

Abstract
Uroporphyrinogen decarboxylase (URO-D), an essential enzyme that functions in the heme biosynthetic pathway, catalyzes decarboxylation of all four acetate groups of uroporphyrinogen to form coproporphyrinogen. Here we report crystal structures of URO-D in complex with the I and III isomer coproporphyrinogen products. Crystallization required use of a novel enzymatic approach to generate the highly oxygen-sensitive porphyrinogen substrate in situ. The tetrapyrrole product adopts a domed conformation that lies against a collar of conserved hydrophobic residues and allows formation of hydrogen bonding interactions between a carboxylate oxygen atom of the invariant Asp86 residue and the pyrrole NH groups. Structural and biochemical analyses of URO-D proteins mutated at Asp86 support the conclusion that this residue makes important contributions to binding and likely promotes catalysis by stabilizing a positive charge on a reaction intermediate. The central coordination geometry of Asp86 allows the initial substrates and the various partially decarboxylated intermediates to be bound with equivalent activating interactions, and thereby explains how all four of the substrate acetate groups can be decarboxylated at the same catalytic center.
Keywords: crystallography/enzyme mechanism/enzyme product complex/protein structure/uroporphyrinogen decarboxylase
Introduction
Uroporphyrinogen decarboxylase (URO-D) catalyzes the fifth step in the heme biosynthetic pathway (Kappas et al., 1995), which is the sequential elimination of carboxyl groups from each of the four acetate side chains of the uroporphyrinogen-III substrate to yield the coproporphyrinogen-III product. This activity is essential for life (Phillips et al., 2001) and deficiency leads to the most common form of porphyria in humans, porphyria cutanea tarda (PCT), which occurs with an estimated frequency of 1 in 20 000 Caucasians (Kappas et al., 1995). PCT results from accumulation of uroporphyrin and partially decarboxylated intermediate porphyrins in the liver, plasma and skin, and is characterized by hyperpigmentation, severe photosensitive dermatosis, and hypertrichosis (Wyckoff and Kushner, 1994; Kappas et al., 1995). Familial PCT is transmitted as an autosomal dominant trait caused by heterozygosity for mutations affecting the URO-D gene, resulting in approximately half normal URO-D activity in all tissues (Kushner et al., 1976; Held et al., 1989).
The two steps preceding URO-D in heme biosynthesis are the polymerization of four porphobilinogen (PBG) molecules by porphobilinogen deaminase (PBG-D) to generate a linear tetrapyrrole, hydroxymethylbilane, followed by cyclization with inversion of the D pyrrole ring by uroporphyrinogen III synthase (U3S) to yield uroporphyrinogen-III (Figure 1). In the absence of U3S, spontaneous cyclization of hydroxymethylbilane occurs to give the symmetric uroporphyrinogen-I isomer. Uroporphy rinogen-III differs from the symmetric I isomer in having reversed positions for propionate and acetate side chains on the D pyrrole ring (Figure 1). Both I and III isomers of uroporphyrinogen are decarboxylated by URO-D, although downstream enzymes are unable to utilize the coproporphyrinogen-I isomer product, which is normally excreted in urine and stool.

Fig. 1. Reaction catalyzed by URO-D and the two preceding steps in the heme biosynthesis pathway. Acetate, propionate and methyl side-chains are denoted A, P and M, respectively. Porphobilinogen deaminase (PBG-D), uroporphyrinogen III synthase (U3S). In the absence of U3S, hydroxymethylbilane cyclizes without inversion of the D-ring, to form uroporphyrinogen I, which differs from the III-isomer shown here by having an identical arrangement of A/P substituents on all four pyrrole rings. Figures 1 and 4 were generated using ChemDraw Pro (CambridgeSoft Corp., Cambridge, MA).
The URO-D mechanism has a number of remarkable features. Unlike most other decarboxylases, URO-D functions independently of a prosthetic group or cofactor (Straka and Kushner, 1983). URO-D also has several substrates, namely the initial uroporphyrinogen-I and -III isomer substrates and the three partially decarboxylated intermediates of each isomer. URO-D is dimeric in solution (Phillips et al., 1997) and the juxtaposition of active-site clefts from each subunit within the dimer suggests the possibility of collaboration between adjacent active sites (Whitby et al., 1998; Martins et al., 2001).
The crystal structure of recombinant human URO-D revealed that the ∼40 kDa protein monomer is comprised of a single domain containing an α/β-barrel with a deep active-site cleft formed by loops at the C-terminal ends of the barrel strands (Whitby et al., 1998). An essentially identical structure was seen for the enzyme from Nicotiana tabacum (Martins et al., 2001). This architecture is consistent with the presence of a single catalytic center, as suggested by stereospecific labeling experiments which show that the chirality of all four acetate Cα atoms is conserved during the decarboxylation reactions (Bernard and Akhtar, 1979).
