

Abstract
Chronic myelogenous leukemia (CML) can progress from an indolent chronic phase to an aggressive blast crisis phase1 but the molecular basis of this transition remains poorly understood. Here we have used mouse models of CML2,3 to show that disease progression is regulated by the Musashi-Numb signaling axis4,5. Specifically, we find that chronic phase is marked by high and blast crisis phase by low levels of Numb expression, and that ectopic expression of Numb promotes differentiation and impairs advanced phase disease in vivo. As a possible explanation for the decreased levels of Numb in blast crisis, we show that NUP98-HOXA9, an oncogene associated with blast crisis CML6,7, can trigger expression of the RNA binding protein Musashi2 (Msi2) which in turn represses Numb. Importantly, loss of Msi2 restores Numb expression and significantly impairs the development and propagation of blast crisis CML in vitro and in vivo. Finally, we show that Msi2 expression is not only highly upregulated during human CML progression but is also an early indicator of poorer prognosis. These data show that the Musashi-Numb pathway can control the differentiation of CML cells, and raise the possibility that targeting this pathway may provide a new strategy for therapy of aggressive leukemias.
Chronic myelogenous leukemia (CML) is initiated by the BCR-ABL translocation, which leads to myeloid cell expansion while allowing differentiation8–11. Secondary translocations such as NUP98-HOXA9 or AML1-EVI1, or mutations in p53 or INK4A/ARF trigger progression through an accelerated phase to a blast crisis phase, with progressive loss of the capacity to differentiate1. While blast crisis CML is in part more aggressive because of arrested differentiation, the pathways that underlie this arrest remain poorly understood. To determine whether CML progression may be driven by reversal of signals that regulate differentiation during normal development, we focused on Numb4, a molecule that can be inherited differentially during asymmetric division and specify a committed fate12–16.
To determine whether Numb regulates leukemia progression we utilized mouse models representing chronic phase and myeloid blast crisis CML. Chronic disease was generated by infecting hematopoietic stem cell enriched populations (c-Kit+Lin−Sca-1+ or KLS) with BCR-ABL and transplanting them into irradiated recipients2,3. Myeloid blast crisis was modeled by transplanting KLS cells transduced with BCR-ABL and NUP98-HOXA96,7,17. Using these we found that Numb was expressed at significantly lower levels in blast crisis compared to chronic phase (Fig. 1a–c). The decreased expression of Numb in blast crisis suggested that keeping Numb at low levels may be essential for maintaining an immature state and that increasing its levels could trigger differentiation and inhibit disease progression. To test this possibility, hematopoietic cells were infected with BCR-ABL and NUP98-HOXA9 together with either control vector or Numb, transplanted and leukemia progression monitored. 83% of control mice developed leukemia compared to 63% of those transplanted with Numb expressing cells (Fig. 1d). Importantly, leukemias that developed in the presence of Numb were more differentiated (Fig. 1e, f), and unable to propagate disease efficiently (93% versus 20%, Fig. 1g) or infiltrate secondary organs (Fig. 1h, i, Supplementary Fig. 1); no signs of leukemia were detected in mice that survived (Fig. 1j, Supplementary Figure 1). Numb also impaired propagation of fully established leukemias and dramatically reduced the frequency of cancer stem cells (Supplementary Fig. 2). These data show that continual repression of Numb is essential for maintenance of blast crisis CML, and that increasing the levels of Numb can inhibit disease.
Figure 1. Expression of Numb impairs blast crisis CML development.

a, CML cells were immunostained with anti-Numb antibody (red) and DAPI (green pseudocolor) and b, fluorescence intensity quantified *p<0.05. c, CML cells were analyzed by Western blot for Numb expression. d, Cells infected with BCR-ABL, NUP98-HOXA9 and either control vector or Numb were transplanted and survival monitored (control, n=18 and Numb, n=19). e, Representative and f, average frequency of Lin− cells from control or Numb expressing leukemias **p<0.001. g, Lin− cells from primary leukemias were serially transplanted and survival monitored (Vector, n=14 and Numb, n=15, **p<0.001). h, Hematoxylin and Eosin stained spleen sections from control vector or i, Numb expressing leukemias or j, surviving mice. Immature myeloid cells (red arrowheads) and lymphoid follicles (black arrowheads). Magnification: 10×. Error bars in all bar graphs are s.e.m. Data shown is representative of three to four independent experiments.
