

Abstract
Helicases are an important class of enzymes involved in DNA and RNA metabolism that couple the energy of ATP hydrolysis to unwind duplex DNA and RNA structures. Understanding the mechanism of helicase action is vital due to their involvement in various biological processes such as DNA replication, repair and recombination. Furthermore, the duplex DNA unwinding property of this class of enzymes is closely related to their single-stranded DNA translocation. Hence the study of its translocation properties is essential to understanding helicase activity. Here we review the methods that are employed to analyze the DNA translocation and unwinding activities of the bacteriophage T4 UvsW and Dda helicases. These methods have been successfully employed to study the functions of helicases from large superfamilies.
Keywords: DNA helicases, Translocation and unwinding, Fluorescence, DNA replication, Recombination and repair
1. Introduction
Helicases use the energy derived from the binding and the hydrolysis of (d)NTPs to unwind duplex DNA and RNA structures [1]. These enzymes are involved in a variety of functions such as DNA replication, repair, recombination and transcription. They are found in viruses, prokaryotes, eukaryotes and archaea. Defects in the function of these ubiquitous enzymes can lead to a variety of disease states, which can be illustrated by mutations in three human helicases WRN, BLM and RecQ4 responsible for Werner's syndrome, Bloom's syndrome and Rothmund–Thomson syndrome, respectively [2]. It is, hence, important to understand the molecular mechanism of this class of enzymes.
The bacteriophage T4 genome encodes all the proteins required for the replication and repair of the 169 kb linear genome except for the RNA polymerase necessary for initiation of replication which is provided by the Escherichia coli host cell [3]. T4 phage has three helicases namely gp41, Dda and UvsW, and are essential for DNA replication and recombination in T4 phage. The characteristics of the T4 helicases are described in Table 1. Gp41 is the hexameric ring-shaped helicase that belongs to the DnaB-like SF4 superfamily of helicases and is the primary replicative helicase involved in DNA synthesis. Gp41 helicase exhibits DNA-dependent ATPase activity that is more strongly stimulated by long segments of single-stranded DNA (ssDNA) than short segments of ssDNA or double-stranded DNA (dsDNA) [4]. The movement of gp41 protein during the ATP-dependent helicase activity and translocation on ssDNA are both in a 5′-to-3′ unidirectional manner [5]. An overhang of 32 nucleotides or more at the 5′-ssDNA of duplex or forked substrate is necessary for the unwinding activity of gp41 helicase [6]. It has been demonstrated that the ATP binding, but not its hydrolysis is necessary to form a stable gp41–ssDNA complex [4]. The rate of translocation of gp41 on ssDNA is measured to be 400 nt/s and the unwinding rate of gp41 is ∼250 bp/s [7]. The latter rate is sufficient to provide a highly processive unwinding of dsDNA during DNA synthesis that occurs at the same rate on the leading strand by the T4 holoenzyme assembly.
Table 1.
Characteristics of the T4 Phage helicases.
| Protein | gp41 helicase | Dda helicase | UvsW helicase |
|---|---|---|---|
| Superfamily | SF4 | SF1 | SF2 |
| Position and length of ssDNA overhang | 5′ overhang and ≥ 32 | 5′ overhang and ≥ 20 | 3′ overhang and ≥ 24 |
| Directionality | Unidirectional 5′-to-3′ |
Unidirectional 5′-to-3′ |
Unidirectional 3′-to-5′ |
| Oligomeric state | Hexameric helicase & translocase | Monomeric helicase | Monomeric translocase |
| Processivity | Highly processive helicase [47] | Moderately processive helicase [40] | Processive translocase |
| SA displacement | Active displacement | Active displacement | None detected |
Dda belongs to the SF1 family of helicases. It is believed to be involved in T4 replication initiation as demonstrated by the delayed DNA synthesis in T4 Dda− mutants [8,9]. Dda also interacts with UvsX and this interaction is essential for the enhancement of the rate of branch migration [10,11]. Thus, Dda is also involved in T4 DNA recombination and repair. Dda is a 5′-to-3′ helicase and also displays a strong directional bias in the 5′-to-3′ ssDNA translocation [12]. Furthermore, Dda is capable of displacing protein blocks in the path of translocation of the enzyme [13–15].