In an effort to understand the mechanism of substrate binding and catalysis we have determined the crystal structure of URO-D in complex with both I and III isomer coproporphyrinogen products. These are the first structures of a protein complex with a porphyrinogen. They reveal a novel mode of binding, including use of an invariant aspartate, Asp86, to coordinate the central pyrrole NH groups. We have also confirmed the importance of Asp86 by biochemical and structural analyses of URO-D proteins mutated at this residue.
Results and discussion
Structure determination
Crystallization of recombinant human URO-D in complex with product ligands required use of an anaerobic chamber and purging of solutions with argon to circumvent the extreme oxygen sensitivity of cyclized tetrapyrroles such as the uroporphyrinogen substrate and coproporphyrinogen product. Initial efforts with either substrate or product prepared by chemical reduction of the oxidized uroporphyrin and coproporphyrin were unsuccessful, probably because of the high pH that results from sodium amalgam-mediated reduction of the porphyrin. An approach was therefore developed that generated the porphyrinogen URO-D substrates in situ by incubation of the upstream substrate PBG with purified recombinant PBG-D enzyme to yield a solution of hydroxymethylbilane that non-enzymatically cyclizes to uroporphyrinogen-I (Figure 1). The same reaction mixture supplemented with purified recombinant U3S enzyme converts the hydroxymethylbilane to uroporphyrinogen-III. These substrate mixtures were added directly to the crystallization solutions to yield crystals of ligand-bound enzyme complexes. It was subsequently determined crystallographically that the bound ligands are actually the corresponding coproporphyrinogen-I or -III products, which are generated by the catalytic activity of URO-D during crystallization.
The URO-D-product co-crystals were isomorphous with the previously determined unliganded structure (Whitby et al., 1998) and were refined at resolutions of 1.7 Å (I isomer) and 1.75 Å (III isomer) to free R-factors of 19.2% and 18.6%, respectively. The URO-D protein structure is essentially identical in the two product complex structures, and is unchanged upon coordinating product, with least squares overlap of the unliganded and wild-type product complexes giving root mean square deviations (r.m.s.ds) of 0.24 Å (I isomer) and 0.23 Å (III isomer) for 354/356 Cα atoms. These structures are also very similar to that of unliganded tobacco URO-D [Protein Data Base (PDB) accession code 1J93] (Martins et al., 2001), which can be superimposed on the human URO-D structures with an r.m.s.d. of 1.63 Å (unliganded) and 1.61 Å/1.63 Å (I/III isomer product complexes) for 317 out of 356 Cα atoms. As described below, we have also determined structures of several mutant URO-D proteins with or without bound product. In all cases these structures were determined at similar resolutions and free R-factors as the wild-type protein, and all of these structures show a similar high degree of structural similarity.
Coordination of product
The I and III isomer products occupy essentially identical positions within the active-site cleft and make equivalent contacts with the protein, with one face buried against a hydrophobic wall of the active-site cleft and one edge buried into the base of the cleft (Figure 2). Even the D pyrrole ring, which has reversed positions for the propionate side chains in the I and III isomers, occupies essentially identical positions in the two structures. The different locations of propionates on the D-rings of I and III isomers are accommodated by altered interactions with protein groups, including Arg41 and Arg50. In general, the products’ various propionate groups are often partially exposed to solvent and usually also make favorable hydrogen-bonding interactions with the protein.




Fig. 2. Structure of the URO-D–product complexes. (A) Stereoview ribbon representation of the dimer, viewed down the 2-fold axis between adjacent monomers. Monomer 1 is blue with an orange beta barrel and a purple flexible loop and flanking beta strands. Monomer 2 is yellow. Coproporphyrinogen-III product is green. Coproporphyrinogen-I (not shown) overlaps almost exactly with the III-isomer. This figure was generated using Molscript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997). (B) Sequence of human URO-D. The additional 21 residues, including a 10-histidine tag, that were inserted before Met1 are not shown explicitly in the figure, although they are present in all the proteins used for structure determination and activity assays performed in this study. The secondary structure shown above has the same color scheme as (A). Disordered residues (at N- and C-termini) are indicated with a thin line. Residues that are invariant in an alignment of 39 sequences (data not shown) are shown on a yellow background. Asp86 is indicated with a red triangle. Residues that contact the product ligand, i.e. have at least one atom within 4.0 Å of the product, are indicated below according to the color scheme indicated. (C) Electron density around the product. Stereoview of the sigmaA-weighted 2Fo–Fc map shown contoured at 1 × r.m.s.d. around the I-isomer product bound to wild-type URO-D. (D) Electrostatic potential surface at the URO-D active site. Stereoview colored on a red/white/blue scale of –0.3/0/+0.3 kT. Coproporphyrinogen-III is shown in green. Hydrogen bonds between the tetrapyrrole NH groups and Asp Oδ1 are indicated with dashed purple lines. (C) and (D) were generated using SwissPDBviewer (http://www.expasy.org/spdbv/) (Guex and Peitsch, 1997).