Since Numb can antagonize Notch signaling in several systems13,18,19, we tested whether Numb and Notch had a reciprocal relationship in CML. Notch signaling was elevated in blast crisis CML (Supplementary Fig. 3), and its inhibition via dnXSu(H) delivery or through conditional deletion of Rbpj paralleled the effects of Numb and led to reduced incidence and propagation of blast crisis CML (Supplementary Fig. 4). In addition, levels of p53, another Numb target20, were higher in Numb-expressing blast crisis CML (Supplementary Fig. 5a). In the absence of p53, Numb was unable to impact leukemic cell growth in vivo or in vitro (Supplementary Fig. 5b–f), indicating that Numb's effects are in part dependent on p53.
The observation that Numb repression was critical for maintenance of blast crisis CML led us to seek the mechanism by which Numb may be downregulated in this context. We focused on the RNA binding protein Musashi (Msi), which has been shown in the nervous system to repress Numb by binding the 3'UTR of the transcript21. Msi was originally identified in Drosophila as a regulator of asymmetric division5,22 and its expression has been associated with stem and progenitor cells in several tissues23,24. In the hematopoietic system we found that Msi2 was expressed at much higher levels than Msi1 (Fig. 2a), and was particularly elevated in stem cells (Fig. 2b). Paralleling this, Msi2 expression was 10-fold higher in the more immature blast crisis CML (Figure 2c); this pattern held true even in matched lineage negative fractions (Fig. 2d) suggesting that Msi2 upregulation in advanced phase is not simply a consequence of altered cellular composition. Finally, expression of Msi2 was most enriched in the lineage negative fraction of blast crisis CML (Fig. 2e). These data indicate that Msi2 expression associates predominantly with normal hematopoietic stem cells and the most immature fraction of leukemic cells.
Figure 2. The RNA binding protein Musashi is highly expressed in immature normal and leukemic cells and is regulated by HoxA9.

a, Musashi (Msi) expression in whole bone marrow (WBM), KLS cells, chronic and blast crisis CML, olfactory bulb (OB), –reverse transcriptase (-RT in OB) and water. Tbp, TATA binding protein. b–e, Realtime RT-PCR analysis of Msi2 expression in b, KLS cells (n=3), Lin+ cells (n=2) **p<0.001, c, blast crisis phase (n=9), chronic phase (n=6) **p<0.001, d, lin− chronic and blast crisis phase cells relative to normal KLS and lin+ cells (lin+, n=2 and others, n=3), e, lin− (n=5) or lin+ (n=5) blast crisis CML cells *p=0.039. Error bars represent s.e.m. f, Control vector- or Msi2-expressing CML cells were stained with anti-Numb antibody (red) and DAPI (green pseudocolor) and g, fluorescence intensity quantified **p<0.001. h, Msi2 expression in KLS cells transduced with either control vector or NUP98-HOXA9 retrovirus along with BCR-ABL *p=0.017. i–l, HoxA9 binds to the Msi2 promoter. Murine Msi2 gene structure: exons (numbered boxes), transcription start site (TSS; +1) and the direction of transcription (flag), putative HOX binding element 5.7kb upstream of TSS (oval) and +110kb site with no HoxA9 binding sequence (open rectangle). i, ChIP was performed either with IgG control or anti-HoxA9 antibody for j, Flt3, a known HoxA9 target gene, as a positive control and k, Msi2 −5.7kb region or l, Msi2 +110kb region. m, KLS cells from Msi2 genetrap reporter mice were transduced with BCR-ABL with either control vector or NUP98-HOXA9 and β-galactosidase reporter activity quantified (n=2 each, *p=0.011).
Because Msi2 and Numb were expressed in a reciprocal pattern, we tested whether Msi2 could repress Numb during leukemogenesis. Expression of Msi2 in chronic phase CML cells led to a downregulation of Numb (Fig. 2f, g). Further, NUP98-HOXA9 could also activate this cascade by increasing expression of Msi2 (Fig. 2h). Since NUP98-HOXA9 initiates transformation through HoxA9 mediated DNA binding and transcription, we tested whether HoxA9 could bind the Msi2 promoter and activate its expression directly. Chromatin immunoprecipitation revealed that HoxA9 was in fact associated with the putative HoxA9 binding element we identified at −5.7kb (Fig. 2i–l). NUP98-HOXA9 expression was also able to induce Msi2 reporter activity in KLS cells (Fig. 2m, Supplementary Fig. 6a). These data show that Msi2 can be upregulated by NUP98-HOXA9 and subsequently contribute to blast crisis CML by repressing Numb.
To test if Msi2 is required for the development of blast crisis CML, we utilized a mouse in which the Msi2 gene was disrupted by a genetrap (Gt) vector25 (Supplementary Fig. 6a, b). Msi2 mutant mice were viable, albeit smaller and less frequent than predicted (+/+:+/Gt:Gt/Gt=38:66:19, p=0.038), and showed a two-three fold reduction in the frequency (Fig. 3a) and absolute numbers (data not shown) of KLS cells. Additionally, the loss of Msi2 led to significantly impaired leukemia growth in vivo (Fig. 3b, 93% for control versus 57% for Gt/Gt).
