

Abstract
Copy number variants and indels in 251 families with evidence of X-linked intellectual disability (XLID) were investigated by array comparative genomic hybridization on a high-density oligonucleotide X chromosome array platform. We identified pathogenic copy number variants in 10% of families, with mutations ranging from 2 kb to 11 Mb in size. The challenge of assessing causality was facilitated by prior knowledge of XLID-associated genes and the ability to test for cosegregation of variants with disease through extended pedigrees. Fine-scale analysis of rare variants in XLID families leads us to propose four additional genes, PTCHD1, WDR13, FAAH2, and GSPT2, as candidates for XLID causation and the identification of further deletions and duplications affecting X chromosome genes but without apparent disease consequences. Breakpoints of pathogenic variants were characterized to provide insight into the underlying mutational mechanisms and indicated a predominance of mitotic rather than meiotic events. By effectively bridging the gap between karyotype-level investigations and X chromosome exon resequencing, this study informs discussion of alternative mutational mechanisms, such as noncoding variants and non-X-linked disease, which might explain the shortfall of mutation yield in the well-characterized International Genetics of Learning Disability (IGOLD) cohort, where currently disease remains unexplained in two-thirds of families.
Introduction
Intellectual disability (ID) comprises a set of clinically and genetically heterogeneous disorders in which brain development and/or function is compromised. ID is defined by substantial limitations in both intellectual functioning and adaptive behavior with onset before the age of 18 years.1,2 ID may be the only consistent clinical feature (termed nonsyndromic ID) or may present in association with dysmorphic, metabolic, neuromuscular, or psychiatric manifestations (syndromic ID). ID is estimated to have a prevalence of 1.5%–2% in resource-rich countries and places a significant demand on healthcare expenditure.3 Nongenetic causes of ID include pre-, peri-, and postnatal infection and perinatal injury, but chromosomal aberrations and single gene defects make a substantial contribution to ID causation.4–7 The identification of causative mutations in familial or idiopathic ID can contribute to the clinical management of affected individuals and their families and can also provide insight into processes of brain development and function.
The observation that males are more commonly affected than females, coupled with the identification of ID families with extended X-linked pedigrees and the relative tractability of X chromosome analysis, has focused gene discovery efforts on the X chromosome (recently reviewed in 8). More than 90 genes have been implicated in syndromic or nonsyndromic forms of X-linked ID (XLID) and these encompass a wide range of biological functions and cellular processes (9 and XLMR update website). Recently, we have reported the results of the large-scale systematic resequencing of the coding X chromosome in order to identify novel genes underlying XLID.10 The coding sequences of 718 X chromosome genes were screened via Sanger sequencing technology in probands from 208 families with probable XLID. This resequencing screen has contributed to the identification of 9 novel XLID-associated genes but identified pathogenic sequence variants in only 35/208 (17%) of the cohort families. Because extensive sequence-based prescreening of known XLID genes had been performed to enrich for the contribution of unknown genes to XLID, this figure underestimates the general contribution of sequence variants to XLID, but nevertheless highlights that disease remains unexplained in the majority of cohort families.
The relatively low yield of pathogenic sequence variants identified by Tarpey et al.10 was surprising and may reflect both technological and experimental limitations, including interpretational difficulties in assigning pathogenicity and the contribution of alternative mutational classes. One mutational class that is inefficiently detected by the direct exon sequencing approach is copy number variation (CNV). CNVs are operationally defined as genomic deletions or amplifications, most commonly duplications, greater than 1 kb in size (variants smaller than 1 kb are termed indels).11 Genomic duplications are undetectable by PCR-based Sanger sequence analysis, which is nonquantitative, unless the duplication breakpoints occur within targeted regions, and here the most likely outcome is failure of PCR amplification. X chromosome deletions in males would also result in PCR failure. Although in principle it would be feasible to use failure to amplify as a marker for CNV discovery, in practice it is difficult to systematically assess PCR failures, which may have multiple causes, in the context of a large-scale screen in which many thousands of PCR amplicons are obtained from hundreds of samples.
Genomic deletions, duplications, and other structural rearrangements have an established role in genetic disease, including many neurological and neurodevelopmental disorders.12,13 A number of studies have used whole-genome microarrays to identify submicroscopic disease-causing variants in ID and such analysis is increasingly incorporated into routine clinical practice.14,15 Studies using X chromosome tiling path BAC arrays with a reported resolution of 80–100 kb have also been effectively employed, identifying pathogenic CNVs in 5%–15% of patient cohorts.16,17 Critically, the ability of these array platforms to detect small aberrations is limited: at the time of study inception, the most feature-dense of the commercially available oligonucleotide-based whole-genome arrays had a theoretical sensitivity of 15 kb, although the pace of technology development in this area is rapid.18
To assess the contribution of genomic rearrangements to disease in the IGOLD cohort, we developed an X chromosome oligonucleotide array with sufficient power to detect single exon imbalances. Our analysis focused on the identification and evaluation of low-frequency, high-penetrance CNVs as an underlying cause of ID.
Material and Methods
Study Cohort
The study cohort consisted of 251 XLID families from Europe, the USA, Australia, and South Africa. Recruitment criteria were as previously described.10 In brief, each family had two or more cases of ID in males with a transmission pattern compatible with X-linked inheritance. Probands had been investigated for cytogenetic abnormalities (karyotype at ≥550 G-band resolution) and FMR1 (MIM 309550) trinucleotide repeat expansion and these had been excluded as underlying causes of disease. Probands from 200 of the 251 families had been resequenced by Tarpey et al.10 The cause of disease in 193 study families (77%) was unknown but the cohort also included 46 families with disease that was attributed to sequence variants and 12 families with pathogenic CNVs identified by parallel analyses (Table S1 available online). These families served as a control group for CNV discovery and interpretation.
Features of the pedigree structure and clinical presentation of the study cohort are summarized in Table S2. The clinical features reflect the heterogeneity of ID, with a broad range of ID severity and a contribution of nonsyndromic and syndromic cases. The families within the cohort ranged from large extended pedigrees with many cases of ID to small nuclear clusters.
Array comparative genomic hybridization (aCGH) and subsequent experiments were performed on genomic DNA extracted directly from peripheral blood or from EBV-transformed lymphoblastoid cell lines (LCLs) via standard methods. CNVs detected in LCL samples were validated in nontransformed samples in parallel with the cell line-derived sample. Hybridization experiments utilized a sample from a single affected male in 246 of 251 families. In the remaining 5 families, there was no available sample from an affected male and so screening was performed on a sample from an obligate carrier female unaffected by ID. All hybridizations were performed against the male reference sample NA10851, obtained from the Coriell Cell Repository. To investigate whether selected CNVs were rare variants that could also be identified in unaffected individuals, a panel of control samples were employed. Control DNAs were obtained either from LCLs derived from adults of European ancestry without ID or from white blood cells from voluntary blood donors. Study protocols were reviewed and approved by local ethics committees and institutional review boards of each collaborating institution, and informed consent for research was obtained for all patients.
Array Design and Hybridization
We employed a custom 385K format Nimblegen oligonucleotide array with a bipartite design providing both backbone coverage of the entire X chromosome and targeting coding regions and phylogenetically conserved elements for high-density probe coverage (Table S3). Probe coverage achieved a median density of 1 probe per 36 bp in the targeted regions and a median gap size of 463 bp across the X chromosome backbone design. The oligonucleotide design, array manufacture, DNA labeling, hybridization, and data normalization were done by the Icelandic Nimblegen Service Laboratory according to recommended and published procedures. 2.5 μg of reference and test DNA were labeled with Cy3 and Cy5, respectively, and cohybridized to the array slides. The intensity of the two fluorescent dyes was extracted with NimbleScan software. The Cy3 and Cy5 signal intensities on each array were normalized to one another via qspline normalization and a log2 ratio calculated for each probe.
CNV Discovery
CNV discovery was performed with the Aberration Detection Method 1 (ADM-1) algorithm implemented in CGH Analytics 3.4 software (Agilent) with centralization and fuzzy zero corrections applied.19 Appropriate analysis thresholds were established with reference to experimental validations (Table S4). Initial settings were permissive in order to maximize the inclusiveness of the primary data set and thus the detection of potentially pathogenic variants. We subsequently employed more-stringent sample-level (Figure S1) and CNV-level QC filtering (Figure S2) to reduce the contribution of false positives to the data set and define a final data set of rare copy number variants. The CNV discovery workflow is outlined in Figure S3.
Experimental Validation of CNVs, Breakpoint Sequencing, and Expression Analysis
Several strategies were employed to validate CNV calls, investigate segregation of variants within XLID families, and characterize breakpoints, including standard PCR and bidirectional sequencing,10 long-range PCR,20 quantitative real-time PCR,21 quantitative multiplex PCR of short fragments,22 reverse transcriptase PCR,23 fluorescent in situ hybridization,24 X inactivation analysis,25 and splinkerette PCR.26 Linkage analysis of family 306 was performed via microsatellite marker panels (Applied Biosystems) and the Infinium II Human Linkage 12 panel (Illumina) and analyzed with MERLIN.27 Full details of experimental methods and primer sequences are available on request.