UvsW, the third helicase of T4 phage, is a member of the SF2 superfamily of helicases. It participates in the transition from origin-dependent to recombination-dependent mode of DNA replication during the late stages of infection by catalyzing the dissociation of R-loop structures [16,17]. UvsW functions in the unwinding of structures that mimic recombination intermediates such as D-loop and Holliday junction structures implicating its importance in branch migration analogous to the Dda enzyme [18,19]. This further suggests that UvsW is responsible for the residual branch resolution activity found in the absence of gp49. Its D-loop unwinding activity and the unwinding of both the leading and lagging strand arms of a model stalled replication fork by UvsW helicase has led to the hypothesis that UvsW participates in both synthesis-dependent strand annealing (SDSA) and replication fork regression DNA repair pathways [18]. Double strand break (DSB) repair in bacteriophage T4 uses the enzymes involved in the DNA recombination pathway [20]. The steps involved in DSB repair are outlined in Fig. 1 and the functions of the associated enzymes include the resection of 5′-end of the broken duplex DNA by gp46/47 and coating of the generated 3′-overhangs by recombinase protein UvsX loaded by UvsY onto the ssDNA. The UvsX-coated ssDNA then invades an intact duplex DNA and forms a D-loop structure upon encountering a homologous sequence of DNA. The D-loop serves to initiate DNA replication forming Holliday junction structures resolved by UvsW helicase and Holliday junction endonuclease, gp49.
Fig. 1.
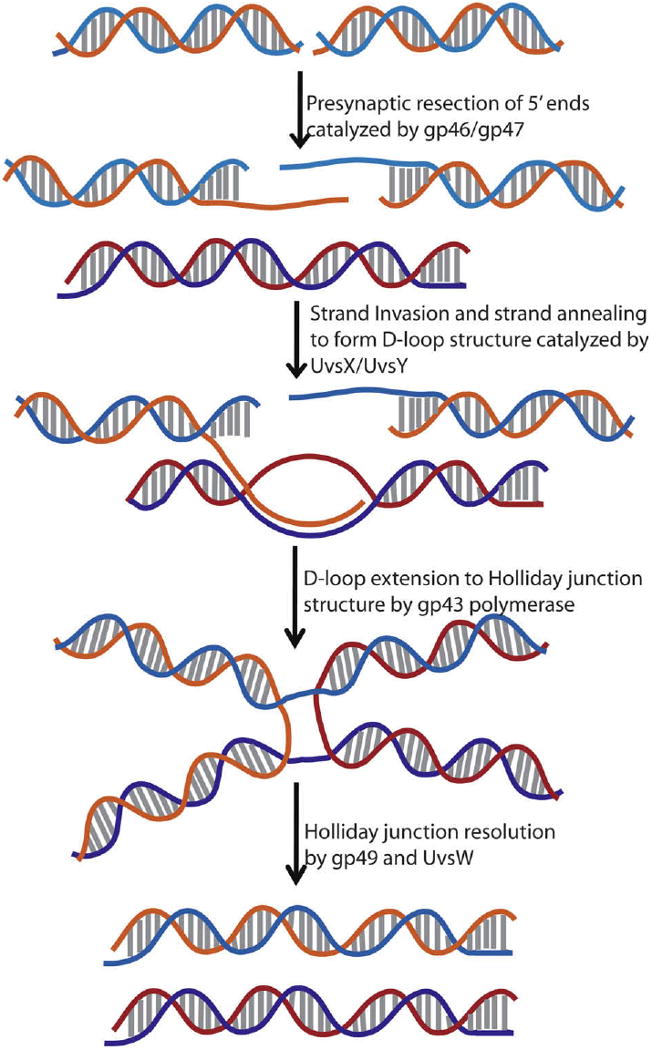
Schematic representation of the homologous recombination-dependent double strand break (DSB) repair pathway in bacteriophage T4. Gp46 and gp47 complex is involved in the recognition of DSBs and resects 5′-ends to generate 3′-overhangs. These 3′-overhangs are coated with UvsX assisted by UvsY followed by the invasion of a homologous strand to form a D-loop structure. The D-loop is extended by polymerase to generate Holliday junction structures that are resolved by UvsW and gp49 endonuclease, thus producing repaired double strand breaks.
Mutants with uvsW knock-out displayed high sensitivity to hydroxyurea and UV light implicating the role of UvsW in processing damaged DNA and stalled replication forks [21–23]. Additionally, due to its ability to unwind R-loops, UvsW has been described as a functional analog of E. coli RecG. This hypothesis has been supported by the ability of UvsW to complement some of the defects found in RecG− E. coli mutants. Unlike gp41 and Dda, UvsW unwinds duplex DNA in a 3′-to-5′ directional manner and also displayes an analogous directional bias of 3′-to-5′ ssDNA translocation. In the following sections, the detailed methods employed in the study of the biochemical properties of Dda and UvsW are described.