Figure 3. Loss of Musashi impairs the development and propagation of blast crisis CML.
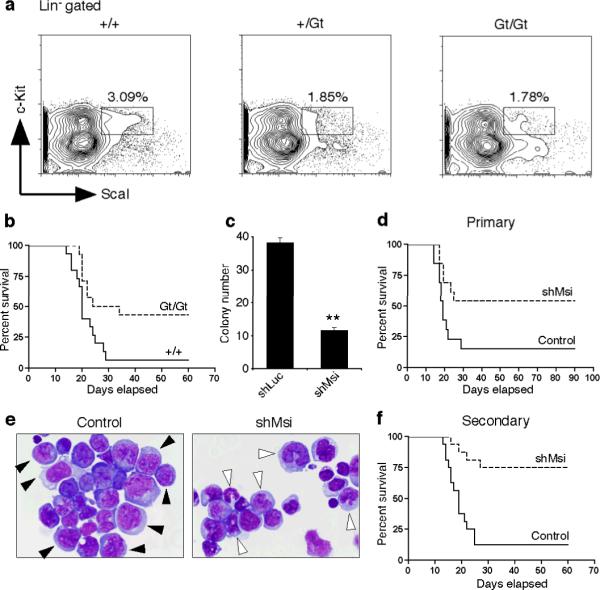
a, Frequency of KLS cells in mice of indicated genotypes (+/+, n=4, +/Gt, n=3 and Gt/Gt, n=4). b, Survival curve of mice transplanted with BCR-ABL and NUP98-HOXA9 infected +/+ or Gt/Gt KLS cells (+/+, n=15 and Gt/Gt, n=14, *p=0.0159). c, Colony-forming ability of blast crisis CML cells transduced with control shRNA (shLuc) or Msi2 shRNA (shMsi) **p<0.001. d, Survival curve of mice transplanted with established blast crisis CML cells infected with control shLuc or shMsi (n=13 each, *p=0.0267). e, Wright's stain of leukemic cells from mice transplanted with control shLuc or shMsi infected blast crisis CML. Immature myeloblasts (closed arrowheads), differentiating myelocytes and mature band cells (open arrowheads). Magnification: 100×. f, Survival curve of mice transplanted with Lin− cells from primary shRNA expressing leukemias (Control, n=15 and shMsi, n=16, **p<0.001). Data shown is representative of two to three independent experiments.
To determine if inhibiting Msi2 could impact the growth of established CML, and to rule out the possibility that the reduced incidence of leukemia in Gt mutants was due to developmental defects, Msi expression was targeted using an alternate shRNA approach (Supplementary Fig. 7a). Delivery of Msi2 shRNAs (shMsi) into established blast crisis CML cells reduced colony growth in vitro (Fig. 3c, Supplementary Fig. 7b–e) and disease incidence in vivo (Fig. 3d). The majority of leukemias that occurred in the presence of shMsi were more differentiated (Fig. 3e), and impaired in their ability to propagate disease (Fig. 3f, 87% control versus 25% shMsi). These data show that Msi2 is important for establishment and continued propagation of blast crisis CML.
Finally, we examined if MSI2 was aberrantly upregulated during human leukemia progression. MSI2 was tracked in 30 patient samples from banks in Korea and the United Kingdom, and found to be expressed at significantly higher levels in blast crisis CML (Fig. 4a, b). To determine if this reflected a general pattern in human CML progression, we examined expression of MSI2 and associated genes in 90 patient samples from banks in the United States26. Microarray analysis revealed a dramatic upregulation of MSI2 in every patient during CML progression (Fig. 4c). In addition, NUMB was downregulated in a majority of blast crisis patients (Fig. 4d). Notably, our mouse model was driven by NUP98-HOXA9 as a second hit, whereas human blast crisis CMLs harbor a variety of secondary mutations. Since Msi2 could be regulated by HoxA9 expression in the mouse model of CML, we examined if HOXA9 was upregulated in blast crisis CML samples. The observation that a majority of patient samples had elevated levels of HOXA9 (Fig. 4e) may explain how MSI2 becomes upregulated in advanced stage disease regardless of the nature of the second hit. Notch signaling targets HES1 and TRIB2 were also elevated in a number of blast crisis patient samples (Fig. 4f, Supplementary Fig. 8), consistent with other independent reports27.
Figure 4. Musashi expression is upregulated during human CML progression.

