

Abstract
The Cu+-ATPase CopA from Archaeoglobus fulgidus belongs to the P1B family of the P-type ATPases. These integral membrane proteins couple the energy of ATP hydrolysis to heavy metal ion translocation across membranes. A defining feature of P1B-1-type ATPases is the presence of soluble metal binding domains at the N-terminus (N-MBDs). The N-MBDs exhibit a conserved ferredoxin-like fold, similar to that of soluble copper chaperones, and bind metal ions via a conserved CXXC motif. The N-MBDs enable Cu+ regulation of turnover rates apparently through Cu-sensitive interactions with catalytic domains. A. fulgidus CopA is unusual in that it contains both an N-terminal MBD and a C-terminal MBD (C-MBD). The functional role of the unique C-MBD has not been established. Here, we report the crystal structure of the apo, oxidized C-MBD to 2.0 Å resolution. In the structure, two C-MBD monomers form a domain-swapped dimer, which has not been observed previously for similar domains. In addition, the interaction of the C-MBD with the other cytoplasmic domains of CopA, the ATP binding domain (ATPBD) and actuator domain (A-domain) has been investigated. Interestingly, the C-MBD interacts specifically with both of these domains, independent of the presence of Cu+ or nucleotides. These data reinforce the uniqueness of the C-MBD and suggest a distinct structural role for the C-MBD in CopA transport.
Keywords: P1B-type ATPase, copper trafficking, copper chaperone, crystal structure, Wilson disease, Menkes syndrome, domain swap
INTRODUCTION
Copper is used as a cofactor in many key enzymes, including cytochrome c oxidase, superoxide dismutase, lysyl oxidase, and dopamine β-monooxygenase.1 Although copper is essential, it can be toxic in excess through the formation of radicals via the Haber-Weiss reaction.2 For these reasons, copper concentrations are tightly regulated inside the cell by a variety of proteins including metallochaperones and copper transporting ATPases.3 The copper transporting ATPases belong to the P1B-1 subgroup of the P-type ATPase family.4,5 P-type ATPases are integral membrane proteins that use the energy of ATP hydrolysis to transport substrates across membranes.6,7 The P1B-1-type ATPases consist of 8 transmembrane helices, an actuator domain between helices 4 and 5 (A-domain), an ATP-binding domain between helices 6 and 7 (ATPBD), and soluble metal binding domains at the N-terminus (N-MBDs). The number of N-MBDs ranges from one or two in bacteria and archaea to six in the human Wilson and Menkes disease Cu+-ATPases.4 Their catalytic mechanism follows the Albers-Post cycle in which metal ion translocation is facilitated by conformational changes in the ATPBD and A-domain coupled to ATP hydrolysis and enzyme phosphorylation.8 Transmembrane metal binding sites involve residues from helices 6, 7, and 8.9 In contrast to the P2-type Ca2+-ATPases for which multiple crystal structures have been solved,10 the molecular basis for metal transport by P1B-type ATPases is not well understood.
CopA from Archaeoglobus fulgidus is a Cu+ efflux pump that has been studied as a model system for the Wilson and Menkes disease proteins.11–16 Besides serving as a simpler model for the human Cu+-ATPases, A. fulgidus CopA is of interest because it has both an N-MBD and an MBD at the C-terminus (C-MBD). The only other P1B-1-type ATPase sequence to contain a C-MBD is one from the termite gut bacteria Sebaldella termitidis ATCC 33386. The MBDs in Cu+-ATPases consist of approximately 70 residues, including a highly conserved CXXC copper binding motif.17,18 A number of these MBDs have been structurally characterized and exhibit a conserved ferredoxin-like βαββαβ fold similar to that of the yeast copper chaperone Atx1 (Atx1-like).3 The copper binding and structural properties of N-MBDs are well understood. Bacterial N-MBDs can exchange metal ions with Atx1-like metallochaperones called CopZ proteins12,19 and may function as Cu+ sensors, regulating transport activity.12,20–22 CopA variants from several species with truncated or mutated N-MBDs are active, although disruption of N-MBDs does affect the enzyme kinetics20–23 and abrogates inhibition at high copper concentrations.21 Co-purification studies indicated that the N-MBD binds the ATPBD of the enzyme.24 This domain-domain interaction is prevented by the presence of either Cu+ or nucleotides. Limited proteolysis25 and cryoelectron microscopy16 data have suggested that the N-MBD may also interact with the A-domain. These observations support a mechanism in which the N-MBD interacts with catalytic components of the enzyme in the absence of metal, slowing the enzyme turnover rate. It is hypothesized that upon Cu+ binding, the interaction is prevented and the enzyme reaches full activity.