Bioinformatics Analysis
For bioinformatics analysis, extensive use was made of the UCSC genome browser and several additional publically available databases including the Database of Genomic Variants, DECIPHER, Genes2Cognition, HapMart, and GNF SymAtlas. Our analysis also used the following tools: Blast2, EMBOSS Blast2 (GeeCee, Fuzznuc, and Palindrome), MFOLD, Primer3, RepeatAround, RepeatMasker, QGRS Mapper, and Z-hunt Online. All genomic coordinates correspond to the human genome assembly build 36 (hg18).
Results
CNV Discovery
The primary goal of our analysis was to maximize our detection of potentially pathogenic rare variants. Informing our analysis settings with experimental validations (Table S4), the first phase of CNV discovery was at low stringency and generated a raw data set of 69,582 calls, of which 33,143 (48%) were deletions and 36,439 (52%) were copy number gains. To assess the performance of our array, we investigated whether the nonpseudoautosomal X chromosome CNVs reported by Conrad et al. were present in our data set.28 Sixty-eight of 107 nonoverlapping CNVs were represented in our cohort. 60% of CNV positions fell within 500 bp of published estimates and in only 5% were there discrepancies of breakpoint estimates greater than 5 kb. Reviewing the 39 CNVs that were absent from our data, 19 were invariant in the European CEU population and 9 CNVs were interrogated by <5 probes on our array.
Because the first phase of CNV discovery prioritized inclusiveness over specificity, the data set was compromised by false positive calls and the presence of overlapping calls within ADM-1 estimates. To determine the extent of the false positive contribution to the data set, four self-self hybridizations were performed, each with a different sample. Across the four control experiments, the median number of CNV calls identified was 39 (range 23–71). For the test hybridizations, the median number of ADM-1 calls per sample was 205 (range 59–1865), giving a false positive rate of 19%.
To generate a more stringent data set, we used outlier analysis to evaluate the performance of probes reporting a CNV in the context of the sample population, via an approach similar to Marioni et al.29 This analysis required the prior exclusion of the poorest-performing samples. Employing sample-level QC, 24 male samples and all 5 female samples were excluded, representing 12% of the study cohort but eliminating 29% of the CNV calls generated via the permissive analysis settings (Figure S3). Although these excluded sample hybridizations were not appropriate for high-resolution analysis, some large CNV calls and potentially pathogenic variants were detected and experimentally validated in these samples (Table S5).
Overlapping raw CNV calls were condensed to a merged data set of 16,583 CNV loci and outlier status was assessed for each locus. This analysis was optimized to capture highly penetrant rare variants and therefore only CNVs with a frequency of <5% were retained. The data set was further reduced to exclude two samples generating an excessive number of apparent calls that were unexplained (Figure S3). The final data set of 952 rare CNVs, corresponding to 861 distinct CNV loci identified in 220 male XLID samples, is summarized in Table 1 and listed in Table S6. The mean number of rare variants per individual was 4.3 (range 0–23). The number of copy number losses (498) and gains (454) were broadly similar, with deletions slightly in excess at the smaller size scale and copy number gains dominating in the larger size categories. 332/952 (35%) CNVs were intergenic and a further 139 CNVs (15%) were contained entirely within introns. The detection of coding variants is skewed by the bias toward array coverage of these regions and 50% of rare CNVs were estimated to have some coding sequence overlap, although in some cases this overlap is minimal and may reflect errors in CNV size estimation.
Table 1.
Summary of Rare X Chromosome CNVs
| All CNVs | <1 kb | 1–10 kb | 10–100 kb | 100–500 kb | >500 kb | |
|---|---|---|---|---|---|---|
| Deletions | 498 (440) | 182 (161) | 224 (202) | 86 (71) | 4 (4) | 2 (2) |
| Duplication | 454 (421) | 150 (146) | 174 (165) | 84 (69) | 31 (26) | 15 (15) |
| Total | 952 (861) | 332 (307) | 398 (367) | 170 (140) | 35 (30) | 17 (17) |
Total call numbers are stated, with numbers in parentheses indicating the number of distinct CNV loci after merging calls across samples.
Experimental Validations and Sources of Array Noise
A subset of CNV calls were experimentally validated by PCR spanning the putative CNV boundaries or by qPCR (Table S7). 40/52 CNV loci were confirmed as deletions or duplications and 7/52 were false positives. Although a proportion of this noise is unexplained, 7 of 12 copy number false-positive calls were associated with underlying sequence variation. We found that single-nucleotide variations could be sufficient to disrupt probe hybridization and to generate artifactual CNV calls where this disrupts a number of overlapping or contiguous probes. The effects of a common SNP and a rare sequence variant on hybridization log2 ratio are shown in Figure 1. Most commonly, sequence variants result in artifactual deletion calls but, rarely, duplication calls were also found to be due to single-nucleotide sequence variants in the test sample.
Figure 1.
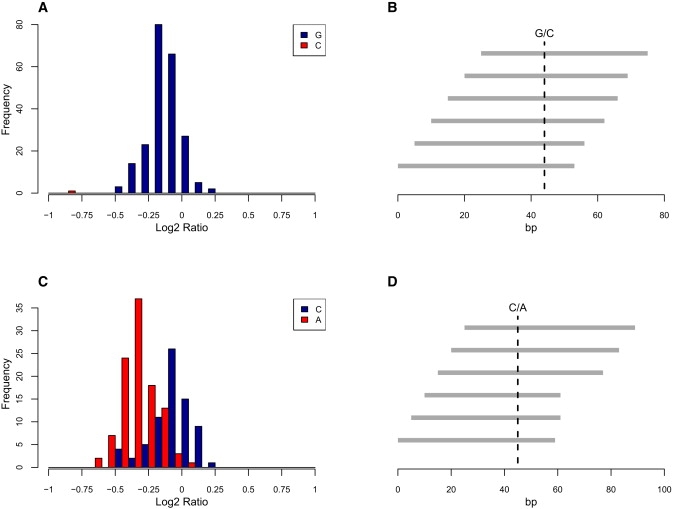
Single-Nucleotide Variants Can Effect Probe Hybridization
(A) Population distribution of mean log2 ratio for six probes overlying the ZDHHC9 IVS1+5G > C point mutation found in a single family.
(B) Relative location of probes and point mutation.
(C) Population distribution of mean log2 ratio for six probes overlying common SNP rs1329546.
(D) Relative location of probes and SNP.
To assess the extent to which SNP-related artifacts confound array analysis, we considered a set of 15 common SNPs that were targeted by five or more probes and where we had previously obtained genotype information by exon resequencing (Table S8). Genotypes at 8 of 15 common SNPs were significantly associated with the average log2 ratio of all overlying probes. In some cases the effect on log2 is minimal, but in 2/15 cases CNVs were called in samples at the extremes of the distribution.
CNVs Affecting Known XLID Genes
One goal was to identify directly pathogenic deletions and duplications down to the scale of single exon abnormalities. In assessing the relevance of a CNV to ID it was necessary to consider both the likely consequence of the CNV on gene structure and function and also, given the extent of benign structural variation, whether the gene(s) are of likely relevance to ID. CNVs were prioritized for detailed evaluation on the basis of (1) overlap with known ID-associated genes, (2) predicted disruption of coding sequence, and (3) not reported in studies of individuals with normal cognitive abilities.
In sum, we detected 25 likely pathogenic CNVs (18 duplications and 7 deletions) affecting known ID-associated genes (Table 2). Twelve of these pathogenic CNVs had previously been identified and were included for control purposes and, in some cases, to refine the boundaries of the rearrangement. The structural variants ranged from 2 kb to >11 Mb in size and varied in content from alterations affecting a single exon to those affecting several genes. PCR or FISH confirmation of these variants was performed, together with segregation analysis where familial samples were available and mRNA expression analysis where appropriate. Clinical summaries of the families and pedigrees with confirmed variant segregation data are in Table 2 and Figure S4, respectively.
Table 2.
