

Abstract
Molecular recognition is central to all biological processes. For the past fifty years, Koshland’s ‘induced fit’ hypothesis has been the textbook explanation for molecular recognition events. However, recent experimental evidence supports an alternative mechanism. ‘Conformational selection’ postulates that all protein conformations pre-exist, and the ligand selects the most favored conformation. Following binding the ensemble undergoes a population shift, redistributing the conformational states. Both conformational selection and induced fit appear to play roles. Following binding by a primary conformational selection event, optimization of side-chain and backbone interactions is likely to proceed by an induced fit mechanism. Conformational selection has been observed for protein-ligand, protein-protein, protein-DNA, protein-RNA and RNA-ligand interactions. These data support a new molecular recognition paradigm for processes as diverse as signaling, catalysis, gene regulation, and protein aggregation in disease, which has the potential to significantly impact our views and strategies in drug design, biomolecular engineering and molecular evolution.
INTRODUCTION
Highly specific and tightly regulated interactions between biological macromolecules are at the basis of all processes in living organisms. An understanding of the fundamental mechanisms of molecular recognition is therefore central to understanding biology at the molecular level. The two textbook mechanistic explanations for molecular recognition are Fischer’s ‘lock-and-key’ model1 and Koshland’s ‘induced fit’ hypothesis2. In the ‘lock-and-key’ model, the conformations of the free and ligand-bound protein are essentially the same, while ‘induced fit’ posits that conformational differences between these two states are the result of the binding interaction driving the protein towards a new conformation which is more complementary to its binding partner. In their simplest forms, both the ‘lock-and-key’ and the ‘induced fit’ models treat the protein as if it exists in a single, stable conformation under given experimental conditions. However, proteins are inherently dynamic and sample a vast ensemble of conformations. Thus, thermally accessible conformational substates other than the ‘native’ (i.e. lowest energy) conformation may play important roles in molecular recognition3 (see Figure 1). The alternative model of conformational selection takes into account this conformational heterogeneity and argues that weakly populated, higher energy conformations are responsible for recognizing and binding to partners with subsequent population shift toward these conformers4–6. Invoked by kinetic data to explain antigen recognition and antibody specificity7–10 and generalized to apply to all binding events, the conformational selection and population shift model4–6,11 challenges the assumption that conformational differences between free and ligand-bound protein automatically implicate an induced-fit type mechanism of molecular recognition. Given the impact that the fifty-year old induced fit hypothesis has had on chemistry and biology and the central role of molecular recognition in all biological processes, an alternative to this decades-old theory based on fundamental physical principles deserves careful consideration.
Figure 1.

Thermodynamic cycle for molecular recognition processes involving induced fit or conformational selection. In conformational selection, the binding competent conformation (red, P2) is pre-existing in solution prior to the addition of ligand (L). The kinetic and thermodynamic rate constants can determine if conformational selection or induced fit is more likely8,99
The conformational selection model derives from the energy landscape theory of protein structure and dynamics3,5,11,12. The energy landscape theory in biology is most familiar in terms of the ‘protein folding funnel’3,5,13, but it also has major consequences in terms of binding interactions and protein function4,5,14. A protein free energy landscape consists of different conformations or ‘substates’ in dynamic equilibrium. The populations of the substates follow statistical thermodynamic distributions and the heights of the energy barriers separating the substates define the timescale of conformational exchange. If the free energy barriers are low relative to the Boltzmann energy (kBT), thermal fluctuations can lead to significant population of more than one conformational state in solution. A ligand may interact with the lowest energy conformation, or with one of a number of higher energy conformational substates that are populated in solution4,5,11. In all cases, the binding interaction does not ‘induce’ a conformational change; it merely leads to a population shift, that is, a redistribution of the relative populations of conformational substates that already pre-exist in solution. Within this context, the ‘lock-and-key’ model is a limiting case of conformational selection when the interaction partner selectively binds to the lowest energy conformation.
Structural differences between the ‘beginning’ (free) and ‘end’ (bound) thermodynamic states, as observed in X-ray and NMR structures, do not by themselves reveal the process of molecular recognition. A structural demonstration of induced fit would require data sampled over the entire course of the binding interaction and would need to show how interaction with the binding partner directly leads to specific structural changes in the protein8. Structural evidence for conformational selection would entail the characterization of alternative conformations in the free state that resemble the final ligand-bound conformation8, and this would need to be complemented by studies demonstrating that the ligand interacts with a small population of a bound substate (Throughout this review we will use the term bound to refer to a conformational substate of the unliganded protein that has a conformation resembling that of the ligand bound state). In the past, structural characterization of conformations other than the one most highly populated in solution or in the crystal has been difficult. Fortunately, single molecule, NMR and other spectroscopic techniques are now beginning to shed more light on the true conformational diversity of proteins in solution15–19, and reveal the presence of conformational substates that resemble the bound state and which form part of the conformational ensemble populated by the unliganded protein; the existence of such bound substates had previously been inferred only through computational protein dynamics20–22. Intriguingly, these results come from a host of molecular recognition events including protein-ligand, protein-protein, protein-DNA and RNA-ligand interactions. In this Perspective, we discuss recent structural support for the conformational selection model and the consequences of a free energy landscape picture of protein structure and function in terms of drug design, biomolecular engineering and molecular evolution.
Conformational Selection in Protein-Ligand Interactions
The energy landscape theory was first used to describe the dynamic behavior of myoglobin as observed by the elegant flash photolysis experiments of Frauenfelder and colleagues3. Experiments that measured the kinetics of light-activated dissociation of oxygen from heme in myoglobin at physiological temperatures suggested a simple one-step process. However, photodissociation experiments below 200 K demonstrated non-exponential kinetics23 and were rationalized by suggesting that myoglobin did not possess a single structure under these conditions, but instead was composed of many conformational substates each with slightly different rebinding rates3. Time-resolved x-ray diffraction24 and subnanosecond spectroscopic techniques25 have provided detailed insights into the complex free energy landscape of myoglobin and other heme-containing proteins. These studies lend strong support to the notion that a protein exists not as a single structure, but rather as a statistical ensemble of conformations that can give rise to complex protein kinetics.