The role of the A. fulgidus CopA C-MBD has not been explored in detail. In addition to its unique location at the C-terminus, the CXXC motif is atypical in that the highly conserved cysteines flank two histidine residues. Although BLAST searches indicate that a CXHC motif occurs fairly frequently, only one other sequence contains a CHHC motif, a soluble metallochaperone homolog from Thermosipho melanesiensis BI429. The CopA C-MBD binds Cu+ with high affinity, but neither mutation of its CXXC motif20 nor its deletion21 significantly affects enzyme activity. Toward understanding the structural role of the CopA C-MBD, we have determined the crystal structure of the metal free (apo) domain. Surprisingly, the structure is a domain-swapped dimer, an arrangement not observed previously for an Atx1-like domain. In addition, we investigated the interactions of the CopA C-MBD with the A-domain and ATPBD. Distinct from the N-MBD, the C-MBD interacts with both catalytic domains in a ligand (Cu+ or nucleotide) insensitive fashion.
MATERIALS AND METHODS
Cloning, expression, and purification
Recombinant C-MBD (residues 736-804) from A. fulgidus was expressed as a C-terminal streptactin tagged protein (tag has sequence SAWSHPQFEK) using the pBR-IBA1 vector (IBA).12 BL21(DE3) pLysS competent cells carrying the rare codon encoding plasmid pSJS124026 were transformed with the C-MBD pBR-IBA1 construct. Colonies were grown in Luria broth containing 100 μg/mL ampicillin, 34 μg/mL chloramphenicol, and 70 μg/mL spectinomycin at 37 °C to an OD600 of 0.6–0.8. Protein expression was induced with 1 mM isopropyl β-D-1-thiogalactopyranoside for 3 hrs at 37°C. Cell cultures were harvested by centrifugation at 5000g for 15 min, flash frozen in liquid nitrogen, and stored at −80°C.
Cells were thawed in a buffer containing 20 mM MOPS, pH 7.0, 20 mM NaCl, 5% glycerol. After addition of 50 units of DNase, 1 mM PMSF, and 1 mM EDTA, the cell paste was resuspended in a metal beaker and lysed by sonication in an ice water bath for 10 min (30 s pulse and 30 s rest). The cell lysate was then centrifuged at 163,000g for 1 hr after which the supernatant was filtered using a 0.22 μm membrane filter. Purification was carried out using a streptactin column (Qiagen) according to the manufacturer’s protocol, and the purified material was exchanged into 10 mM MOPS, pH 7.0, 10 mM NaCl, 5% glycerol. A Centriprep device (Millipore) with a cutoff of 5 kDa was used to concentrate the protein to 21.7 mg/mL. The protein was diluted to 20 mg/mL for crystallization screening. Protein concentration was determined by UV-spectroscopy using a calculated ε280 = 8605 M−1 cm−1.
Crystallization, data collection, and structure determination
Crystallization screens in 96-well sitting drop trays gave an initial hit using 0.1 M citrate, pH 5.0, 30% PEG 6000 as a precipitant. Optimization in 24 well hanging drop trays at room temperature using 1 μL of 20 mg/mL C-MBD and 1 μL precipitant was performed. Long rod-like crystals were obtained using 0.1 M citrate, pH 4.5, 30% PEG 6000. Crystals were flash frozen in liquid nitrogen using 0.1 M sodium citrate, pH 4.5, 30% PEG 6000, 25% glycerol as a cryoprotectant. Diffraction data were collected at DND-CAT beamline 5ID at the Advanced Photon Source at a wavelength of 1.376 Å.
The data were processed in space group P2 with unit cell dimensions a = 40.13, b = 35.99, c = 51.26, and β = 104.89° (Table I). The data were integrated and scaled using HKL2000.27 The program MrBUMP28 was used to obtain a molecular replacement solution using MerP from Ralstonia metallidurans CH34 (PDB accession code 1OSD)29 as a search model. An initial model of the CopA C-MBD was built with Coot30 and refinements were carried out with CNS (Table I).31 Refinement of the domain swapped model gave values of R and Rfree of 0.223 and 0.238 as compared to 0.249 and 0.259 for a backswapped model. Ramachandran plots calculated with PROCHECK32 indicate that 95.7% of the residues are in the favored regions and 4.3% are in the allowed regions. Structural figures were generated with PyMOL.33
Table I.