2. Materials and methods
The oligonucleotides substrates were purchased from Integrated DNA Technologies (Coralville, IA) or Amersham Pharmacia Biotech. [γ-32P]ATP and [α-32P]ATP were obtained from Perkin Elmer Life Sciences. Unlabeled ribonucleotides were purchased from Roche Applied Sciences. The pET28a and pET26b, pTYB3 and pGEM vectors were obtained from Novagen (now Merck Biosciences), New England Biolabs and Promega, respectively. E. coli RecA protein, T4 polynucleotide kinase (PNK), Bovine Serum Albumin (BSA), proteinase K, T7 RNA polymerasewere purchased from NEB. Single-stranded M13 DNA (ssM13) was prepared from M13mp18 infected E. coli cells using standard protocols [24]. Phosphoenolpyruvate (tricyclohexyl ammonium salt) (PEP), adenosine triphosphate (ATP), reduced nicotinamide adenine dinucleotide (NADH), Sephadex G-25, streptavidin, a buffered aqueous glycerol solution of phosphoenolpyruvate kinase (PK)/lactate dehydrogenase (LDH), bromophenol blue and xylene cyanol FF were obtained from Sigma.
3. Purification of recombinant UvsW and Dda Helicases
3.1. UvsW helicase
The gene encoding the T4 UvsW helicase was amplified using T4 genomic DNA as the template with the following primers, forward primer: 5′-GCGGCAGCCATATGGA TATTAA AGTACATTTTCACGA C-3′, reverse primer: 5′-GTCGGATCCCGTAAATTAACTGTTTTCA TTAC-3′. The amplified gene was inserted into the NdeI and BamHI sites of a pET28b vector. The pET28 vector carrying the gene for UvsW with a N-terminal His6-tag was transformed into BL21(DE3) and selected on agar plates containing 50 μg/ml kanamycin. A single colony was selected from the agar plate and grown overnight at 37 °C in LB containing kanamycin (50 μg/ml). A 10 ml of the overnight inoculation was diluted into 1 L of LB with kanamycin. The cells were grown at 37 °C until the OD600nm reached ∼0.5. The temperature of the culture was dropped to 18 °C and the protein expression was induced by the addition of 0.2 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG). The cells were then grown for an additional 10–16 h at 18 °C after which the cells were pelleted by centrifugation in a Beckman centrifugation device at 5000 rpm at 4 °C for 15 min. The cells were resuspended in 10 ml of 20 mM Tris–HCl, 500 mM NaCl, and 5 mM imidazole (pH 8.0) buffer for every gram of the cell pellet in the presence of EDTA-free Complete protease inhibitor cocktail from Roche Diagnostics. The resuspended cells were then lysed using sonication, and centrifuged at 15,000 rpm for 45 min at 4 °C The lysate was then loaded onto a nickel nitrilotriacetic acid (Ni–NTA)-agarose column, washed with 50 column volumes of lysis buffer containing 20 mM imidazole and 1 M NaCl and the protein eluted with lysis buffer containing 100 mM imidazole. The eluted protein was diluted 3-fold with a buffer containing 20 mM Tris–HCl (pH 7.5), 400 mM NaCl and loaded onto a 20 ml P11 phosphocelluose column. The column was washed with 10 column volumes of the same buffer before eluting UvsW with a linear gradient of 0.4–1.2 M NaCl. The fractions were analyzed on 12% SDS–PAGE and the fractions containing UvsW were pooled together and concentrated to 22 μM using an Amicon centrifugation device with a 30 kD cut-off and flash-frozen in liquid N2 in 10 μl aliquots at −80 °C. Protein concentration was calculated based on an extinction coefficient of 73,920 M−1 cm−1. The K141A active-site mutant of UvsW was expressed and purified identically to that of the wild-type enzyme using the pET28b vector carrying the K141A mutant gene of UvsW generated by site-directed mutagenesis using the following mutagenic primer: 5′-CTTCCAACATCTGCAGGTGCGTCTTTAATTCAAGCTT TGC-3′ (the bold underlined letters here indicate the mutation site). The reverse primer for the mutagenesis is the reverse complement of the above mentioned forward primer.