Although time-resolved Laue diffraction can provide a wealth of information about conformational heterogeneity, application of the method is restricted to proteins that can be photoactivated. This limits the structural information that can be obtained about the conformational ensembles of other proteins. Classical X-ray crystallography usually reveals only a single protein conformation under a given set of conditions. However, different protein conformations are sometimes observed when the same protein complex is crystallized in other space groups under different conditions26. Alternative conformations for distinct protein molecules in the asymmetric unit of the same crystal may also be observed27,28. In one noteworthy case, the monoclonal antibody SPE7 was crystallized in two different conformations in the absence of antigen. Structural analyses of the free and antigen-bound conformations suggest that each conformation is responsible for binding particular antigens29. Pre-steady state kinetics further support pre-binding conformational isomerism and conformational selection in the function of SPE729. Similar kinetic results support conformational selection in other antibody-antigen interactions7,9,10,30 and in other types of protein interactions31–33 (see Box 1 for a kinetic and thermodynamic comparison of conformational selection vs. induced fit).
Box 1. Kinetics and thermodynamics of ‘induced fit’ vs. ‘conformational selection’.
Simple induced fit and conformational selection processes can be viewed in terms of a thermodynamic cycle (Figure 1). The key difference between these two extreme mechanisms is that in the induced-fit model the bound protein conformation forms only after interaction with a binding partner, whereas in the conformational selection model it pre-exists in the ensemble of conformations sampled by the free protein in the absence of ligand. The kinetic rate constants describing the thermodynamic cycle can dictate which molecular recognition pathway will dominate. According to this model, if the concentration of the higher energy bound conformation ([P2]) is larger than the concentration of the induced-fit intermediate ([P1L]), conformational selection will be the preferred kinetic pathway99. This means that for induced fit to be operative, there must be some initial favorable interaction between the ligand and the protein (K1) prior to conformational change, and/or the ligand concentration needs to be sufficiently high. Of course, depending on the rate constants and protein/ligand concentrations both recognition mechanisms may be feasible in a given system. In conformational selection, the rate of formation of the final bound complex (P2L) depends linearly on the concentration of the higher energy bound conformation ([P2]) and nonlinearly on the total concentration of the protein ([P1+P2])8. Unfortunately, if the population of the higher energy conformation (P2) is very low (<5%), it may be difficult to kinetically distinguish induced fit vs. conformational selection processes. It should be noted that the thermodynamic box as presented is an oversimplification of the binding process, considering that many substates within the conformation ensemble could have some affinity for the ligand.
NMR is a powerful technique for studying conformational heterogeneity and the free energy landscape of macromolecules34. NMR provides atomic resolution insights into both protein structure and dynamics over a large range of timescales (ps-s) and, unlike time-resolved techniques that perturb chemical and structural equilibria, directly monitors dynamics under steady-state conditions. Importantly, NMR methods developed over the past decade have allowed the structural characterization of weakly populated states (as little as 1% population) in the conformational ensemble that may play a role in the molecular recognition process34. In particular, relaxation dispersion techniques provide kinetic and thermodynamic information about exchange between two or more conformational substates on the μs-ms timescale and provide information about chemical shifts in the higher energy state that can be used to characterize its structure (see Figure 2). Advances toward directly incorporating chemical shifts in NMR structure refinement35 and newer R2 relaxation dispersion experiments that report on inter-nuclear vector orientations in higher energy substates36,37 provide structural constraints that may allow for direct characterization of higher energy protein conformations38.
Figure 2.

Conformational selection in protein-ligand interactions observed by NMR R2 relaxation dispersion experiments. (a) Locations of conformational exchange are indicated as spheres on the structure of DHFR (pdb 1rx5). (b) A linear correlation between Δω (chemical shift difference between ground-state and higher energy conformations) from R2 relaxation dispersion experiments of the product binary complex of DHFR (enzyme bound with tetrahydrofolate (E:THF)) and Δδ from ground-state chemical shift differences between product binary and ternary (enzyme bound with tetrahydrofolate and NADPH cofactor (E:THF:NADPH)) complexes indicate that the higher energy conformation of the product binary complex is structurally similar to the ground-state of the product ternary complex (i.e. chemical shifts of the higher energy conformation of the product binary complex are similar to the chemical shifts of the ground-state conformation of the product ternary complex) (data taken from ref41). (c) The binding of the NADPH cofactor changes the free energy landscape of the enzyme. Structurally similar conformations are colored alike.
Conformational selection processes are implicated in a number of enzymes, including ribonuclease A39, adenylate kinase27,40 and dihydrofolate reductase41,42. The results with adenylate kinase are especially significant since this enzyme has been used as a textbook example for induced fit. NMR analyses of mesophilic (Escherichia coli) and thermophilic (Aquifex aeolicus) adenylate kinases are consistent with two-state conformational exchange between the open and closed states observed in x-ray crystal structures of free and ligand-bound adenylate kinase respectively27,40. Chemical shift values from R2 relaxation dispersion experiments (Δω) obtained during catalytic turnover show a strong linear correlation between the chemical shift differences (Δδ) between the open and closed conformational states. Moreover, there is a strong correlation between the lid opening rates (44 s−1 and 286 s−1 for E.coli and A. aeolicus, respectively) measured by R2 relaxation dispersion and catalytic turnover (kcat= 30 s−1 and 263 s−1 for E.coli and A.aeolicus respectively)40. These results suggest that both adenylate kinases fluctuate between open and closed conformations during catalysis, and that product release depends on the closed-to-open conformational transition. Conformational exchange between open and closed conformations is not solely the function of catalytic turnover considering that similar motions are also observed in the free enzyme27. In the crystal structure of the free A. aeolicus enzyme, there are three molecules in the asymmetric unit each with a slightly different conformation27. These conformations lie along the trajectory between fully open and fully closed conformations27. Moreover, R2 relaxation dispersion NMR spectroscopy, single molecule FRET and paramagnetic NMR relaxation experiments are all consistent with the fluctuation of the enzyme into a bound conformation in the absence of substrate27,43.