Data Collection and Refinement Statistics
| CopA C-MBD | |
|---|---|
| Data collection | |
| Resolution (Å) | 27.3-2.0A |
| No. reflections | 62387 |
| Unique reflections | 9568 |
| Rsym | 0.076 (0.245) |
| I/0I | 24.0(4.13) |
| Completeness (%) | 97.3(81.7) |
| Redundancy | 6.5(3.9) |
| Refinement | |
| Rwork | 0.22 |
| Rfree | 0.24 |
| No. atoms | 1149 |
| Water | 44 |
| Ligand/ion | 1 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.016 |
| Bond angles (°) | 1.47 |
Gel filtration chromatography
Analytical size exclusion chromatography was performed on a 111 ml prepacked HiLoad 16/60 Superdex 75 pg column equilibrated with 20 mM MOPS, pH 8.0, 0.2 M NaCl, 1 mM EDTA, 5 mM DTT. Samples of the C-MBD were injected in 1 ml aliquots at ~50mg/ml, and the molecular masses were determined using the following standards: blue dextran (void volume), conalbumin, 75 kDa; ovalbumin, 43 kDa; carbonic anhydrase, 29 kDa; RNase A, 13.7 kDa; aprotinin, 6.5 kDa.
Crosslinking experiments
The crosslinking assay was performed as described with some modifications.34 Cross-linking reactions were carried out in a buffer containing 15 mM sodium phosphate, pH 7.5, 150 mM NaCl, 0.125 mM tris(2,2′-bipyridyl)ruthenium(II) chloride. C-MBD were used at a concentration of 40 μM. Ammonium persulfate (APS) was added to final a concentration of 2.5 mM immediately before irradiation. The reaction mixture was irradiated for 30 sec with a 100 W white lamp. Reactions were quenched with 10 μl of 100 mM TrisCl, pH 6.8, 5% SDS, 2% β-mercaptoethanol, 20% glycerol, 0.2% bromophenol blue. Samples were subjected to SDS-PAGE and protein bands were visualized by Coomassie Brilliant Blue staining. To carboxymethylate the cysteines, C-MBD (1 mg/ml) was incubated with 10 mM dithiothreitol in 100 mM Tris, pH 8.5, 150 mM NaCl buffer for 20 min at room temperature to reduce disulfide bonds. The reduced protein was incubated with 20 mM iodoacetic acid (IAA) for 1 hr at room temperature in the dark. The protein was then washed in an Amicon Ultra-5 Centricon (Millipore) with 15 volumes 100 mM Tris, pH 8.0. The remaining free thiol was measured by the DTNB colorimetric assay as described previously.35
Domain-domain co-purification assays
Interactions between the C-MBD and the ATPBD and A-domain were investigated by batch affinity chromatography as described previously for the CopA N-MBD.24 Samples of the ATPBD and A-domain were prepared according to published protocols.14,15 The C-MBD was loaded with Cu+ using the same method as for the N-MBD.24 The effects of nucleotide binding were tested by including 5 mM ADP-MgCl2 in the assay media. For experiments in the absence of Cu+, 200 μM bathocuproine disulfonic acid (BCS) was added to the reaction. Briefly, samples were incubated with 20 μL Ni2+-nitrilotriacetic acid resin (Qiagen) for 10 min at room temperature and centrifuged at 14,000 rpm for 5 min to collect the unbound proteins in the supernatant (FT fraction). The bound proteins were eluted with imidazole (E fraction), and the fractions analyzed by SDS-PAGE. Control experiments using the individual domains were performed in an identical fashion.
Additional control experiments included co-purification of the C-MBD with BSA and the Na+/K+-ATPase ATPBD. For BSA, interactions were characterized as described above except that samples were incubated with 100 μL of pre-equilibrated streptactin resin for 10 min at room temperature with gentle agitation and then centrifuged at 14,000 rpm for 5 min to collect the unbound proteins in the supernatant (FT fraction). The proteins bound to the resin were washed with 1 mL of 100 mM Tris, pH 8.0, 150 mM NaCl followed by elution with 500 μl of 2.5 mM desthiobiotin in the same buffer (E fraction). The vector carrying the coding sequence for the His-tagged ATPBD of the Na+/K+-ATPase was a gift from Dr. Craig Gatto (Illinois State University, Normal, IL).36 Co-purification of this domain with the C-MBD was attempted as described above for the CopA ATPBD and C-MBD.
RESULTS AND DISCUSSION
Domain-swapped dimer
The apo C-MBD structure was determined to 2.0 Å resolution (Table I) and contains two molecules per asymmetric unit. Most of both chains were visible in the electron density maps with the exception of part of the C-terminal streptactin tag used for purification. The overall βαββαβ fold resembles that of other MBDs and Atx1-like chaperones.3 There are extensive interactions between the two monomers mediated by a domain swap of the N-terminal β strand (residues 1-15) [Fig. 1(a)]. The electron density clearly indicates the presence of this domain swap [Figs. 1(c), (d)]. This domain swap is stabilized by salt bridges and hydrogen bonding between monomers. Specifically, residues 5-12 from one monomer interact via regular β sheet hydrogen bonds with residues 64-68 and 40-46 from the second monomer. In addition, residues 14 and 16 from each monomer form a short antiparallel β sheet interaction. A salt bridge between residue Glu5 on one monomer and Lys48 from the second provides additional stabilization. The cysteines from the CXXC motifs are oxidized and form intramonomer disulfide bonds, resulting in a ring-like structure for residues 16-19 [Fig. 1(b)]. A citrate molecule derived from the crystallization solution interacts with the amide nitrogen atom of His 17 and the carbonyl and side chain oxygen atoms of Ser 15 from one monomer, further locking the swapped conformation in place. Ordered water molecules also link the citrate molecule to the protein via a network of hydrogen bonds. The core regions of the two monomers superpose well with an rmsd of 0.67 Å for residues 12-68. There is significant divergence in the position of the swapped strand and also at the C-terminus, which includes the first four residues of the streptactin tag.