a and b, PCR analysis of MSI2 expression in chronic and blast crisis CML patient samples from a, the Korean Leukemia Bank, Korea (n=9 per cohort, Mann-Whitney U test **p<0.001) and b, the Hammersmith MRD Lab Sample Archive, United Kingdom (n=6 per cohort, Mann-Whitney U test, **p<0.001). c–f, Microarray analysis of expression of c, MSI2 d, NUMB e, HOXA9 (all p<.001) and f, HES1 (p=.68) in bone marrow and peripheral blood samples from 42 chronic (red), 17 accelerated (green) and 31 blast crisis phase (blue) patients in the United States. g, Proposed model for the role of Musashi and Numb in CML progression.
Because the highest MSI2 expression was observed in blast crisis patients, where treatment outcomes are extremely poor, and because a range of expression was observed in both chronic and accelerated phase CML, we tested whether MSI2 expression correlated with outcomes after allogeneic transplantation. Patients were divided into two groups based on median expression of MSI2. Among 37 chronic phase patients with available outcomes (9 relapses) increased MSI2 expression was associated with a higher risk of relapse (hazard ratio = 4.35; 95% CI, 0.90 – 21.06, p=0.07). Additionally, among 13 accelerated phase patients with available outcomes (6 deaths and 3 relapses), increased MSI2 expression was not only associated with higher risk of relapse (all relapses occurred in the increased MSI2 group, p=0.06), but also with higher risk of death (hazard ratio = 6.76; 95% CI, 0.78 – 58.57, p=0.08). The association of MSI2 with poorer outcomes suggests that Msi2 may be an early marker of advanced CML disease.
Our work identifies the Musashi-Numb axis as an important regulator of myeloid leukemia and indicates that maintenance of the immature state is dependent on reversal of classic differentiation cues. Specifically, we find that Msi2 is upregulated and Numb downregulated as chronic phase CML progresses to blast crisis, and that modulation of this pathway can inhibit disease (Fig. 4g). Although previous work has implicated Musashi and Numb in normal development13,14,23,24, to our knowledge this is the first demonstration that this pathway is required for hematologic malignancy.
Our previous work showing that while BCR-ABL cannot affect the choice between asymmetric and symmetric division, NUP98-HOXA9 can trigger a bias toward symmetric renewal15 had led us to propose that regulators of asymmetric division might regulate leukemic differentiation, and could thus be targets for therapy in advanced myeloid leukemia. Our current work supports this and shows that Numb, which drives commitment and differentiation, can impair blast crisis CML establishment and propagation. It should be noted that just as Numb's influence may be mediated through p53 and/or Notch signaling12,13,20, Musashi may act through Numb as well as other targets such as p21WAF121,28.
Because blast crisis CML is uniformly resistant to current treatments, it is critical to identify new pathways that drive this aggressive disease. In that light, our work is important because it shows that specific differentiation cues associated with the Musashi-Numb cascade can unlock the differentiation potential of blast crisis CML and impair its growth. These data, together with the fact that Musashi appears to be an early marker of advanced CML, suggest that its expression could serve as a prognostic tool, and that targeting it might represent a new approach to therapy. Finally, reports of increased expression of Musashi in glioblastoma29 and decreased expression of Numb in high grade breast cancer30 raise the possibility that this pathway may also be relevant in solid cancers.
Methods Summary
Mouse models of CML were generated by transducing bone marrow stem and progenitor cells with retroviruses carrying BCR-ABL (chronic phase), or BCR-ABL and NUP98-HOXA9 (blast crisis phase) and transplanted into irradiated recipient mice. The development of CML was confirmed by flow cytometry and histopathology. For Msi2 knockdown experiments, lineage negative blast crisis CML cells were infected with Msi2 or control Luciferase shRNA retroviral constructs and leukemia incidence monitored. ChIP assays were performed using the myeloid leukemia cell line M1. DNA was crosslinked and immunoprecipitated with control or anti-HOXA9 antibodies and analyzed by PCR for regions of interest. CML patient samples were obtained from the Korean Leukemia Bank (Korea), the Hammersmith MRD Lab Sample Archive (United Kingdom), the Fred Hutchinson Cancer Research Center (United States) and the Singapore General Hospital (Singapore). Gene expression in human chronic and blast crisis CML was analyzed by PCR or by DNA microarrays.
