

Abstract
Infection of human dendritic cells (DCs) by negative-strand RNA viruses, such as Newcastle disease virus, leads to the induction of the IFNβ gene, IFNB1, through the activation of the RNA helicase RIG-I, which is encoded by DDX58. Expression levels of IFNB1 and DDX58 in infected DCs showed positive correlations at the population and the single-cell levels. DDX58 has a common and potentially functional single nucleotide polymorphism, rs10813831 (A/G), encoding an Arg7Cys amino acid change in the RIG-I protein caspase recruitment domain (CARD). Quantitative RT-PCR analysis on Newcastle disease virus-infected primary DCs from 130 individuals revealed a significant association of the Arg7Cys single nucleotide polymorphism with increased IFNB1 and DDX58 transcription. Allelic imbalance analysis ruled out allele-specific DDX58 message levels and suggested that the observed association between Arg7Cys and IFNB1 and DDX58 transcription originated from a functional change in RIG-I due to the amino acid substitution in the CARD. DDX58 transfection experiments in 293T cells confirmed a biological functional difference between RIG-I 7Cys and the more common RIG-I 7Arg. Taken together, these data indicate that the innate immune response to viral infection of human cells is modified by a functional polymorphism in the RIG-I CARD.
Dendritic cells (DCs) are considered the “first responders” to viral infection by the innate immune system. They also play a crucial role in the development of viral Ag-specific adaptive immunity (1). A key step in the DC response to viral infection is the induction of IFNβ (IFNB1), a secreted cytokine that complexes with type 1 IFNRs to initiate a coordinated signaling pathway leading to widespread viral resistance (2, 3). Induction of IFNB1 can be triggered through TLRs (4) or through TLR-independent pathways, which are activated by the caspase recruitment domain (CARD) family RNA helicases RIG-I (RIG-I, encoded by DDX58) or MDA5 (encoded by MDA5) (5, 6). RIG-I and MDA5 proteins contain two CARDs and an RNA helicase domain.
RIG-I undergoes a conformational change upon interacting with short, double-stranded, 5′-triphosphate-containing viral RNA (6–8), an interaction that may depend on sequence context (9). Its CARDs unwind to form RIG-I homomultimers (10) and bind to another CARD family protein mitochondrial antiviral signaling protein (MAVS), also called IPS-1, Cardif, and VISA, at the mitochondrial membrane (11–15). RIG-I binding to mitochondrial antiviral signaling protein leads to the activation of several transcription factors, including AP1, IFN regulatory factors, and NF-κB, to assemble the enhanceosome of IFNB1 and, thus, initiate its transcription (16–18). The RIG-I gene DDX58 is constitutively expressed at a low level in DCs and is inducible through autocrine and paracrine signaling triggered by IFN binding to the type 1 IFNR (6). Thus, in a DC culture infected by a virus at a low multiplicity ofinfection (MOI), the level of RIG-I in all DCs may be affected by IFN signaling, whereas the activation of RIG-I leading to IFN induction occurs only in the virus-infected DCs (10).
Single nucleotide polymorphisms (SNPs) can have a major impact on how humans respond to disease, including environmental insults, such as viral infection (19, 20). Because RIG-I is such a crucial component of the type-1 IFN pathway, and the RIG-I-CARD plays an essential role in its activity, a polymorphism in the CARD might impact RIG-I function and further modify the response to viral infection. CARD is a functional domain of RIG-I that can act by itself to activate the signaling cascade leading to IFNB1 expression (6). A common nonsynonymous SNP in DDX58, rs10813831 (A/G), introduces an Arg7Cys amino acid change to the first CARD of its corresponded protein RIG-I. This polymorphism was shown to be functional in two transfection studies using murine and human cells (21, 22). We investigated the effect of this SNP on DDX58 and IFNB1 gene expression in Newcastle disease virus (NDV)-infected primary human DCs derived from a population of 130 blood donors. NDV is an avian virus that lacks the ability to prevent induction of IFN in human cells (23–25). Because NDV infection efficiently stimulates the innate immune response and the maturation of human DCs mainly through RIG-I, it provides an ideal experimental perturbation to study RIG-I–mediated DC responses to viral infection. IFNB1 expression is a good indicator of the antiviral state of the DCs. We observed a significant association between this RIG-I SNP and expression of the DDX58 and IFNB1 genes.
mRNA levels can be affected by polymorphisms involved in mRNA synthesis (e.g., promoter polymorphisms) or degradation (often in the 3′ untranslated region [UTR]). If such a functional polymorphism were in linkage disequilibrium with the RIG-I Arg7Cys polymorphism, it could lead to a spurious association. To rule out this possibility, we used the method of mRNA allelic imbalance (AI) (26). In individuals heterozygous for another common unlinked RIG-I SNP, rs12006123 in the 3′UTR, we observed no effect of the RIG-I Arg7Cys polymorphism on rs12006123 allele-specific mRNA levels, indicating that the RIG-I Arg7Cys polymorphism in the protein was indeed the cause of the observed association. To confirm the functionality of the RIG-I Arg7Cys polymorphism, we transfected 293T cells with RIG-I Arg7 and RIG-I Cys7 expression vectors. Following influenza virus A/PR/8/34 ΔNS1 or NDV infection, neither of which blocks IFN induction, we observed a significant RIG-I Arg7Cys-dependent difference in the IFNB1 induction profile.