Figure 1.

Structure of CopA C-MBD. (A) The domain swapped dimer. One monomer is shown in ligth green and the other is shown in purple. (B) Close up view of the hinge region and CXXC motif. (C) Superposition of the 2Fo−Fc (blue, contoured at 2σ) and Fo−Fc electron density maps (positive density in green, negative density in red, both contoured at 3σ) in the region of the domain swap for the domain swapped dimer and (D) for a model in which the domains are not swapped.
Domain-swapped structures have been observed for a number of proteins, although this is the first of an Atx1-like MBD. Domain swapping is believed to occur at hinge regions that unfold locally prior to complete unfolding.37 The hinge is the region of the domain swapped structure that differs from the non-swapped monomer and links the exchanging domains.38,39 Structural weakness, defined as a sequence that is not optimal with respect to the fold, is typically located in or near the hinge region.38 In the C-MBD structure, residues 14-16 comprise the hinge loop. By deleting these residues and reconnecting the appropriate secondary structure elements, it is possible to construct a “backswapped” monomer that closely resembles other MBD and copper chaperone proteins. This monomer superposes with oxidized apo MerP with an rmsd of 1.3 Å for 64 Cα coordinates and with oxidized apo Atx1 with an rmsd of 1.9 Å for 59 Cα coordinates (Fig. 2).
Figure 2.

Superposition of backswapped C-MBD model (light green) with the apo oxidized structures of R. metallidurans CH34 MerP (pink, PDB accession code 1OSD) and yeast Atx1 (blue, PDB accession code 1CC7).
The relationship between sequence and the propensity to domain swap is not well understood, but several factors likely contribute to the observed structure. Oxidation of the two cysteine residues may impart significant steric strain. However, three other structures exhibit intramonomer disulfide bonds that are not accompanied by a domain swap. These structures include apo oxidized yeast Atx140, oxidized R. metallidurans CH34 MerP,29 and the Atx1-like domain of the yeast copper chaperone for superoxide dismutase (yCCS).41 One difference between the C-MBD and these structures is strong interaction between a citrate molecule from the crystallization buffer and residue Ser15 in the hinge region. The sequence of the CXXC metal binding motif also differs. In the C-MBD, the two cysteines are separated by two histidine residues (CHHC) whereas the corresponding sequences are CSGC in yeast Atx1, CSAC in MerP, and CENC in yCCS. The SG and SA sequences may be more flexible than the two histidines and thus able to accommodate disulfide formation without drastic conformational rearrangement. In the case of yCCS, a domain swap is precluded by the presence of additional domains. Interestingly, a Ramachandran plot of the backswapped C-MBD model shows that residues 15 and 16 fall within the disallowed regions. These two residues would thus be under extreme stress in the back swapped structure if an orientation similar to that of previously characterized MBDs was maintained. Given that the domain swap is probably related to the presence of exogenous citrate and to cysteine oxidation, which would not occur in the cell, it is likely an artifact of crystallization. Nevertheless, it is intriguing that such an arrangement is possible. There may be conditions in vivo that favor domain swapping, perhaps involving the multiple MBDs of the human Cu+ ATPases.
Oligomerization state of C-MBD in solution
To assess whether the C-MBD dimerizes in solution, gel filtration chromatography and chemical crosslinking experiments were performed. The theoretical molecular mass of the C-MBD with the streptactin tag is 8579.7 Da. Three peaks eluted from the gel filtration column, one with the void volume, one at 27,247 Da and one at 11,672 Da (Fig. 3). The latter two peaks likely correspond to the dimeric and monomeric forms of the C-MBD. The elution volume is smaller than expected, likely due to the unstructured 10 amino acid streptactin tag. The peak at high molecular mass is not always observed and indicates some aggregation at the high protein concentration used. The observation that the dimer molecular mass is somewhat higher than twice the monomer molecular mass is consistent with the non-globular, elongated shape of the domain-swapped dimer [Fig. 1(a)]. Oligomerization is also observed upon crosslinking (Fig. 4). Treatment with crosslinker yields aggregation, probably due to reaction with the cysteine resides. However, carboxymethylation of the cysteine residues gives two distinct crosslinked products. These products likely correspond to dimers and tetramers of the C-MBD and are consistent with protein-protein interactions in solution.