Methods
Mice
C57BL6/J and BA (C57BL/Ka-Thy1.1) were used as transplant donors, and B6-CD45.1 (B6.SJL-PtprcaPepcb/BoyJ) and HZ (C57BL/Ka-Thy1.1-CD45.1) mice were used as transplant recipients. All mice were 8–16 weeks of age. Msi2 mutant mice, B6;CB-Msi2Gt(pU-21T)2Imeg, was made and established by genetrap mutagenesis (CARD, Kumamoto University). Floxed Rbpj mice, B6.Cg-Rbpsuhtm3Kyo, were from RIKEN BioResource Center (RBRC01071), and crossed with Vav-cre transgenic mice31,32. Mice were bred and maintained in the animal care facility at Duke University Medical Center. All animal experiments were performed according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
Cell isolation and FACS analysis
HSCs were sorted from mouse bone marrow essentially as described31. c-Kit positive cells were enriched by staining whole bone marrow with anti-CD117/c-Kit microbeads and isolating positively labeled cells with autoMACS cell separation (Miltenyi Biotec). For lineage analysis peripheral blood cells were obtained by submandibular bleeding and diluted in 0.5 ml of 10 mM EDTA in PBS. 1 ml of 2% dextran was then added to each sample, and red blood cells depleted by sedimentation for 45 minutes at 37°C. Red blood cells were lysed using RBC Lysis Buffer (eBioscience) before staining for lineage markers. The following antibodies were used to define the lineage positive cells in leukemic samples: 145-2C11 (CD3ε), GK1.5 (CD4), 53-6.7 (CD8), RB6-8C5 (Ly-6G/Gr-1), M1/70 (CD11b/Mac-1), TER119 (Ly-76/TER119), and 6B2 (CD45R/B220). Other antibodies used for HSC sorts included 2B8 (CD117/c-Kit) and D7 (Ly-6A/E/Sca-1). All antibodies were purchased from BD Pharmingen or eBioscience. Analysis and cell sorting were carried out on a FACSVantage SE, FACStar, FACSCanto II, or FACSDiva (all from Becton Dickinson) at the Duke Comprehensive Cancer Center Flow Core Facility, and data were analyzed with FlowJo software (Tree Star Inc.).
Retroviral constructs and production
MIG-BCR-ABL was cloned into MSCV-IRES-YFP or -CFP retroviral vector. MSCV-NUP98-HOXA9-IRES-YFP was cloned into the MSCV-IRES-tNGFR vector. Numb cDNA (p65 isoform, NCBI Accession number BC033459) was cloned into the MSCV-IRES-GFP vector. Msi2 cDNA (IMAGE clone ID 40045350) was purchased from Open Biosystems, and its protein coding region was cloned into MSCV-IRES-GFP or -CFP. Short hairpin RNA (shRNA) constructs were designed and cloned in MSCV/LTRmiR30-PIG (LMP) vector from Open Biosystems according to their instructions. The target sequences are 5'-CCCAGATAGCCTTAGAGACTAT-3' for Msi2 and 5'-CTGTGCCAGAGTCCTTCGATAG-3' for firefly luciferase as a negative control. MSCV-IRES-CFP with Msi2 mutant cDNA resistant to shMsi was constructed by inverse PCR strategy using primers with silent mutations (underlined) in the shMsi target sequence; 5'-CCTGACTCTCTGAGGGACTATTTTAGCAAATTTGG-3'. Lentiviral shRNA construct with the alternate Msi2 target sequence, 5'-AGTTAGATTCCAAGACGA-3', was cloned in FG12 as described previously33. Virus was produced in 293T cells transfected with viral constructs along with gag-pol, VSV-G and Rev (in case of FG12) constructs. Viral supernatants were collected for three to five days and concentrated by ultracentrifugal at 50,000×g for 3h.
In vitro methylcellulose colony formation assays
Lineage negative (Lin−), NUP98-HOXA9-IRES-YFP positive cells from blast crisis CML were sorted and infected retrovirally with either Vector-IRES-GFP or Numb-IRES-GFP. After 48 hours of infection, cells were sorted and serially plated in complete methylcellulose medium (Methocult GF M3434 from StemCell Technologies). For knockdown experiments, Lin− population in blast crisis CML were sorted and infected with the indicated retroviruses for 48h. Infected cells were sorted based on their fluorescent protein expression and plated as above. Colonies were counted 5–7 days after plating.
