

Abstract
Neopeltolide, a potent cytotoxin from a Carribean sponge, was synthesized through a brief sequence that highlights the use of ethers as oxocarbenium ion precursors. Other key steps include an acid-mediated etherification and sequence that features a Sonogashira reaction, an intramolecular alkyne hydrosilylation reaction, and a Tamao oxidation. The alkene that is required for the oxidative cyclization can be hydrogenated to provide access to the natural product or an epimer, or can be epoxidized or dihydroxylated to form polar analogs.
Keywords: Synthesis, Cations, Macrocycle, Oxidation, Analogs
1. Introduction
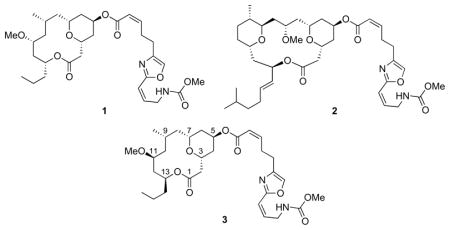
In 2007 Wright and co-workers reported1 the isolation of neopeltolide from the sponge Daedolapelta of the Neopeltodae family off the Jamaican coast. This compound was initially assigned as structure 1 based on extensive NMR analysis. Neopeltolide exhibits potent cytotoxic and anti-fungal activity, with IC50 values of <10 nM against a number of cancer cell lines and an MIC of 0.62 μg/mL (1 μM) against Candida albicans. Although neopeltolide is structurally less complex than the related macrolide leucascandrolide A (2),2 a well studied synthetic target,3 its biological activity is comparable or superior. Neopeltolide’s interesting biological activity and accessible structure have inspired extensive interest in its total synthesis. Panek4 and Scheidt5 independently reported total syntheses of neopeltolide, and through these efforts showed that the correct structure is 3. Since these reports a number of elegant total and formal syntheses have been reported.6
Synthesis has allowed for further biological studies of neopeltolide. Kozmin and co-workers identified cytochrome bc1,6d a mitochondrial enzyme that is involved in ATP synthesis, as a viable cellular target for 2 and 3. Crews, Scheidt, and coworkers reported6h that neopeltolide exhibits cell line selectivity for its cytotoxic activity. Several groups have prepared neopeltolide analogs and studied their cytotoxicity. These efforts have shown that the oxazole-containing side chain, while showing no activity on its own, is essential for activity and significant changes to this unit are generally not tolerated.6g,h Several stereochemical changes in the lactone core are tolerated, though 1 is nearly two orders of magnitude less active than 3.6g,h,i Removing the C9 methyl group or the C11 methoxy group does not dramatically alter activity.6k This is significant because these analogs can be prepared more readily than neopeltolide. No efforts toward altering the physical properties of neopeltolide through introducing hydrophilic functional groups have been reported.
Our interest in neopeltolide arose from a desire to apply our recently-developed oxidative cyclization protocol7 in the context of natural product synthesis. This method proceeds through DDQ-mediated carbon–hydrogen bond cleavage of benzylic or allylic ethers to form oxocarbenium ions. These electrophiles react with appended nucleophiles to form tetrahydropyrans with excellent stereocontrol. The use of oxidative carbon hydrogen bond cleavage also allows for the formation of macrocyclic oxocarbenium ions through cyclic ether oxidation (Scheme 1).8 These intermediates, which can be utilized for stereoselective transannular reactions, have been prepared previously through conventional intramolecular condensation reactions between alcohols or silyl ethers and aldehydes.5,6a,l,9 The generality of this approach, however, has not been established. Rychnovsky and co-workers, for example, conducted computational studies to show that the efficient formation of the macrocyclic oxocarbenium ion in their kendomycin synthesis arose from conformational preorganization of the hydroxyl and aldehyde groups.9d Oxidizing a pre-formed macrocycle represents a potentially more general approach to this type of intermediate.
Scheme 1.

Oxidative cyclization through a macrocyclic oxocarbenium ion.
The formation of the neopeltolide macrolactone from a macrocyclic allylic ether precursor is advantageous from a strategic perspective since etherification can be used as an early stage fragment coupling reaction. The stable ether linkage obviates the need for protecting groups during the sequence and for the introduction of activating groups for electrophile formation. In this manuscript we report the total synthesis of neopeltolide through an oxidatively generated macrocyclic oxocarbenium ion. Key steps in the sequence include fragment coupling through a BrØnsted acid-mediated etherification process and a Sonogashira reaction, and a regioselective alkyne hydration through hydrosilylation. We also report that the alkene in the cyclization product can be functionalized with good to excellent stereochemical control en route to the natural product and analogs.
2. Results and Discussion
2.1 Retrosynthetic analysis
We designed our synthesis of 3 to proceed through 4, a known precursor5,6e to the natural product (Scheme 2). This bridged bicycle can be accessed from 5 through oxidative cyclization and stereoselective alkene reduction. The macrocycle is derived from diester 6, which in turn can be prepared from readily-accessible subunits 7, 8, and 9.
Scheme 2.

Retrosynthetic analysis of neopeltolide.
2.2 Neopeltolide synthesis
The synthesis of 7 (Scheme 3) proceeded through 2-butyn-1-ol hydroalumination with Red-Al10 followed by quenching with I2. The desired trichloroacetimidate was formed by exposing the iodoallylic alcohol to NaH and Cl3CCN. Epoxide 8 is commercially available in enantiomerically pure form, though cost constraints mandate that it be synthesized rather than purchased. Epoxidation of ethyl 3-butenoate with m-CPBA followed by hydrolytic kinetic resolution11 with Jacobsen’s (S,S)-salenCo(III) catalyst provided 8 in suitable yield. The epoxide was opened with lithium trimethylsilylacetylide and Et2AlCl12 to provide alcohol 10 in 85% yield. This unit was prepared in >99% ee as determined by Mosher ester analysis.13 Epoxide 9 is also commercially available in enantiomerically pure form but expense again dictated that it be prepared through hydrolytic kinetic resolution of the less expensive racemic epoxide with the (S,S)-salenCo(III) catalyst. Opening the epoxide with the ethylene diamine complex of lithium acetylide14 yielded 11 in 71% yield and in >99% ee (as determined by Mosher ester analysis). Attempts to prepare 11 directly through the addition of allenyl tributyltin to butanal in the presence of (R)-BINOL and Ti(OiPr)415 were productive with respect to overall yield, though the enantiomeric excess of 67% was not suitable for the synthesis.
Scheme 3.

Subunit preparation.
We initially attempted to use TMSOTf to promote the ether linkage formation16 between 7 and 10 (Scheme 4) to avoid the use of harshly basic Williamson ether synthesis conditions that could cause 10 to undergo a retro aldol reaction. Significant alkene isomerization was observed, however, leading to an inseparable 4:1 mixture of geometrical isomers 12 and 13 in 74% yield. During the course of an extensive optimization effort that focused on concentration, stoichiometry, and temperature variations, we observed that the addition of 2,6-di-tert-butylpyridine completely suppressed the etherification reaction. This indicated that adventitious TfOH was the real catalyst for the reaction. Combining 10 with an excess of 7 in the presence of 15 mol% TfOH resulted in the isolation of a 7.3:1 mixture of 12 and 13 in 77% yield.