In the case of DHFR, the dynamics of five different complexes, representing all the intermediates formed in the catalytic cycle, were measured using NMR relaxation dispersion techniques41,42. The conformational dynamics of DHFR are ligand-dependent, in marked contrast to the ligand-independent fluctuations observed in other proteins27,44. The binary complexes, with cofactor or product bound, both fluctuate into conformations resembling the ternary complex (i.e. bound with both cofactor and substrate or product), as suggested by the linear correlation between the dynamic chemical shift differences between lowest and higher energy conformations for the binary complexes (Δωbinary) and the ground-state chemical shift differences between the binary and ternary complexes (Δδ (binary-ternary))41 (Figure 2). Single molecule experiments also support the fluctuation of DHFR into other conformations in the presence of substrate or cofactor that may assist binding of the second ligand45. The dynamics observed in complexes of DHFR appear to be functionally relevant in that each complex fluctuates into a conformation resembling the next and/or previous step in the catalytic cycle41. Thus, population redistributions, mediated by the interactions between enzyme and ligand(s), play roles in substrate binding, product release and catalytic turnover. This suggests that the free energy landscape of E.coli DHFR is dynamic11, in the sense that ligand binding (or release) can alter the nature of the thermally accessible substates in the conformational ensemble and the kinetic and thermodynamic parameters governing the conformational equilibria41,46–48.
Conformational Selection in Protein Interactions
Studies of enzyme-substrate and enzyme-product complexes demonstrate that proteins can fluctuate into conformations that resemble those of the bound state, even in the absence of ligand. These results and others49–54 provide structural support for conformational selection in molecular recognition between proteins and small molecules. Similar experimental support exists for conformational selection mechanisms in protein-protein55–60 and protein-nucleic acid interactions61–63.
NMR provides a number of observables, other than relaxation dispersion, that are used to gain insights into the nature of the conformational ensemble. An especially powerful approach is to use NMR observables as restraints in molecular dynamic simulations, or alternatively, NMR observables can be used to bias populations of pre-generated conformations representing potential substates within the conformational ensemble (for more comprehensive discussion of the methodology, see reviews64–66 and references therein). In one approach, NMR order parameters and nuclear Overhauser effects (NOE) were used to characterize the conformational ensembles of ubiquitin67 and calmodulin55. NOEs are commonly used as restraints in NMR structure determination, while Lipari-Szabo order parameters (S2)68 give an indication of the amplitude of protein motion on a ps-ns timescale. Intriguingly, the calculated conformational ensemble of Ca2+-calmodulin include structures that are very similar to the conformation of calmodulin bound to myosin light chain kinase55. Calmodulin consists of N-terminal and C-terminal domains separated by an interdomain linker. The NMR-derived conformational ensemble of Ca2+-calmodulin suggests that the unliganded C-terminal domain adopts a bound conformation much more frequently than does the unliganded N-terminal domain. Based on these results and other biophysical evidence55, the authors postulate a molecular recognition process in which myosin light chain kinase first interacts with the C-terminal domain, followed by population shifts within the conformational ensemble such that the bound conformations of the N-terminal domain become more populated55. Although a multistep process, the mechanism of binding involves shifting of populations within an ensemble of pre-existing conformations4–6,8,11 rather than ‘induction’ of a new conformation not observed in the free protein. Recent single molecule atomic force microscopy experiments further support a population shift mechanism for myosin light chain kinase binding to Ca2+-calmodulin57.
One shortcoming of the classical Lipari-Szabo NMR order parameters is that they only report on ps-ns timescale protein dynamics and therefore do not give information about dynamic processes on slower timescales that might also be relevant for molecular recognition. Newer approaches utilize structural data from residual dipolar couplings (RDCs)65 and/or paramagnetic relaxation enhancement (PRE)66 to characterize conformational ensembles on slower timescales. For example, RDCs that report on internuclear vector orientations can be used both in NMR structure determination and to identify protein motions on the ps-ms timescale65,69. Thus, RDC-derived order parameters that are smaller in magnitude than the Lipari-Szabo order parameters suggest additional motions on the ns-ms timescale that are not captured by the classical order parameters. By measuring RDCs resulting from partial alignment in a large number of media, the conformational ensemble of ubiquitin was calculated up to the microsecond timescale60. The most striking feature of the conformational ensemble is the presence of bound conformations in the free state of ubiquitin (Figure 3). In fact, the backbone conformations in all forty-six of the known x-ray crystal structures of ubiquitin in complex with various partner proteins are represented within the conformational ensemble, despite the absence of any crystallographic information in ensemble refinement60. This gives strong evidence that bound conformations of ubiquitin are present in the free protein, in the absence of protein interaction partners, and that conformational selection-type processes are important for molecular recognition in ubiquitin. However, subsequent conformational changes may be induced, especially in the sidechains, after the initial binding interaction60. Kinetic studies implicate conformational selection in Alzheimer’s Aβ amyloidosis70, which can also be considered a protein-protein recognition event71.
Figure 3.