The interplay between IFNB1 and DDX58 is complicated. RIG-I is required to trigger IFNB1 induction in the DC response to NDV infection (27), whereas induction of the RIG-I–encoding gene DDX58 is dependent on IFN signaling (5, 6, 28). Because a significant association was observed between the RIG-I Arg7Cys polymorphism and expression of the DDX58 and IFNB1 genes, we examined the IFNB1–DDX58 circuit in greater detail. We developed a single-cell assay for the study of innate immunity and maturation of NDV-infected primary human DCs (29). Using this single-cell assay, we measured the expression of both genes in individual DCs, with and without Abs to block IFN signaling required for DDX58 induction. The expression levels were quantified using a nested real-time quantitative PCR procedure (see Materials and Methods). These studies demonstrated that the correlation between expression of the DDX58 and IFNB1 genes extended to the single-cell level.
Materials and Methods
DDX58 genotyping
All human research protocols for this work were reviewed and approved by the Institutional Review Board of the Mount Sinai School of Medicine. The Arg7Cys SNP (rs10813831) (A/G) and the 3′UTR SNP (rs12006123) (A/G) contained in the DDX58 transcripts were screened in 130 samples from anonymous blood donors. Genomic DNA was extracted from buffy coats using a DNA Isolation Kit (Roche Applied Science, Indianapolis, IN). Genotyping of the Arg7Cys SNP was performed using a TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA). The 3′UTR SNP was genotyped by RFLP. The amplified 515-bp fragments were digested with AciI (New England Biolabs, Beverly, MA) overnight at 37°C and analyzed by electrophoresis on a 2% agarose gel. The fragments containing an A allele were not digested, whereas those containing a G allele were digested and generated two fragments: 228 bp and 287 bp. PCR primers are given in Supplemental Table I.
Differentiation of human DCs
Monocyte-derived DCs were obtained from the same anonymous human blood donors following a standard protocol (30). Briefly, human PBMCs were isolated from buffy coats by Ficoll density-gradient centrifugation (Histopaque; Sigma-Aldrich, St. Louis, MO) at 2300 rpm, and CD14+ monocytes were immunomagnetically purified using a MACS CD14 isolation kit (Miltenyi Biotec, Auburn, CA). Monocytes (0.7 × 106 cells/ml) were differentiated into immature myeloid DCs after a 5–6-d incubation in DC growth media with RPMI Medium 1640 (Life Technologies, Carlsbad, CA) supplemented with 10% FCS (Hyclone, Logan, UT), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies), 500 U/ml human GM-CSF (PeproTech, Rocky Hill, NJ), and 1000 U/ml human IL-4 (PeproTech).
Virus preparation and viral infection
As previously described (24), the recombinant Hitchner B1 strain of NDV was prepared, and aliquots of allantoic fluid from embryonated chicken eggs were harvested, snap-frozen, and stored at −80°C. All virus preparations were free of bacterial contamination, as tested by the inoculation of blood agar plates. NDV was titered by infection of Vero cell plates and identification of viral growth by the addition of mAbs specific for NDV hemagglutinin-neuraminindase protein (Mount Sinai Hybridoma Core Facility), followed by the addition of anti-mouse IgG-FITC and visualization using fluorescent microscopy. Titered NDV stock was diluted 40 times in DMEM and added directly into pelleted DCs at an MOI of 0.5, as previously described (31). After incubation for 40 min at 37°C, fresh DC growth medium was added back to the infected DCs (1 × 106 cells/ml). Virus-free allantoic fluid was added to additional tubes of cells to serve as a negative control. Influenza A/PR/8/34 ΔNS1 was prepared in the same way, except the embryonated eggs were 7 d old.
Ab blockage
To block IFN signaling, DCs (1 × 106 cells/ml) were pretreated for 30 min at 37°C before NDV infection with a mixture of Abs, including polyclonal sheep anti-human type I IFN-α neutralizing Abs (4000 U/ml; PBL Biomedical Laboratories), polyclonal sheep anti-human type I IFN-β neutralizing Abs (4000 U/ml; PBL Biomedical Laboratories, Piscataway, NJ), and monoclonal mouse anti-human type 1 IFN-α/β–chain 2 neutralizing Abs (10 μg/ml; Antigenix, Burlingame, CA).
Measurement of IFNB1, DDX58, and RNF135 mRNA expression in DCs
NDV-infected DCs and uninfected (rested) controls from the 130 donors were pelleted at 10 h. Total RNA was isolated using a Total RNA Extraction Kit (Roche Applied Science). The total RNAs were converted to amplified cDNA by using the Nugen WT-Ovation RNA Amplification System (San Carlos, CA). Real-time PCR results showed those cDNAs that preserved the expression level differences among selected genes (data not shown). These amplified cDNAs were used as templates for real-time PCR to determine corresponding mRNA expression levels. Real-time PCR was performed on a Roche LightCycler 480, based on the manufacturer’s recommended standard protocol, except for cycling conditions. After 95°C for 10 min, as required for activation of AmpliTaq Gold (Applied Biosystems), amplification was carried out for 50 cycles using 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The real-time PCR data were normalized to β-actin (ACTB). IFNB1, DDX58, and RNF135 long- and short-isoform expression levels were calculated using crossing point (Cp) values obtained from the amplification curves. PCR primers are given in Supplemental Table I. With the same protocols, the PCR measurements for IFNB1 and DDX58 in the same samples were performed on a different platform using an ABI 7900 system (Applied Biosystems).
Measurement of NDV RNA in DCs
NDV-infected DCs from the 130 donors were pelleted at 10 h. Total RNA was extracted using a total RNA Extraction Kit (Roche Applied Science). The total RNAs were directly used as RT-PCR templates in the one-step real-time RT-PCR reaction performed on a Roche LightCycler 480 and based on the protocol described elsewhere (29). The real-time PCR data were normalized to β-actin (ACTB). NDV RNA expression level was calculated using Cp values obtained from the amplification curves. PCR primers are given in Supplemental Table I.