Scheme 4.

TfOH-mediated etherification.
The remaining carbons for the neopeltolide macrolactone could, in principle, be introduced through a Sonogashira coupling17 between 11 and 12 (Scheme 5). The yield of enyne 14, however, never exceeded 35% despite repeated attempts to optimize the reaction conditions with respect to palladium catalyst, solvent, and amine. We postulated that the inefficiency derived from the free hydroxyl group in 11, and this was confirmed when we subjected silyl ether 15 to standard Sonogashira conditions with 12 and isolated enyne 16 in 89% yield. While this yield was acceptable, the use of a silyl ether solely as a protecting group was not desirable. In consideration our plan to perform a regioselective alkyne hydration reaction through intramolecular hydrosilylation, we studied the possibility of using dialkylsilyl ethers in the coupling reaction. While the dimethylsilyl and diphenylsilyl ethers of 11 proved to be too labile to survive the coupling, diisopropylsilyl ether 17, prepared in 77% yield by treating 11 with iPr2Si(H)Cl and imidazole, coupled with 12 to form enyne 18 in 89% yield.
Scheme 5.

Sonogashira coupling.
The regioselective alkyne hydration reaction required that the sequence18 of intramolecular hydrosilylation and Tamao oxidation proceed without isomerization of the product β,γ-unsaturated ketone to the conjugated isomer. The hydrosilylation of 18 proceeded quite smoothly with Pt(DVDS)19 (DVDS = divinyl dimethyldisoloxane) to form siloxane 19. The isopropyl groups in 19 create greater steric congestion than the methyl groups that are generally used in these reactions, dictating that the Tamao oxidation20 be conducted under than unusually harsh conditions. Extensive optimization led to the identification of a suitable protocol in which 19 was exposed to a mixture of Bu4NF, KF, KHCO3, and aqueous H2O2 in THF and DMF at 40°C to yield 20 in 57% yield as an acceptable 11:1 mixture with conjugated isomer 21. These conditions also led to concomitant cleavage of the alkynylsilane group. The alkene stereoisomer that had been carried through the sequence since the etherification reaction was easily be removed at this stage. Attempts to improve the yield of the hydration by conducting a trans-hydrosilylation with [(C6H6)RuCl2]221 were unsuccessful.
The remainder of the sequence to form the macrolactone substrate (Scheme 7) proceeded with little incident. Reduction of 20 under Evans-Tischenko22 conditions provided hydroxy ester 22 in 77% yield as a single stereoisomer. Conjugated isomer 21 was unreactive in this reaction and was easily separated. This transformation is ideally suited for neopeltolide synthesis, as observed in most approaches to this structure, because it establishes the appropriate stereochemical relationship between C11 and C13 while selectively protecting the C13 hydroxyl group. Selective protection allows for efficient methylation of the C11 hydroxyl group to form 22. Cleaving both ester groups and lactonizing under Yamaguchi’s conditions23 provided 23 in 71% overall yield. Exposing 23 to HOAc and [(p-cymene)RuCl2]224 provided cyclization substrates 5 along with regioisomer 24 as an inseparable 5:1 mixture in 82% yield. We have modified the reported protocol for enol acetate formation to include 1-decyne. This promotes greater reproducibility for the reaction by using a sacrificial alkyne to aid in the generation of the catalytically relevant ruthenium phosphine complex.
Scheme 7.

Completion of the cyclization substrate synthesis.
The oxidative cyclization of 5 (Scheme 8) was significantly slower than previous cyclization reactions of acyclic allylic ethers that contain trisubstituted alkenes.7 Two factors could lead to the slow kinetics. The proximity of the ether oxygen to the electron withdrawing carboxyl group reduces its capacity to stabilize the intermediate oxocarbenium ion. We have observed that these reactions are dependent upon the oxidation potential of the substrate and the stability of the intermediate carbocation.25 Inductively destabilizing the cation, therefore, slows the reaction. The macrocycle also could adapt a conformation that inhibits proper alignment between the cleaving carbon–hydrogen bond and the π-bond of the alkene. Stereoelectronic effects have been established as an important kinetic factor in other oxidative cleavage reactions.26 The reaction proceeded to completion after 18 h at rt by using 3 eq DDQ. This provided a 57% yield of bicyclic tetrahydropyran 25. In consideration of the fact that the starting material was an 84:16 mixture of enol acetate regioisomers, this yield could be extrapolated to 65% if pure 5 were used in the reaction. No 5-endo-cyclization product from the reaction of 24 was isolated.
Scheme 8.

Oxidative cyclization.
Completing the synthesis (Scheme 9) required that the Δ8,9 alkene be reduced with high facial control. A crystal structure27 of 25 showed that the molecule prefers a curved conformation in which the sterically more-accessible convex face is the should react with H2 to provide the desired stereoisomer. Indeed, subjecting 25 to H2 and Pd/C provided 4 in 74% yield. A small amount (<10%, impure) of the stereoisomer at C9 was also isolated. The conversion of 4 to neopeltolide has been reported,5,6f but we completed the synthesis to have the natural product for subsequent biological studies. Reduction of 4 with NaBH4 in MeOH followed by Mitsunobu esterification28 with acid 26 (prepared according to the protocol of Wipf and Graham29) provided 3.
Scheme 9.

Synthesis of neopeltolide.
2.3 Analog synthesis
The presence of the Δ8,9 alkene in 25 creates opportunities for analog synthesis. Simply reducing the C5 carbonyl group of 25 with NaBH4 followed by appending the side chain provided dehydroneopeltolide 27 (Scheme 10). Although this structure is not likely to differ from neopeltolide with respect to physical properties or biological activity, the removal of one step in the sequence improves analog accessibility.
Scheme 10.

Synthesis of dehydroneopeltolide.
We subjected 25 to hydrogenation in the presence of Crabtree’s catalyst30 in an effort to determine whether we could exploit reagent coordination to the tetrahydropyranyl oxygen to functionalize the alkene from the convex face (Scheme 11). While the diastereoselectivity for the reaction was low (~1.4:1), major product 28, the C9-epimer of 4, was isolated in a reasonable 54% yield. Despite the low selectivity, this result suggests that future analogs can be accessed through coordinated delivery of reagents31 to the less accessible face of the alkene. Reduction of 28 with NaBH4 followed by esterification with 26 provided C9-epi-neopeltolide 29.
Scheme 11.

Synthesis of 9-epi-neopeltolide.
The alkene provides opportunities for introducing polar groups that could improve physical properties such as water solubility. Exposing 25 to OsO4 and pyridine, followed by osmate ester cleavage with NMO yielded diol 30 in 75% yield as a single diastereomer. This reaction required stoichiometric OsO4 to proceed at a reasonable rate, though at a larger scale the OsO4 loading could conceivably be lowered. Ketone reduction and acylation provided a 50% yield of dihydroxyneopeltolide 31. We approached the epoxide analog by reducing 25 with NaBH4 followed by exposing the resulting alcohol to m-CPBA to provide 32. Conducting the reduction prior to epoxidation averted the potential for 25 to undergo a competitive Bayer-Villiger reaction. Acylation gave epoxyneopeltolide 33 in 66% yield.