Generation and analysis of leukemic mice
Bone marrow c-Kit+ or KLS cells were sorted and cultured overnight in X-Vivo15 media (BioWhittaker) supplemented with 50 μM 2-mercaptoethanol, 10% fetal bovine serum, stem cell factor (SCF, 100 ng/ml, R&D Systems), and thrombopoietin (TPO, 20 ng/ml, R&D Systems). Subsequently, cells were infected with the retroviruses. Viruses used were as follows: MSCV-BCR-ABL-IRES-YFP (or CFP or GFP) to generate chronic phase leukemia, or MSCV-BCR-ABL-IRES-YFP (or CFP or GFP) and MSCV-NUP98-HOXA9-IRES-YFP (or tNGFR) to generate blast crisis CML. Cells were harvested 48 hours after infection and transplanted retro-orbitally into groups of B6-CD45.1 mice. Recipients were lethally irradiated (10Gy) for chronic phase leukemia, and sublethally (7Gy) or non-irradiated for blast crisis CML. For Numb overexpression, cells were infected with either MSCV-Numb-IRES-GFP or MSCV-IRES-GFP along with MSCV-BCR-ABL-IRES-YFP (or CFP) and MSCV-NUP98-HOXA9-IRES-tNGFR (or YFP) and 20,000 to 100,000 infected cells were transplanted per mouse. For secondary transplantation, cells from primary transplanted mice were sorted for either MSCV-Numb-IRES-GFP and MSCV-NUP98-HOXA9-IRES-YFP or MSCV-IRES-GFP and MSCV-NUP98-HOXA9-IRES-YFP, and 7,000 to 8,000 cells were transplanted per mouse. For Msi2 knockdown by retroviral shRNA transduction, Lin− population from blast crisis CML were sorted and infected with either control shLuc (against luciferase) or shMsi (against Msi2) retrovirus for 48h. Infected cells were sorted based on their GFP expression, and 1,000 to 3,000 cells were transplanted in sublethally irradiated B6-CD45.1 recipients. After transplantation, recipient mice were maintained on antibiotic water (sulfamethoxazole and trimethoprim) and evaluated daily for signs of morbidity, weight loss, failure to groom, and splenomegaly. Premorbid animals were sacrificed and relevant tissues were harvested and analyzed by flow cytometry and histopathology.
Immunofluorescence staining
For immunofluorescence, relevant leukemic cell populations were sorted, cytospun and fixed in 4% paraformaldehyde for 5 minutes. Samples were then blocked using 20% normal donkey serum in PBS with 0.1% Tween 20, and stained at 4°C overnight with an antibody followed by Alexafluor-conjugated secondary antibody (Molecular probe) and DAPI. Slides were mounted using mounting media (Fluoromount-G SouthernBiotech) and viewed on the Axio Imager (Zeiss). Antibodies used were as follows: anti-Numb, Ab4147 (Abcam) or C29G11 (Cell Signaling Technology); anti-p53, DO-1 (Thermo Scientific); anti-cleaved Notch1, Val1744 (Cell Signaling). Fluorescence intensity was analyzed using Metamorph software (Molecular Devices).
Chromatin immunoprecipitation (ChIP) assays
To identify potential HOX binding sites in the Msi2 gene upstream promoter region, we employed an algorithm ConCise Scanner34 using a combination of the following matrices V$HOXA9.01, V$HOXB9.01, V$PBX_HOXA9.01, V$HOX_PBX.01, V$MEIS1A_HOXA9.01 and V$MEIS1B_HOXA9.01. The myeloid leukemia cell line M1 was maintained in RPMI1640 media supplemented with 10% fetal bovine serum, and 1×107 cells were subjected to DNA-protein cross-linking. ChIP assays were performed according to a modified protocol based on ChIP-IT Express kit (Active Motif). PCR primer sequences are as follows; for Msi2 (−5.7kb site), 5'-TGGACAGCCTCATCCACAGAGCA-3' and 5'-ACTGTGCTACATTCCCAGCCGCT-3'; for Msi2 (+110kb site), 5'-GTTCTTAGCTGCCTCTCTCAGA-3' and 5'-GAACAATGTCTCTGTCAGGCCT-3'; for Flt3, 5'-AGTCAGAAGGGACTGGCTCC-3' and 5'-GAGTGCTGCTTAGCAGATTACC-3'.
β-galactosidase reporter gene assays
KLS cells were isolated from Msi2 genetrap heterozygote bone marrow and infected with MSCV-BCR-ABL-IRES-YFP and MSCV-NUP98-HOXA9-IRES-GFP. GFP and YFP double positive cells were sorted 48 hours after infection, and cultured in X-Vivo15 media supplemented with SCF and TPO as described. After 4 days, cells were harvested in reporter lysis reagent, and β-galactosidase activities were analyzed by using β-Gal reporter gene assay, Chemiluminescent (Roche Diagnostics).
Realtime and standard RT-PCR analysis
RNA was isolated using RNAqueous-Micro (Ambion), equal amounts of RNAs were converted to cDNA using Superscript II reverse transcriptase (Invitrogen). Quantitative realtime PCRs were performed using an iCycler (BioRad) by mixing cDNAs, iQ SYBRGreen Supermix (BioRad) and gene specific primers. Results were normalized to the level of beta-2 microglobulin (B2m, mouse) or beta-actin (ACTB, human). Primer sequences are as follows: Numb-F, ATGAGTTGCCTTCCACTATGCAG; Numb-R, TGCTGAAGGCACTGGTGATCTGG; Msi1-F, ATGGATGCCTTCATGCTGGGT; Msi1-R, CTCCGCTCTACACGGAATTCG; Msi2-F, TGCCATACACCATGGATGCGT; Msi2-R, GTAGCCTCTGCCATAGGTTGC; B2m-F, ACCGGCCTGTATGCTATCCAGAA; B2m-R, AATGTGAGGCGGGTGGAACTGT; MSI2-F, GTTATCTGCGAACACAGTAGTG; MSI2-R, ACCCTCTGTGCCTGTTGGTAG; ACTB-F, AAGCCACCCCACTTCTCTCTAA; ACTB-R, AATGCTATCACCTCCCCTGTGT. Human HES1 (Hs00172878_m1) and TRIB2 (Hs00222224_m1) gene levels were analyzed with TaqMan Gene Expression Assays.
