubMed] [Google Scholar]
- 22.Marala RB, Sitaramayya A, Sharma RK. Dual regulation of atrial natriuretic factor-dependent guanylate cyclase activity by ATP. FEBS Lett. 1991;281:73–76. doi: 10.1016/0014-5793(91)80361-6. [DOI] [PubMed] [Google Scholar]
- 23.Sharma RK, Yadav P, Duda T. Allosteric regulatory step and configuration of the ATP-binding pocket in atrial natriuretic factor receptor guanylate cyclase transduction mechanism. Can J Physiol Pharmacol. 2001;79:682–691. [PubMed] [Google Scholar]
- 24.Duda T, Venkataraman V, Ravichandran S, Sharma RK. ATP-regulated module (ARM) of the atrial natriuretic factor receptor guanylate cyclase. Peptides. 2005;26:969–984. doi: 10.1016/j.peptides.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 25.Foster DC, Garbers DL. Dual role for adenine nucleotides in the regulation of the atrial natriuretic peptide receptor, guanylyl cyclase-A. J Biol Chem. 1998;273:16311–16318. doi: 10.1074/jbc.273.26.16311. [DOI] [PubMed] [Google Scholar]
- 26.Potter LR, Hunter T. Phosphorylation of the kinase homology domain is essential for activation of the A-type natriuretic peptide receptor. Mol Cell Biol. 1998;18:2164–2172. doi: 10.1128/mcb.18.4.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burczynska B, Duda T, Sharma RK. ATP signaling site in the ARM domain of atrial natriuretic factor receptor guanylate cyclase. Mol Cell Biochem. 2007;301:193–207. doi: 10.1007/s11010-006-9400-7. [DOI] [PubMed] [Google Scholar]
- 28.Joubert S, Jossart C, McNicoll N, de Lean A. Atrial natriuretic peptide-dependent photolabeling of a regulatory ATP-binding site on the natriuretic peptide receptor-A. FEBS J. 2005;272:5572–5580. doi: 10.1111/j.1742-4658.2005.04952.x. [DOI] [PubMed] [Google Scholar]
- 29.Duda T, Bharill S, Wojtas I, Yadav P, Gryczynski I, Gryczynski Z, Sharma RK. Atrial natriuretic factor receptor guanylate cyclase signaling: new ATP-regulated transduction motif. Mol Cell Biochem. 2009;324:39–53. doi: 10.1007/s11010-008-9983-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duda T, Yadav P, Jankowska A, Venkataraman V, Sharma RK. Three dimensional atomic model and experimental validation for the ATP-Regulated Module (ARM) of the atrial natriuretic factor receptor guanylate cyclase. Mol Cell Biochem. 2001;217:165–172. doi: 10.1023/a:1007236917061. [DOI] [PubMed] [Google Scholar]
- 31.Meggio F, Donella Deana A, Ruzzene M, Brunati AM, Cesaro L, Guerra B, Meyer T, Mett H, Fabbro D, Furet P. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur J Biochem. 1995;234:317–322. doi: 10.1111/j.1432-1033.1995.317_c.x. [DOI] [PubMed] [Google Scholar]
- 32.Prade L, Engh RA, Girod A, Kinzel V, Huber R, Bossemeyer D. Staurosporine-induced conformational changes of cAMP-dependent protein kinase catalytic subunit explain inhibitory potential. Structure. 1997;5:1627–1637. doi: 10.1016/s0969-2126(97)00310-9. [DOI] [PubMed] [Google Scholar]
- 33.Lamers MB, Antson AA, Hubbard RE, Scott RK, Williams DH. Structure of the protein tyrosine kinase domain of C-terminal Src kinase (CSK) in complex with staurosporine. J Mol Biol. 1999;285:713–725. doi: 10.1006/jmbi.1998.2369. [DOI] [PubMed] [Google Scholar]
- 34.Kinoshita T, Matsubara M, Ishiguro H, Okita K, Tada T. Structure of human Fyn kinase domain complexed with staurosporine. Biochem Biophys Res Commun. 2006;346:840–844. doi: 10.1016/j.bbrc.2006.05.212. [DOI] [PubMed] [Google Scholar]
- 35.Rossmann MG. The molecular replacement method. Acta Cryst A. 1990;46:73–82. doi: 10.1107/s0108767389009815. [DOI] [PubMed] [Google Scholar]
- 36.Goraczniak RM, Duda T, Sharma RK. A structural motif that defines the ATP-regulatory module of guanylate cyclase in atrial natriuretic factor signalling. Biochem J. 1992;282:533–537. doi: 10.1042/bj2820533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh S, Lowe DG, Thorpe DS, Rodriguez H, Kuang WJ, Dangott LJ, Chinkers M, Goeddel DV, Garbers DL. Membrane guanylate cyclase is a cell-surface receptor with homology to protein kinases. Nature. 1988;334:708–712. doi: 10.1038/334708a0. [DOI] [PubMed] [Google Scholar]
- 38.Duda T, Goraczniak RM, Sharma RK. Site-directed mutational analysis of a membrane guanylate cyclase cDNA reveals the atrial natriuretic factor signaling site. Proc Natl Acad Sci USA. 1991;88:7882–7886. doi: 10.1073/pnas.88.17.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marala R, Duda T, Goraczniak RM, Sharma RK. Genetically tailored atrial natriuretic factor-dependent guanylate cyclase. Immunological and functional identity with 180 kDa membrane guanylate cyclase and ATP signaling site. FEBS Lett. 1992;296:254–258. doi: 10.1016/0014-5793(92)80298-u. [DOI] [PubMed] [Google Scholar]
- 40.Ogawa H, Qiu Y, Ogata CM, Misono KS. Crystal structure of hormone-bound atrial natriuretic peptide receptor extracellular domain: rotation mechanism for transmembrane signal transduction. J Biol Chem. 2004;279:28625–28631. doi: 10.1074/jbc.M313222200. [DOI] [PubMed] [Google Scholar]
- 41.De Léan A, McNicoll N, Labrecque J. Natriuretic peptide receptor A activation stabilizes a membrane-distal dimer interface. J Biol Chem. 2003;278:11159–11166. doi: 10.1074/jbc.M212862200. [DOI] [PubMed] [Google Scholar]
- 42.Duda T, Sharma RK. Two membrane juxtaposed signaling modules in ANF-RGC are interlocked. Biochem Biophys Res Commun. 2005;332:149–156. doi: 10.1016/j.bbrc.2005.04.102. [DOI] [PubMed] [Google Scholar]
- 43.Venkataraman V, Duda T, Ravichandran S, Sharma RK. Neurocalcin delta Modulation of ROS-GC1, a New Model of Ca(2+) Signaling. Biochemistry. 2008;47:6590–6601. doi: 10.1021/bi800394s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Ruoho AE, Rao VD, Hurley JH. Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc Natl Acad Sci USA. 1997;94:13414–1341. doi: 10.1073/pnas.94.25.13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sambrook MJ, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 46.Nambi P, Aiyar NV, Sharma RK. Adrenocorticotropin-dependent particulate guanylate cyclase in rat adrenal and adrenocortical carcinoma: comparison of its properties with soluble guanylate cyclase and its relationship with ACTH-induced steroidogenesis. Arch Biochem Biophys. 1982;217:638–646. doi: 10.1016/0003-9861(82)90545-8. [DOI] [PubMed] [Google Scholar]
- 47.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 48.Bowen JP, Allinger NL. Molecular Mechanics: The arts and science of parametrization. Rev Compt Chem. 1991;3:81–87. [Google Scholar]