3. Summary and Conclusions
We have shown that our method for oxidative carbocation formation in a macrocycle can be applied to the total synthesis of the potent cytotoxin neopeltolide. The use of a relatively inert ether as a precursor to an oxocarbenium ion obviates the need for step investment with respect to protecting groups and/or activating groups, leading to a brief overall sequence. This work also highlights the versatility of alkynes in complex molecule synthesis32 (coupling through the Sonogashira reaction, hydration to form a ketone, and acetoxylation to form an enol acetate). While neopeltolide does not contain the alkenyl group that is required to conduct the oxidative cyclization, the unsaturation can be exploited to provide a number of interesting analogs. Hydrogenation can be conducted with Pd/C or Crabtree’s catalyst provides the natural product or an epimer. Retaining the alkene provide a structure that can be accessed in one fewer step. Dihydroxylation and epoxidation yield analogs that are more polar than the natural product. We have initiated studies to evaluate the biological activities of these and other analogs. The results of these studies will be reported elsewhere.
4. Experimental Section
4.1. General experimental
Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded on Bruker Avance 300 spectrometer at 300 MHz and 75 MHz, respectively. The chemical shifts are given in parts per million (ppm) on the delta (δ) scale. Tetramethylsilane (TMS) or the solvent peak was used as a reference value, for 1H NMR: TMS (in CDCl3) = 0.00 ppm, CD3OD =3.31, for 13C NMR: TMS (in CDCl3) = 0.00, CD3OD = 49.00. Data are reported as follows: (s = singlet; d = doublet; t = triplet; q = quartet; dd = doublet of doublets; dt = doublet of triplets; br = broad). High resolution and low resolution mass spectra were recorded on a VG 7070 spectrometer. Infrared (IR) spectra were collected on a Mattson Cygnus 100 spectrometer. Optical rotations were measured on a Perkin-Elmer 241 polarimeter. Samples for IR were prepared as a thin film on a NaCl plate by dissolving the compound in CH2Cl2 and then evaporating the CH2Cl2. Analytical TLC was performed on E. Merck pre-coated (25 mm) silica gel 60F-254 plates. Visualization was done under UV (254 nm). Flash chromatography was done using ICN SiliTech 32–63 60 Å silica gel. Methylene chloride was distilled under N2 from CaH2. Reagent grade ethyl acetate, diethyl ether, pentane and hexanes (commercial mixture) were purchased from EM Science and used as is for chromatography. Benzene was dried with 4A molecular sieves. THF was distilled from sodium. Other reagents were obtained from commercial source without further purification. All reactions were performed in oven or flame-dried glassware with magnetic stirring unless otherwise noted. The synthesis and characterization of compounds 4, 5, 7, 10, 12, 18, 20, 22, 23, and 25 have previously been reported.
4.2. General procedure for macrocyclic ketone reduction
To the macrocyclic ketone (0.020 mmol) in MeOH (0.5 mL) at 0 °C was added NaBH4 (0.040 mmol) was added. After 10 min, AcOH (0.40 mmol) was added and the reaction was concentrated under vacuum. The resulting residue was purified by flash column chromatography to afford the desired macrocyclic alcohol.
4.3. General procedure for Mitsunobu acylation
Tohe macrocyclic alcohol (0.019 mmol), 26 (0.078 mmol), and PPh3 (0.086 mmol) in benzene (0.5 mL) was added DIAD (0.086 mmol). After ten minutes the reaction was concentrated under vacuum. The resulting residue was purified by flash column chromatography to afford the desired product.
4.4 Procedures and characterization data
4.4.1. (1R,5S,7S,9S,11R,13S)-13-hydroxy-7-methoxy-9-methyl-5-propyl-4,15-dioxabicyclo[9.3.1]pentadecan-3-one
The general ketone reduction protocol was followed with 4 (6.5 mg, 0.020 mmol) and NaBH4 (1.6 mg, 0.042 mmol). Flash column chromatography (50% EtOAc in hexanes) yielded the desired product (6.2 mg, 95%). 1H NMR (300 MHz, CDCl3) δ 5.12–5.21 (m, 1H), 3.75–3.87 (m, 1H), 3.73 (ddd, J = 2.0, 4.4, 10.9 Hz, 1H), 3.59 (t, J = 9.4 Hz, 1H), 3.32 (s, 3H), 3.18 (t, J =9.3 Hz, 1H), 2.63 (dd, J = 4.4, 14.5 Hz, 1H), 2.44 (dd, J = 10.7, 14.5 Hz, 1H), 1.99 (ddd, J = 1.8, 1.8, 11.5 Hz, 1H), 1.81–1.92 (m, 2H), 1.43–1.78 (m, 5H), 1.12–1.41 (m, 6H), 1.0 (d, J = 6.7 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 170.8, 78.6, 75.6, 73.3, 72.3, 68.1, 56.2, 44.1, 42.2, 42.2, 41.9, 40.7, 40.0, 36.9, 31.2, 25.5, 19.0, 13.9; IR (film) 3416, 2918, 2871, 1730, 1650, 1459, 1087 cm−1; HRMS (ESI) calcd. for C18H32O5Na ([M+Na]+) 351.2147, found 351.2159; [α]D25 = +17.1 (CHCl3, c = 0.2).
4.4.2. Synthesis of neopeltolide (3)
The general Mitsunobu acylation procedure was followed with the macrocyclic alcohol (6.2 mg, 0.019 mmol), 26 (22.0 mg, 0.078 mmol), Ph3P (11.4 mg, 0.086 mmol) and DIAD (16.8 μL, 0.086 mmol). Flash column chromatography (30% EtOAc in hexanes) yielded neopeltolide (10.5 mg, 93%). 1H NMR (300 MHz, CD3OD) δ 7.66 (s,1H), 6.32–6.41 (m, 1H), 6.28 (dt, J = 2.1, 13.8 Hz, 1H), 6.03 (pentet, J = 6.0 Hz, 1H), 5.88 (d, J = 11.6 Hz, 1H), 5.20 (t, J = 2.8 Hz, 1H), 5.12–5.22 (m, 1H), 4.30 (dd, J = 1.4, 4.2 Hz, 2H), 4.06 (t, J = 9.3 Hz, 1H), 3.70 (s, 1H), 3.65 (s, 3H), 3.56 (t, J = 9.0 Hz, 1H), 3.28 (s, 3H), 3.01 (dd, J = 7.6, 15.1 Hz, 2H), 2.71 (s, 1H), 2.70 (dt, J = 4.4, 14.7 Hz, 1H), 2.23 (dd, J = 11.0, 14.7 Hz, 1H), 1.73–1.90 (m, 2H), 1.65–1.73 (m, 2H), 1.44–1.60 (m, 5H), 1.26–1.44 (m, 6H), 1.07–1.18 (m, 1H), 0.97(d, J = 6.3, 3H), 0.92 (d, J = 13.6 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 173.1, 166.9, 161.9, 159.5, 150.0, 142.3, 139.3, 136.0, 121.7, 116.0, 77.2, 77.1, 74.0, 71.4, 69.2, 56.4, 52.6, 45.3, 43.5, 43.3, 41.1, 38.0, 37.6, 37.4, 36.2, 32.6, 29.0, 26.4, 26.0, 20.0, 14.2; IR (film) 3357, 2954, 2922, 2854, 1719, 1646, 1537, 1458, 1376, 1342, 1249, 1178, 1064, 994, 777 cm−1; HRMS (ESI) calcd. for C31H46N2O9Na ([M+Na]+) 613.3101, found 613.3076; [α]D25 = +17.5 (MeOH, c = 0.24).