Likely Pathogenic CNVs Affecting Known ID-Associated Genes
|
CNV |
Clinical Presentation |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Family | Type | Gene(s) | Genomic Coordinatesa | Cytoband | Extent (kb) | XIb | ID Severity | S/NSc | Reference |
| 57 | Dup +Trip | several, including MECP2 | Dup:152 786 182-153 278 914 (E) Trip:152 953 000-153 218 000(M) |
Xq28 | 492 | nd | severe | S | |
| 164 | Dup | several, including MECP2 | 152 021 972-153 401 219 (E) | Xq28 | 1379 | nd | severe | S | 42,79 K8300 |
| 185 | Dup | several, including MECP2 | 152 487 808-153 062 828 (M) | Xq28 | 575 | nd | severe | S | 42,79 K8210 |
| 241 | Dup | several, including MECP2 | 152 824 478-153 167 906 (E) | Xq28 | 343 | nd | severe | S | |
| 340 | Dup | several, including MECP2 | 152 575 696-153 262 980 (E) | Xq28 | 687 | 4:96 | severe | S | 80 family 1 |
| 344 | Dup | several, including MECP2 | 152 788 677-153 287 392 (E) | Xq28 | 499 | 1:99 | severe | S | - |
| 389 | Dup | several, including MECP2 | 152 780 000-153 220 753 (M) | Xq28 | 441 | nd | severe | S | - |
| 495 | Dup | several, including MECP2 | 152 466 800-154 426 868 (M) | Xq28 | 1960 | 10:90 | severe | S | 80 family 2 |
| 509 | Dup +Trip | several, including MECP2 | Dup:152 757 505-153 246 869 (E) Trip:153 183 000-153 218 000 (M) |
Xq28 | 489 | nd | severe | S | - |
| 77 | Dup | several, including HUWE1 and HSD1710B | 53 240 015-53 999 700 (E) | Xp11.22 | 759 | nd | moderate | N | 81 |
| 304 | Dup | several, including HUWE1 and HSD1710B | 53 409 042-53 786 049 (E) | Xp11.22 | 377 | nd | moderate | N | 81 |
| 359 | Dup | several, including HUWE1 and HSD1710B | 53 004 979-53 729 979 (E) | Xp11.22 | 715 | nd | moderate | N | 81 |
| 538 | Dup | several, including HUWE1 and HSD1710B | 53 232 828-54 256 595 (E) | Xp11.22 | 1024 | 2:98 | moderate | N | |
| 376 | Dup | several, including RPS6KA3, MBTPS2, and SMS | 18 985 933-22 751 175 (E) | Xp22.13-Xp22.11 | 3765 | 8:92 | moderate | N | |
| 317 | Dup | several, including GRIA3 | 119 698 636-125 699 533 (S) | Xq24-Xq25 | 6001 | nd | moderate | ||
| 422 | Dup | several, including MED12, NLGN3, SLC16A2, KIAA2022, ATRX, and BRWD3 | 70 134 868-81 653 582 (E) | Xq13.1-q21.1 | 11519 | nd | severe | S | |
| 505 | Dup | ARX, POLA1 (partial) | 24 902 835-24 943 900 (S) | Xp21.3 | 41 | 12:88 | moderate | N | |
| 110 | Dup | AFF2 (partial) | 147 547 319-147 757 141 (S) | Xq28 | 210 | nd | mild | N | |
| 32 | Del | IL1RAPL1 (partial) | 28 939 863-29 497 216 (E) | Xp21.3-21.2 | 557 | 46:54 | moderate | N | |
| 121 | Del | Il1RAPL1 (partial) | 28 922 932-29 253 959 (S) | Xp21.3 | 331 | nd | moderate | N | |
| 398 | Del | SLC16A2 (partial) | 73 666 964-73 669 304 (S) | Xq13.2 | 2 | 48:52 | severe | N | |
| 399 | Del | SLC16A2 (partial) | 73 552 449-73 567 609 (S) | Xq13.2 | 15 | 18:82 | severe | N | |
| 115 | Del | SLC9A6 (partial) | 134 934 236-134 943 268 (E) | Xq26.3 | 9 | 48:52 | severe | ||
| 147 | Del | MAOA and MAOB (partial) | 43 426 228-43 666 586 (S) | Xp11.3 | 240 | 41:59 | severe | N | |
| 506 | Del | CUL4B (noncoding) | 119 578 701-119 584 448 (S) | Xq24 | 6 | nd | moderate | N | |
Letters in parentheses after genomic coordinates specify whether CNV bounds are ADM-1 estimates (E) or have been adjusted after breakpoint sequencing (S) or qPCR and manual inspection of probe log2 ratios (M).
X inactivation status (percentages of each allele in an obligate female carrier).
S, syndromic; N, nonsyndromic; further information on clinical presentation is available on request. nd, not determined.
Sixteen families had duplications >100 kb that are considered to cause disease as a consequence of altered dosage of intact genes, although there may be some contribution from disrupted genes, particularly close to the duplication boundaries. Of these, nine families had Xq28 duplications, which included MECP2 (MIM 300005), and four families had duplications on Xp11.22 including the HUWE1 (MIM 300697) and HSD17B10 (MIM 300256) genes. These two “hotspots” of duplication contained large common regions of overlap (238 kb for Xq28 and 321 kb for Xp11.22), but the breakpoint coordinates of each rearrangement were different. A recent study of MECP2 duplications emphasized the occurrence of rearrangement complexity in one quarter of cases.30 In keeping with this finding, two of nine MECP2-containing duplications had segments of triplication embedded within the duplicated region.
The three remaining large duplications affecting multiple genes were each found in a single family. A 3.8 Mb duplication of Xp22.11-p22.13 was detected in family 376. This region contains 18 RefSeq genes, of which 3 are known XLID-associated genes (RPS6KA3 [MIM 300075], MBTPS2 [MIM 300294], and SMS [MIM 300105]) and 3 (SH3KBP1 [MIM 300374], PDHA1 [MIM 300502], and CNKSR2 [MIM 300724]) are orthologs of mouse postsynaptic density (PSD) components (Genes2Cognition database). The duplicated region overlies a smaller duplicated region reported in DECIPHER (case 00249293). Family 317 has a 6 Mb duplication containing the cancer testis antigen CT47 cluster (MIM 300780-90) and a further 9 RefSeq genes, of which only GRIA3 (MIM 305915) has a known association with XLID and is also a PSD component. Family 422 has an 11.5 Mb duplication containing 70 RefSeq genes, including the XLID-associated genes MED12 (MIM 300188), NLGN3 (MIM 300336), SLC16A2 (MIM 300095), KIAA2022 (MIM 300524), ATRX (MIM 300032), and BRWD3 (MIM 300553).
Two further duplications were identified that are predicted to disrupt expression levels or function of a single XLID-associated gene. In family 505, a 41 kb duplication extending from the terminal exon of POLA1 (MIM 312040) across the entire coding sequence of ARX (MIM 300382) was identified. The duplication was maternally inherited and not present in an unaffected brother. The duplication was in direct tandem orientation, but the absence of ARX expression in available tissue material precluded further dissection of the cellular consequences of the rearrangement. In family 110, a 210 kb region including exons 3–7 of AFF2 (MIM 300806) were found to be duplicated. The direct tandem orientation of the duplicated segment is predicted to introduce a premature termination codon into the transcript or to trigger aberrant splicing, but experimental verification of this was not possible because of the absence of AFF2 expression in available tissues.31 Segregation studies revealed that the duplication was absent from the youngest of three brothers, all of whom were considered to be affected.
The seven deletions likely to be pathogenic included two families with intragenic deletions of IL1RAPL1 (MIM 300206) (families 32 and 121), two single exon deletions within SLC16A2 (MIM 300095) (families 398 and 399), and one intragenic deletion of SLC9A6 (MIM 300231) (family 115). In each case, the clinical presentation was compatible with published reports of loss-of-function sequence variants in these genes.32–34 In family 147, we identified a 240 kb deletion that eliminated the function of both monoamine oxidase genes (MAOA [MIM 309850] and MAOB [MIM 309860]) without affecting the adjacent Norrie's disease gene (NDP [MIM 300658]), permitting further dissection of the phenotype associated with monoamine oxidase dysfunction.20
We were also able to ascribe pathogenicity to a 2 kb noncoding deletion in family 506 that disrupts the 5′UTR of the widely expressed CUL4B (MIM 30304) isoform 2 (Figure 2A). The deletion cosegregated with disease (Figure 2B), and qPCR analysis of mRNA from patient LCLs revealed a complete loss of CUL4B expression (Figure 2C). This is one of several noncoding variants in proximity to the coding sequences of XLID-associated genes. Some noncoding variants, such as the intronic 5 kb IL1RAPL1 deletion in family 329, could be discounted because of the presence of more convincing causative variants, in this case a CUL4B sequence variant.23 There remain other noncoding variants where the significance remains unclear; examples include the 9 kb intronic deletion of NLGN4X (MIM 300427) in family 126 and the 182 kb duplication of PCDH11X (MIM 300246) intronic sequence in family 335 (Table 3).
Figure 2.
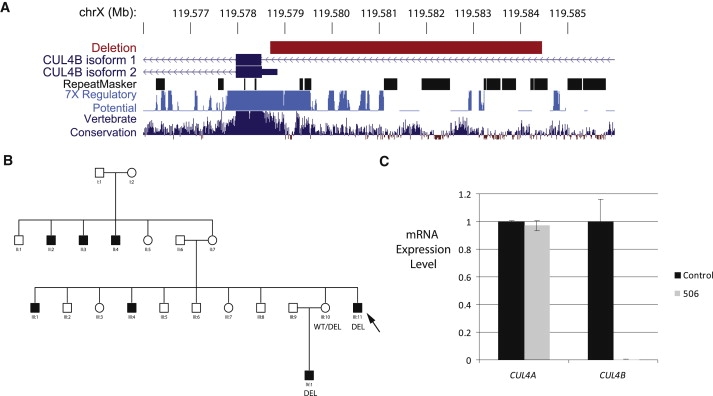
A Noncoding Deletion Eliminates CUL4B Expression
(A) UCSC genome browser snapshot showing 6 kb deletion location relative to CUL4B RefSeq transcripts, repetitive elements, and regions with regulatory potential and vertebrate conservation.
(B) Cosegregation of deletion and disease in family 506 could be tested in only a subsection of the extended X-linked pedigree because of limited sample availability. Arrow indicates individual analyzed by aCGH.
(C) qPCR analysis indicates absence of CUL4B mRNA, but not control CUL4A transcript, in patient LCLs relative to a wild-type control. Expression analysis was performed as described in Kerzendorfer et al.78 with Applied Biosystems TaqMan gene expression assays Hs00757716_m1 (CUL4A) and Hs00186086_m1 (CUL4B).
Table 3.