Figure 3.

Gel filtration analysis of the C-MBD. Inset shows plot of Kav as a function of log(molecular weight).
Figure 4.
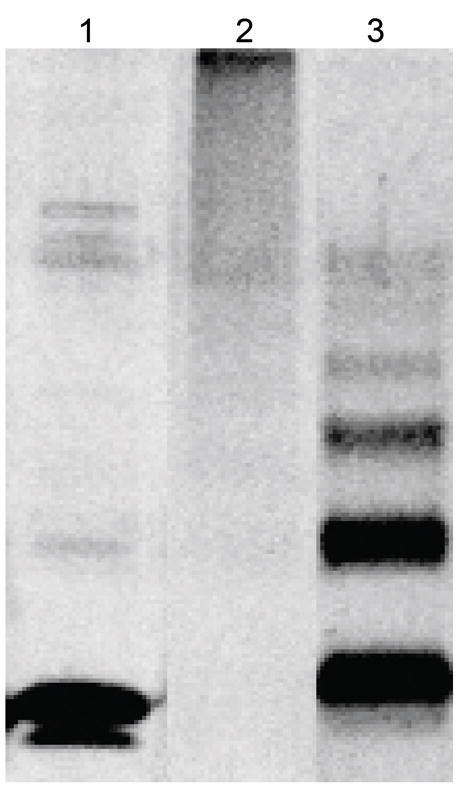
SDS-PAGE of a representative crosslinking assay of the C-MBD. Lane 1, 2 μg purified C-MBD. Lane 2, crosslinking products of 2 μg C-MBD. Lane 3, crosslinking products of 2 μg C-MBD after treating with iodoacetic acid to modify all the cysteines.
Metal binding loop
Although the C-MBD is structurally and sequentially similar to copper metallochaperones and N-MBDs, there are some notable differences in the metal binding region. As mentioned above, the CHHC sequence is unusual, and His 18 is oriented such that it could provide a third ligand to Cu+. Coordination by both histidine and cysteine could account for the higher affinity of the C-MBD for Cu+ as compared to the N-MBD as well as the high efficiency of Cu+ transfer from CopZ to the C-MBD.12 Residue Tyr63 is within hydrogen bonding distance of the cysteine residues and may further stabilize the metal binding site. Instead of a methionine at position 14 as observed in a number of homologous structures, the C-MBD contains a leucine which fulfills the same role in hydrophobic packing. Electrostatic surface complementarity is believed to play a role in copper chaperone-MBD interactions.40,42 There are a number of lysine residues that create positively charged patches on the C-MBD surface, including Lys24, Lys25, Lys33, and Lys36. There are also several regions of negative charge (Fig. 5). These patches may interact with the CopZ chaperone, which is predicted to have a negatively charged surface.19
Figure 5.

Surface of the backswapped C-MBD model colored according to electrostatic potential: red, −60 kT; white, 0 kT; blue, +60 kT. The view on the right is rotated 180° around the vertical axis from the view on the left.
Domain-domain interactions
Domain-domain interaction studies were carried out with the C-MBD to probe its role in CopA function. A batch affinity chromatography method24 was used to assess co-purification of the C-MBD with the ATPBD and A-domain (Fig. 6). The C-MBD interacts with both the ATPBD and A-domain. By contrast, the N-MBD only interacts with the ATPBD using the same assay.24 The association of the C-MBD with the ATPBD and A-domain is specific since control experiments performed with the unrelated proteins BSA and the ATPBD from the Na+,K+-ATPase do not show co-purification (Supporting Information Figs. S1 and S2). In the 17 Å resolution cryoelectron microscopy structure of CopA lacking the C-MBD, the C-terminus of the eighth transmembrane helix is hypothesized to lie near the ATPBD,16 and extension of this helix into the C-MBD would place it in position to interact with the ATPBD. The molecular basis for the observed interaction of C-MBD with the A-domain is not obvious from the cryoelectron microscopy model, however. Domain interactions within A. fulgidus CopA have been proposed to involve complementary electrostatic surfaces14,15 and the interactions observed here may be mediated by the aforementioned acidic or basic patches on the C-MBD (Fig. 3)
Figure 6.

Interactions of C-MBD with the other cytoplasmic domains of CopA. (A) SDS-PAGE of a representative co-purification assay between ATPBD and C-MBD or Cu+-loaded C-MBD. (B) SDS-PAGE of a representative co-purification assay between C-MBD and ATPBD in the absence and in the presence of 5 mM ADP-Mg2+. (C) SDS-PAGE of a representative co-purification assay between A-domain and C-MBD or Cu+-loaded C-MBD. 40% of unbound protein (FT) or 40% of the bound protein (E) fractions were loaded in each lane.