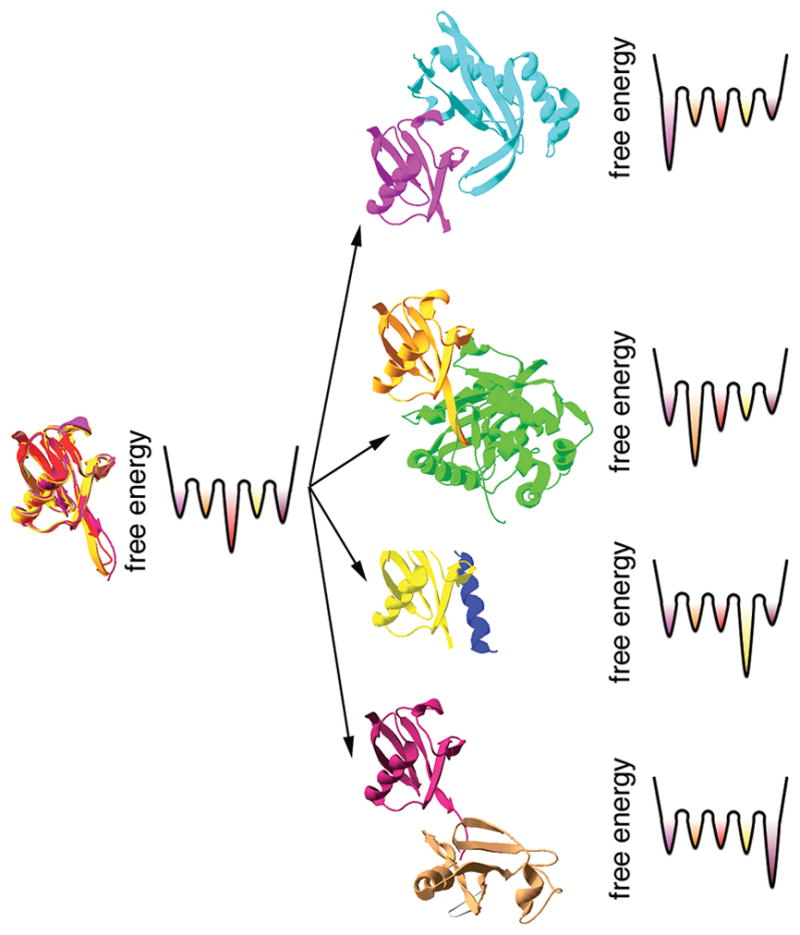
A schematic illustration of molecular recognition processes involving ubiquitin. The NMR-derived conformational ensemble of ubiquitin indicates that all bound conformations exist in the absence of protein binding partners66 (left). Although the conformational ensemble encompasses all forty six of the known crystal structures of ubiquitin, only five are shown here for clarity (pdb 1f9j, 1s1q, 1xd3, 2d36 and 2g45). The free energy landscapes are hypothetical considering that the relative population of each conformation in the ensemble and the energy barriers separating the conformations are not known.
Conformational Selection in RNA/DNA Interactions
The principles of energy landscape theory can also be applied to protein-nucleic acid recognition and to the structure and dynamics of RNA and DNA63,72. For example, the specificity of lac repressor headpiece binding to DNA can be explained by describing differences in the protein free energy landscape when bound to either cognate or noncognate DNA61,72. NMR studies indicate that there are both structural and dynamic differences between noncognate and cognate DNA - lac repressor complexes61 (Figure 4). Protein motions on the μs-ms timescale that are observed in the DNA-free lac repressor headpiece are significantly increased when it binds to nonspecific DNA, especially for residues at the protein-DNA interface61. However, upon binding the cognate DNA sequence, the μs-ms timescale motions observed by NMR are effectively quenched61. Many of the residues that display changes in μs-ms timescale dynamics adopt alternative conformations in the NMR-derived structural ensemble of the nonspecific complex61. Together, these results suggest that the lac repressor headpiece samples many conformations in the DNA-free form and when bound to nonspecific DNA, but that a single protein conformation is selected when protein binds to its specific DNA recognition sequence61,72. The molecular recognition mechanism described for the lac repressor would allow the protein to bind initially to nonspecific DNA and allow for a fast one-dimensional diffusional search for the cognate recognition site73,74, while also providing a mechanism for tight binding with the target sequence.
Figure 4.

DNA recognition by the lac repressor headpiece. Differences in structure (left) and dynamics (middle) between (A) lac repressor headpiece in the free state (PDB code 1lqc), (B) lac repressor headpiece bound to noncognate DNA (pdb 1osl) and (C) lac repressor headpiece bound to cognate DNA (1l1m). The middle column shows Rex, the contribution to the NMR R2 transverse rate constant from μs-ms time scale conformational fluctuations, mapped onto the structures of the lac repressor protein. (Adapted from ref72 with permission from the American Chemical Society). The free energy landscape (shown schematically on the right) is rough, with many interconverting substates in the complex of lac repressor headpiece with noncognate DNA, but a single dominant conformation is formed in the complex with cognate DNA.
The structural and dynamic diversity of RNA rivals that of proteins63. Many RNA molecules also exist as conformational ensembles containing interconverting substates and binding interactions can be rationalized based on population shifts and conformational selection processes63. For example, the RDC-derived dynamics of the transactivation response (TAR) RNA from the human immunodeficiency virus type-1 demonstrate that the RNA samples bound conformations even in the absence of ligand75. The motions of the two TAR RNA helices appear to be highly correlated and they trace out a dynamic trajectory that encompasses the conformations observed in nearly all of the ligand-bound structures75. Thus, conformational selection appears to be universal in molecular recognition processes involvingbiomolecules.
Conformational Selection in Protein Regulation
Conformational selection is closely related to the Monod-Wyman-Changeux (MWC) theory of allostery76. The MWC theory also envisions the presence of two, or more, protein conformations in solution each with different binding or functional characteristics. For example, the enzyme aspartate transcarbamoylase is suggested to exist in two conformations, a fully active R state and a less active T state77. Although the crystal structures of the R and T states of aspartate transcarbamoylase are well characterized, only recently has it been possible to detect both conformations in solution by NMR and directly monitor the population redistributions upon the addition of allosteric ligands78,79. Results from the NMR studies are fully consistent with the MWC model.
Population redistributions of active and inactive protein conformations can also explain other regulatory processes80. Phosphorylation of the bacterial signaling protein NtrC leads to a large conformational change81. NMR studies demonstrate that unphosphorylated NtrC accesses a conformation similar to that of phosphorylated Ntrc, suggesting that phosphorylation results in a population redistribution between active and inactive conformations that pre-exist in solution81. Likewise, NMR relaxation dispersion experiments demonstrate that the photoswitch LOV2-Jα fluctuates between ‘dark’ inactive and ‘light’ active conformations82. Shining light on the protein results in a population redistribution that favors the active conformational substate. Thus, changes in protein populations represent a general mechanism for protein function that interconnects molecular recognition and protein regulation.