Experimentally Verified CNVs of Unknown Significance or Excluded from Disease Causation by Segregation Analysis
| Family | CNV Type | Genomic Coordinatesa | Extent (kb) | Genes | Present in DGV? | Cosegregates with Disease? | Comments |
|---|---|---|---|---|---|---|---|
| 126 | Del | 6 027 992-6 037 317 (S) | 9 | NLGN4X (noncoding) | no | yes (limited pedigree) | |
| 335 | Dup | 91 132 100-91 314 578 (E) | 182 | PCDH11X (noncoding) | no | possibly (absent from mildly affected female) | |
| 329 | Del | 28 779 866-28 784 639 (E) | 5 | IL1RAPL1 (noncoding) | no | nd | pathogenic CUL4B mutation in family |
| 62 | Del | 69 363 478-69 378 892 (M) | 15 | AWAT1 | no | no | pathogenic UPF3B mutation in family |
| 10 | Dup | 148 156 928-148 867 255 (E) | 710 | several including IDS | no (partial overlap) | no | |
| 93 | Dup | 122 797 004-123 203 499 (E) | 406 | XIAP and STAG2 | no (partial overlap) | no | |
| 93 | Dup | 134 077 508-134 635 763 (E) | 558 | CXorf48, ZNF75, ZNF449, DDX26B | no (partial overlap) | no | |
| 507 | Dup | 514 451-1 394 128 (E) | 880 | SHOX, CRLF2, CSF2RA | no (partial overlap) | no | |
| 25 | Dup | 2 128 149-2 498 028 (E) | 370 | DHRSX, ZBED1 | no (partial overlap) | no |
Letters in parentheses after genomic coordinates specify whether CNV bounds are ADM-1 estimates (E) or have been adjusted after breakpoint sequencing (S) or qPCR and manual inspection of probe log2 ratios (M). Pedigrees and CNV segregation data in Figure S5. nd, not determined.
Identification of Novel XLID Genes
In addition to affecting known XLID-associated genes, some CNVs may disrupt genes not previously known to cause XLID. Here, the key challenge is to distinguish clinically significant and thus pathogenic CNVs from benign but rare variation. We used several criteria to assess likely significance: (1) cosegregation of CNV with disease within the pedigree; (2) absence of CNV from public databases and control populations; and (3) expression pattern and predicted gene function compatible with neurological disease. There were four genes (PTCHD1, WDR13 [MIM 300512], FAAH2 [MIM 300654], and GSPT2 [MIM 300418]) that met these criteria and warrant further analysis (Figure 3 and Table 4).
Figure 3.
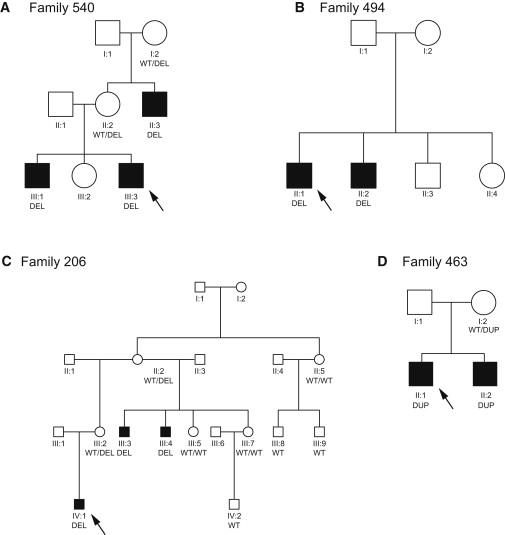
Cosegregation of CNVs and Disease for Putative Novel XLID Genes
Arrows indicate the individual subjected to aCGH analysis. Genotypes of all tested individuals are reported with alleles described as WT (wild-type), del (deletion), or dup (duplication).
(A) Family 540, PTCHD1 deletion.
(B) Family 494, FAAH2 intragenic deletion.
(C) Family 206, WDR13 intragenic deletion.
(D) Family 463, GSPT2 duplication.
Table 4.
Candidate Novel XLID-Associated Genes
| Family | CNV Type | Genomic Coordinatesa | Genomic Extent (kb) | Genes | Present in DGV? | Present in Controls? | Brain Expressionb? |
|---|---|---|---|---|---|---|---|
| 540 | Del | 23 239 008-23 329 210 (S) | 90 | PTCHD1 | no | 0/447 | yes |
| 206 | Del | 48 345 024-48 348 048 (S) | 3 | WDR13 | no: overlaps single report of large variant (7789) | 0/615 | yes |
| 494 | Del | 57 487 858-57 493 349 (E) | 5 | FAAH2 | no | 0/450 | yes |
| 463 | Dup | 51 469 871-51 509 041 (S) | 39 | GSPT2 | no | 0/181 | yes |
Letters in parentheses after genomic coordinates specify whether CNV bounds are ADM-1 estimates (E) or have been adjusted after breakpoint sequencing (S) or qPCR and manual inspection of probe log2 ratios (M).
GNF symatlas data.
A 90 kb deletion spanning the entire PTCHD1 gene was identified in family 540. Intragenic deletions removing multiple exons were detected in WDR13 (family 206) and FAAH2 (family 494). In LCLs from affected males in family 494, who lack FAAH2 exons 9 and 10, two misspliced FAAH2 transcript species were detected: exon 8 spliced to exon 11 and exon 7 spliced to exon 11. Both mRNA species introduce frame shift mutations and are predicted to disrupt the amidase domain.
In family 463, we identified a whole-gene duplication of GSPT2 and confirmed the direct tandem orientation of the duplicated region.
Analysis of a Retrotransposition Event
Family 306 was found to have a duplication corresponding to the coding exons only of LUZP4 (MIM 300616) (Figure 4). The presence of an insertion of the LUZP4 transcript, consistent with a retrotransposition event, was confirmed by PCR amplification and sequencing of the spliced transcript from genomic DNA template in the proband. The inserted LUZP4 copy cosegregated with disease through the pedigree. Splinkerette PCR to identify the sequence flanking the insertion revealed that the retrotransposed copy was embedded in intron 5 of the IQCE gene on chromosome 7p22.2. Microsatellite and SNP markers flanking the insertion site also segregated in a manner consistent with the retroinsertion data. No retrotransposed copy of LUZP4 was detected in 450 control males. Furthermore, linkage analysis via microsatellites and SNPs excluded the nonpseudoautosomal X chromosome from association with disease in this pedigree.
Figure 4.
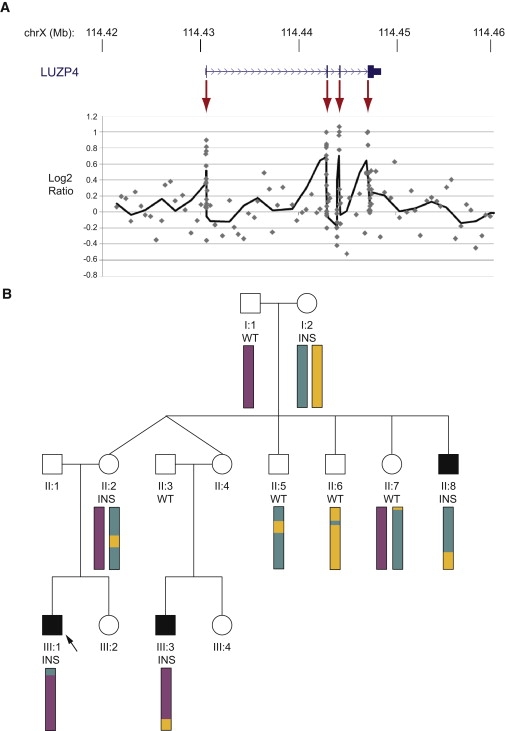
Retroinsertion of LUZP4
(A) Increased log2 ratios were observed for LUZP4 exonic probes (highlighted by red arrows) but not for surrounding noncoding regions. Scatter plot shows individual probes, solid line shows six probe sliding average.
(B) The retroinsertion (INS) is found in all three affected males and was inherited from their mothers. Both unaffected males did not have the retroinsertion (WT). The arrow indicates the individual analyzed by aCGH. Colored bars summarize nonpseudoautosomal X chromosome microsatellite and SNP linkage data and reveal no common region of haploidentity between the three affected males.
Variants Not Associated with Disease
Segregation studies allowed us to exclude a primary role in disease causation for a number of CNVs, some of which would appear a priori to be good candidates for XLID association. Cautious interpretation of cosegregation data is required because ID is sufficiently common for phenocopies to exist within families. Consequently, the presence of a variant in an unaffected male is more compelling evidence that the variant does not cause disease than not finding the variant in an affected male. CNVs that failed to cosegregate with disease are listed in Table 3, with accompanying segregation data in Figure S5. For example, a 710 kb duplication encompassing the IDS (MIM 309900) gene was identified in family 10. Loss of function of IDS causes Hunter Syndrome, a lysosomal storage disease, which can have central nervous system manifestations, and a similar duplication has been proposed as potentially significant in an autistic spectrum disorder (ASD) study.35 In this study, we can discount the variant from disease causation as indicated by the fact that it is absent from an affected male (III:3) and obligate female carrier (II:4) and present in an unaffected male (II:2).