The presence of Cu+ or ADP in the assay media does not prevent interactions between the C-MBD and the ATPBD or A-domain (Fig. 6). Thus, conformational changes upon substrate (nucleotide to ATPBD) or regulatory ligand (Cu+ to C-MBD) binding do not strongly affect domain-domain interactions. By contrast, Cu+ binding to the N-MBD prevents interaction with the ATPBD24 as does Cu+ binding to the N-MBDs of the Wilson disease protein.43 It is also possible that the ligands modulate interactions differently when the C-MBD is tethered to the transmembrane domain of CopA. C-terminal metal binding domains are present in eukaryote P1B-2-type Zn2+ ATPases,4 including a novel regulatory metal binding domain at the C-terminus of the Arabidopsis Zn2+-ATPase HMA2.35 Although these appear to play a similar autoinhibitory role in the absence of the transported metal ion, they are structurally different from the Cu+-ATPase N-MBDs. Perhaps more relevant is the autoinhibitory C-terminal domains present in plasma membrane Ca+- and H+-ATPases.44,45 In both cases, interactions with the ATPBD and A-domain have been suggested as a mechanism of inhibition.45–47 Interestingly, enzyme activation is dependant on the interaction of the C-terminal domains with regulatory proteins: 14-3-3 for the H+-ATPase and Ca-calmodulin for the Ca2+-ATPase. The CopA C-MBD could play a similar role, modulating activity via interaction with regulatory proteins rather than Cu+ binding, particularly taking into account that the high affinity of this domain for Cu+ precludes “sensing” of available Cu+.12 The absence of in vitro kinetic effects upon mutation or removal of the C-MBD, although puzzling, does not exclude this possibility.
Taken together, the structural and biochemical data reveal several unusual properties of the CopA C-MBD. The observed domain swap is novel and suggests that MBDs and copper chaperones may be able to access conformations beyond what has been observed previously. The interaction of the C-MBD with the ATPBD and A-domain is consistent with a role in the CopA function. However, it is likely that this putative function is mediated by other factors rather than by Cu+ binding. Since A. fulgidus is an anaerobic, sulfur-metabolizing organism that grows at > 80 °C,48 additional regulation or functionality may be required in vivo.
Supplementary Material
Acknowledgments
NIH; Grant number: GM58518 (A. C. R.). NSF; Grant number: MCB-0743901 (J. M. A). NIH; Grant number: GM8382 (S. A). NIH; Grant number: GM073457 (M. H. S).
Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company and the State of Illinois. Use of the APS was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The atomic coordinates (code 3FRY) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/)
Abbreviations
- A-domain
actuator domain of A. fulgidus CopA
- ATPBD
ATP binding domain of A. fulgidus CopA
- BCS
bathocuproine disulfonic acid
- C-MBD
C-terminal metal binding domain of A. fulgidus CopA
- N-MBD
N-terminal metal binding domain of A. fulgidus CopA
Footnotes
SUPPORTING INFORMATION AVAILABLE
Figures of control domain-domain co-purification assays.
References
- 1.Mirica LM, Ottenwaelder X, Stack TDP. Structure and spectroscopy of copper-dioxygen complexes. Chem Rev. 2004;104:1013–1045. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 2.Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219:1–14. doi: 10.1042/bj2190001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boal AK, Rosenzweig AC. Structural biology of copper trafficking. Chem Rev. 2009;109:4760–4779. doi: 10.1021/cr900104z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Argüello JM. Identification of ion-selectivity determinants in heavy-metal transport P1B-type ATPases. J Membr Biochem. 2003;195:93–108. doi: 10.1007/s00232-003-2048-2. [DOI] [PubMed] [Google Scholar]
- 5.Barry AN, Shinde U, Lutsenko S. Structural organization of human Cu-transporting ATPases: learning from building blocks. J Biol Inorg Chem. 2010;15(1):47–59. doi: 10.1007/s00775-009-0595-4. [DOI] [PubMed] [Google Scholar]
- 6.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 7.Moller JV, Juul B, Lemaire M. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta. 1996;1286:1–51. doi: 10.1016/0304-4157(95)00017-8. [DOI] [PubMed] [Google Scholar]