Measurement of DDX58 mRNA AI in DCs
DDX58 amplicons containing the 3′UTR polymorphism (rs12006123) were obtained by PCR amplification using amplified cDNA as template. These amplicons were used as templates to quantify the amplification from the two alleles using allele-specific PCR (ASPCR) (32). The ASPCR assay was performed on a Roche LightCycler 480 system with ΔZ05 GOLD DNA Polymerase (a hot-start DNA polymerase with improved discrimination against misextension kindly provided by Roche Molecular Systems; research samples of this polymerase may be obtained from Dr. Thomas Myers [ thomas.myers@roche.com]), as previously described (29). The cycling conditions were 95°C for 12 min to activate the polymerase and 50 cycles of 15 s at 95°C, 25 s at 55°C, and 25 s at 72°C. ASPCR of DDX58 produced 85-bp amplicons, the quality of which was verified by melting-curve analysis. All assays were replicated. PCR primers are given in Supplemental Table I. AI was calculated using the difference in Cp (ΔCp) between the two ASPCR reactions normalized to heterozygous genomic control DNA, as previously described (33).
Single DC sorting
Single DCs were directly sorted into 384-well PCR plates, as previously described (29). Briefly, NDV-infected DCs were resuspended on ice in PBS at a concentration of 2–5 × 105 cells/ml. Cells were filtered prior to FACS sorting to remove aggregates. DCs were screened and sorted by visual light scatter (MoFlo high-speed cell sorter) directly into 384-well bar-coded PCR plates (Roche LC480), which contained 5 μl cell lysis buffer (4 mM magnesium acetate [Sigma-Aldrich], 0.05% Nonidet P-40 [Sigma-Aldrich], 0.8 U/μl Protector RNase Inhibitor [Roche Applied Science]) in each well. Sorted DCs were immediately placed on dry ice and stored at −70°C to prevent RNA degradation.
Measurement of IFNB1 and DDX58 mRNA expression in single DCs
Two-step multiplexed nested real-time RT-PCR reactions were performed in 384-well PCR plates on a Roche LightCycler 480 system. The first step was a reverse transcription at 65°C for 30 min, followed by eight cycles of amplification (denaturing at 94°C for 15 s and extension at 60°C for 50 s). This high-temperature RT-PCR was made possible by using AccuRT plus aptamer, a hot-start, magnesium-activated thermostable DNA polymerase that extends a primer on an RNA or DNA template (34). Five microliters of 2× PCR reaction mix (2× buffer, 4 mM magnesium acetate, 0.2 μM each IFNB1 and DDX58 primer sets, IFNB1 F1/R1, and DDX58 F1/R1, 0.4 mM each 2′-deoxynucleoside 5′-triphosphate [0.8 mM deoxyuridine triphosphate replacing deoxythymidine triphosphate]) and 0.375 U/μl AccuRT enzyme (provided by Roche Molecular Systems; research samples of this polymerase may be obtained from Dr. Thomas Myers [ thomas.myers@roche.com]) were added to each well of a FACS single-cell–sorted 384-well PCR plate using a multichannel pipettor. When finished, 2 μl each of the RT-PCR products from 192 wells were split into two wells containing 8 μl second PCR reaction mix (5 μl 2×LightCycler 480 Probe Master Mix, 0.2 μM allele-specific primer set IFNB1MF/R1 or DDX58MF/R1, 0.1 μM Roche UPL probe #79 and #6, 3 μl nuclease-free water). The second PCR amplification was carried out for 50 cycles using 94°C for 10 s, 62°C for 20 s, and 72°C for 10 s. PCR data from serial dilutions of a DNA standard (4000 copies, 400 copies, 40 copies, 4 copies) carried through the same steps were used to determine the numbers of transcripts for each corresponding gene in each individual DC. All sequences for PCR primers and competitive oligonucleotides are given in Supplemental Table I.
Cells transfection and plasmids
Transient transfection of 293T cells was performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions. Flag-tagged RIG-I or RIG-I–CARD cDNA encoding arginine 7 or cysteine 7 were cloned into pCAGGS mammalian expression vectors. 293T cells (2 × 106 cells) were transfected with 50 ng pCAGGS-RIG-I-7Cys, 50 ng pCAGGS-RIG-I-7Arg, or 50 ng empty plasmid as control. At 12 h posttransfection, cells were mock infected or infected with NDV at an MOI of 2. Cells were harvested before or 3, 6, 9, 12, or 24 h after virus infection. Cells were collected, and total RNAs were extracted. IFNB1, DDX58, and ACTB expression was quantified by real-time RT-PCR with the primers given in Supplemental Table I.
RIG-I reporter assay
293T cells (2 × 106) were transfected with 100 ng the IFNβ driven firefly luciferase reporter vector pGL4-IFNβ-firefly luciferase and 100 ng the constitutive expression plasmid Renilla luciferase reporter vector pRL-TK (Promega), together with RIG-I–expressing plasmids or control. Twelve hours posttransfection, cells were infected for 1 h with influenza A/PR/8/34 ΔNS1 at an MOI of 2 or were mock infected. Twelve hours postinfection, the cells were harvested and lysed in dual-reporter lysis buffer following Dual Luciferase Reporter Assay (Promega) specifications. IFN reporter firefly luciferase values were normalized using Renilla luciferase values. The same procedure was used to test the effect of the CARD Arg7Cys polymorphism.