Tarpey et al.10 identified a number of genes, totalling approximately 1% of all X chromosome genes, which were deemed nonessential on the basis of the presence of polymorphic truncating variants or other null variants, which failed to cosegregate with a disease phenotype and were found in clinically normal individuals. In this study, we identified an additional gene that may not be required for normal existence or cognitive function: AWAT1, an acyl-CoA wax alcohol acyltransferase that functions in lipid esterification.36 The proband in family 62 has a deletion encompassing the entire AWAT1 gene. This inherited deletion does not cosegregate with disease, in contrast to a truncating sequence variant in UPF3B mutation to which XLID causation has been attributed.37
Adopting a model often applied in complex disease studies, a number of recent reports have identified sequence or copy number variants at elevated frequencies in neuropsychiatric disease cases relative to controls and have suggested that these variants may have reduced penetrance, modify the clinical presentation, or function as risk factors, predisposing to disease but not sufficient in themselves to cause disease.38,39 These variants may be found in combination with other deleterious variants or do not cosegregate fully with disease as in the case of the TSPAN7 duplications identified by Froyen et al.16 CNVs identified in the IGOLD cohort that have been purported to have disease associations, either in ID or ASD or schizophrenia cases, are detailed in Table S9. Interpretation of the significance of these findings will require deep analyses with cohorts and matched controls at least an order of magnitude larger than the present study.
Cohort Comparisons
The utility of comparison to control data sets, such as those collated in the Database of Genomic Variants, is limited by the small number of studies that have captured rare X chromosome structural variants at a comparable resolution. Instead, to assess whether there is evidence for the contribution of unidentified CNVs to disease, we performed intrastudy comparisons of CNV burden between families where the cause of disease has been identified to those families where the cause of disease remains unidentified (Table 5). The rare CNV burden of families with pathogenic point mutations compared to those with no identified cause of disease is broadly similar.
Table 5.
Comparison of CNV Burden in XLID Cohort Subdivided by Cause of Disease
| Disease Attributed to CNV (n = 23) | Disease Attributed to Sequence Variant (n = 43) | Cause of Disease Unknown (n = 154) | ||
|---|---|---|---|---|
| Number of rare variants | Deletions | 54 (2.3) | 86 (2.0) | 358 (2.3) |
| Duplications | 63 (2.7) | 82(1.9) | 309 (2.0) | |
| Total | 117 (5.1) | 168 (3.9) | 667 (4.3) | |
| Mean extent of rare variants (kb) | Deletions | 27.3 | 11.7 | 7.8 |
| Duplications | 306.2 | 26.2 | 39.2 | |
| Total | 177.5 | 18.8 | 22.4 |
Only samples that passed QC for high-resolution analysis were compared (n = 220). Numbers in parentheses indicate mean values per sample.
We also compared the clinical features of cases in which ID-causing mutations have been identified to those in which the basis of disease causation remains unknown (Figure S6). Overall, pathogenic mutations, i.e., sequence variants (point mutations and small indels) and CNVs, have been identified in 30% of families with >3 affected males compared to 19% of sibling pairs. In general, ID was more severe in cases with structural rearrangements compared to those with pathogenic sequence variants or unknown disease causation. This finding is likely to be skewed by the high number of probands with MECP2 duplications detected in the cohort, all of whom have a severe phenotype. In other respects, the pathogenic CNV and sequence variant subgroups were similar. For example, epilepsy, neurological involvement, and dysmorphism were found in a similar proportion of families with pathogenic CNVs and sequence variants and at levels slightly higher than in the unresolved families.
Breakpoints and Rearrangement Mechanisms
Analysis of CNV junction sequences and features of the local genomic architecture can provide insight into the mechanism of CNV formation. In some genomic disorders, the critical region is flanked by highly homologous low-copy repeats (LCRs), which can mediate nonallelic homologous recombination (NAHR), resulting in recurrent deletion or duplication of the intervening DNA segment.24,40,41 LCRs and genomic architecture have also been associated with nonrecurrent rearrangements, particularly those in which some breakpoint grouping is apparent, such as MECP2 and PLP1 duplications.30,42–44 However, although global surveys of CNV genomic distribution in clinically normal populations suggest an association between CNV-rich regions and segmental duplications, pathogenic rearrangements often consist of rare nonrecurrent CNVs with no association with LCRs.45–49 In some cases there may be underlying sequence features, such as the presence of repetitive elements or non-B DNA structure-associated sequences (reviewed in 50), which may confer genomic instability.42,49,51
We obtained junction sequences for 12 pathogenic CNVs (8 deletions and 4 duplications). Alignments of breakpoint sequences are provided in Figure S7 and their key characteristics are summarized in Table S10. All sequenced rearrangement events were simple, with intronic or intragenic breakpoints. Nine of 12 breakpoints had microhomology of 2–9 bp, two had a single nucleotide that could have been derived from either breakpoint, and one appeared to be a blunt-ended join with no microhomology. In addition to microhomology at the deletion breakpoint in family 206, a 12 bp duplication was detected that could be explained by a serial replication slippage event. Each of the four sequenced duplication events were found to be tandem and directly orientated. This arrangement was also suggested by FISH characterization of the duplication in family 376, one of four cases where no junction sequence was obtained by PCR approaches. The other two cases where the junction sequence was not identified were deletions in families 115 and 494. In both cases, analysis of mRNA confirmed that the orientation of exons flanking the deletion was unaltered, and PCR amplicons just outside the deletion breakpoints were successfully amplified. In combination, these results suggest that the failure to obtain junction PCR products in these cases may reflect the presence of short sequences refractory to PCR or more complex rearrangements with additional novel sequence at the breakpoint.
Ten of 12 breakpoint regions had either unilateral or bilateral involvement of repetitive elements, but in no case was the class of repeat shared between breakpoint pairs and no significant sequence similarity between any breakpoint pairs was detected by Blast2 analysis. Furthermore, no significant enrichment of specific repeat elements or LCRs was detected (Table S11). We also evaluated the recombination and breakpoint-associated motifs listed by Abeysinghe et al.52 but found no significant overrepresentation of any of these motifs (data not shown). However, we did detect significant enrichment of GC content in breakpoint regions compared to a simulated data set of 500 random X chromosome 150 bp sequences. The mean GC content of breakpoint regions was 47%, compared to 39% in controls (two-tailed t test, t = −4.059, p < 0.001). Non-B DNA-associated structures such as G-quartet and Z-DNA were also found at elevated frequency (p < 0.05).
Breakpoint sequencing also enabled us to assess array performance in terms of both the precision of the ADM-1 array estimates and probe performance. Despite the position of breakpoints in noncoding regions, which had a median spacing of 1 probe per 515 bp, 21 of 24 sequenced breakpoints fell within 1 kb of the array estimates. Analysis of sequence-characterized events also revealed that log2 ratio deviations in deletions were reported more strongly than duplications, in part reflecting the greater impact of a change from 1 to 0 copies (in a male deletion) compared to moving from 1 to 2 copies (in a male duplication). For example, whereas 85% of probes with a deletion had a log2 value of <−0.25 (with 70% <−0.5), only 73% of probes in duplications attained log2 values of more than 0.25 (with 48% >0.5). Although we did not sequence Xq28 duplication breakpoints, qPCR was used to validate array estimates of duplication extent and copy number state. In six of nine cases, there was good agreement between previous analyses, estimated array bounds, and qPCR data, but in three cases the extent of the duplication is underreported (families 185, 495, and 389). The discrepancies here are more extensive than for other evaluated CNVs and may reflect the notoriously complex architecture and sequence variation in Xq28.
Discussion
We report the investigation of X chromosome copy number variation in a cohort of individuals with familial intellectual impairment suggestive of X-linked disease. The use of a custom-designed X chromosome-specific high-density oligonucleotide array enabled us to obtain high-resolution and targeted coverage of functional regions of the genome down to the resolution of a single exon while maintaining sufficient coverage of the noncoding X for accurate definition of rearrangement boundaries, aiding interpretation and downstream analysis. We could confidently assign pathogenicity to CNVs on the X chromosome in 25 cases, which corresponds to 10% of the cohort (pathogenic variants were detected in 25/251 sample hybridizations and 23/220 samples analyzed at high resolution). This is higher than the 5% reported by Froyen et al.16 because of the combination of higher resolution of the analysis platform and selection of familial cases. The breakpoints of the deletions and duplications were accurately predicted from the array analysis, which facilitated rapid confirmations of CNVs or indels by independent methods such as FISH or qPCR. On the basis of this, the beneficial use of higher-resolution array CGH is clear. With the advent of higher probe capacity for a single-array experiment, the use of this method to investigate CNVs or indels throughout the whole genome is attractive for investigation of all intellectual disability in the future. The capacity to examine samples at high resolution is, however, contingent on hybridization quality: 12% (31/251) of samples were excluded from high-resolution analysis because of poor performance, although in some of these samples large pathogenic variants were readily detected and more robust lower resolution arrays would have been sufficient to detect these. Similar to Sharp et al., we observed that probe performance was related to the presence of SNPs within the sample underlying the probes.53 Although improved iterative probe design (to avoid common SNP-rich regions) will reduce the noise of high-resolution CGH, some noise is unavoidable as shown by the fact that, for example, we found a rare SNP underlying a series of contiguous probes, suggesting a deletion, but was due to a pathogenic truncating mutation in the sample.10 In reporting the CNV calls from this analysis, it is likely that a significant proportion of the calls reflect underlying sequence variants and not real copy number changes. The SNP-associated calls often achieved smaller log2 ratio deviations than genuine deletions but are nonetheless difficult to tease out in the context of considerable regional and sample-level variability in log2 ratios.