- 8.Argüello JM, Eren E, González-Guerrero M. The structure and function of heavy metal transport P1B-type ATPases. Biometals. 2007;20:233–248. doi: 10.1007/s10534-006-9055-6. [DOI] [PubMed] [Google Scholar]
- 9.González-Guerrero M, Eren E, Rawat S, Stemmler TL, Argüello JM. Structure of the two transmembrane Cu+ transport sites of the Cu+-ATPases. J Biol Chem. 2008;283:29753–29759. doi: 10.1074/jbc.M803248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toyoshima C, Inesi G. Structural basis of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Ann Rev Biochem. 2004;73:269–292. doi: 10.1146/annurev.biochem.73.011303.073700. [DOI] [PubMed] [Google Scholar]
- 11.Mandal AK, Cheung WD, Argüello JM. Characterization of a thermophilic P-type Ag+/Cu+-ATPase from the extremophile Archaeglobus fulgidus. J Biol Chem. 2002;277:7201–7208. doi: 10.1074/jbc.M109964200. [DOI] [PubMed] [Google Scholar]
- 12.González-Guerrero M, Argüello JM. Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc Natl Acad Sci USA. 2008;105:5992–5997. doi: 10.1073/pnas.0711446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mandal AK, Yang Y, Kertesz TM, Argüello JM. Identification of the transmembrane metal binding site in Cu+-transporting PIB-type ATPases. J Biol Chem. 2004;279:54802–54807. doi: 10.1074/jbc.M410854200. [DOI] [PubMed] [Google Scholar]
- 14.Sazinsky MH, Agarwal S, Argüello JM, Rosenzweig AC. Structure of the actuator domain from the Archaeoglobus fulgidus Cu+-ATPase. Biochemistry. 2006;45:9949–9955. doi: 10.1021/bi0610045. [DOI] [PubMed] [Google Scholar]
- 15.Sazinsky MH, Mandal AK, Argüello JM, Rosenzweig AC. Structure of the ATP binding domain from the Archaeoglobus fulgidus Cu+-ATPase. J Biol Chem. 2006;281:11161–11166. doi: 10.1074/jbc.M510708200. [DOI] [PubMed] [Google Scholar]
- 16.Wu CC, Rice WJ, Stokes DL. Structure of a copper pump suggests a regulatory role for its metal-binding domain. Structure. 2008;16(6):976–985. doi: 10.1016/j.str.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnesano F, Banci L, Bertini I, Ciofi-Baffoni S, Molteni E, Huffman DL, O’Halloran TV. Metallochaperones and metal-transporting ATPases: A comparative analysis of sequences and structures. Genome Res. 2002;12:255–271. doi: 10.1101/gr.196802. [DOI] [PubMed] [Google Scholar]
- 18.Singleton C, LeBrun NE. Atx1-like chaperones and their cognate P-type ATPases: copper-binding and transfer. Biometals. 2007;20:275–289. doi: 10.1007/s10534-006-9068-1. [DOI] [PubMed] [Google Scholar]
- 19.Sazinsky MH, LeMoine B, Orofino M, Davydov R, Bencze KZ, Stemmler TL, Hoffman BM, Argüello JM, Rosenzweig AC. Characterization and structure of a Zn2+ and [2Fe-2S]-containing copper chaperone from Archaeoglobus fulgidus. J Biol Chem. 2007;282:25950–25959. doi: 10.1074/jbc.M703311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandal AK, Argüello JM. Functional roles of metal binding domains of the Archaeoglobus fulgidus Cu+-ATPase CopA. Biochemistry. 2003;42:11040–11047. doi: 10.1021/bi034806y. [DOI] [PubMed] [Google Scholar]
- 21.Rice WJ, Kovalishin A, Stokes DL. Role of metal-binding domains of the copper pump from Archaeoglobus fulgidus. Biochem Biophys Res Commun. 2006;348:124–131. doi: 10.1016/j.bbrc.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 22.Hatori Y, Hirata A, Toyoshima C, Lewis D, Pilankatta R, Inesi G. Intermediate phosphorylation reactions in the mechanism of ATP utilization by the copper ATPase (CopA) of Thermotoga maritima. J Biol Chem. 2008;283(33):22541–22549. doi: 10.1074/jbc.M802735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan B, Rosen BP. Biochemical characterization of CopA, the Escherichia coli Cu(I)-translocating P-type ATPase. J Biol Chem. 2002;277(49):46987–46992. doi: 10.1074/jbc.M208490200. [DOI] [PubMed] [Google Scholar]
- 24.González-Guerrero M, Hong D, Argüello JM. Chaperone-mediated Cu+ Delivery to Cu+ transport ATPases. Requirement of nucleotide binding. J Biol Chem. 2009;284(31):20804–20811. doi: 10.1074/jbc.M109.016329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatori Y, Majima E, Tsuda T, Toyoshima C. Domain organization and movements in heavy metal ion pumps - papain digestion of CopA, a Cu+-transporting ATPase. J Biol Chem. 2007;282(35):25213–25221. doi: 10.1074/jbc.M703520200. [DOI] [PubMed] [Google Scholar]