Western blotting
Total protein concentration from cell lysates was quantified by the Bradford method, and 10–20 μg/well was mixed with the loading buffer and loaded into the gel. Proteins were separated by NaDodSO4-PAGE (SDS-PAGE 4–20%) (Bio-Rad, Hercules, CA) and then transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). Membrane blocking with 1× PBS containing 0.1% Tween-20 and 5% nonfat milk was followed by incubation with the indicated Abs. After several washes with PBS containing 0.1% Tween-20, the membranes were incubated with the secondary HRP-conjugated anti-mouse or anti-rabbit Ig Ab (Amersham Biosciences, Piscataway, NJ). The HRP immunocomplexes were detected using an ECL kit (catalog no. NEL101) from PerkinElmer (Wellesley, MA). Protein expression levels were tested by Western blot using specific Abs against the Flag-tag (anti-FLAG M2 Ab; Sigma-Aldrich), endogenous tubulin (anti-β tubulin; Abcam, Boston, MA), and influenza polymerase acidic subunit A proteins (rabbit polyclonal sera anti-PA). The ubiquitination of RIG-I CARDs was examined with HA-tag ubiquitin-specific polyclonal Ab (polyclonal anti-HA Ab; Sigma-Aldrich). For RING finger protein leading to RIG-I activation (RIPLET) (also known as REUL or RNF135) detection we used rabbit pAb to RNF135 (ab28636; Abcam). For tripartite motif protein 25 (TRIM25), we used rabbit pAb to TRIM25 (ab86365; Abcam).
Immunoprecipitation
Transfected cells were washed, collected, and resuspended in PBS. Cells were collected by centrifugation and lysed in immunoprecipitation buffer (Tris-HCl pH 7.6, 150 mM NaCl and 1% Nonidet P-40, 1 mM EDTA) and Complete Protease Inhibitor (Roche), according to the manufacturer’s instructions. The lysates were incubated with anti-FLAG M2 agarose beads (Sigma-Aldrich) at 4°C for 12 h. The beads were washed seven times with the immunoprecipitation buffer; 2× SDS Laemmli buffer was added to the beads and boiled to elute the proteins. After centrifugation, the immune-precipitated proteins eluted from the beads were subjected to SDS-PAGE and Western blotting, as described.
Statistical analysis
All statistical analyses were performed using SPSS software (version 11.0) (SPSS, Chicago, IL). Linear regression was used to study the association between DDX58 SNPs and DDX58 and IFNB1 expression levels, as well as AI. Trend tests were used to calculate possible dose effects of DDX58 genotypes by using ordinal values (0, 1, 2) to represent the three genotypes, with homozygous wild type assigned to be 0. Log-transformed values were used in regression analyses. Correlations between noninfected and NDV-infected samples and between genes were determined by Pearson correlation analysis. The significance of AI measurements of heterozygotes was determined by the Kolmogorov–Smirnov test. The significance between the luciferase activities in Arg7 or Cys7 constructs was determined by the paired two-sample t test.
Results
Correlation between DDX58 and IFNB1 expression levels in a population and individual DCs
We observed that the expression levels of IFNB1 and DDX58 in 130 NDV-infected donor DCs were correlated (r2 = +0.53 by Pearson correlation analysis; p < 0.005) (Fig. 1). Because RIG-I (DDX58) is inducible by IFN signaling, to further address how this autocrine and paracrine IFN feedback loop affects the correlation between DDX58 and IFNB1 expression, we measured the expression level of these two genes in individual DCs 10 h after NDV infection, with or without Ab blockage of the IFN feedback loop (see Materials and Methods). Ab blockage retards the innate immune response. For example, IFNB1 and DDX58 mRNA levels at 7 h postinfection were reduced > 90% in the presence of blocking Abs. Because infection is required to trigger the DC response leading to expression of IFNB1, we used NDV at a low MOI (0.5) to partially infect DCs and partially activate IFNB1 induction in a DC population. Thus, DDX58 induction in uninfected DCs would fully depend on the paracrine feedback loop from the virus-infected DCs. The results showed induction of DDX58 in almost the entire cell population (Fig. 2A) without blocking Abs. When coincubated with Abs to block the IFNβ-triggered Jak-Stat signaling pathway, a lower DDX58 induction (p < 0.001) was observed (Fig. 2B), and it was limited to NDV-infected DCs. Interestingly, a much stronger correlation between IFNB1 and DDX58 expression was seen in the presence of blocking Abs than without (r2 = +0.60 versus +0.28).
FIGURE 1.
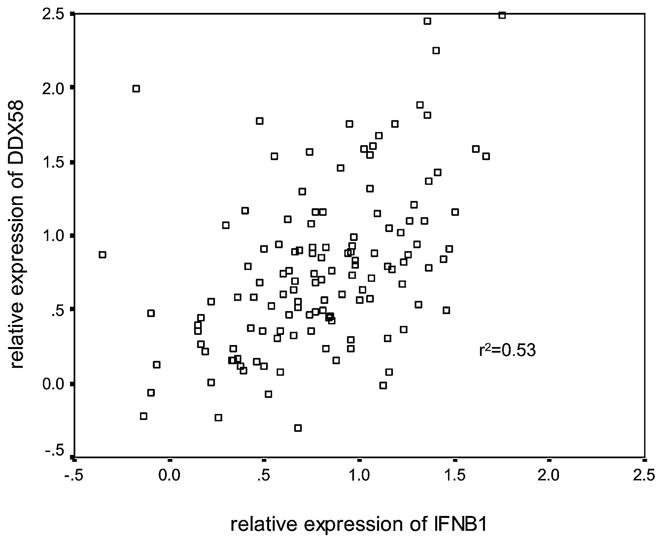
Expression covariation between IFNB1 and DDX58 in NDV-infected DCs. Ten hours after NDV infection at an MOI of 0.5, total RNA was extracted, and relative expression levels were determined for IFNB1 and DDX58 mRNA in each DC sample. The expression levels of DDX58 and IFNB1 are plotted on a logarithmic scale. These data showed a strong correlation between IFNB1 and DDX58 expression in this population (r2 = +0.53; p < 0.005).
FIGURE 2.