We found that duplications in Xq28 and Xp11.2 were detected at relatively high frequency, had family-specific breakpoints and, in some cases, rearrangement complexity, consistent with the findings of others.30,42 The reciprocal deletions were not observed. The elevated frequency of duplications at these loci may reflect features of genomic architecture predisposing to rearrangement, as has been postulated for the complex LCR architecture surrounding MECP2. In examining sequenced rearrangement breakpoints, we identified an enrichment of GC content, as has also been noted by others.28,42 Because of our small sample size, we combined our analysis of deletions and duplications but the underlying mechanisms may differ for different types of rearrangement, as is suggested by the identification of certain sequence features that were associated with duplications but not deletions by Conrad et al.28 The detection of two IL1RAPL1 deletions, in combination with other published reports,54,55 may further support this hypothesis: the absence of reciprocal IL1RAPL1 intragenic duplications in the published literature is intriguing because these would be predicted to result in the same loss-of-function phenotype as intragenic deletions. The mechanism of IL1RAPL1 deletion may be related to proximity to the fragile site FRAXC and this mechanism may favor deletion.
Overall, pathogenic duplications were more frequently identified than deletions, with duplications particularly dominant at the larger size scale. These observations can be explained by biological and methodological factors. Although analysis of de novo mutations at four NAHR hotspots identified an imbalance in the rates of deletion and duplication occurrence,56 it is not clear whether there is an intrinsic bias in rearrangements formed by mitotic mechanisms, such as microhomology-mediated break-induced repair, which appear to be the predominant mutational mechanism in this study. Deletions in males on the X chromosome are frequently lethal or may be associated with a well-defined clinical syndrome. By comparison, duplications may be milder in effect that might lead to their enrichment in the cohort. Consistent with this, population studies in Drosophila indicate that purifying selection against large variants is weaker for duplications than for deletions.57 Deletions are also more readily identified by alternative experimental methods and by low-resolution array CGH. Notably, the contribution of small duplications is limited and probably reflects the increased difficulty in identification.28
The substantial coincidence of pathogenic CNVs with known XLID-associated genes may indicate that we are approaching a saturation point of XLID gene discovery. However, by current estimates ∼10% of the genes on the X chromosome can harbor mutations causing ID and these are distributed throughout the chromosome.9 Consequently, large deletions or duplications will, in all likelihood, include at least one XLID-associated gene. For smaller variants, the bias toward known genes is influenced by challenges in interpreting rare variant significance. Cautious interpretation of variants is necessary, as indicated by the fact that resequencing analysis has revealed that loss of function of ∼1% of X chromosome genes is not associated with a disease phenotype.10 Variants in the four candidate genes (PTCHD1, FAAH2, WDR13, and GSPT2) were each identified in only a single family. Although segregation analysis has provided an invaluable aid, as was illustrated by analysis of the IDS duplication, more conclusive association of a gene with disease causation requires the identification of multiple independent cases and would be facilitated by the analysis of larger cohorts.
The involvement of PTCHD1 in neurological disease is supported by the report of a partial gene deletion in a family with autism and by further mutation screening and functional characterization.21,35 Further analysis also demonstrates PTCHD1 expression in fetal and adult brain, largely confined to the cerebellum, and provides additional evidence for a role of PTCHD1 in neurodevelopmental and neuropsychiatric disorders.21 PTCHD1 also serves as an illustration of the emerging genetic overlap between neurological disorders including ID, autistic spectrum disorders, and schizophrenia.58,59 One explanation for this is that abnormalities in the same genes can manifest as clinically distinct disorders. Alternatively, the observation may reflect ascertainment bias and clinical overlap between cohorts. Collection and analysis of large, carefully phenotyped patient cohorts would facilitate gene discovery and would also power statistical analysis of the significance of so-called risk factor associations.
Both the WDR13 and FAAH2 deletions identified are predicted to render the respective proteins nonfunctional. WDR13 is a widely expressed WD-repeat containing protein.60 Although the specific function of WDR13 is not known, WD-repeat domains are involved in a wide range of cellular functions and commonly mediate protein-protein interactions.61 Interestingly, upregulation of WDR13 in rat hippocampal neurons has been noted after traumatic injury,62 and we hypothesize that this role in reactive synaptogenesis may recapitulate a neurodevelopmental function. FAAH2 encodes a fatty acid amide hydrolase found in primates and other vertebrates but not murids.63 Fatty acid hydrolases degrade, and thus inactivate, several endogenous lipid messengers, including oleamide and the endocannabinoid anandamide.64 The capacity of FAAH enzymes to modulate neurobehavioral processes, including pain, sleep pattern, and feeding behavior, has led to considerable interest in FAAH inhibitors as therapeutic agents (reviewed in 65). The extent of functional redundancy between the other human fatty acid amide hydrolase, FAAH, and FAAH2 is unclear. On the basis of relative expression levels, it has been suggested that FAAH2 may function predominantly in peripheral tissues, but FAAH2 is also expressed in the central nervous system.63 Further studies will be required to determine whether the partial deletion of FAAH2 underlies the ID and degenerative eye disease in family 494 or is a coincidental finding.
In family 463, we identified a whole gene duplication of GSPT2 and confirmed the direct tandem orientation of the duplicated region. GSPT2 is a single exon gene that has been postulated to have arisen by retrotransposition of GSPT1 mRNA.66 GSPT2 is believed to function as a polypeptide chain releasing factor during protein synthesis, thus acting as an important regulator of translational fidelity. Functional studies in mouse have indicated that GSPT2, which is widely expressed but relatively abundant in brain, but not GSPT1 can functionally complement the orthologous yeast gene SUP35.67,68 GSPT2 upregulation has been detected in rat in response to in utero treatment with a neurotoxic agent associated with deficits in memory and learning.69 Whether GSPT2 duplication results in altered expression levels at significant developmental stages, and whether this may explain the ID phenotype in family 463, remains to be determined.
Focusing on just the 179 families where we have completed both X chromosome exon resequencing10 and high-resolution analysis of the array CGH data, the cause of disease has been identified in 58 families (32%), which can be subdivided into 38 (21%) by sequence variants and 20 (11%) by CNVs. The cause of disease in the majority of families remains unexplained. A number of explanations should be considered. For example, difficulties in assigning pathogenicity to coding and noncoding rare CNVs, and nonsynonymous and synonymous sequence variants, in the face of considerable benign variation may account for some of the mutation shortfall. Also, the presence of variable penetrant alleles and also individuals who are phenocopies of the disease within pedigrees makes the interpretation of the data into simple monogenic models of disease within family complex (AFF2 in family 110). In addition, both the sequencing and array approaches are imperfect methodologies and gaps in the coverage and sequencing annotations may harbor additional mutations.
Two findings in particular, the noncoding deletion affecting CUL4B expression in family 506 and the X-autosome retrotransposition event in family 306, provide tantalizing hints toward the contribution of additional disease mechanisms. The depth of our analysis of noncoding regions by resequencing and array CGH is markedly inferior to that of coding regions. Analysis of noncoding variants is also complicated by our relatively limited knowledge of functional noncoding elements and CNV interpretation can be clouded by long-range position effects.70–73 In the future, the increasing availability of whole-genome sequences, genome-wide expression data, and computational tools will aid our ability to detect and interpret noncoding mutations.
The X chromosome has been reported to be enriched as both a donor and recipient of retrotransposition events, perhaps indicating that this mechanism may contribute disproportionately to X-linked disease.74 Retrotransposition events would be inefficiently detected by PCR-based sequencing and probe-based analysis strategies employed thus far. The result in family 306 also brings into question the contribution of non-X-linked causes in pedigrees with segregation patterns that are classically associated with X-linked disease. In family 306, the disease tracks in the family with the presence of an insertion located on chromosome 7p22.2 and linkage analysis excludes the X chromosome despite a pedigree with several affected males and unaffected female “carriers.” The cohort analyzed were all familial cases where males were more severely affected than females, suggesting X-linked inheritance. The distribution of resolved to unresolved families is skewed toward the larger pedigrees but there remains a substantial shortfall of mutations in some of the largest families. The question of whether all families with disease are truly monogenic with identifiable high penetrance mutations or whether other modes of inheritance should be considered remains to be answered. In the meantime, this work shows that where there are two or more males with intellectual impairment in a family, high-resolution array CGH will identify the cause of disease in ∼10% of cases. This adds to the recently established data that sequence analysis for coding mutations in exons on the X chromosome will yield results in a further ∼20% of cases.10 A comprehensive assessment of the total contribution of X-linked disease genes to familial ID in males awaits the power of whole genome next generation sequence analysis75–77 and the development of robust methods to determine pathogenic significance.
Acknowledgments
We would like to thank both the families and referring clinicians for their long-term cooperation. This work was supported by grants from the Wellcome Trust, Action Medical Research, Baily Thomas Charitable Trust, NIHR, The Evelyn Trust, Australian NH&MRC Program Grant #400121, the State of NSW Health Department through their support of the NSW GOLD Service, a NICHD grant (HD26202), and a grant from the South Carolina Department of Disabilities and Special Needs (SCDDSN). We wish to acknowledge Jenny Moon, Josep Parnau, and Richard Francis for their technical expertise in the early stages of the project and Guy Froyen for sharing unpublished data.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
DECIPHER, http://decipher.sanger.ac.uk
EMBOSS, http://emboss.sourceforge.net/
Genes2Cognition, http://www.genes2cognition.org
GNF SymAtlas, http://biogps.gnf.org
Merlin, http://www.sph.umich.edu/csg/abecasis/Merlin/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/primer3/
RepeatAround, http://www.portugene.com/repeataround.html
RepeatMasker, http://www.repeatmasker.org/
QGRS Mapper, http://bioinformatics.ramapo.edu/QGRS/
UCSC genome browser, http://genome.ucsc.edu/
XLMR Update Website, http://www.ggc.org/xlmr.htm
Z-hunt Online, http://gac-web.cgrb.oregonstate.edu/zDNA/
Accession Numbers
Primary microarray data is available for all samples on request from appropriate researchers under a managed data access agreement in order to preserve patient confidentiality. Applications for access to the data should be made to F.L.R., flr24@cam.ac.uk.