Although the product binds in a deep active-site cleft, binding of substrate and release of product do not appear to require significant conformational changes of the protein. A relatively mobile loop (residues 100–106), which adopts the same conformation and similar thermal parameters in the unliganded and product-bound structures, partially covers the opening to the active site (Figure 2A). The narrowest separation between atomic centers of URO-D atoms across the opening to the active site is ∼9 Å and a minor change in the conformation of the mobile loop would increase this distance, suggesting that binding and release of the flexible substrate and product occur with only minimal changes in URO-D conformation.
URO-D has a significant overall negative charge (43 Asp/Glu and 36 Arg/Lys per monomer). Consistent with the negative charge of URO-D substrates, however, electrostatic potential at the active site is relatively positive (Figure 2D). This results in large part from a number of arginine side chains, including Arg37, Arg41 and Arg50, which are invariant or highly conserved and generally coordinate the products’ propionate groups. These side chains display flexibility in their interactions with product, and in many cases occupy different conformations between the I and III isomer complexes or display alternative, partially occupied conformations. It appears, therefore, that positively charged side chains contribute to binding the negatively charged substrates by locally ameliorating some of the protein’s overall negative electrostatic potential, but do not make a major contribution to precisely orienting the substrate on the enzyme surface (Figure 2D).
Role of Asp86 in binding and catalysis
Extensive interactions are formed between one face of the tetrapyrrole product and a ring of invariant or conserved hydrophobic side chains of URO-D (Figures 2B, and 3A and B). About half of URO-D’s invariant residues contact product, while all the other invariant residues perform obvious structural roles. The enzyme surface molds the tetrapyrrole into a highly non-planar, domed conformation that presents the central tetrapyrrole amides for hydrogen-bonding interactions with the Oδ1 atom of the invariant residue Asp86. This residue provides the only negatively charged side chain in the URO-D active-site cleft, and is held in a precise orientation by a hydrogen bond between the Oδ2 atom and the main chain NH of Leu88. Three of the pyrrole units are tilted approximately 35–60° from the plane defined by the four central pyrrole nitrogen atoms, with the result that their NH groups point directly at Asp86 Oδ1 for optimal hydrogen-bonding geometry (Table I). In contrast, the C pyrrole ring (Figure 3A and B) lies approximately in the plane of the central nitrogen atoms, with resulting suboptimal hydrogen bonding geometry and long (3.5 Å) N–Oδ1 distance.



Fig. 3. Interactions at the active site. (A) Uroporphyrinogen I product (green), wild-type URO-D (yellow). Protein residues are shown if one atom from the residue lies within 4.0 Å of the product in at least one of the structures. Also shown is Leu88. The pyrrole rings are denoted A, B, C and D, with the D-ring being the site where acetate and propionate groups are reversed in the III-isomer product. Van der Waals’ surfaces are shown around the protein atoms, with residues nearer the viewer given a more transparent surface. Hydrogen bonds are indicated with dashed lines. The apparent hydrogen bond seen between Ala39 O and the C-ring propionate indicates that this carboxylate is protonated. (B) Same as (A), but for the III-isomer product complex. The I- and III-isomer products superimpose very closely following overlap on the protein Cα atoms. The major differences are the conformations of Arg37 and Arg 41 side chains. (C) Comparison of product bound to various URO-D variants. Structures were superimposed by overlap on the protein Cα atoms. The water molecules that lie roughly in the position of the wild-type Asp86 side chains are shown explicitly. Color scheme is indicated and is the same as in Figure 2B. This figure was generated using Molscript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997).
Table I. Geometry of pyrrole coordination by Asp86.
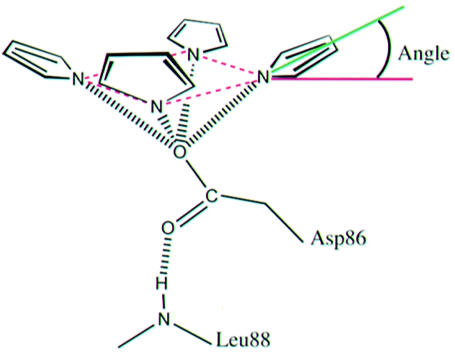
As illustrated in the figure, distances are between pyrrole N atoms and Asp86 Oδ1 (or Glu86 Oε1 or equivalent water molecule in Asp86Glu/Gly proteins). Angles are between the plane of the four tetrapyrrole N atoms (dashed purple lines) and a line that passes through the center of the pyrrole ring and the pyrrole N atom (green line). Note that an angle of ∼45° corresponds to a linear NH–O hydrogen bond. The tetrapyrrole rings are designated A, B, C and D, as shown in Figure 3A.