4.4.3. (1R,5S,7S,11S,13R,Z)-13-hydroxy-7-methoxy-9-methyl-5-propyl-4,15-dioxabicyclo[9.3.1]pentadec-9-en-3-one
The general ketone reduction protocol was followed with 25 (14.9 mg, 0.046 mmol) and NaBH4 (3.6 mg, 0.095 mmol). Flash column chromatography (50% EtOAc in hexanes) yielded the desired product (12.6 mg, 84%). 1H NMR (300 MHz, CDCl3) δ 5.31–5.37 (m, 1H), 5.30 (d, J =7.2 Hz, 1H), 3.82–3.93 (m, 3H), 3.49–3.56 (m, 1H), 3.36 (s, 3H), 2.63 (dd, J = 3.8, 15.1 Hz, 1H), 2.51 (dd, J = 11.1, 15.1 Hz, 1H), 2.33 (d, J = 13.3 Hz, 1H), 1.95–2.05 (m, 3H), 1.86 (d, J = 0.6 Hz, 3H), 1.81–1.89 (m, 1H), 1.64–1.74 (m, 1H), 1.47–1.59 (m, 2H), 1.44 (s, 1H), 1.18–1.40 (m, 3H), 0.92 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 170.3, 146.9, 125.1, 81.8, 74.1, 72.4, 72.0, 68.1, 57.9, 43.3, 42.1, 41.8, 40.5, 40.3, 37.4, 25.2, 18.7, 13.9; IR (film) 3422, 2959, 2923, 2854, 1732, 1654, 1454, 1373, 1315, 1264, 1195, 1080, 1035, 981, 939, 842, 755 cm−1; HRMS (ESI) calcd. for C18H30O5Na ([M+Na]+) 349.1991, found 349.1992; [α]D25 = −47.0 (CHCl3, c = 0.38).
4.4.4. Dehydroneopeltolide (27)
The general Mitsunobu acylation protocol was followed with the macrocyclic alcohol (6.8 mg, 0.021 mmol), 26 (23.0 mg, 0.083 mmol), Ph3P (23.5 mg, 0.090 mmol) and DIAD (17.8 μL, 0.090mmol). Flash column chromatography (30% EtOAc in hexanes) yielded dehydro-neopeltolide (8.6 mg, 70%). 1H NMR (300 MHz, CD3OD) δ 7.67 (s, 1H), 6.34–6.43 (m, 1H), 6.28 (dt, J = 2.0, 12.0 Hz, 1H), 6.05 (pentet, J = 6.0 Hz, 1H), 5.90 (dt, J = 1.5, 11.5 Hz, 1H), 5.25–5.33 (m, 1H), 5.27 (t, J = 2.8 Hz, 1H), 5.22 (d, J = 6.7 Hz, 1H), 4.31 (d, J = 4.7 Hz, 2H), 4.19–4.28 (m, 2H), 3.66 (s, 3H), 3.58–3.65 (m, 1H), 3.34 (s, 3H), 3.03 (ddd, J = 1.4, 7.7, 7.7 Hz, 2H), 2.73(t, J = 7.0 Hz, 2H), 2.69(dd, J = 3.4, 10.2 Hz, 1H), 2.27–2.38 (m, 2H), 1.95 (dd, J = 10.4, 13.6 Hz, 1H), 1.84 (s, 3H), 1.62–1.88 (m, 5H), 1.49–1.58 (m, 3H), 1.23–1.30 (m, 3H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 172.7, 166.9, 162.0, 159.7, 150.1, 148.0, 142.3, 139.3, 136.0, 127.0, 121.8, 116.0, 83.4, 75.2, 71.3, 71.1, 69.1, 58.2, 52.7, 44.2, 43.3, 42.5, 41.1, 38.5, 36.3, 35.9, 29.1, 26.5, 25.8, 19.9, 14.3; IR (film) 3351, 2957, 2923, 1719, 1521, 1458, 1268, 1170, 1097, 1072, 892, 818, 776 cm−1; HRMS (ESI) calcd. for C31H44N2O9Na ([M+Na]+) 611.2945, found 611.2997; [α]D25 = −44.1 (MeOH, c = 0.56).
4.4.5. (1S,5S,7S,9R,11R)-7-methoxy-9-methyl-5-propyl-15-oxabicyclo[9.3.1]pentadecane-3,13-dione (28)
To 25 (6.1 mg, 0.019 mmol) in CH2Cl2 (0.2 mL) was added Crabtree’s catalyst (3.2 mg, 0.004 mmol). The flask was quickly evacuated and backfilled with H2, and this process was repeated three times. The reaction mixture was stirred at room temperature for 30 min., then was filtered through Celite, concentrated, and purified by flash chromatography (5% Et2O in CH2Cl2) to afford the desired product (3.3 mg, 54%). 1H NMR (300 MHz, CDCl3) δ 5.14–5.22 (m, 1H), 3.93–4.02 (m, 1H), 3.69 (m, 1H), 3.32 (s, 3H), 3.23–3.31 (m, 1H), 2.72 (dd, J = 4.5, 14.4 Hz, 1H), 2.46 (dd, J = 8.5, 14.4 Hz, 1H), 2.42 (dd, J = 4.1, 7.0 Hz, 1H), 2.31 (d, J = 7.8 Hz, 2H), 1.92 (dt, J = 2.2, 14.8 Hz, 1H), 1.74–1.83 (m, 1H), 1.43–1.73 (m, 4H), 1.16–1.39 (m, 6H), 0.94 (d, J = 6.8 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 206.4, 179.8, 80.4, 75.0, 73.8, 72.6, 56.3, 48.2, 46.8, 41.7, 41.6, 40.1, 39.0, 37.7, 28.6, 20.9, 18.7, 13.9; IR (film) 2956, 2923, 2853, 1723, 1459, 1370, 1330, 1252, 1185, 1105, 1081, 844, 799 cm−1; HRMS (ESI) calcd. for C18H30O5Na ([M+Na]+) 349.1991, found 349.1974; [α]D25 = −7.9 (CHCl3, c = 0.17).