Implications and Future Directions
The studies described above clearly indicate that proteins and RNA can access bound conformations even in the absence of a binding partner. However, the mere fact that the bound conformation is present in the ensemble of conformations sampled by the free protein does not in itself implicate this conformation in the recognition process. The Dbl homology (DH) domain of the oncoprotein Vav1 provides a striking example of the role of a higher energy conformational substate in biological function83. NMR relaxation dispersion experiments show that the DH domain fluctuates between a ground-state conformation, in which an autoinhibitory helix binds to and blocks the active-site, and a weakly populated higher energy conformation in which the autoinhibitory helix is dissociated from the catalytic surface83. Mutations that alter the equilibrium distributions of the two states change the catalytic activity of the DH domain and the rate of phosphorylation of the autoinhibitory helix by the Lck kinase; the catalytic activity and the phosphorylation rate are linearly dependent on the population of the higher energy ‘helix dissociated’ conformation83.
Although in the preceding sections we emphasized the importance of conformational selection mechanisms, it is likely that both conformational selection and induced fit play important roles in molecular recognition. Following initial binding through a conformational selection mechanism, it is probable that further changes to the protein structure and underlying free energy landscape are required to optimize the intermolecular interactions; such conformational rearrangements constitute an induced fit process. The co-existence of both mechanisms is evident in interactions of the maltose-binding protein. The ligand-free protein fluctuates between a predominantly open form and a minor partially closed species49. Maltose could potentially interact with both conformations, but further structural changes would need to take place to form the final ligand-bound, fully closed conformation49.
The energy landscape view of protein folding and function has numerous practical consequences in areas such as drug design, protein engineering and molecular evolution. For example, small molecule inhibitors can be designed against higher energy conformations present in the conformational ensemble to target different conformations of the binding pocket84–86. Along these lines, several molecular docking procedures have previously been designed to account for protein flexibility in molecular recognition86–89. In one procedure, a conformational ensemble is pre-generated using experimental information (e.g. different x-ray crystal structures, NMR-derived conformational ensembles, etc.) and/or computational data (e.g. molecular dynamics simulation, normal mode analyses, principal component analyses, etc.); similar conformations are clustered and binding partners are then docked to representative target conformations88. Further energy minimization procedures and/or molecular dynamics simulations can account for additional small-scale motions in the interaction complex. The two stages of the procedure (initial docking of the conformational ensemble, followed by refinement and/or molecular dynamics) essentially represent conformational selection and induced fit processes, respectively. However, a large hurdle in accounting for protein target flexibility in the docking procedure is the increased computation time. The experimental information presented in this review suggests that low-lying conformational substates are primarily responsible for molecular recognition; consequently, biased docking to highly populated conformational clusters (i.e. the lowest energy conformational substates) may significantly diminish the computational time. Experimental techniques such as those described here can be used to further decrease the size of the conformational ensemble required for docking.
Nonetheless, applying energy landscape theory to drug design is not straightforward. Not all conformations in the ensemble are likely to be equally ‘druggable’ reflecting key differences in energetics and/or accessibility of the potential binding pocket in different members of the conformational ensemble. In such cases, enhanced exploration of the conformational space of the target through additional computational procedures may be required86. An alternative strategy for incorporating the conformational ensemble in drug design is the search for allosteric inhibitors (or activators) that would alter the distribution of the conformational ensemble. While considerable progress has been made towards this goal90, and allosteric drugs are already in the market or are currently under development85, unfortunately, no general strategy is currently available for developing allosteric inhibitors to interact with binding pockets that exist only in a subset of substates in the conformational ensemble. A better understanding of protein dynamics and the role of conformational entropy in determining inhibitor binding affinity is urgently required. The complexity of the interactions that determine the overall binding affinity is evident from recent studies of calmodulin, which show clearly that changes in protein dynamics before and after peptide binding have a significant entropic effect on peptide binding affinity91. Thus, the notion that higher affinity binding always results in a more stable, rigid complex may not be entirely correct, and attention must also be paid to the nature of the conformational ensemble populated by the protein in the ligand-bound state.
Considerations of conformational heterogeneity and conformational selection could potentially aid greatly in the design of protein function and engineering of enzymes92,93. Despite impressive successes, initial attempts to design artificial enzymes resulted in relatively poor catalysts, compared to naturally occurring enzymes. Similarly, catalytic antibodies raised against transition-state analogs are also inefficient catalysts, in general, compared to their natural enzyme counterparts94. This suggests that either we do not have a firm enough understanding of the chemistry and transition-state complexes involved, or that we ignore and do not select for other critical parameters that are essential for efficient enzyme catalysis. In the case of one newly engineered enzyme, in vitro evolution was able to select for catalysts with activities similar to naturally-occurring enzymes92. While this result argues that a combined computational design and directed evolution approach can yield effective biocatalysts, not all mutations introduced by in vitro evolution can be readily explained. Current enzyme design efforts tend to consider only the chemical steps in an enzyme’s catalytic cycle, overlooking essential steps such as substrate binding, product release, and conformational change that could severely limit the catalytic turnover rate in a natural enzyme. Studies of enzyme such as dihydrofolate reductase41, adenylate kinase27 and ribonuclease A39 suggest that conformational selection-type processes occur throughout the catalytic cycle and can govern steady-state turnover41. Hence, we argue that in the future, consideration of low-lying conformational ensembles and conformational changes (that is, re-distribution of the conformer populations) in parallel with optimization of chemistry, substrate binding and product release, will be essential for the design of more effective catalysts that rival naturally occurring enzymes in catalytic activity. Incorporation of such elements into the design strategy could also permit the engineering of regulatory elements that modify newly evolved enzyme activities via remote-site allosteric modulation. In turn, these studies will enhance comprehension of conformational ensembles in ligand interactions, enzyme catalysis and molecular evolution.