Coordination of the pyrrole NH groups by Asp86 is consistent with a mechanism of catalysis that proceeds through protonation of a pyrrole Cα atom (Figure 4). In this scheme, the negatively charged aspartate would stabilize the positive charge of the protonated pyrrole, which can be delocalized onto the pyrrole nitrogen. The protonated pyrrole is an attractive hypothetical intermediate because it could serve as an ‘electron sink’, in a manner analogous to the well known reaction of pyridoxal 5′-phosphate-dependent decarboxylases by accepting the electron retained when the acetate carboxylate is released as carbon dioxide (Boeker and Snell, 1972; Akhtar, 1994). The final steps in this reaction scheme are protonation of the methylene carbon and deprotonation of the pyrrole Cα. An analogous role for an aspartate in stabilizing positive charge on a pyrrole NH appears to be important for the catalytic mechanism of porphobilinogen deaminase (Woodcock and Jordan, 1994). A URO-D reaction pathway via a protonated intermediate was suggested previously (Akhtar, 1994) and was discussed in conjunction with modeling calculations using the unliganded tobacco URO-D structure (Martins et al., 2001), although the role now implied for Asp86 by the product complex crystal structures was not suggested earlier and the geometry of ligand binding is very different from the previous proposal (Martins et al., 2001).

Fig. 4. Proposed reaction pathway. The second figure in the series shows how the positive charge from protonation can be delocalized to the pyrrole nitrogen and explains how Asp86 promotes formation of the proposed protonated intermediate. The positively charged intermediate serves as an electron sink for release of the acetate carboxylate as carbon dioxide. Two protons are introduced in this proposed pathway, in the first and third panels.
Structure and activity of Asp86 mutants
In order to probe the importance of Asp86 for binding and catalysis further, we have performed structural and biochemical studies on the Asp86Gly, Asp86Asn and Asp86Glu URO-D proteins (Tables I–III) (Figure 3C). As expected, mutation of Asp86 to asparagine resulted in very low activity (0.1–0.6% of wild type), and efforts to obtain product co-crystals yielded URO-D structures that had empty active-site clefts. The inability of this mutant protein to bind substrate/product with detectable occupancy, despite structures that are essentially identical to wild type, is explained by the loss of favorable interactions with Asp86. In the case of Asp86Asn, the hydrogen bond between the side chain oxygen and the main chain NH of Leu88 (also seen in the wild-type structures) dictates that the Asn86 Nδ2 atom is in the same place as Asp86 Oδ1 of wild-type URO-D, thereby introducing a repulsive interaction with the pyrrole NH groups.
Table III. Crystallographic data and refinement.
| URO-D (product) | URO-D (I) | URO-D (III) | D86N | D86G (I) | D86G (III) | D86E (I) | Y164F (III) |
|---|---|---|---|---|---|---|---|
| Crystallographic data | |||||||
| Wavelength (Å) | 1.10 | 1.10 | 1.5418 | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Data collection a | ALS | ALS | RA | RA | RA | RA | RA |
| Resolution (Å) | 30.0–1.70 (1.76–1.70) | 30.0–1.75 (1.81–1.75) | 20.0–1.85 (1.92–1.85) | 30.0–1.65 (1.71–1.65) | 30.0–1.70 (1.76–1.70) | 20.0–1.90 (1.97–1.90) | 20.0–1.70 (1.76–1.70) |
| No. of reflections measured | 270 866 | 225 047 | 175 858 | 260 521 | 230 692 | 218 142 | 290 721 |
| No. of unique reflections | 50 593 | 45 954 | 36 928 | 51 852 | 47 218 | 35 021 | 49 087 |
| Complete (%) | 99.6 (99.8) | 99.8 (99.8) | 96.0 (79.8) | 96.6 (75.2) | 95.7 (87.5) | 98.2 (95.2) | 98.9 (99.2) |
| <I/σI> | 13 (2.8) | 11 (2.8) | 9 (1.9) | 10 (1.8) | 13 (2.7) | 9 (2.5) | 12 (1.8) |
| Mosaicity (°) | 0.25 | 0.47 | 0.83 | 0.86 | 0.49 | 0.36 | 0.64 |
| Rsymb (%) | 0.092 (0.485) | 0.091 (0.423) | 0.083 (0.437) | 0.072 (0.236) | 0.096 (0.232) | 0.134 (0.399) | 0.071 (0.390) |
| Refinement statistics | |||||||
| Resolution (Å) | 30.0–1.700 (1.79–1.70) | 30.0–1.75 (1.85–1.75) | 20.0–1.85 (1.95–1.85) | 30.0–1.65 (1.74–1.65) | 30.0–1.70 (1.79–1.70) | 20.0–1.90 (2.00–1.90) | 20.0–1.70 (1.79–1.70) |