References
- 1.World Health Organization; Geneva: 1992. The ICD-10 Classification of Mental and Behavioural Disorders. [Google Scholar]
- 2.American Psychiatric Association; Washington, D.C.: 1994. Diagnostic and Statistical Manual of Mental Disorders DSM-IV. [Google Scholar]
- 3.Leonard H., Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 4.Ropers H.H. Genetics of intellectual disability. Curr. Opin. Genet. Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Schreppers-Tijdink G.A., Curfs L.M., Wiegers A., Kleczkowska A., Fryns J.P. A systematic cytogenetic study of a population of 1170 mentally retarded and/or behaviourly disturbed patients including fragile X-screening. The Hondsberg experience. J. Genet. Hum. 1988;36:425–446. [PubMed] [Google Scholar]
- 6.Raymond F.L., Tarpey P. The genetics of mental retardation. Hum. Mol. Genet. 2006;15(Spec No 2):R110–R116. doi: 10.1093/hmg/ddl189. [DOI] [PubMed] [Google Scholar]
- 7.Ravnan J.B., Tepperberg J.H., Papenhausen P., Lamb A.N., Hedrick J., Eash D., Ledbetter D.H., Martin C.L. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J. Med. Genet. 2006;43:478–489. doi: 10.1136/jmg.2005.036350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gecz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Chiurazzi P., Schwartz C.E., Gecz J., Neri G. XLMR genes: Update 2007. Eur. J. Hum. Genet. 2008;16:422–434. doi: 10.1038/sj.ejhg.5201994. [DOI] [PubMed] [Google Scholar]
- 10.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O'Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scherer S.W., Lee C., Birney E., Altshuler D.M., Eichler E.E., Carter N.P., Hurles M.E., Feuk L. Challenges and standards in integrating surveys of structural variation. Nat. Genet. 2007;39:S7–S15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lupski J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 13.Lee J.A., Lupski J.R. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 14.Stankiewicz P., Beaudet A.L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr. Opin. Genet. Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Koolen D.A., Pfundt R., de Leeuw N., Hehir-Kwa J.Y., Nillesen W.M., Neefs I., Scheltinga I., Sistermans E., Smeets D., Brunner H.G. Genomic microarrays in mental retardation: A practical workflow for diagnostic applications. Hum. Mutat. 2009;30:283–292. doi: 10.1002/humu.20883. [DOI] [PubMed] [Google Scholar]
- 16.Froyen G., Van Esch H., Bauters M., Hollanders K., Frints S.G., Vermeesch J.R., Devriendt K., Fryns J.P., Marynen P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes. Hum. Mutat. 2007;28:1034–1042. doi: 10.1002/humu.20564. [DOI] [PubMed] [Google Scholar]
- 17.Madrigal I., Rodriguez-Revenga L., Armengol L., Gonzalez E., Rodriguez B., Badenas C., Sanchez A., Martinez F., Guitart M., Fernandez I. X-chromosome tiling path array detection of copy number variants in patients with chromosome X-linked mental retardation. BMC Genomics. 2007;8:443. doi: 10.1186/1471-2164-8-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coe B.P., Ylstra B., Carvalho B., Meijer G.A., Macaulay C., Lam W.L. Resolving the resolution of array CGH. Genomics. 2007;89:647–653. doi: 10.1016/j.ygeno.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 19.Lipson D., Aumann Y., Ben-Dor A., Linial N., Yakhini Z. Efficient calculation of interval scores for DNA copy number data analysis. J. Comput. Biol. 2006;13:215–228. doi: 10.1089/cmb.2006.13.215. [DOI] [PubMed] [Google Scholar]
- 20.Whibley A., Urquhart J., Dore J., Willatt L., Parkin G., Gaunt L., Black G., Donnai D., Raymond F.L. Deletion of MAOA and MAOB in a male patient causes severe developmental delay, intermittent hypotonia and stereotyped hand movements. Eur. J. Hum. Genet. 2010 doi: 10.1038/ejhg.2010.41. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noor A., Whibley A., Marshall C.R., Gianakopoulos P.J., Piton A., Carson A.R., Orlic M., Lionel A.C., Sato D., Pinto D. Disruption at the PTCHD1 locus on Xp22.11 in autism spectrum disorder and intellectual disability. Science Translational Medicine. 2010 doi: 10.1126/scitranslmed.3001267. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yau S.C., Bobrow M., Mathew C.G., Abbs S.J. Accurate diagnosis of carriers of deletions and duplications in Duchenne/Becker muscular dystrophy by fluorescent dosage analysis. J. Med. Genet. 1996;33:550–558. doi: 10.1136/jmg.33.7.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tarpey P.S., Raymond F.L., O'Meara S., Edkins S., Teague J., Butler A., Dicks E., Stevens C., Tofts C., Avis T. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 2007;80:345–352. doi: 10.1086/511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willatt L., Cox J., Barber J., Cabanas E.D., Collins A., Donnai D., FitzPatrick D.R., Maher E., Martin H., Parnau J. 3q29 microdeletion syndrome: Clinical and molecular characterization of a new syndrome. Am. J. Hum. Genet. 2005;77:154–160. doi: 10.1086/431653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plenge R.M., Hendrich B.D., Schwartz C., Arena J.F., Naumova A., Sapienza C., Winter R.M., Willard H.F. A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat. Genet. 1997;17:353–356. doi: 10.1038/ng1197-353. [DOI] [PubMed] [Google Scholar]
- 26.Horn C., Hansen J., Schnutgen F., Seisenberger C., Floss T., Irgang M., De-Zolt S., Wurst W., von Melchner H., Noppinger P.R. Splinkerette PCR for more efficient characterization of gene trap events. Nat. Genet. 2007;39:933–934. doi: 10.1038/ng0807-933. [DOI] [PubMed] [Google Scholar]
- 27.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 28.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marioni J., Thorne N., Valsesia A., Fitzgerald T., Redon R., Fiegler H., Andrews T.D., Stranger B., Lynch A., Dermitzakis E. Breaking the waves: Improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;8:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carvalho C.M., Zhang F., Liu P., Patel A., Sahoo T., Bacino C.A., Shaw C., Peacock S., Pursley A., Tavyev Y.J. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum. Mol. Genet. 2009;18:2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gecz J., Oostra B.A., Hockey A., Carbonell P., Turner G., Haan E.A., Sutherland G.R., Mulley J.C. FMR2 expression in families with FRAXE mental retardation. Hum. Mol. Genet. 1997;6:435–441. doi: 10.1093/hmg/6.3.435. [DOI] [PubMed] [Google Scholar]
- 32.Tabolacci E., Pomponi M.G., Pietrobono R., Terracciano A., Chiurazzi P., Neri G. A truncating mutation in the IL1RAPL1 gene is responsible for X-linked mental retardation in the MRX21 family. Am. J. Med. Genet. A. 2006;140:482–487. doi: 10.1002/ajmg.a.31107. [DOI] [PubMed] [Google Scholar]
- 33.Friesema E.C., Grueters A., Biebermann H., Krude H., von Moers A., Reeser M., Barrett T.G., Mancilla E.E., Svensson J., Kester M.H. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- 34.Gilfillan G.D., Selmer K.K., Roxrud I., Smith R., Kyllerman M., Eiklid K., Kroken M., Mattingsdal M., Egeland T., Stenmark H. SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am. J. Hum. Genet. 2008;82:1003–1010. doi: 10.1016/j.ajhg.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turkish A.R., Henneberry A.L., Cromley D., Padamsee M., Oelkers P., Bazzi H., Christiano A.M., Billheimer J.T., Sturley S.L. Identification of two novel human acyl-CoA wax alcohol acyltransferases: Members of the diacylglycerol acyltransferase 2 (DGAT2) gene superfamily. J. Biol. Chem. 2005;280:14755–14764. doi: 10.1074/jbc.M500025200. [DOI] [PubMed] [Google Scholar]
- 37.Tarpey P.S., Raymond F.L., Nguyen L.S., Rodriguez J., Hackett A., Vandeleur L., Smith R., Shoubridge C., Edkins S., Stevens C. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hannes F.D., Sharp A.J., Mefford H.C., de Ravel T., Ruivenkamp C.A., Breuning M.H., Fryns J.P., Devriendt K., Van Buggenhout G., Vogels A. Recurrent reciprocal deletions and duplications of 16p13.11: The deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J. Med. Genet. 2009;46:223–232. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Girirajan S., Rosenfeld J.A., Cooper G.M., Antonacci F., Siswara P., Itsara A., Vives L., Walsh T., McCarthy S.E., Baker C. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang F., Carvalho C.M., Lupski J.R. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lupski J.R., Stankiewicz P. Genomic disorders: Molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bauters M., Van Esch H., Friez M.J., Boespflug-Tanguy O., Zenker M., Vianna-Morgante A.M., Rosenberg C., Ignatius J., Raynaud M., Hollanders K. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res. 2008;18:847–858. doi: 10.1101/gr.075903.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.del Gaudio D., Fang P., Scaglia F., Ward P.A., Craigen W.J., Glaze D.G., Neul J.L., Patel A., Lee J.A., Irons M. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- 44.Lee J.A., Inoue K., Cheung S.W., Shaw C.A., Stankiewicz P., Lupski J.R. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum. Mol. Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- 45.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong K.K., deLeeuw R.J., Dosanjh N.S., Kimm L.R., Cheng Z., Horsman D.E., MacAulay C., Ng R.T., Brown C.J., Eichler E.E. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koolen D.A., Sistermans E.A., Nilessen W., Knight S.J., Regan R., Liu Y.T., Kooy R.F., Rooms L., Romano C., Fichera M. Identification of non-recurrent submicroscopic genome imbalances: the advantage of genome-wide microarrays over targeted approaches. Eur. J. Hum. Genet. 2008;16:395–400. doi: 10.1038/sj.ejhg.5201975. [DOI] [PubMed] [Google Scholar]