Expression covariation between IFNB1 and DDX58 in single DCs. Ten hours after NDV infection at an MOI of 0.5, DDX58 and IFNB1 mRNA in each individual DC from a single donor was quantified by a nested real-time quantitative RT-PCR method described in Materials and Methods. The expression levels of DDX58 and IFNB1 are plotted on a logarithmic scale. Straight lines join duplicate measurements. A, The data showed that without Ab blockage, DDX58 induction was seen in the entire cell population, but with a narrower distribution than that seen for IFNB1 expression. B, With Ab blockage, a much stronger association was observed between DDX58 and IFNB1 expression (r2 = +0.60 versus +0.28).
We also measured NDV RNA levels in the same 130 donor DCs 10 h postinfection. Pearson correlation analysis showed no significance between NDV replication and IFNB1 expression (p = 0.44) or DDX58 expression (p = 0.56), consistent with our results at the single DC level previously reported (29). Because RIG-I is a major player in induction of IFNB1 by NDV, it is expected that DCs having higher levels of RIG-I will induce higher levels of IFNB1. Fig. 2B shows that this is indeed the case. Alternatively, IFN regulatory factor 3 activation of IFNB1 and DDX58 expression may explain the correlation seen between the expressions of these two genes in Fig. 2B.
Association of DDX58 polymorphisms with DDX58 and IFNB1 expression level
The same 130 human blood samples were genotyped for the Arg7Cys SNP rs10813831 and the 3′UTR SNP rs12006123, two of the most common polymorphisms in RIG-I transcripts. The genotype frequencies for both SNPs (Table I) were in Hardy–Weinberg equilibrium (p = 0.34 for Arg7Cys SNP; p = 0.89 for 3′UTR SNP). Gene expression was measured by quantitative RT-PCR 10 h after NDV infection of monocyte-derived DCs. By linear-regression analysis (Fig. 3), heterozygous Cys7 allele carriers showed higher IFNB1 and DDX58 expression levels (p = 0.050 for IFNB1; p = 0.067 for DDX58) than those having the Arg7Arg7 genotype. In particular, homozygous Cys7 allele carriers had significantly higher levels of IFNB1 (2.2-fold) and DDX58 (2.4-fold) expression compared with those homozygous for Arg7 (p = 0.019 for IFNB1; p = 0.034 for DDX58). Also, a trend was significant, suggesting an allele dose effect (ptrend = 0.021 for IFNB1; ptrend = 0.022 for DDX58). A repeated measurement with a different platform was consistent, showing a similar higher level of IFNB1 and DDX58 expression in Cys7 homozygotes. In contrast, the SNP in the 3′UTR region was not associated with IFNB1 or DDX58 expression levels (p = 0.765 for IFNB1; p = 0.465 for DDX58). Resting (uninfected) cells from the same population showed no association between uninduced DDX58 expression levels and the Arg7Cys polymorphism (p = 0.65). Also, regression analysis did not show a significant association between NDV replication and the DDX58 Arg7Cys SNP (p = 0.19). Thus, the Arg7Cys SNP is unique among the most common polymorphisms in RIG-I mRNA in its association with the expression of these two genes central to innate immunity.
Table I.
Genotype frequencies for DDX58 polymorphisms
| Population | Arg7Cys (rs18313831) |
3′UTR (rs12006123) |
||||
|---|---|---|---|---|---|---|
| GG(RR) % (n) | GA(RC) % (n) | AA(CC) % (n) | GG % (n) | GA % (n) | AA % (n) | |
| DC samples | 61.5 (80) | 30.8 (40) | 7.7 (10) | 56.3 (72) | 38.3 (49) | 5.4 (7) |
FIGURE 3.

DDX58 and IFNB1 mRNA expression in DCs derived from 130 individuals were examined 10 h after NDV infection. DDX58, IFNB1, and ACTB (internal control) mRNA were quantified by real-time PCR. The relative IFNB1 (left panel) and DDX58 (right panel) expression levels (normalized to ACTB) were converted to a logarithmic scale for comparison among genotypes (0 = GG, ArgArg homozygote; 1 = GA, ArgCys heterozygote; 2 = AA, CysCys homozygote) of the DDX58 Arg7Cys polymorphism. The box plots show median (solid bar) and interquartile ranges (boxes).
DDX58 SNPs and DDX58 AI
The relative expression level of identical genes from two alleles can be characterized by mRNA AI, which is defined as the difference in the number of transcripts from the two alleles (M1−M2) divided by the total transcripts (M1+M2), expressed as a percentage. This difference can be calculated based on the differences in Cp values (ΔCp) obtained from ASPCR (see Materials and Methods) for the two allele-specific primers:
We investigated whether the association between the Arg7Cys polymorphism and expression levels could be explained by a differential allelic expression of DDX58. DDX58 AI was distinguished using the 3′UTR SNP rs12006123 in 128 DC samples with or without NDV infection measured by ASPCR. Fig. 4A and 4B shows allele frequencies for homozygotes (0, 2) and heterozygotes (1) and a preference in amplification of one allele versus the other in RNA compared with heterozygous DNA controls (3) (p = 0.001 for infected samples, p = 0.004 for rested [uninfected] samples). This preference could be explained by allele-specific mRNA levels or by preferential reverse transcription and amplification of one allele. We did not explore this phenomenon further because the 3′UTR polymorphism did not have any association with DDX58 and IFNB1 expression. The heterozygotes in Fig. 4A and 4B were analyzed by Arg7Cys polymorphism genotype in Fig. 4C and 4D. AI was not associated with the Arg7Cys polymorphism in infected or uninfected samples (p = 0.89 for infected samples; p = 0.63 for uninfected samples) (Fig. 4C, 4D). Therefore, we ruled out linkage disequilibrium of the Arg7Cys polymorphism to an unknown functional polymorphism affecting mRNA levels and concluded that the observed association between Arg7Cys and IFNB1 and DDX58 transcription originated from the structural change in the RIG-I CARD.