3.2. Dda helicase
Recombinant Dda was overexpressed and purified as previously reported [25]. Briefly, the protein was expressed in E. coli BL21/DE3 cells using the plasmid pET26b-Dda. Induction of the overexpression was carried out at 15 °C overnight by addition of 0.5 mM IPTG and 0.2% dextrose. A 30 g cell pellet harvested by centrifugation was then suspended in 175 ml of lysis buffer containing 25 mM Tris acetate (pH 7.6), 500 mM NaCl, 1 mM EDTA, 5 mM 2-mercapto ethanol (BME), 10% glycerol, 2 mM phenylmethylsulfonyl fluoride, 4 μg/ml pepstatin A, and 0.2 mg/ml lysozyme. The cell lysis was achieved by one cycle of freezing and thawing followed by sonication. The lysed cells were clarified by centrifugation in a JA-25.50 rotor (Beckman) at 20,000 rpm for 20 min at 4 °C. The lysate was further cleared by centrifugation in a 50.2 TI rotor (Beckman) at 45,000 rpm for 2 h at 4 °C. The supernatant thus obtained was applied to a 30 ml column of Macro Prep High Q anion exchange resin (Bio-Rad) equilibrated with lysis buffer. The proteins that eluted in the flow-through from this column contain Dda and were precipitated with 55% ammonium sulfate, and the precipitate was pelleted by centrifugation at 20,000 rpm for 1 h at 4 °C on a JA-25.50 rotor. The pellet was resuspended in a buffer containing 25 mM Tris acetate (pH 7.6), 50 mM NaCl, 1 mM EDTA, 5 mM BME, 10% glycerol and 1.5 M ammonium sulfate. Protein was loaded onto a 40 ml column of Methyl-HIC resin (Bio-rad) and eluted with a linear gradient of 1.5–0 M ammonium sulfate in the above buffer. Dda eluted between 0.9 and 0.6 M ammonium sulfate and the fractions containing Dda as determined by analytical PAGE (12 %) were dialyzed into a buffer containing 25 mM Tris acetate (pH 7.6), 50 mM NaCl, 1 mM EDTA, 5 mM BME, and 10% glycerol. The dialyzed protein was then loaded onto a 20 ml ssDNA cellulose column (Sigma). Dda was eluted using a linear gradient of 0.05–2 M NaCl and the fractions containing Dda were pooled, concentrated using an Amicon centrifugation device with a 10 kD cutoff and dialyzed into a buffer containing 25 mM Tris acetate (pH 7.6), 50 mM NaCl, 1 mM EDTA, 5 mM BME, and 10% glycerol. The dialyzed protein was applied onto a 15 ml Macro Prep High Q anion exchange column equilibrated with the dialysis buffer. Dda was eluted using a linear gradient from 50 to 500 mM of NaCl and Dda eluted from 250 to 320 mM NaCl as determined by analytical PAGE. Dda fractions were concentrated to ∼0.5 mg/ml using an Amicon concentrator and then dialyzed into the storage buffer containing 25 mM HEPES (pH 7.5), 50 mM NaCl, 2 mM BME, 1 mM EDTA, and 20% glycerol. Protein concentration was measured using a calculated extinction coefficient of 59,010 cm−1 M−1. Aliquots were frozen in liquid N2 prior to storing at −80 °C.
4. Unwinding activity of UvsW and Dda helicases
The unwinding activity of helicases, in general, can be evaluated by two major methods namely radiometric and fluorescence-based assays. The radiometric assay involves the use of radioactively labeled oligonucleotides to make the substrates for the unwinding reactions, which allows to detect the products of helicase catalyzed unwinding. The fluorescent-based assay utilizes oligonucleotides that contain fluorescent probes. Helicase-mediated unwinding of the duplex strand containing the probe causes changes in its fluorescent probe to monitor unwinding reactions in real-time. These two methods have been used successfully to study the helicase activity of UvsW and Dda helicases.
4.1. Preparation of substrates for unwinding activity of UvsW and Dda helicases
Simple DNA structures that were used in the analysis of unwinding activities and the directionality of unwinding are outlined in Fig. 2A–E. These structures resemble DNA structures found in vivo such as replication-origin bubble DNA structure or a replication fork. Although the bubble DNA substrate has not been assessed as a substrate for UvsW and Dda, it acts as a substrate for the RecQ family of helicases [26–30]. RecQ helicases have strand annealing activity and is suggested to be physiologically important, as replication fork regression is catalyzed only by RecQ helicases with strand annealing activity [31,32]. UvsW has a strong strand annealing activity [18]. Furthermore, the C-terminal four helix bundle resembles the HRDC (Helicase and RNase D C-terminal) domain from RecQ helicases [33]. Hence, UvsW is suggested to be functionally similar to the RecQ helicases that are members of the SF2 family of helicases and involved in DNA replication, recombination and repair. DNA structures such as the duplex DNA Y and the static synthetic Holliday junction structures shown in Fig. 2F and G mimic the recombination intermediates and have been evaluated as substrates of UvsW helicase [19]. The collective structures were formed by the following procedure. Select DNA strands were annealed by mixing the complementary oligonucleotides in a 1:1 ratio followed by heating to 95 °C for 3 min in a water bath, and then cooled to room temperature over a period of 1 h. These annealed duplex DNA substrates were radiolabeled at the 5′-ends with [γ-32P]ATP by T4 PNK. The reaction involved 160 nM of oligonucleotides, 31 μl of H2O, 5 μl of 10× T4 kinase buffer, 4 μl of [γ-32P]ATP and 2 μl of T4 PNK. The reaction mixture was mixed thoroughly and maintained at 37 °C for 60 min. The enzyme was then inactivated by heating the contents to 65 °C for 10 min. The synthetic Holliday junction and duplex DNA Y substrates used by the Kruezer laboratory were prepared by first radiolabeling one of the substrate strands followed by annealing the labeled strand to its complementary strand [19].