4.4.6. (1R,5S,7S,9R,11R,13S)-13-hydroxy-7-methoxy-9-methyl-5-propyl-15-oxabicyclo[9.3.1]pentadecan-3-one
The general reduction protocol was followed with 28 (3.3 mg, 0.010 mmol) and NaBH4 (1.1 mg, 0.029 mmol). Flash column chromatography (35% EtOAc in hexanes) yielded the desired product (3.2 mg, 96%). 1H NMR (300 MHz, CDCl3) δ 5.11–5.20 (m, 1H), 3.80–3.86 (m, 1H), 3.69 (tdd, J = 1.9, 4.6, 11.0 Hz, 1H), 3.78 (tt, J = 2.4, 8.7 Hz, 1H), 3.31 (s, 3H), 3.27 (dt, J = 2.0, 9.1 Hz, 1H), 2.64 (dd, J = 4.6, 14.3 Hz, 1H), 2.41 (dd, J = 9.0, 14.3 Hz, 1H), 1.90–1.99 (m, 2H), 1.83–1.88 (m, 2H), 1.60–1.80 (m, 3H), 1.43–1.60 (m, 3H), 1.13–1.40 (m, 4H), 0.89–0.93 (m, 6H); 13C NMR (75 MHz, CDCl3) δ 170.5, 80.5, 74.6, 72.4, 71.5, 68.4, 56.4, 41.8, 41.7, 41.5, 40.3, 39.6, 39.4, 37.7, 28.7, 21.1, 18.7, 13.9; IR (film) 3363, 3296, 2916, 2852, 1718, 1490, 1457, 1364, 1274, 1256, 1220, 1160, 1108, 1074, 1036, 931 cm−1; HRMS (ESI) calcd. for C18H32O5Na ([M+Na]+) 351.2147, found 351.2166; [α]D25 = −9.6 (CHCl3, c = 0.32).
4.4.7. 9-epi-neopeltolide (29)
The general Mitsunobu acylation protocol was followed with the alcohol (3.1 mg, 0.0095 mmol), 26 (10.5 mg, 0.038 mmol), Ph3P (10.7 mg, 0.041 mmol) and DIAD (8.1 μL, 0.041 mmol). Flash column chromatography (30% EtOAc in hexanes) yielded C9-epi-neopeltolide (3.8 mg, 67%). 1H NMR (300 MHz, CD3OD) δ 7.65 (s, 1H), 6.36 (dt, J = 7.4, 11.6 Hz, 1H), 6.26 (dt, J = 2.0, 11.8 Hz, 1H), 6.02 (pentet, J = 6.1 Hz, 1H), 5.86 (dt, J = 1.6, 11.5 Hz, 1H), 5.22 (t, J = 2.8 Hz, 1H), 5.11–5.20 (m, 1H), 4.29 (dd, J = 1.5, 5.8 Hz, 2H), 3.95–4.05 (m, 1H), 3.76 (t, J = 6.0 Hz, 1H), 3.64 (s, 3H), 3.29 (s, 3H), 2.99 (dd, J = 7.1, 7.1 Hz, 2H), 2.69 (t, J = 7.7 Hz, 2H), 2.65 (dd, J = 4.4, 9.4 Hz, 1H), 2.24 (dd, J = 9.2, 14.4 Hz, 1H), 1.90 (d, J = 8.9 Hz, 1H), 1.75–1.84 (m, 3H), 1.39–1.68 (m, 4H), 1.23–1.39 (m, 6H), 1.22 (t, J = 7.2 Hz, 2H), 1.09–1.19 (m, 1H), 0.91 (t, J = 7.3 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 172.6, 166.9, 161.9, 159.6, 150.0, 142.2, 139.2 135.9, 121.7, 115.9, 82.1, 75.8, 70.8, 70.6, 69.4, 68.9, 56.5, 52.6, 42.6, 41.0, 40.9, 40.5, 38.8, 37.0, 35.8, 30.0, 29.0, 26.4, 21.5, 19.8, 14.2; IR (film) 2923, 2854, 1716, 1519, 1458, 1375, 1249, 1179, 1103, 817 cm−1; HRMS (ESI) calcd. for C31H46N2O9Na ([M+Na]+): 613.3101, found 613.3075; [α]D25 = +0.2 (MeOH, c = 0.20).
4.4.8 (1R,5S,7R,9R,10S,11S)-9,10-dihydroxy-7-methoxy-9-methyl-5-propyl-4,15-dioxa-bicyclo[9.3.1]pentadecane-3,13-dione (30)
To OsO4 (6.1 mg, 0.024 mmol) and pyridine (61μL, 0.76 mmol) in THF (0.2 mL) at 0 °C was added 25 (5.3 mg, 0.016 mmol) in THF (0.2 mL). After 10 min, NMO•H2O (4.3 mg, 0.032 mmol) was added and the reaction was allowed to room temperature over 30 min. The reaction was quenched by 1 mL saturated Na2S2O3 at 0 °C and extracted with three 2 mL portions of EtOAc. The organic layer was concentrated under vacuum and purified by flash column chromatography (5% MeOH in CH2Cl2) to afford the diol (4.4 mg, 75%). 1H NMR (300 MHz, CDCl3) δ 5.03–5.14 (m, 1H), 4.04 (tt, J = 3.4, 11.2 Hz, 1H), 3.66 (tt, J = 3.0, 11.2 Hz, 1H), 3.59 (s, 1H), 3.54–3.58 (m, 1H), 3.39 (s, 3H), 3.28 (t, J = 7.4 Hz, 1H), 2.84 (d, J = 7.2 Hz, 1H), 2.71 (dt, J = 1.8, 15.0, 1H), 2.65 (dd, J = 3.8, 14.5 Hz, 1H), 2.50 (dd, J = 11.1, 14.3 Hz, 2H), 2.40 (dd, J = 12.2, 15.1 Hz, 1H), 2.27 (dd, J = 11.5, 13.5 Hz, 1H), 2.15 (ddd, J = 1.3, 3.5, 14.4 Hz, 1H), 1.92 (dd, J = 2.9, 15.2 Hz, 1H), 1.63 (dd, J = 10.5, 14.8 Hz, 2H), 1.42–1.62 (m, 2H), 1.15–1.39 (m, 5H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 205.5, 169.5, 77.8, 77.7, 73.3, 73.0, 72.9, 56.2, 47.1, 45.0, 42.7, 41.5, 38.2, 37.5, 24.8, 18.4, 13.9; IR (film) 3441, 2923, 1721, 1555, 1460, 1377, 1267, 1183, 1100, 1069, 964, 934, 817 cm−1; HRMS (ESI) calcd. for C18H30O7Na ([M+Na]+) 381.1889, found 381.1902; [α]D25 = −28.5 (MeOH, c = 0.13).