FIGURE 4.

AI of DDX58 expression. DDX58 amplicons containing the 3′UTR polymorphism rs12006123 were amplified by PCR from preamplified total cDNAs to serve as templates for ASPCR. AI was determined by ASPCR. The box plots show median (solid bar) and interquartile ranges (boxes). The AI value of each sample without NDV infection (A) or with NDV infection (B) was compared for the three genotypes (0 = GG, 1 = GA, 2 = AA) of SNP rs12006123 and a genomic DNA control from 13 randomly selected heterozygous individuals (3 = genomic DNA). The heterozygotes in A and B were characterized by the Arg7Cys polymorphism genotype (0, 1, 2) in C and D. There was no AI associated with the Arg7Cys SNP in uninfected (C) or infected (D) DC samples (p = 0.63 without infection; p = 0.89 with infection).
Transfection with RIG-I Cys7 and RIG-I Arg7
To verify biochemically that the RIG-I Arg7Cys polymorphism affects IFNB1 expression, we created expression plasmids for the two alleles. These plasmids were transfected into 293T cells with an IFNB1 promoter reporter plasmid expressing luciferase and a control plasmid expressing Renilla luciferase. Postinfection by A/PR/8/34 ΔNS1, the measurements of the reporter assay suggested that RIG-I Arg7 was more effective than RIG-I Cys7 (p < 0.05; Fig. 5A). Although expression was lower, the same trend was observed in the mock-infected controls. To further explore this difference, the time course for expression of the endogenous IFNB1 gene was examined in triplicate in 293T cells transfected with the same RIG-I expression plasmids (Fig. 5B). The Arg7 isoform induced IFNB1 expression more robustly and at earlier times, with the largest difference seen 6–9 h postinfection (p < 0.03 for all time points after 6 h by the t test). Because all of the measurements were performed in cells with a functioning endogenous DDX58 gene, we also examined the function of the CARD in the absence of virus infection. Two plasmids were constructed expressing aa 1–211 of RIG-I containing the two CARD-like domains of the molecule, with Cys or Arg at position 7; their function was examined using the luciferase reporter assay at 24 h after transfection (Fig. 5C). The results highlight the differences between the polymorphic RIG-I proteins, with RIG-I Arg7 being significantly more active (p = 0.01). Because RIG-I was reported to be ubiquitinated in the CARD region (35), we looked for possible allele-specific differences in protein ubiquitination in RIG-I 1–211 proteins in the same cells (Fig. 5D, 5E). Immunoprecipitation and Western blotting data were consistent, with greater RIG-I 1–211 Arg7 protein ubiquitination, possibly leading to greater activity.
FIGURE 5.

RIG-I position 7 polymorphism phenotypes in transiently transfected 293T cells. A and B, Twelve hours after transfection with RIG-IArg7, RIG-I Cys7, or empty plasmids, 293T cells were infected with influenza A/PR/8/34 ΔNS1 at an MOI of 3 or with NDV at an MOI of 2. A, Relative IFNB1 induction was analyzed using an IFNβ-luciferase reporter assay to compare the two polymorphic forms of RIG-I. Experiments were performed in triplicate, and error bars show the SD variation within constructs. B, The mRNA expression levels of IFNB1 from 0–24 h after NDV infection were measured by real-time PCR. C, Comparison of the effect of Arg7 and Cys7 on the relative IFNB1 induction by the RIG-I CARD region (aa 1–211 from RIG-I). D and E, RIG-I Arg7 and Cys7 CARD region modifications detected by Western blot. D, Arrowheads denote the modified forms of the proteins. E, RIG-I Arg7 CARD shows significantly more polyubiquitination than the Cys7 CARD in 293T cells. HC, Ab H chain; Influenza PA, influenza polymerase acidic subunit A; LC, Ab L chain.
Expression of RIG-I and the ubiquitin ligases TRIM25 and RNF135 (RIPLET) in 293T cells and human monocyte-derived DCs
Western blot time-course analysis demonstrated differences in RIG-I induction and ubiquitin ligase downregulation kinetics between 293T cells and human monocyte-derived DCs following NDV infection (Fig. 6A, 6B). Transfected 293T cells showed low basal levels of endogenous RIG-I and insignificant induction following infection, whereas DCs had a high basal level and showed significant induction. 293T cells and DCs showed expression of the long isoform of RIPLET (NP_115698.3) (Fig. 6A, 6B); however, the short isoform (NT_010799.15) was only detected in DCs (Fig. 6B). The relative amount of the short form observed in DCs varied with the donor (Fig. 6B, 6C) We further measured the gene expression ratio of RNF135 short- and long-isoform mRNAs in DCs and observed high correlations in expression level between the two isoforms in uninfected and NDV-infected DCs from the population of donors (Supplemental Fig. 1A). We also observed that the distribution of the RNF135 expression ratio (short isoform/long isoforms) was broad, with the mean ~0.1 (Supplemental Fig. 1B). The ratio was independent of NDV infection and RIG-I R7C genotype. Fig. 6C shows higher expression of TRIM25 in DCs than in 293T cells. RIPLET and TRIM25 showed less stability in 293T cells than in human DCs 12 h after NDV infection (Fig. 6A, 6B). Thus, 293T cells and DCs differ in the kinetics of RIG-I and RIG-I–associated ubiquitin ligase expression and in the expressed isoforms of RIPLET.
FIGURE 6.