Conformational ensembles are also important in the evolution of molecular interactions. The bound conformation observed in free enzymes such as adenylate kinase and dihydrofolate reductase, can be regarded as the second most highly populated conformation in solution; other conformations even higher in energy and of much lower population are not detectable by NMR relaxation dispersion techniques. This suggests that the intrinsic fluctuations of a protein (or RNA) are highly ‘tuned’ and have evolved to be functionally relevant84, and that it is the lowest energy subset of conformational states that are most relevant to biological function. In the case of ubiquitin, the conformational ensemble contains a very large number of different conformational substates; these may have evolved to allow ubiquitin to interact with a diverse range of binding partners, or conversely, protein binding partners may have taken advantage of pre-existing conformational diversity. Both processes may be important in the evolution of molecular recognition. This is similar to a 70-year old proposal for antigen-antibody interactions that suggested that both conformational and amino acid sequence diversity would allow antibodies to interact with the greatest variety of antigens95. Darwinian evolution requires that a function exists to some extent prior to natural selection6,11, thus protein conformational diversity and functional promiscuity are closely connected and are a potential means of molecular evolution96,97. Conformational selection processes may also be more resilient to evolutionary forces than induced fit-type mechanisms11. In the case of conformational selection, deleterious mutations may significantly alter the free energy landscape but binding-competent conformers may still be present in the conformational ensemble. This is in contrast to an induced-fit type mechanism where an unfavorable mutation can critically disrupt the step-wise conformational changes required for competent binding11. Except for a few studies of the maturation process of antibody-antigen interactions7,29,98, there is very little information regarding the role of the conformational ensemble in directing molecular evolution. This situation is unfortunate, since a better understanding of the evolutionary aspects of conformational heterogeneity could have an immediate and positive impact upon efforts to design and engineer de novo proteins with novel functions.
Acknowledgments
This work was supported by the National Institutes of Health (grants GM75995 and CA96865 to PEW) and by the Skaggs Institute for Chemical Biology. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Contributor Information
David D. Boehr, Email: ddb12@psu.edu.
Ruth Nussinov, Email: ruthnu@helix.nih.gov.
Peter E. Wright, Email: wright@scripps.edu.
References
- 1.Fischer E. Einfluss der configuration auf die Wirkung der Enzyme. Ber Dtsch Chem Ges. 1894;27:2984–2993. [Google Scholar]
- 2.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 4.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–20. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 5.Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–90. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsai CJ, Ma B, Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc Natl AcadSci U S A. 1999;96:9970–2. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci USA. 1994;91:10370–4. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bosshard HR. Molecular recognition by induced fit: how fit is the concept? News Physiol Sci. 2001;16:171–3. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 9.Berger C, et al. Antigen recognition by conformational selection. FEBS Lett. 1999;450:149–53. doi: 10.1016/s0014-5793(99)00458-5. [DOI] [PubMed] [Google Scholar]
- 10.Leder L, et al. Spectroscopic, calorimetric, and kinetic demonstration of conformational adaptation in peptide-antibody recognition. Biochemistry. 1995;34:16509–18. doi: 10.1021/bi00050a035. [DOI] [PubMed] [Google Scholar]
- 11.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 2000;9:10–9. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller DW, Dill KA. Ligand binding to proteins: the binding landscape model. Protein Sci. 1997;6:2166–79. doi: 10.1002/pro.5560061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–9. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 14.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–97. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenleaf WJ, Woodside MT, Block SM. High-resolution, single-molecule measurements of biomolecular motion. Annu Rev Biophys Biomol Struct. 2007;36:171–90. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parak FG. Proteins in action: the physics of structural fluctuations and conformational changes. Curr Opin Struct Biol. 2003;13:552–7. doi: 10.1016/j.sbi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Hinterdorfer P, Dufrene YF. Detection and localization of single molecular recognition events using atomic force microscopy. Nat Methods. 2006;3:347–55. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- 18.Busenlehner LS, Armstrong RN. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch Biochem Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Palmer AG., 3rd Nmr probes of molecular dynamics: overview and comparison with other techniques. Annu Rev Biophys Biomol Struct. 2001;30:129–55. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 20.Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci U S A. 2005;102:18908–13. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keskin O. Binding induced conformational changes of proteins correlate with their intrinsic fluctuations: a case study of antibodies. BMC Struct Biol. 2007;7:31. doi: 10.1186/1472-6807-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grunberg R, Leckner J, Nilges M. Complementarity of structure ensembles in protein-protein binding. Structure. 2004;12:2125–36. doi: 10.1016/j.str.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Austin RH, Beeson KW, Eisenstein L, Frauenfelder H, Gunsalus IC. Dynamics of ligand binding to myoglobin. Biochemistry. 1975;14:5355–73. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 24.Schotte F, et al. Watching a protein as it functions with 150-ps time-resolved x-ray crystallography. Science. 2003;300:1944–7. doi: 10.1126/science.1078797. [DOI] [PubMed] [Google Scholar]
- 25.Vos MH. Ultrafast dynamics of ligands within heme proteins. Biochim Biophys Acta. 2008;1777:15–31. doi: 10.1016/j.bbabio.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Lee AY, Gulnik SV, Erickson JW. Conformational switching in an aspartic proteinase. Nat Struct Biol. 1998;5:866–71. doi: 10.1038/2306. [DOI] [PubMed] [Google Scholar]
- 27.Henzler-Wildman KA, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–44. doi: 10.1038/nature06410. This study provides experimental support from X-ray crystallography, NMR and single molecule fluorescence that adenylate kinase fluctuates between open and closed states in the absence of ligand. [DOI] [PubMed] [Google Scholar]