4.4.9. (1R,5S,7R,9R,10S,11S,13R)-9,10,13-trihydroxy-7-methoxy-9-methyl-5-propyl-4,15-dioxabicyclo[9.3.1]pentadecan-3-one
The general reduction protocol was followed with 30 (3.6 mg, 0.010 mmol) and NaBH4 (1.1 mg, 0.029 mmol). Flash column chromatography (5% MeOH in CH2Cl2) yielded the desired triol (3.3 mg, 92%). 1H NMR (300 MHz, CD3OD) δ 5.09–5.18 (m, 1H), 3.70–3.83 (m, 2H), 3.49–3.55 (m, 1H), 3.46 (ddd, J = 1.7, 7.1, 11.1, 1H), 3.32 (s, 3H), 3.28 (s, 1H), 2.76 (dd, J = 4.6, 14.1 Hz, 1H), 2.30 (dd, J = 8.8, 14.1 Hz, 1H), 2.12 (dt, J = 2.2, 12.4 Hz, 1H), 1.97 (t, J = 14.8 Hz, 2H), 1.91–1.95 (m, 1H), 1.68 (dd, J = 10.8, 14.7 Hz, 2H), 1.50–1.65 (m, 3H), 1.20–1.40 (m, 8H), 0.97 (t, J =7.2 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 171.2, 77.0, 76.6, 76.5, 74.1, 74.0, 72.0, 67.5, 55.0, 45.4, 41.2, 40.6, 39.9, 37.6, 37.4, 25.6, 18.2, 12.8; IR (film) 3397, 2924, 1729, 1457, 1373, 1263, 1195, 1099, 1044, 937, 731 cm−1; HRMS (ESI) calcd. for C18H32O7Na ([M+Na]+): 383.2046, found 383.2034; [α]D25 = −8.6 (MeOH, c = 0.32).
4.4.10. Dihydroxyneopeltolide (31)
The general Mitsunobu acylation protocol was followed with the triol (3.5 mg, 0.01 mmol), 26 (4.1 mg, 0.015 mmol) and Ph3P (4.2 mg, 0.016 mmol), and DIAD (3.2 μL, 0.016 mmol) in benzene (0.2 mL). Flash chromatography (5% MeOH in CH2Cl2) yielded the desired product (3.0 mg, 50%). 1H NMR (300 MHz, CDCl3) δ 7.40 (s, 1H), 6.26–6.34 (m, 2H), 6.09 (pentet, J = 6.4 Hz, 1H), 5.90 (dt, J = 1.7, 11.5 Hz, 1H), 5.57 (br, 1H), 5.32 (t, J = 2.8 Hz, 1H), 5.06–5.14 (m, 1H), 4.31 (t, J = 6.1 Hz, 2H), 4.00 (tdd, J = 1.9, 1.9, 9.3 Hz, 1H), 3.69 (s, 3H), 3.59–3.67 (m, 2H), 3.40 (s, 1H), 3.37 (s, 3H), 3.35 (s, 1H), 3.16 (dd, J = 5.7, 8.4 Hz, 1H), 3.04 (ddt, J = 1.9, 7.4, 7.4 Hz, 2H), 2.90 (d, J = 6.0 Hz, 1H), 2.72 (t, J = 7.2 Hz, 2H), 2.54 (dd, J = 3.8, 14.4 Hz, 1H), 2.36 (dd, J = 11.2, 14.4 Hz, 1H), 2.01–2.11 (m, 3H), 1.83 (d, J = 9.6 Hz, 1H), 1.68 (dt, J = 3.0, 11.5 Hz, 1H), 1.50–1.65 (m, 5H), 1.35 (t, J = 7.3 Hz, 2H), 1.27 (s, 3H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 171.5, 165.5, 148.6, 140.8, 137.8, 134.6, 120.3, 114.5, 77.3, 76.8, 74.0, 73.9, 73.6, 69.5, 67.6, 54.9, 45.1, 41.1, 40.1, 37.4, 34.5, 32.5, 27.5, 25.6, 24.9, 18.2, 12.8; IR (film) 3358, 2957, 2922, 2853, 2500, 1715, 1635, 1553, 1460, 1396, 1262, 1169, 1095, 1052, 1019, 816, 795, 780 cm−1; HRMS (ESI) calcd. for C31H46N2O11Na ([M+Na]+) 645.2999, found 645.2996; [α]D25 = −2.1 (MeOH, c = 0.16).
4.4.12. (1S,2S,4R,6R,8S,12R,14R)-14-Hydroxy-6-methoxy-4- methyl-8-propyl-3,9,16-trioxatricyclo[10.3.1.02,4]hexadecan-10- one (32)
To the unsaturated lactone (5.9 mg, 0.018 mmol) and NaHCO3 (5.8 mg, 0.069 mmol) in CH2Cl2 (1 mL) at 0 °C was added recrystallized m-CPBA (4.5 mg, 0.026 mmol). The reaction stirred at 0 °C for 4 hours and then warmed to room temperature and stirred overnight. The reaction was quenched by the addition of 0.7 mL saturated Na2S2O3 and 0.7 mL saturated NaHCO3 at 0 °C and extracted with three 1 mL portions of ethyl acetate. Flash chromatography (50% EtOAc in hexanes) afforded the epoxy alcohol (3.4 mg, 55%). 1H NMR (300 MHz, CDCl3) δ 5.32–5.42 (m, 1H), 3.8 (br, 1H), 3.76 (tdd, J = 1.5, 3.8, 10.9 Hz, 1H), 3.65 (dd, J = 5.4, 8.9 Hz, 1H), 3.06 (ddd, J = 2.0, 8.0, 10.9 Hz, 1H), 2.71 (d, J = 7.8 Hz, 1H), 2.58 (dd, J = 3.8, 15.1 Hz, 1H), 2.50 (dd, J = 10.8, 15.2 Hz, 1H), 2.25 (dt, J = 2.4, 12.4 Hz, 1H), 2.12 (dd, J = 11.2, 14.6 Hz, 1H), 1.86–2.00 (m, 3H), 1.43–1.62 (m, 5H), 1.34 (s, 3H), 1.24–1.39 (m, 6H), 0.91 (t, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 170.2, 73.9, 73.4, 67.4, 64.6, 61.8, 57.2, 44.4, 41.8, 41.6, 39.8, 39.3, 37.3, 23.3, 22.7, 18.7, 13.9; IR (film) 3435, 2924, 2854, 1732, 1459, 1374, 1262, 1200, 1078, 942, 799cm−1; HRMS (ESI) calcd. for C18H30O6Na ([M+Na]+) 365.1940, found 365.1932; [α]D25 = −23.5 (CHCl3, c = 0.33).