- 49.Vissers L.E., Bhatt S.S., Janssen I.M., Xia Z., Lalani S.R., Pfundt R., Derwinska K., de Vries B.B., Gilissen C., Hoischen A. Rare pathogenic microdeletions and tandem duplications are micro homology-mediated and stimulated by local genomic architecture. Hum. Mol. Genet. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 50.Wells R.D. Non-B DNA conformations, mutagenesis and disease. Trends Biochem. Sci. 2007;32:271–278. doi: 10.1016/j.tibs.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 51.de Smith A.J., Walters R.G., Coin L.J., Steinfeld I., Yakhini Z., Sladek R., Froguel P., Blakemore A.I. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS ONE. 2008;3:e3104. doi: 10.1371/journal.pone.0003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abeysinghe S.S., Chuzhanova N., Krawczak M., Ball E.V., Cooper D.N. Translocation and gross deletion breakpoints in human inherited disease and cancer I: Nucleotide composition and recombination-associated motifs. Hum. Mutat. 2003;22:229–244. doi: 10.1002/humu.10254. [DOI] [PubMed] [Google Scholar]
- 53.Sharp A.J., Itsara A., Cheng Z., Alkan C., Schwartz S., Eichler E.E. Optimal design of oligonucleotide microarrays for measurement of DNA copy-number. Hum. Mol. Genet. 2007;16:2770–2779. doi: 10.1093/hmg/ddm234. [DOI] [PubMed] [Google Scholar]
- 54.Jin H., Gardner R.J., Viswesvaraiah R., Muntoni F., Roberts R.G. Two novel members of the interleukin-1 receptor gene family, one deleted in Xp22.1-Xp21.3 mental retardation. Eur. J. Hum. Genet. 2000;8:87–94. doi: 10.1038/sj.ejhg.5200415. [DOI] [PubMed] [Google Scholar]
- 55.Nawara M., Klapecki J., Borg K., Jurek M., Moreno S., Tryfon J., Bal J., Chelly J., Mazurczak T. Novel mutation of IL1RAPL1 gene in a nonspecific X-linked mental retardation (MRX) family. Am. J. Med. Genet. A. 2008;146A:3167–3172. doi: 10.1002/ajmg.a.32613. [DOI] [PubMed] [Google Scholar]
- 56.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emerson J.J., Cardoso-Moreira M., Borevitz J.O., Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320:1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- 58.Merikangas A.K., Corvin A.P., Gallagher L. Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends Genet. 2009;25:536–544. doi: 10.1016/j.tig.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Guilmatre A., Dubourg C., Mosca A.L., Legallic S., Goldenberg A., Drouin-Garraud V., Layet V., Rosier A., Briault S., Bonnet-Brilhault F. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singh B.N., Suresh A., UmaPrasad G., Subramanian S., Sultana M., Goel S., Kumar S., Singh L. A highly conserved human gene encoding a novel member of WD-repeat family of proteins (WDR13) Genomics. 2003;81:315–328. doi: 10.1016/s0888-7543(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 61.Smith T.F. Diversity of WD-repeat proteins. Subcell. Biochem. 2008;48:20–30. doi: 10.1007/978-0-387-09595-0_3. [DOI] [PubMed] [Google Scholar]
- 62.Price M., Lang M.G., Frank A.T., Goetting-Minesky M.P., Patel S.P., Silviera M.L., Krady J.K., Milner R.J., Ewing A.G., Day J.R. Seven cDNAs enriched following hippocampal lesion: Possible roles in neuronal responses to injury. Brain Res. Mol. Brain Res. 2003;117:58–67. doi: 10.1016/s0169-328x(03)00285-7. [DOI] [PubMed] [Google Scholar]
- 63.Wei B.Q., Mikkelsen T.S., McKinney M.K., Lander E.S., Cravatt B.F. A second fatty acid amide hydrolase with variable distribution among placental mammals. J. Biol. Chem. 2006;281:36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 64.Alexander S.P., Kendall D.A. The complications of promiscuity: Endocannabinoid action and metabolism. Br. J. Pharmacol. 2007;152:602–623. doi: 10.1038/sj.bjp.0707456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seierstad M., Breitenbucher J.G. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J. Med. Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 66.Zhouravleva G., Schepachev V., Petrova A., Tarasov O., Inge-Vechtomov S. Evolution of translation termination factor eRF3: Is GSPT2 generated by retrotransposition of GSPT1's mRNA? IUBMB Life. 2006;58:199–202. doi: 10.1080/15216540600686862. [DOI] [PubMed] [Google Scholar]
- 67.Hoshino S., Imai M., Mizutani M., Kikuchi Y., Hanaoka F., Ui M., Katada T. Molecular cloning of a novel member of the eukaryotic polypeptide chain-releasing factors (eRF). Its identification as eRF3 interacting with eRF1. J. Biol. Chem. 1998;273:22254–22259. doi: 10.1074/jbc.273.35.22254. [DOI] [PubMed] [Google Scholar]
- 68.Le Goff C., Zemlyanko O., Moskalenko S., Berkova N., Inge-Vechtomov S., Philippe M., Zhouravleva G. Mouse GSPT2, but not GSPT1, can substitute for yeast eRF3 in vivo. Genes Cells. 2002;7:1043–1057. doi: 10.1046/j.1365-2443.2002.00585.x. [DOI] [PubMed] [Google Scholar]
- 69.Royland J.E., Kodavanti P.R. Gene expression profiles following exposure to a developmental neurotoxicant, Aroclor 1254: Pathway analysis for possible mode(s) of action. Toxicol. Appl. Pharmacol. 2008;231:179–196. doi: 10.1016/j.taap.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 70.Henrichsen C.N., Chaignat E., Reymond A. Copy number variants, diseases and gene expression. Hum. Mol. Genet. 2009;18:R1–R8. doi: 10.1093/hmg/ddp011. [DOI] [PubMed] [Google Scholar]
- 71.Henrichsen C.N., Vinckenbosch N., Zollner S., Chaignat E., Pradervand S., Schutz F., Ruedi M., Kaessmann H., Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 72.Reymond A., Henrichsen C.N., Harewood L., Merla G. Side effects of genome structural changes. Curr. Opin. Genet. Dev. 2007;17:381–386. doi: 10.1016/j.gde.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 73.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Emerson J.J., Kaessmann H., Betran E., Long M. Extensive gene traffic on the mammalian X chromosome. Science. 2004;303:537–540. doi: 10.1126/science.1090042. [DOI] [PubMed] [Google Scholar]
- 75.Tuzun E., Sharp A.J., Bailey J.A., Kaul R., Morrison V.A., Pertz L.M., Haugen E., Hayden H., Albertson D., Pinkel D. Fine-scale structural variation of the human genome. Nat. Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 76.Korbel J.O., Urban A.E., Grubert F., Du J., Royce T.E., Starr P., Zhong G., Emanuel B.S., Weissman S.M., Snyder M. Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome. Proc. Natl. Acad. Sci. USA. 2007;104:10110–10115. doi: 10.1073/pnas.0703834104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kerzendorfer C., Whibley A., Carpenter G., Outwin E., Chiang S.C., Turner G., Schwartz C., El-Khamisy S., Raymond F.L., O'Driscoll M. Mutations in Cullin 4B result in a human syndrome associated with increased camptothecin-induced topoisomerase I-dependent DNA breaks. Hum. Mol. Genet. 2010;19:1324–1334. doi: 10.1093/hmg/ddq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Friez M.J., Jones J.R., Clarkson K., Lubs H., Abuelo D., Bier J.A., Pai S., Simensen R., Williams C., Giampietro P.F. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:1687–1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- 80.Clayton-Smith J., Walters S., Hobson E., Burkitt-Wright E., Smith R., Toutain A., Amiel J., Lyonnet S., Mansour S., Fitzpatrick D. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur. J. Hum. Genet. 2009;17:434–443. doi: 10.1038/ejhg.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Froyen G., Corbett M., Vandewalle J., Jarvela I., Lawrence O., Meldrum C., Bauters M., Govaerts K., Vandeleur L., Van Esch H. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008;82:432–443. doi: 10.1016/j.ajhg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.