- 28.Muller YA, Kelley RF, de Vos AM. Hinge bending within the cytokine receptor superfamily revealed by the 2.4 A crystal structure of the extracellular domain of rabbit tissue factor. Protein Sci. 1998;7:1106–15. doi: 10.1002/pro.5560070504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–7. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 30.Hanes J, Jermutus L, Weber-Bornhauser S, Bosshard HR, Pluckthun A. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc Natl Acad Sci U S A. 1998;95:14130–5. doi: 10.1073/pnas.95.24.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stella L, et al. Flexibility of helix 2 in the human glutathione transferase P1-1. time-resolved fluorescence spectroscopy. J Biol Chem. 1998;273:23267–73. doi: 10.1074/jbc.273.36.23267. [DOI] [PubMed] [Google Scholar]
- 32.Xu J, Root DD. Conformational selection during weak binding at the actin and myosin interface. Biophys J. 2000;79:1498–510. doi: 10.1016/S0006-3495(00)76401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao Y, Musah RA, Wilcox SK, Goodin DB, McRee DE. Protein conformer selection by ligand binding observed with crystallography. Protein Science. 1998;7:72–78. doi: 10.1002/pro.5560070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–8. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 35.Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc Natl Acad Sci U S A. 2007;104:9615–20. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vallurupalli P, Hansen DF, Stollar E, Meirovitch E, Kay LE. Measurement of bond vector orientations in invisible excited states of proteins. Proc Natl Acad Sci U S A. 2007;104:18473–7. doi: 10.1073/pnas.0708296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Igumenova TI, Brath U, Akke M, Palmer AG., 3rd Characterization of chemical exchange using residual dipolar coupling. J Am Chem Soc. 2007;129:13396–7. doi: 10.1021/ja0761636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansen DF, Vallurupalli P, Kay LE. Using relaxation dispersion NMR spectroscopy to determine structures of excited, invisible protein states. J Biomol NMR. 2008;41:113–20. doi: 10.1007/s10858-008-9251-5. [DOI] [PubMed] [Google Scholar]
- 39.Beach H, Cole R, Gill ML, Loria JP. Conservation of mus-ms enzyme motions in the apo-and substrate-mimicked state. J Am Chem Soc. 2005;127:9167–76. doi: 10.1021/ja0514949. [DOI] [PubMed] [Google Scholar]
- 40.Wolf-Watz M, et al. Linkage betweendynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat Struct Mol Biol. 2004;11:945–9. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 41.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–42. doi: 10.1126/science.1130258. This study suggests that every functional intermediate of dihydrofolate reductase fluctuates into a higher energy conformation that is structurally similar to the next and/or previous complex in the catalytic cycle. [DOI] [PubMed] [Google Scholar]
- 42.McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc Natl Acad Sci U S A. 2005;102:5032–7. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanson JA, et al. Illuminating the mechanistic roles of enzyme conformational dynamics. Proc Natl Acad Sci U S A. 2007;104:18055–60. doi: 10.1073/pnas.0708600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–21. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 45.Antikainen NM, Smiley RD, Benkovic SJ, Hammes GG. Conformation coupled enzyme catalysis: single-molecule and transient kinetics investigation of dihydrofolate reductase. Biochemistry. 2005;44:16835–43. doi: 10.1021/bi051378i. [DOI] [PubMed] [Google Scholar]
- 46.Boehr DD, Dyson HJ, Wright PE. Conformational relaxation following hydride transfer plays a limiting role in dihydrofolate reductase catalysis. Biochemistry. 2008;47:9227–33. doi: 10.1021/bi801102e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kitahara R, et al. High pressure NMR reveals active-site hinge motion of folate-bound Escherichia coli dihydrofolate reductase. Biochemistry. 2000;39:12789–95. doi: 10.1021/bi0009993. [DOI] [PubMed] [Google Scholar]
- 48.Mauldin RV, Carroll MJ, Lee AL. Dynamic dysfunction in dihydrofolate reductase results from antifolate drug binding: modulation of dynamics within a structural state. Structure. 2009;17:386–94. doi: 10.1016/j.str.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–82. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 50.Lu ZL, Coetsee M, White CD, Millar RP. Structural determinants for ligand-receptor conformational selection in a peptide G protein-coupled receptor. J Biol Chem. 2007;282:17921–9. doi: 10.1074/jbc.M610413200. [DOI] [PubMed] [Google Scholar]
- 51.Fenwick R, et al. Solution structure and dynamics of the small GTPase RalB in its active conformation: significance for effector protein binding. Biochemistry. 2009 doi: 10.1021/bi802129d. [DOI] [PubMed] [Google Scholar]
- 52.Saitoh T, et al. Tom20 recognizes mitochondrial presequences through dynamic equilibrium among multiple bound states. EMBO J. 2007;26:4777–87. doi: 10.1038/sj.emboj.7601888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brath U, Akke M. Differential response of the backbone and side chain conformational dynamics in FKBP12 upon binding the transition state analog FK506: Implications for transition state stabilization and target protein recognition. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.01.047. [DOI] [PubMed] [Google Scholar]
- 54.Keramisanou D, et al. Disorder-order folding transitions underlie catalysis in the helicase motor of SecA. Nat Struct Mol Biol. 2006;13:594–602. doi: 10.1038/nsmb1108. [DOI] [PubMed] [Google Scholar]
- 55.Gsponer J, et al. A coupled equilibrium shift mechanism in calmodulin-mediated signal transduction. Structure. 2008;16:736–46. doi: 10.1016/j.str.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nevo R, et al. A molecular switch between alternative conformational states in the complex of Ran and importin beta1. Nat Struct Biol. 2003;10:553–7. doi: 10.1038/nsb940. [DOI] [PubMed] [Google Scholar]
- 57.Junker JP, Ziegler F, Rief M. Ligand-dependent equilibrium fluctuations of single calmodulin molecules. Science. 2009;323:633–7. doi: 10.1126/science.1166191. [DOI] [PubMed] [Google Scholar]
- 58.Koglin A, et al. Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science. 2006;312:273–6. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 59.Koglin A, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–11. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lange OF, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–5. doi: 10.1126/science.1157092. This report presents an NMR-derived conformational ensemble of ubiquitin that encompasses all the crystallographically determined conformations of ubiquitin. [DOI] [PubMed] [Google Scholar]