Human leukemia specimens and microarray gene expression studies
Chronic and blast crisis CML samples were obtained from the Korean Leukemia Bank (Seoul, Korea), the Hammersmith MRD Lab Sample Archive (London, United Kingdom), the Fred Hutchinson Cancer Research Center (Seattle, United States) and the Singapore General Hospital (Singapore) from Institutional Review Board approved protocols with written informed consent in accordance with the Declaration of Helsinki. Gene expression profiles of CML patient samples have been described previously35. This published data set has been reanalyzed to examine expression of MSI2, NUMB, HES1, and HOXA9 in bone marrow and peripheral blood samples from 42 chronic phase, 17 accelerated phase and 31 blast crisis CML patients. The procedures for RNA extraction, amplification, labeling, and hybridization, as well as statistical analysis methods for the Rosetta platform are as previously published35. GenePlus™ software (Enodar Biologic, Seattle, WA) was used to determine differential expression between groups (i.e. by disease phase); p-values were calculated using gene-by-gene ANOVA and estimating equation techniques were used to calculate the number of false discoveries (NFD)36.
Statistical analysis
The statistical analysis was carried out using the R language version 2.6.2 (http://www.r-project.org/) and GraphPad Prism software version 4.0c (GraphPad software Inc.).
Supplementary Material
Acknowledgements
We thank Ann Marie Pendergast, John Chute, Koji Itahana, Luiz Penalva and Lee Grimes for advice and reagents, Ken-ichi Yamamura for the Msi2 genetrap mice, Tasuku Honjo for the Rbpj conditional mice, Nicholas Gaiano for the TNR mice, David Baltimore for the lentiviral shRNA constructs and Anthony Means and Brigid Hogan for comments on the manuscript. We also thank Mike Cook, Beth Harvat and Lynn Martinek for cell sorting, Mark Fereshteh for advice on analysis of patient samples, Donald McDonnell and Hillary Wade for advice on ChIP experiments, and Alan Chen and Sam Honeycutt for excellent technical help. The BCR-ABL construct was a gift from Warren Pear and the NUP98-HOXA9 construct a gift from Gary Gilliland. TI is the recipient of a postdoctoral fellowship from the Astellas Foundation for Research on Metabolic Disorders, KLC is the recipient of an American Heart Association predoctoral award, BZ received support from T32 GM007184-33 and TR is the recipient of a Leukemia and Lymphoma Society Scholar Award. This work was also supported by an LLS Translational Research grant, an ASH Junior Faculty Award to VGO, as well as NIH grants CA18029 to JPR, CA140371 to VGO, CA122206 to C.T.J. and DK63031, DK072234, AI067798, HL097767, DP1OD006430 and an Alexander and Margaret Stewart Fund grant to TR. We are grateful for the support received from the Lisa Stafford Research Prize and dedicate this work to the Stafford family and to the memory of Lisa Stafford.
Footnotes
Reprints and permissions information is available at npg.nature.com/reprint and permissions. The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com.