| Rcrystd (%) | 0.167 (0.217) | 0.158 (0.228) | 0.161 (0.254) | 0.163 (0.318) | 0.169 (0.147) | 0.151 (0.218) | 0.157 (0.220) |
| Rfreee (%) | 0.192 (0.275) | 0.186 (0.265) | 0.192 (0.290) | 0.190 (0.312) | 0.200 (0.226) | 0.187 (0.281) | 0.188 (0.245) |
| Protein residuesc | 10–366 | 11–366 | 11–366 | 11–366 | 11–366 | 10–366 | 11–366 |
| No. of water molecules | 326 | 329 | 381 | 384 | 361 | 387 | 359 |
| No. of Cop’gen mols | 1 | 1 | – | 1 | 1 | 1 | 1 |
| No. of CO2/BME mols | 1 CO2 | – | – | – | 1 BME | ||
| R.m.s.d. bonds | 0.016 | 0.016 | 0.020 | 0.014 | 0.015 | 0.017 | 0.015 |
| R.m.s.d. angles | 1.633 | 1.627 | 1.690 | 1.845 | 1.547 | 1.630 | 1.503 |
| φ/ψ anglesf | |||||||
| Most favored (%) | 95.7 | 95.3 | 94.0 | 94.7 | 94.7 | 94.4 | 94.0 |
| Additional allowed (%) | 4.3 | 4.7 | 6.0 | 5.3 | 5.3 | 5.6 | 6.0 |
| <B>protein (Å2) | 28.2 | 27.1 | 22.5 | 25.2 | 24.6 | 18.4 | 26.2 |
| <B>main-chain (Å2) | 26.4 | 25.2 | 21.0 | 23.7 | 23.3 | 16.9 | 24.8 |
| <B>water (Å2) | 41.9 | 41.2 | 38.4 | 39.2 | 38.9 | 31.5 | 38.5 |
| <B>Cop’gen (Å2) | 30.9 | 33.3 | – | 69.0 | 53.6 | 26.7 | 51.9 |
| <B>CO2/BME (Å2) | 45.2 | – | – | – | 42.4 | ||
All structures are isomorphous with the unliganded wild-type crystal structure (Whitby et al., 1998). Values in parentheses refer to the high-resolution shell. Cop’gen (coproporphyrinogen), BME (β-mercaptoethanol).
aData were collected at the ALS synchrotron, beamline 5.0.2 on a Quantum IV detector (ALS) or on a Rotating Anode source with an Raxis-IV detector (RA).
bRsym = Σ|I–<I>|/ΣI, where I is the intensity of an individual measurement and <I> is the corresponding mean value.
cFor each structure, eight to 21 of the residues were modeled in two alternative 0.5 occupied conformations.
dRcryst = Σ||Fo|–|Fc||/Σ|Fo|, where |Fo| is the observed and |Fc| the calculated structure factor amplitude.
eRfree is the same as Rcryst calculated with a randomly selected test set of reflections that were never used in refinement calculations. In each case the free set included 1122–1404 reflections, corresponding to 2.5–4.0% of the unique data.
fFor non-Gly and non-Pro residues only.
Despite its low (0.1–2.4%) activity, Asp86Gly URO-D binds the I- and III-isomer products with similar geometry as wild type, although in these structures the density for the ligands is much less well defined than for the other URO-D product complexes, and the III isomer is displaced ∼0.5 Å from the wild-type position and shows altered coordination geometry (Figure 3C and Table I). Binding of product is accommodated by replacement in the structure of the Asp86 side chain with two water molecules, one of which lies 0.6/1.1 Å (I/III isomers) from the Asp86 Oδ1 position and makes similar contacts to the tetrapyrrole NH groups. These observations support the model stating that, in addition to its role in binding, Asp86 also plays a direct role in catalysis through electrostatic stabilization of a protonated intermediate.
The Asp86Glu URO-D showed approximately 5–10% activity and, although we did not obtain crystals of the III-isomer product complex, the I-isomer product structure is very similar to the wild-type product complexes, with the Glu86 side chain adopting a conformation that preserves the hydrogen bond between Oε2 and the main chain NH of Leu88, and positions the Oε1 atom only 1.1/1.3 Å (I/III isomer) from the wild-type Oδ1 of Asp86. The tetrapyrrole adjusts primarily by a translation equal to that of the Asp86 Oδ1–Glu86 Oε1 displacement. Consequently, the interactions of carboxylate and tetrapyrrole are preserved, with similar hydrogen-bonding interactions with Glu86 Oε1 and pyrrole angles with respect to the plane of pyrrole nitrogen atoms. The reduced activity, despite maintenance of coordination geometry about the carboxylate of residue 86, indicates that this does not provide all of the interactions necessary for optimal catalysis. Possible explanations for the diminished activity include reduced binding affinity due to suboptimal interactions of the translated tetrapyrrole, for example with the surrounding hydrophobic residues, or distortion of the sites required for protonation of the pyrrole Cα atom or decarboxylation of the acetates.