RIG-I, RIPLET, and TRIM25 expression in uninfected and NDV-infected 293T cells and human monocyte-derived DCs. Western blot showing the level of protein expression for RIG-I, two isoforms of RIPLET (gene RNF135) and TRIM25 (gene TRIM25) at 0, 6, and 12 h after NDV infection in 293T cells and human DCs. A, Time course after NDV infection for RIG-I, RIPLET, and TRIM25 expression in transfected 293T cells. Normalized to tubulin. B, Same time course for donor 1 DCs. Arrowheads denote TRIM25 and the two isoforms of RIPLET. C, Combined assay of transfected 293T cells and DCs from a second donor with (+) or without (−) NDV infection (10 h). C7, RIG-I Cys7 transfection; Ctr, no transfection; R7, RIG-I Arg7 transfection.
Discussion
In summary, our data revealed that the Arg7Cys polymorphism (rs10813831) is associated with the expression level of the RIG-I gene DDX58 (Fig. 3). This SNP is also associated with the expression level of IFNB1, which is induced by NDV-triggered RIG-I activation. IFNB1 expression correlated with DDX58 expression level at the population level, as well as at the single-DC level (Figs. 1, 2A). After blocking paracrine signaling, the single-cell correlation between IFNB1 and DDX58 increased (Fig. 2B), consistent with induction of RIG-I dependent upon IFN signaling. AI analysis showed that this SNP had no association with mRNA transcription or degradation differences between alleles, pointing to the Arg7Cys polymorphism as the functional polymorphism affecting DDX58 and IFNB1 expression (Fig. 4). IFNB1 mRNA induction in human DCs mainly occurs 6 h after NDV infection, whereas NDV replication starts much earlier (29). The lack of association between IFNB1 mRNA and NDV RNA at 10 h post-infection is consistent with our previous observation that these RNA levels are not correlated in single-cell experiments (29).
Two recent transfection studies examined the common RIG-I Arg7Cys polymorphism in the context of loss of function and other rare variants (21, 22). The first study showed greater activation of endogenous or reporter IFNB1 promoters by RIG-I Arg7 expression plasmids, compared with Cys7 expression plasmids, in RIG-I knockout mouse embryo fibroblasts when triggered using 5′-triphosphate-ssRNA or Sendai virus infection. The second study showed the same trend in human cell lines BEAS-B and HEK 293T with RNA and viral inducers. Our transfection and reporter experiments also showed that the proteins encoded by the two RIG-I alleles exhibited different signaling behavior (Fig. 5A, 5B), again showing greater activation with the RIG-I Arg7 expression constructs. Although all three sets of reporter assays showed an effect of this SNP on IFNB1 induction to be opposite to our population analysis result, these assays confirmed a functional role of the Arg7Cys polymorphism in RIG-I, a key virus-sensing protein in regulating the innate immune response of virus-infected cells. Differences in outcome of IFN induction upon viral infection, depending on the experimental system used, have been reported for the expression of the RIG-I–like helicase LGP2. Overexpression of LGP2 results in lower levels of IFN induction, whereas LGP2 knockout mice are, in contrast, deficient in IFN induction after picornavirus infection (36). In addition to transfection and over-expression, the different responses to the polymorphism may be due to the difference between the lower basal levels of RIG-I in DCs compared with the transfected cell lines. It also may be due to the difference between the components involved in regulation of RIG-I sensing in DCs versus other cell lines. All of these possible mechanisms need to be explored.
The reporter assay with RIG-I 1–211 (Fig. 5C) showed that the effect of the polymorphism on the IFN induction through RIG-I may be attributed to this CARD region. The different effect of the polymorphism on IFNB1 expression between DCs and 293T cells could be explained more specifically by differences in the cellular CARD-modification machinery in different cells. Small differences in the posttranslational modification of CARD could influence a differential regulation of the IFN pathway activation, as suggested by Fig. 5D and 5E. Two CARD E3 ubiquitin ligases were reported to interact with RIG-I and are required for the activation of the protein, TRIM25 and RIPLET (35, 37). One known modification is ubiquitination in the second CARD by both enzymes but with a different pattern of ubiquitination for the two ligases. We note that TRIM25 interacts with the first CARD containing the Arg7Cys polymorphism (38), and the first CARD is required for the ubiquitin binding (39). Fig. 6 shows the differential expression of RIG-I and the two ligases in 293T cells versus human monocyte-derived DCs. Of particular interest is the expression of the second isoform of RIPLET, which was only observed in DCs. The function of the RIPLET short isoform needs additional exploration. The Arg7Cys polymorphism may affect the biological function of only one of the ubiquitin E3 ligases or one of the isoforms, which could reconcile the unexpected observations in the population study.
Taken together, we conclude that the innate immune response of human DCs to infection by NDVand many other viruses is strongly dependent on the level of DDX58 expression and is modified by a common functional polymorphism in the RIG-I CARD. The existence of polymorphic alleles of RIG-I with differential activities may reflect the delicate balance between the amounts of IFN induction needed for an efficient antiviral response versus too much IFN induction leading to autoimmune disorders, such as systemic lupus erythematosus (40). Different selection pressures resulting from exposure to different pathogens might have led to the selection of RIG-I alleles with greater or lesser IFN-inducing ability. It will be interesting to investigate whether the presence of the different alleles correlates with a better survival postinfection by important pathogenic viruses and/or with an increased incidence of autoimmune disorders.
Supplementary Material
Acknowledgments
We thank the Mount Sinai Flow Cytometry Shared Research Facility for single-cell sorting and the Mount Sinai Microarray, PCR, Bioinformatics Shared Research Facility for genotyping. We thank Roche Molecular Systems for generously providing AccuRT plus aptamer and ΔZ05 GOLD polymerases. We thank Richard Cadagan for technical support.
This work was supported by Contract HHSN266200500021C and Grants U19AI06231, U19AI083025, P01AI082325, and R01AI046954 from the National Institute of Allergy and Infectious Diseases.