4.4.13. Epoxyneopeltolide (33)
The general Mitsunobu acylation protocol was followed with 32 (3.1 mg, 0.009 mmol), 26 (11.2 mg, 0.040 mmol), Ph3P (11.4 mg, 0.044 mmol) and DIAD (8.6 μL, 0.044 mmol). Flash column chromatography (30% EtOAc in CH2Cl2) yielded epoxyneopeltolide (3.6 mg, 66%). 1H NMR (300 MHz, CD3OD) δ 7.65 (s, 1H), 6.36 (dt, J = 4.1, 7.3 Hz, 1H), 6.27 (dt, J = 2.1, 11.9 Hz, 1H), 6.03 (pentet, J = 6.0 Hz, 1H), 5.89 (dt, J = 1.6, 11.6 Hz, 1H), 5.27–5.39 (m, 1H), 5.28 (t, J = 3.0 Hz, 1H), 4.30 (dd, J = 1.8, 6.1 Hz, 2H), 4.08–4.18 (m, 1H), 3.72 (dd, J = 5.3, 10.0 Hz, 1H), 3.65 (s, 3H), 3.34–3.42 (m, 1H), 3.37 (s, 3H), 3.00 (ddd, J = 1.7, 7.4, 7.4 Hz, 2H), 2.71 (t, J = 7.1 Hz, 2H), 2.63–2.70 (m, 2H), 2.33 (dd, J = 11.6, 15.2 Hz, 1H), 2.04 (dd, J = 10.4, 14.7 Hz, 2H), 1.74–1.96 (m, 4H), 1.47–1.66 (m, 4H), 1.26–1.40 (m, 4H), 1.30 (s, 3H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (75 MHz, CD3OD) δ 172.4, 166.7, 161.9, 150.2, 142.2, 139.1, 136.0, 121.6, 116.0, 77.8, 74.9, 72.2, 68.2, 66.1, 63.0, 57.0, 44.8, 42.7, 42.0, 38.4, 35.4, 34.9, 29.0, 26.4, 23.7, 19.7, 14.1; IR (film) 3352, 2960, 2924, 1724, 1644, 1521, 1440, 1380, 1266, 1253, 1173, 1126, 1077, 1006, 968, 778 cm−1; HRMS (ESI) calcd. for C31H46N2O10Na ([M+Na]+) 627.2894, found 627.2910; [α]D25 = −11.3 (MeOH, c = 0.34).
Supplementary Material
Scheme 6.

Alkyne hydration through hydrosilylation.
Scheme 12.

Synthesis of dihydroxy- and epoxyneopeltolide.
Acknowledgments
We thank the National Institutes of Health (GM062924) for generous support of this work. We thank Dr. Steve Geib for crystallographic analysis.
Footnotes
Copies of 1H and 13C NMR spectra for all new compounds can be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Wright AE, Botelho JC, Guzman E, Harmody D, Linley P, McCarthy PJ, Pitts TP, Pomponi SA, Reed JK. J Nat Prod. 2007;70:412. doi: 10.1021/np060597h. [DOI] [PubMed] [Google Scholar]
- 2.a) D’Ambrosio M, Guerriero A, Debitus C, Pietra F. Helv Chim Acta. 1996;79:51. [Google Scholar]; b) D’Ambrosio M, Tató M, Poscfalvi G, Debitus C, Pietra F. Helv Chim Acta. 1999;82:347. [Google Scholar]
- 3.a) Hornberger KR, Hamblett CL, Leighton JL. J Am Chem Soc. 2000;122:12894. [Google Scholar]; b) Kopecky DJ, Rychnovsky SD. J Am Chem Soc. 2001;123:8420. doi: 10.1021/ja011377n. [DOI] [PubMed] [Google Scholar]; c) Wipf P, Reeves JT. Chem Commun. 2002:2066. doi: 10.1039/b207383h. [DOI] [PubMed] [Google Scholar]; d) Fettes A, Carreira EM. Angew Chem, Int Ed. 2002;41:4098. doi: 10.1002/1521-3773(20021104)41:21<4098::AID-ANIE4098>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; e) Fettes A, Carreira EM. J Org Chem. 2003;68:9274. doi: 10.1021/jo034964v. [DOI] [PubMed] [Google Scholar]; f) Wang Y, Janjic J, Kozmin SA. J Am Chem Soc. 2002;124:13670. doi: 10.1021/ja028428g. [DOI] [PubMed] [Google Scholar]; g) Wang Y, Janjic J, Kozmin SA. Pure Appl Chem. 2005;77:1161. [Google Scholar]; h) Paterson I, Tudge M. Angew Chem, Int Ed. 2003;42:343. doi: 10.1002/anie.200390112. [DOI] [PubMed] [Google Scholar]; i) Paterson I, Tudge M. Tetrahedron. 2003;59:6833. [Google Scholar]; j) Williams DR, Plummer SV, Patnaik S. Angew Chem, Int Ed. 2003;42:3934. doi: 10.1002/anie.200351817. [DOI] [PubMed] [Google Scholar]; k) Williams DR, Patnaik S, Plummer SV. Org Lett. 2003;5:5035. doi: 10.1021/ol036071v. [DOI] [PubMed] [Google Scholar]; l) Crimmins MT, Siliphaivanh P. Org Lett. 2003;5:4641. doi: 10.1021/ol035797o. [DOI] [PubMed] [Google Scholar]; m) Su Q, Panek JS. Angew Chem, Int Ed. 2005;44:1223. doi: 10.1002/anie.200462408. [DOI] [PubMed] [Google Scholar]; n) Su Q, Dakin LA, Panek JS. J Org Chem. 2007;72:2. doi: 10.1021/jo0610412. [DOI] [PubMed] [Google Scholar]; o) Ferrié L, Reymond S, Capdevielle P, Cossy J. Org Lett. 2007;9:2461. doi: 10.1021/ol070670a. [DOI] [PubMed] [Google Scholar]; p) Van Orden LJ, Patterson BD, Rychnovsky SD. J Org Chem. 2007;72:5784. doi: 10.1021/jo070901r. [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Jung HH, Seiders JR, II, Floreancig PE. Angew Chem, Int Ed. 2007;46:8464. doi: 10.1002/anie.200702999. [DOI] [PubMed] [Google Scholar]; r) Ferrié L, Boulard L, Pradaux F, Bouzbouz S, Reymond S, Capdevielle P, Cossy J. J Org Chem. 2008;73:1864. doi: 10.1021/jo701315h. [DOI] [PubMed] [Google Scholar]; s) Evans PA, Andrews WJ. Angew Chem, Int Ed. 2008;47:5426. doi: 10.1002/anie.200801357. [DOI] [PubMed] [Google Scholar]
- 4.Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Angew Chem, Int Ed. 2007;46:9211. doi: 10.1002/anie.200704122. [DOI] [PubMed] [Google Scholar]
- 5.Custar DW, Zabawa TP, Scheidt KA. J Am Chem Soc. 2008;130:804. doi: 10.1021/ja710080q. [DOI] [PubMed] [Google Scholar]
- 6.a) Woo SK, Kwon MS, Lee E. Angew Chem, Int Ed. 2008;47:3242. doi: 10.1002/anie.200800386. [DOI] [PubMed] [Google Scholar]; b) Vintonyak VV, Maier ME. Org Lett. 2008;10:1239. doi: 10.1021/ol8001255. [DOI] [PubMed] [Google Scholar]; c) Fuwa H, Naito S, Goto T, Sasaki M. Angew Chem, Int Ed. 2008;47:4737. doi: 10.1002/anie.200801399. [DOI] [PubMed] [Google Scholar]; d) Ulanovskaya OA, Janjic J, Suzuki M, Sabharwal SS, Schumaker PT, Kron SJ, Kozmin SA. Nature Chem Biol. 2008;4:418. doi: 10.1038/nchembio.94. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Paterson I, Miller NA. Chemical Commun. 2008:4708. doi: 10.1039/b812914b. [DOI] [PubMed] [Google Scholar]; f) Kartika R, Gruffi TR, Taylor RE. Org Lett. 2008;10:5047. doi: 10.1021/ol802254z. [DOI] [PubMed] [Google Scholar]; g) Vintonyak VV, Kunze B, Sasse F, Maier ME. Chem Eur J. 2008;14:11132. doi: 10.1002/chem.200801398. [DOI] [PubMed] [Google Scholar]; h) Custar DW, Zabawa TP, Hines J, Crews CM, Scheidt KA. J Am Chem Soc. 2009;131:12406. doi: 10.1021/ja904604x. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Kim H, Park Y, Hong J. Angew Chem, Int Ed. 2009;48:7577. doi: 10.1002/anie.200903690. [DOI] [PubMed] [Google Scholar]; j) Guinchard X, Roulland E. Org Lett. 2009;11:4700. doi: 10.1021/ol902047z. [DOI] [PubMed] [Google Scholar]; k) Fuwa H, Saito A, Naito S, Konoki K, Yotsu-Yamashita M, Sasaki M. Chem Eur J. 2009;15:12807. doi: 10.1002/chem.200901675. [DOI] [PubMed] [Google Scholar]; l) Yadav JS, Krishna GG, Kumar SN. Tetrahedron. 2010;66:480. [Google Scholar]; m) Gallon J, Reymond S, Cossy J. Comp Rend Chim. 2008;11:1463. [Google Scholar]
- 7.a) Tu W, Liu L, Floreancig PE. Angew Chem, Int Ed. 2008;47:4184. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; b) Liu L, Floreancig PE. Org Lett. 2009;11:3152. doi: 10.1021/ol901188q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a preliminary account of a portion of the material in this paper, see: Tu W, Floreancig PE. Angew Chem, Int Ed. 2009;48:4567. doi: 10.1002/anie.200901489.