Possible sites of decarboxylation
Although the product complex structures indicate a role for Asp86 in coordination and stabilization of an intermediate, no protein groups appear well positioned to serve as proton donor for any of the four pyrrole rings, suggesting that the proton may be transferred from solvent. It is also currently unclear which of the pyrrole rings of the product complex structures occupies the site where decarboxylation takes place. The conservation of Tyr164 throughout evolution suggested that this residue might play a critical role in catalysis (Whitby et al., 1998; Martins et al., 2001), with one possibility being to facilitate decarboxylation in the site occupied by the product B ring pyrrole (Figure 3A and B). The product methyl lies just 2.9/3.3 Å (I/III isomer) from the phenolic oxygen of Tyr164, which is therefore a candidate to protonate the methyl carbon that is generated by decarboxylation of the acetate. A role had also been suggested for this residue in performing the initial protonation (Martins et al., 2001), although this possibility does not seem consistent with the product complex crystal structures. Tyr164 is not critical for catalysis, however, since mutation to phenylalanine retains approximately 25–30% activity (Table II). One possibility is that Tyr164 does make direct contributions to the decarboxylation step, but that catalysis is primarily driven by Asp86-promoted formation of the proposed protonated intermediate.
Table II. Activity of URO-D mutants.
| Protein | Activity(%)a |
|
|---|---|---|
| I-isomer | III-isomer | |
| Asp86Gly | 2.4 | 0.1 |
| Asp86Asn | 0.6 | 0.1 |
| Asp86Glu | 9.2 | 5.8 |
| Tyr164Phe | 29.1 | 24.3 |
aActivity is expressed as a percentage of the wild-type protein (see Materials and methods).
An alternative potential decarboxylation site is occupied by the C ring pyrrole. Simple modeling suggests that the acetate in this position may occupy a restricted environment that is relatively hydrophobic. Thus, one possibility is that the energy of substrate binding is used to drive this acetate into a more hydrophobic environment, thereby promoting conversion to the hydrophobic carbon dioxide product. An analogous ground state destabilization or ‘Circe-effect’ mechanism has been proposed for other decarboxylases (Gallagher et al., 1989; Grishin et al., 1999), including orotidine 5′-monophosphate decarboxylase (Appleby et al., 2000; Feng et al., 2000; Wu et al., 2000), although this interpretation has been disputed (Warshel et al., 2001). Unfortunately, a mutagenic approach to testing this possible decarboxylation site is likely to be complicated by the apparent role of the surrounding hydrophobic residues in stabilizing the protein fold. In view of the finding that decarboxylation of the pyrrole ring acetates occurs in the sequence D, A, B, C (Jackson et al., 1976), it is notable that this possible decarboxylation site is occupied by the product C-ring, i.e. by the last pyrrole to be decarboxylated.
Curiously, the wild-type I-isomer product structure displays electron density above the tetrapyrrole that has the appearance of a bound carbon dioxide molecule. While it is provocative to find this feature suggestive of a reaction product, it is not clear how one of the acetates could be decarboxylated by displacement of CO2 into this position, and this feature is not seen in any of the other structures. Indeed, the density is quite variable in this region, with density suggestive of a β-mercaptoethanol molecule in the Asp86Glu I-isomer product structure and the guanidinium of Arg37 approaching this site in all of the other structures.
The structures and activity assays reported here reveal a critical role for Asp86 in binding the product and, by inference, the substrate pyrrole NH groups. These interactions contribute to coordination of the substrate in a domed conformation against a hydrophobic surface of the enzyme and likely activate the substrate by stabilizing the positive charge on a protonated intermediate. Although the site of decarboxylation is not currently known, and given the dominant role of Asp86 may even occur at more than one (perhaps all four) site(s), biochemical data are most consistent with the reaction occurring at a unique site on the enzyme surface (Bernard and Akhtar, 1979). In this regard, the central binding of Asp86 with the pyrrole NH groups is attractive, since the same interactions will be made with protein as the substrate, and partially decarboxylated intermediates bind and rebind in orientations that are rotated by 90° about the contact point with Asp86. In this way, essentially identical modes of binding could be accommodated for each of the decarboxylation reactions.
Materials and methods
Reagents
Recombinant histidine-tagged human URO-D was produced in BL21-pLysS using standard growth and induction procedures. The protein was affinity purified using metal chelate affinity chromatography on Ni2+-NTA, dialyzed into 50 mM Tris pH 6.8, 10% glycerol, and concentrated for crystallization to 7–16 mg/ml (Phillips et al., 1997). Mutations were introduced into the wild-type URO-D cDNA using the pALTER (Promega, Madison, WI) site-directed mutagenesis system. Recombinant human uroporphyrinogen III synthase (U3S) and Rhodopseudomonas spheroides PBG-D used in the porphyrinogen generating reactions were expressed and purified as described previously (Phillips and Kushner, 1999; Mathews et al., 2001).
The uroporphyrinogen-I reaction solution was generated in an anaerobic chamber by adaptation of an enzymatic protocol to produce the URO-D substrate uroporphyrinogen I (Phillips and Kushner, 1999), in which 150 µg/ml PBG-D and 750 µM PBG (Porphyrin Products, Logan, UT) were mixed in a buffer of 50 mM Tris pH 7.6, 7.5 mM DTT. Incubation for 30 min then allowed non-enzymatic cyclization of hydroxymethylbilane to uroporphyrinogen-I. Uroporphyrinogen-III was generated in a similar way, but with inclusion of 30 ug/ml of U3S in the reaction buffer.