- 26.Kim R, Sandler SJ, Goldman S, Yokota H, Clark AJ, Kim S-H. Overexpression of archaeal proteins in Escherichia coli. Biotechnol Lett. 1998;20:207–210. [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.Keegan RM, Winn MD. Automated search-model discovery and preparation for structure solution by molecular replacement. Acta Cryst. 2007;D63:447–457. doi: 10.1107/S0907444907002661. [DOI] [PubMed] [Google Scholar]
- 29.Serre L, Rossy E, Pebay-Peyroula E, Cohen-Addad C, Coves J. Crystal structure of the oxidized form of the periplasmic mercury-binding protein MerP from Ralstonia metallidurans CH34. J Mol Biol. 2004;339:161–171. doi: 10.1016/j.jmb.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular crystallography. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski RA. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 33.Delano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 34.Fancy DA, Dension C, Kim K, Xie Y, Holdeman T, Amini F, Kodakek T. Scope, limitations and mechanistic aspects of the photo-induced cross-linking of proteins by water-soluble metal complexes. Chem Biol. 2000;7:697–708. doi: 10.1016/s1074-5521(00)00020-x. [DOI] [PubMed] [Google Scholar]
- 35.Eren E, Kennedy DC, Maroney MJ, Arguello JM. A novel regulatory metal binding domain is present in the C terminus of Arabidopsis Zn2+-ATPase HMA2. J Biol Chem. 2006;281(45):33881–33891. doi: 10.1074/jbc.M605218200. [DOI] [PubMed] [Google Scholar]
- 36.Gatto C, Wang AX, Kaplan JH. The M4M5 cytoplasmic loop of the Na,K-ATPase, overexpressed in Escherichia coli, binds nucleoside triphosphates with the same selectivity as the intact native protein. J Biol Chem. 1998;273(17):10578–10585. doi: 10.1074/jbc.273.17.10578. [DOI] [PubMed] [Google Scholar]
- 37.Ding F, Prutzman KC, Campbell SL, Dokholyan NV. Topological determinants of protein domain swapping. Structure. 2006;14(1):5–14. doi: 10.1016/j.str.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 38.Dehouck Y, Biot C, Gilis D, Kwasigroch JM, Rooman M. Sequence-structure signals of 3D domain swapping in proteins. J Mol Biol. 2003;330(5):1215–1225. doi: 10.1016/s0022-2836(03)00614-4. [DOI] [PubMed] [Google Scholar]
- 39.Gronenborn AM. Protein acrobatics in pairs - dimerization via domain swapping. Curr Op Struct Biol. 2009;19(1):39–49. doi: 10.1016/j.sbi.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenzweig AC, Huffman DL, Hou MY, Wernimont AK, Pufahl RA, O’Halloran TV. Crystal structure of the Atx1 metallochaperone protein at 1.02 Å resolution. Structure. 1999;7:605–617. doi: 10.1016/s0969-2126(99)80082-3. [DOI] [PubMed] [Google Scholar]
- 41.Lamb AL, Wernimont AK, Pufahl RA, O’Halloran TV, Rosenzweig AC. Crystal structure of the copper chaperone for superoxide dismutase. Nature Struct Biol. 1999;6:724–729. doi: 10.1038/11489. [DOI] [PubMed] [Google Scholar]
- 42.Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nature Chem Biol. 2006;2(7):367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- 43.Tsivkovskii R, MacArthur BC, Lutsenko S. The Lys1010-Lys1325 fragment of the Wilson’s disease protein binds nucleotides and interacts with the N-terminal domain of this protein in a copper-dependent manner. J Biol Chem. 2001;276:2234–2242. doi: 10.1074/jbc.M003238200. [DOI] [PubMed] [Google Scholar]
- 44.Palmgren MG. Plant plasma membrane H+-ATPases: powerhouses for nutrient uptake. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:817–845. doi: 10.1146/annurev.arplant.52.1.817. [DOI] [PubMed] [Google Scholar]
- 45.Di Leva F, Domi T, Fedrizzi L, Lim D, Carafoli E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch Biochem Biophys. 2008;476(1):65–74. doi: 10.1016/j.abb.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 46.Pedersen BP, Buch-Pedersen MJ, Morth JP, Palmgren MG, Nissen P. Crystal structure of the plasma membrane proton pump. Nature. 2007;450(7172):1111–1114. doi: 10.1038/nature06417. [DOI] [PubMed] [Google Scholar]
- 47.Kühlbrandt W, Zeelen J, Dietrich J. Structure, mechanism, and regulation of the neurospora plasma membrane H+-ATPase. Science. 2002;297(5587):1692–1696. doi: 10.1126/science.1072574. [DOI] [PubMed] [Google Scholar]
- 48.Stetter KO. Extremophiles and their adaptation to hot environments. FEBS Lett. 1999;452:22–25. doi: 10.1016/s0014-5793(99)00663-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.