- 9.a) Schulteelte KH, Hauser A, Ohloff G. Helv Chim Acta. 1979;62:2673. [Google Scholar]; b) Wender PA, De Brabander J, Harran PG, Jimenez JM, Koehler MFT, Lippa B, Park CM, Shiozaki M. J Am Chem Soc. 1998;120:4534. [Google Scholar]; c) Wender PA, DeChristopher BA, Schrier AJ. J Am Chem Soc. 2008;130:6658. doi: 10.1021/ja8015632. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bahnck KB, Rychnovsky SD. J Am Chem Soc. 2008;130:13177. doi: 10.1021/ja805187p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denmark SE, Jones TK. J Org Chem. 1982;47:4595. [Google Scholar]
- 11.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 12.Shen R, Lin CT, Porco JA., Jr J Am Chem Soc. 2002;124:5650. doi: 10.1021/ja026025a. [DOI] [PubMed] [Google Scholar]
- 13.Dale JA, Dull DL, Mosher HS. J Org Chem. 1969;34:2543. [Google Scholar]
- 14.Schmidt DR, Park PK, Leighton JL. Org Lett. 2003;5:3535. doi: 10.1021/ol035431b. [DOI] [PubMed] [Google Scholar]
- 15.Yu CM, Choi HS, Yoon SK, Jung WH. Synlett. 1997:889. [Google Scholar]
- 16.a) Wei SY, Tomooka K, Nakai T. J Org Chem. 1991;56:5973. [Google Scholar]; b) Maleczka RE, Jr, Geng F. Org Lett. 1999;1:1111. doi: 10.1021/ol9909132. [DOI] [PubMed] [Google Scholar]; c) Clark JS, Fessard TC, Wilson C. Org Lett. 2004;6:1773. doi: 10.1021/ol049483s. [DOI] [PubMed] [Google Scholar]
- 17.a) Sonogashira K, Todah Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]; b) Chinchilla R, Nájera C. Chem Rev. 2007;107:874. doi: 10.1021/cr050992x. [DOI] [PubMed] [Google Scholar]
- 18.a) Marshall JA, Yanik MM. Org Lett. 2000;2:2173. doi: 10.1021/ol0061182. [DOI] [PubMed] [Google Scholar]; b) Burova SA, McDonald FE. J Am Chem Soc. 2004;126:2495. doi: 10.1021/ja039618+. [DOI] [PubMed] [Google Scholar]; c) Trost BM, Ball ZT, Laemmerhold KM. J Am Chem Soc. 2005;127:10028. doi: 10.1021/ja051578h. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Robles O, McDonald FE. Org Lett. 2008;10:1811. doi: 10.1021/ol800659z. [DOI] [PubMed] [Google Scholar]
- 19.Denmark SE, Wang Z. Org Lett. 2001;3:1073. doi: 10.1021/ol0156751. [DOI] [PubMed] [Google Scholar]
- 20.Tamao K, Maeda K, Tanaka T, Ito Y. Tetrahedron Lett. 1988;29:6955. [Google Scholar]
- 21.Denmark SE, Pan W. Org Lett. 2002;4:4163. doi: 10.1021/ol026933c. [DOI] [PubMed] [Google Scholar]
- 22.Evans DA, Hoveyda AH. J Am Chem Soc. 1990;112:6447. [Google Scholar]
- 23.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 24.a) Goossen LJ, Paetzold J, Koley D. Chem Commun. 2003:706. [PubMed] [Google Scholar]; b) Neveux M, Bruneau C, Dixneuf PH. J Chem Soc, Perkin Trans 1. 1991:1197. [Google Scholar]
- 25.Jung HH, Floreancig PE. Tetrahedron. 2009;65:10830. doi: 10.1016/j.tet.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Tolbert LM, Khanna RK, Popp AE, Gelbaum L, Bottomley LA. J Am Chem Soc. 1990;112:2373. [Google Scholar]; b) Perrott AL, de Lijser HJP, Arnold DR. Can J Chem. 1997;75:384. [Google Scholar]; c) Freccero M, Pratt A, Albini A, Long C. J Am Chem Soc. 1998;120:284. [Google Scholar]; d) Floreancig PE. Synlett. 2007:191. [Google Scholar]
- 27.CCDC721650 contains the supplimentary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 28.Mitsunobu O. Synthesis. 1981:1. [Google Scholar]
- 29.Wipf P, Graham TH. J Org Chem. 2001;66:3242. doi: 10.1021/jo005787q. [DOI] [PubMed] [Google Scholar]
- 30.Crabtree RH, Davis MW. J Org Chem. 1986;51:2655. [Google Scholar]
- 31.Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307. [Google Scholar]
- 32.For recent discussions, see: Trost BM, Weiss AH. Angew Chem, Int Ed. 2007;46:7664. doi: 10.1002/anie.200702637.Trost BM, Sieber JD, Qian W, Dhawan R, Ball ZT. Angew Chem, Int Ed. 2009;48:5478. doi: 10.1002/anie.200901907.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.