Enzyme activity assay
All assays were performed in triplicate and in parallel with wild-type protein, with enzymatic activity expressed as a percentage of wild type, as described previously (Phillips and Kushner, 1999). Briefly, 10 µl of purified recombinant URO-D at 2 µg/ml (2 ng total protein) were added to 70 µl 50 mM phosphate pH 6.8. This solution was then mixed with 120 µl of the uroporphyrinogen-I or -III isomer reaction solution (above). After incubation for 30 min at 37°C, the reaction was stopped by the addition of 200 µl of 3 N HCl and the porphyrinogens oxidized under a UV lamp for 30 min prior to HPLC separation. The HPLC peaks were assigned using porphyrin standard mix (Porphyrin Products), and enzyme activity estimated from the peak areas as described previously (Phillips and Kushner, 1999).
Crystallization
URO-D was crystallized at room temperature (∼21 °C) by sitting drop vapor diffusion as described previously (Phillips et al., 1997), with the following modifications to permit co-crystallization of URO-D product complexes. All procedures were performed in an anaerobic chamber, and all crystallization solutions were equilibrated in the oxygen-free environment for 7 days prior to use. The reservoir (500 µl) comprised 1.5 M sodium citrate, pH 6.5. The crystallization drop was prepared by mixing 5 µl of the URO-D protein solution (15–20 mg/ml) with 15 µl of the uroporphyrinogen-I or -III reaction solution (above), incubating for 5 min, and adding 8 µl of the reservoir solution. Crystallization trials remained in the anaerobic chamber during the crystallization process. Crystals typically grew in 3–10 days.
Crystallographic analysis
URO-D product complexes crystallized isomorphously with recombinant human apo enzyme. The crystals belong to the space group P3121, with one molecule in the asymmetric unit and unit cell dimensions a = b = 102.9 Å, c = 74.5 Å. For data collection, crystals were suspended in Rayon loops, transferred to 1.7 M Na citrate pH 7.0, 5% glycerol for 2 min and plunged into liquid nitrogen. Crystals were maintained at 100 K during data collection, which was performed at beamline 5.0.2 of the Advanced Light Source (ALS) at Lawrence Berkeley Laboratories or on an image plate detector mounted on a rotating anode source. Data were processed using DENZO and SCALEPACK (Otwinowski and Minor, 1997), and phased directly using a model of the URO-D apo-enzyme (PDB accession code 1URO). The model was subjected to rigid-body minimization, positional and B-factor refinement, and the resulting difference electron density maps examined for evidence of substrate or product bound to the enzyme. The protein, ligand and solvent models were subsequently improved by several rounds of model rebuilding (Jones et al., 1991) and refinement. Crystallographic refinements were performed with REFMAC (CCP4, 1994) using the maximum likelihood target function (Murshudov et al., 1997). Coordinates and diffraction data have been deposited at the PDB under accession codes 1R3Q (URO-D-I), 1R3Y (URO-D-III), 1R3R (D86N), 1R3S (D86G-I), 1R3T (D86G-III), 1R3V (D86E-I) and 1R3W (Y164F-III).
Acknowledgments
Acknowledgements
We thank Heidi Schubert for critical comments on the manuscript. We thank Gerry McDermott and the beamline personnel of ALS beamline 5.0.2 for assistance with data collection. Operations of the Advanced Light Source are supported by the U.S. Department of Energy, Office of Basic Energy Sciences and by the National Institutes of Health. This work was supported by NIH grants CA24014, DK20503, and GM56775.