- 61.Kalodimos C, et al. Structure and flexibility adaptation in nonspecific and specific protein-DNA complexes. Science. 2004;305:386–389. doi: 10.1126/science.1097064. This study compares the structure and dynamics of the lac repressor headpiece when bound to cognate versus noncognate DNA, and demonstrates that the underlying energy landscapes are distinct. [DOI] [PubMed] [Google Scholar]
- 62.Zhang Q, Stelzer AC, Fisher CK, Al-Hashimi HM. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–7. doi: 10.1038/nature06389. This study demonstrates that TAR RNA fluctuates into multiple “bound” conformations in the absence of ligands. [DOI] [PubMed] [Google Scholar]
- 63.Al-Hashimi HM, Walter NG. RNA dynamics: it is about time. Curr Opin Struct Biol. 2008;18:321–9. doi: 10.1016/j.sbi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr Opin Struct Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 65.Lakomek NA, et al. Residual dipolar couplings as a tool to study molecular recognition of ubiquitin. Biochem Soc Trans. 2008;36:1433. doi: 10.1042/BST0361433. [DOI] [PubMed] [Google Scholar]
- 66.Clore GM, Tang C, Iwahara J. Elucidating transient macromolecular interactions using paramagnetic relaxation enhancement. Curr Opin Struct Biol. 2007;17:603–16. doi: 10.1016/j.sbi.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–32. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 68.Lipari G, Szabo A. Nuclear magnetic resonance relaxation in nucleic acid fragments: models for internal motion. Biochemistry. 1981;20:6250–6. doi: 10.1021/bi00524a053. [DOI] [PubMed] [Google Scholar]
- 69.Lakomek NA, Carlomagno T, Becker S, Griesinger C, Meiler J. A thorough dynamic interpretation of residual dipolar couplings in ubiquitin. J Biomol NMR. 2006;34:101–15. doi: 10.1007/s10858-005-5686-0. [DOI] [PubMed] [Google Scholar]
- 70.Esler WP, et al. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000;39:6288–95. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 71.Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–61. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalodimos C, Boelens R, Kaptein R. Toward an integrated model of protein DNA recognition as inferred from NMR studies on the lac repressor system. Chem Rev. 2004;104:3567–3586. doi: 10.1021/cr0304065. [DOI] [PubMed] [Google Scholar]
- 73.von Hippel PH, Berg OG. Facilitated target location in biological systems. J Biol Chem. 1989;264:675–8. [PubMed] [Google Scholar]
- 74.Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15:768–74. doi: 10.1038/nsmb.1441. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Q, Stelzer A, Fisher C, Al-Hashimi H. Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature. 2007;450:1263–1267. doi: 10.1038/nature06389. [DOI] [PubMed] [Google Scholar]
- 76.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 77.Kantrowitz ER, Lipscomb WN. Escherichia coli aspartate transcarbamoylase: the molecular basis for a concerted allosteric transition. Trends Biochem Sci. 1990;15:53–9. doi: 10.1016/0968-0004(90)90176-c. [DOI] [PubMed] [Google Scholar]
- 78.Velyvis A, Yang YR, Schachman HK, Kay LE. A solution NMR study showing that active site ligands and nucleotides directly perturb the allosteric equilibrium in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2007;104:8815–20. doi: 10.1073/pnas.0703347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Velyvis A, Schachman HK, Kay LE. Application of methyl-TROSY NMR to test allosteric models describing effects of nucleotide binding to aspartate transcarbamoylase. J Mol Biol. 2009 doi: 10.1016/j.jmb.2009.01.066. [DOI] [PubMed] [Google Scholar]
- 80.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–43. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 81.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–33. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 82.Yao X, Rosen MK, Gardner KH. Estimation of the available free energy in a LOV2-J alpha photoswitch. Nat Chem Biol. 2008;4:491–7. doi: 10.1038/nchembio.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li P, Martins IR, Amarasinghe GK, Rosen MK. Internal dynamics control activation and activity of the autoinhibited Vav DH domain. Nat Struct Mol Biol. 2008;15:613–8. doi: 10.1038/nsmb.1428. This study presents a linear correlation between biological activity and the population of the Vav DH higher energy conformation. This provides both structural and functional evidence for the involvement of a higher energy conformation in biological function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem Rev. 2006;106:3055–79. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 85.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324:213–5. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bursavich MG, Rich DH. Designing non-peptide peptidomimetics in the 21st century: inhibitors targeting conformational ensembles. J Med Chem. 2002;45:541–58. doi: 10.1021/jm010425b. [DOI] [PubMed] [Google Scholar]
- 87.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–84. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andrusier N, Mashiach E, Nussinov R, Wolfson HJ. Principles of flexible protein-protein docking. Proteins. 2008;73:271–89. doi: 10.1002/prot.22170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chaudhury S, Gray JJ. Conformer selection and induced fit in flexible backbone protein-protein docking using computational and NMR ensembles. J Mol Biol. 2008;381:1068–87. doi: 10.1016/j.jmb.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lindsley CW, Emmitte KA. Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr Opin Drug Discov Devel. 2009;12:446–57. [PubMed] [Google Scholar]
- 91.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–9. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–5. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 93.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–91. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hilvert D. Critical analysis of antibody catalysis. Annu Rev Biochem. 2000;69:751–93. doi: 10.1146/annurev.biochem.69.1.751. [DOI] [PubMed] [Google Scholar]
- 95.Pauling L, Delbruck M. The nature of the intermolecular forces operative in biological processes. Science. 1940;92:77–79. doi: 10.1126/science.92.2378.77. [DOI] [PubMed] [Google Scholar]
- 96.James LC, Tawfik DS. Conformational diversity and protein evolution--a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28:361–8. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 97.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–7. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 98.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276:1665–9. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 99.Weikl T, Von Deuster C. Selected-fit versus induced-fit protein binding: Kinetic differences and mutational analysis. Proteins. 2009;75:104–110. doi: 10.1002/prot.22223. [DOI] [PubMed] [Google Scholar]