References
- 1.Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–4022. doi: 10.1182/blood-2003-12-4111. [DOI] [PubMed] [Google Scholar]
- 2.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 3.Pear WS, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 4.Uemura T, Shepherd S, Ackerman L, Jan LY, Jan YN. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell. 1989;58:349–360. doi: 10.1016/0092-8674(89)90849-0. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura M, Okano H, Blendy JA, Montell C. Musashi, a neural RNA-binding protein required for Drosophila adult external sensory organ development. Neuron. 1994;13:67–81. doi: 10.1016/0896-6273(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 6.Mayotte N, Roy DC, Yao J, Kroon E, Sauvageau G. Oncogenic interaction between BCR-ABL and NUP98-HOXA9 demonstrated by the use of an in vitro purging culture system. Blood. 2002;100:4177–4184. doi: 10.1182/blood-2002-04-1244. [DOI] [PubMed] [Google Scholar]
- 7.Dash AB, et al. A murine model of CML blast crisis induced by cooperation between BCR/ABL and NUP98/HOXA9. Proc Natl Acad Sci U S A. 2002;99:7622–7627. doi: 10.1073/pnas.102583199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witte O. The role of Bcr-Abl in chronic myeloid leukemia and stem cell biology. Semin Hematol. 2001;38:3–8. doi: 10.1016/s0037-1963(01)90111-8. [DOI] [PubMed] [Google Scholar]
- 9.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 10.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 11.Goldman JM, Melo JV. BCR-ABL in chronic myelogenous leukemia--how does it work? Acta Haematol. 2008;119:212–217. doi: 10.1159/000140633. [DOI] [PubMed] [Google Scholar]
- 12.Knoblich JA. Mechanisms of asymmetric cell division during animal development. Curr Opin Cell Biol. 1997;9:833–841. doi: 10.1016/s0955-0674(97)80085-3. [DOI] [PubMed] [Google Scholar]
- 13.Spana EP, Doe CQ. Numb antagonizes Notch signaling to specify sibling neuron cell fates. Neuron. 1996;17:21–26. doi: 10.1016/s0896-6273(00)80277-9. [DOI] [PubMed] [Google Scholar]
- 14.Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- 15.Wu M, et al. Imaging hematopoietic precursor division in real time. Cell Stem Cell. 2007;1:541–554. doi: 10.1016/j.stem.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Ouyang Y, Somers WG, Chia W, Lu B. Polo inhibits progenitor self-renewal and regulates Numb asymmetry by phosphorylating Pon. Nature. 2007;449:96–100. doi: 10.1038/nature06056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neering SJ, et al. Leukemia stem cells in a genetically defined murine model of blast-crisis CML. Blood. 2007;110:2578–2585. doi: 10.1182/blood-2007-02-073031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Justice N, Roegiers F, Jan LY, Jan YN. Lethal giant larvae acts together with numb in notch inhibition and cell fate specification in the Drosophila adult sensory organ precursor lineage. Curr Biol. 2003;13:778–783. doi: 10.1016/s0960-9822(03)00288-4. [DOI] [PubMed] [Google Scholar]
- 19.Wakamatsu Y, Maynard TM, Jones SU, Weston JA. NUMB localizes in the basal cortex of mitotic avian neuroepithelial cells and modulates neuronal differentiation by binding to NOTCH-1. Neuron. 1999;23:71–81. doi: 10.1016/s0896-6273(00)80754-0. [DOI] [PubMed] [Google Scholar]
- 20.Colaluca IN, et al. NUMB controls p53 tumour suppressor activity. Nature. 2008;451:76–80. doi: 10.1038/nature06412. [DOI] [PubMed] [Google Scholar]
- 21.Imai T, et al. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol Cell Biol. 2001;21:3888–3900. doi: 10.1128/MCB.21.12.3888-3900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okabe M, Imai T, Kurusu M, Hiromi Y, Okano H. Translational repression determines a neuronal potential in Drosophila asymmetric cell division. Nature. 2001;411:94–98. doi: 10.1038/35075094. [DOI] [PubMed] [Google Scholar]
- 23.Sakakibara S, et al. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci U S A. 2002;99:15194–15199. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okano H, et al. Function of RNA-binding protein Musashi-1 in stem cells. Exp Cell Res. 2005;306:349–356. doi: 10.1016/j.yexcr.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 25.Taniwaki T, et al. Characterization of an exchangeable gene trap using pU-17 carrying a stop codon-beta geo cassette. Dev Growth Differ. 2005;47:163–172. doi: 10.1111/j.1440-169X.2005.00792.x. [DOI] [PubMed] [Google Scholar]
- 26.Radich JP, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahara F, et al. Hes1 immortalizes committed progenitors and plays a role in blast crisis transition in chronic myelogenous leukemia. Blood. 2010;115:2872–2881. doi: 10.1182/blood-2009-05-222836. [DOI] [PubMed] [Google Scholar]
- 28.Battelli C, Nikopoulos GN, Mitchell JG, Verdi JM. The RNA-binding protein Musashi-1 regulates neural development through the translational repression of p21WAF-1. Mol Cell Neurosci. 2006;31:85–96. doi: 10.1016/j.mcn.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 29.Liu G, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pece S, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao C, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12:528–541. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han H, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14:637–645. doi: 10.1093/intimm/dxf030. [DOI] [PubMed] [Google Scholar]
- 33.Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jegga AG, et al. Detection and visualization of compositionally similar cis-regulatory element clusters in orthologous and coordinately controlled genes. Genome Res. 2002;12:1408–1417. doi: 10.1101/gr.255002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radich JP, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao LP, Prentice R, Breeden L. Statistical modeling of large microarray data sets to identify stimulus-response profiles. Proc Natl Acad Sci U S A. 2001;98:5631–5636. doi: 10.1073/pnas.101013198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.