References
- Akhtar M. (1994) The modification of acetate and propionate side chains during the biosynthesis of haem and chlorophylls: mechanistic and stereochemical studies. In Ciba Foundation Symposium (ed.) The Biosynthesis of the Tetrapyrrole Pigments. John Wiley and Sons Ltd, Chichester, NY, USA. Vol. 180, pp. 131–155. [DOI] [PubMed] [Google Scholar]
- Appleby T.C., Kinsland,C., Begley,T.P. and Ealick,S.E. (2000) The crystal structure and mechanism of orotidine 5′-monophosphate decarboxylase. Proc. Natl Acad. Sci. USA, 97, 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard G.F. and Akhtar,M. (1979) Stereochemical and mechanistic studies on the decarboxylation of uroporphyrinogen III in haem biosynthesis. J. C. S. Perkin I, 10, 2354–2360. [DOI] [PubMed] [Google Scholar]
- Boeker E.A. and Snell,E.E. (1972) Amino acid decarboxylases. In Boyer,P.D. (ed.) The Enzymes. Academic Press, New York, Vol. 6, pp. 217–254. [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Feng W.Y., Austin,T.J., Chew,F., Gronert,S. and Wu,W. (2000) The mechanism of orotidine 5′-monophosphate decarboxylase: catalysis by destabilization of the substrate. Biochemistry, 39, 1778–1783. [DOI] [PubMed] [Google Scholar]
- Gallagher T., Snell,E.E. and Hackert,M.L. (1989) Pyruvoyl-dependent histidine decarboxylase. Active site structure and mechanistic analysis. J. Biol. Chem., 264, 12737–12743. [PubMed] [Google Scholar]
- Grishin N.V., Osterman,A.L., Brooks,H.B., Phillips,M.A. and Goldsmith,E.J. (1999) X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with α-difluoromethylornithine. Biochemistry, 38, 15174–15184. [DOI] [PubMed] [Google Scholar]
- Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- Held J.L., Sassa,S., Kappas,A. and Harber,L.C. (1989) Erythrocyte uroporphyrinogen decarboxylase activity in porphyria cutanea tarda: a study of 40 consecutive patients. J. Invest. Dermatol., 93, 332–334. [PubMed] [Google Scholar]
- Jackson A.H., Sancovich,H.A., Ferrmola,A.M., Evans,N., Games,D.E., Matlin,S.A., Elder,G.H. and Smith,S.G. (1976) Macrocyclic intermediates in the biosynthesis of porphyrins. Phil. Trans. R. Soc. Lond. Series B, 273, 191–206. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.-Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kappas A., Sassa,S., Galbraith,R.A. and Nordmann,Y. (1995) The porphyrias. In Scriver,C.R., Beaudet,A.L., Sly,W.S. and Valle,D. (eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, pp. 2103–2160. [Google Scholar]
- Kraulis P.J. (1991) Molscript: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kushner J.P., Barbuto,A.J. and Lee,G.R. (1976) An inherited enzymatic defect in porphyria cutanea tarda: decreased uroporphyrinogen decarboxylase activity. J. Clin. Invest., 58, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins B.M., Grimm,B., Mock,H.P., Huber,R. and Messerschmidt,A. (2001) Crystal structure and substrate binding modeling of the uroporphyrinogen-III decarboxylase from nicotiana tabacum: implications for the catalytic mechanism. J. Biol. Chem., 276, 44108–44116. [DOI] [PubMed] [Google Scholar]
- Mathews M.A.A., Schubert,H.L., Whitby,F.G., Alexander,K.J., Schadick,K., Bergonia,H.A., Phillips,J.D. and Hill,C.P. (2001) Crystal structure of human uroporphyrinogen III synthase. EMBO J., 20, 5832–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. In Carter,C.W.J. and Sweet,R.M. (eds) Methods in Enzymology. Academic Press, Vol. 277, pp. 505–524. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin,A.A. and Dodson,E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. In Carter,C.W.J. and Sweet,R.M. (eds) Methods in Enzymology. Academic Press, Vol. 276, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Phillips J.D. and Kushner,J.P. (1999) Measurement of uroporphyrinogen decarboxylase activity. In Current Protocols in Toxicology. John Wiley and Sons, Inc., New York, pp. 8.4.1–8.4.13. [DOI] [PubMed] [Google Scholar]
- Phillips J.D., Whitby,F.G., Kushner,J.P. and Hill,C.P. (1997) Characterization and crystallization of human uroporphyrinogen decarboxylase. Protein Sci., 6, 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J.D., Jackson,L.K., Bunting,M., Franklin,M.R., Thomas,K.R., Levy,J.E., Andrews,N.C. and Kushner,J.P. (2001) A mouse model of familial porphyria cutanea tarda. Proc. Natl Acad. Sci. USA, 98, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straka J.G. and Kushner,J.P. (1983) Purification and characterization of bovine hepatic uroporphyrinogen decarboxylase. Biochemistry, 22, 4664–4672. [DOI] [PubMed] [Google Scholar]
- Warshel A., Florian,J., Strajbl,M. and Villa,J. (2001) Circe effect versus enzyme preorganization: what can be learned from the structure of the most proficient enzyme? Chembiochem, 2, 109–111. [DOI] [PubMed] [Google Scholar]
- Whitby F.G., Phillips,J.D., Kushner,J.P. and Hill,C.P. (1998) Crystal structure of human uroporphyrinogen decarboxylase. EMBO J., 17, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock S.C. and Jordan,P.M. (1994) Evidence for participation of aspartate-84 as a catalytic group at the active site of porphobilinogen deaminase obtained by site-directed mutagenesis of the hemC gene from Escherichia coli. Biochemistry, 33, 2688–2695. [DOI] [PubMed] [Google Scholar]
- Wu N., Mo,Y., Gao,J. and Pai,E.F. (2000) Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl Acad. Sci. USA, 97, 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff E.E. and Kushner,J.P. (1994) Heme biosynthesis, the porphyrias and the liver. In Arias,I.M., Boyer,J.L., Fausto,N., Jakoby,W.B., Schachter,D.A. and Shafritz,D.A. (eds) The Liver: Biology and Pathobiology. Raven Press Ltd, New York, pp. 505–527. [Google Scholar]