Abbreviations used in this paper
- AI
allelic imbalance
- ASPCR
allele-specific PCR
- C7
RIG-I Cys7 transfection
- CARD
caspase recruitment domain
- Cp
crossing point
- Ctr
no transfection
- DC
dendritic cell
- HC
Ab H chain
- Influenza PA
in-fluenza polymerase acidic subunit A
- LC
Ab L chain
- MOI
multiplicity of infection
- NDV
Newcastle disease virus
- R7
RIG-I Arg7 transfection
- RIPLET
RING finger protein leading to RIG-I activation
- SNP
single nucleotide polymorphism
- TRIM25
tripartite motif protein 25
- UTR
untranslated region
Footnotes
The online version of this article contains supplemental material.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 3.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 4.Samuel CE. Innate immunity minireview series: making biochemical sense of nucleic acid sensors that trigger antiviral innate immunity. J Biol Chem. 2007;282:15313–15314. doi: 10.1074/jbc.R700013200. [DOI] [PubMed] [Google Scholar]
- 5.Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci USA. 2002;99:637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci USA. 2009;106:12067–12072. doi: 10.1073/pnas.0900971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity. 2009;31:25–34. doi: 10.1016/j.immuni.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M., Jr Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M., Jr Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci USA. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 12.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 13.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 14.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24:633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 16.Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D. Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell. 2000;103:667–678. doi: 10.1016/s0092-8674(00)00169-0. [DOI] [PubMed] [Google Scholar]
- 17.Panne D, Maniatis T, Harrison SC. Crystal structure of ATF-2/c-Jun and IRF-3 bound to the interferon-beta enhancer. EMBO J. 2004;23:4384–4393. doi: 10.1038/sj.emboj.7600453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–1123. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berndt SI, Huang WY, Chatterjee N, Yeager M, Welch R, Chanock SJ, Weissfeld JL, Schoen RE, Hayes RB. Transforming growth factor beta 1 (TGFB1) gene polymorphisms and risk of advanced colorectal adenoma. Carcinogenesis. 2007;28:1965–1970. doi: 10.1093/carcin/bgm155. [DOI] [PubMed] [Google Scholar]
- 20.Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, et al. NCI-NHGRI Working Group on Replication in Association Studies. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 21.Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem. 2009;284:13348–13354. doi: 10.1074/jbc.M809449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pothlichet J, Burtey A, Kubarenko AV, Caignard G, Solhonne B, Tangy F, Ben-Ali M, Quintana-Murci L, Heinzmann A, Chiche JD, et al. Study of human RIG-I polymorphisms identifies two variants with an opposite impact on the antiviral immune response. PLoS ONE. 2009;4:e7582. doi: 10.1371/journal.pone.0007582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, et al. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci USA. 2003;100:10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park MS, Garciá-Sastre A, Cros JF, Basler CF, Palese P. Newcastle disease virus V protein is a determinant of host range restriction. J Virol. 2003;77:9522–9532. doi: 10.1128/JVI.77.17.9522-9532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.López CB, Yount JS, Moran TM. Toll-like receptor-independent triggering of dendritic cell maturation by viruses. J Virol. 2006;80:3128–3134. doi: 10.1128/JVI.80.7.3128-3134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pastinen T, Hudson TJ. Cis-acting regulatory variation in the human genome. Science. 2004;306:647–650. doi: 10.1126/science.1101659. [DOI] [PubMed] [Google Scholar]
- 27.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 28.Yount JS, Moran TM, López CB. Cytokine-independent upregulation of MDA5 in viral infection. J Virol. 2007;81:7316–7319. doi: 10.1128/JVI.00545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J, Sealfon SC, Hayot F, Jayaprakash C, Kumar M, Pendleton AC, Ganee A, Fernandez-Sesma A, Moran TM, Wetmur JG. Chromosome-specific and noisy IFNB1 transcription in individual virus-infected human primary dendritic cells. Nucleic Acids Res. 2007;35:5232–5241. doi: 10.1093/nar/gkm557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang WL, Baumgarth N, Yu D, Barry PA. Human cytomega-lovirus-encoded interleukin-10 homolog inhibits maturation of dendritic cells and alters their functionality. J Virol. 2004;78:8720–8731. doi: 10.1128/JVI.78.16.8720-8731.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.López CB, Garciá-Sastre A, Williams BR, Moran TM. Type I interferon induction pathway, but not released interferon, participates in the maturation of dendritic cells induced by negative-strand RNA viruses. J Infect Dis. 2003;187:1126–1136. doi: 10.1086/368381. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Germer S, Higuchi R, Berkowitz G, Godbold J, Wetmur JG. Kinetic polymerase chain reaction on pooled DNA: a high-throughput, high-efficiency alternative in genetic epidemiological studies. Cancer Epidemiol Biomarkers Prev. 2002;11:131–136. [PubMed] [Google Scholar]
- 33.Wetmur JG, Kumar M, Zhang L, Palomeque C, Wallenstein S, Chen J. Molecular haplotyping by linking emulsion PCR: analysis of paraoxonase 1 haplotypes and phenotypes. Nucleic Acids Res. 2005;33:2615–2619. doi: 10.1093/nar/gki556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith ES, Li AK, Wang AM, Gelfand DH, Myers TW. PCR Primer: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2003. Amplification of RNA: High-Temperature Reverse Transcription and DNA Amplification with a Magnesium-Activated Thermostable DNA Polymerase; pp. 211–219. [DOI] [PubMed] [Google Scholar]
- 35.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 36.Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178:6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- 37.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J Biol Chem. 2009;284:807–817. doi: 10.1074/jbc.M804259200. [DOI] [PubMed] [Google Scholar]
- 38.Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, Myong S, Ha T, Hopfner KP, Jung JU. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci USA. 2008;105:16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–153. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.