Fig. 2.
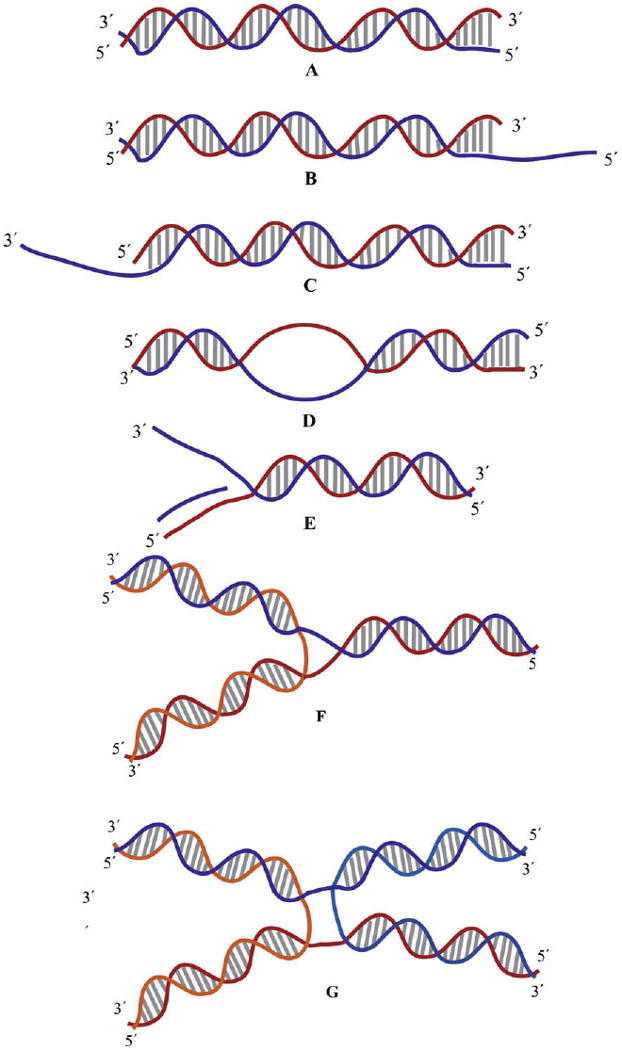
Schematic representation of the substrates used in the study of helicases. (A) Blunt ended duplex DNA. (B and C) Partial duplex with 5′ and 3′ ssDNA overhangs, respectively. (D) Bubble DNA structure. (E) Replication fork structure with a primer on the leading strand. (F) Duplex DNA Y substrate. (G) Synthetic Holliday junction structure.
4.2. Unwinding reactions of Dda and UvsW helicase
The unwinding reactions were carried out at 37 °C in helicase buffer (20 mM Tris acetate (pH:7.8), 125 mM potassium acetate, 10 mM magnesium acetate) containing 5 mM ATP, 2 nM substrate. The reactions were initiated by the addition of 200 nM protein. The unwinding reactions were quenched at various times with a quench solution containing 20 mM EDTA, proteinase K and 0.2 % sodium dodecyl sulfate (SDS). The aliquots then were mixed with loading buffer (50% glycerol, 1 μg/ml bromophenol blue, 1 μg/ml xylene cyanol FF). The products of the unwinding reactions were analyzed on a 10% PAGE eluted in 1× TBE (Tris (90 mM), borate (90 mM), EDTA (1 mM)) buffer at 25 mA for 3 h at room temperature. The unwinding reactions of the duplex DNA Y and synthetic Holliday junction substrates were accomplished under similar conditions. The gels were then exposed to a Phosphoimager screen overnight, imaged using Phosphoimager and quantitated using ImageQuant Software both from GE Healthcare Life Sciences. The products of a representative unwinding reaction catalyzed by UvsW and Dda helicase and resolved on a PAGE gel are shown in Fig. 3.
Fig. 3.
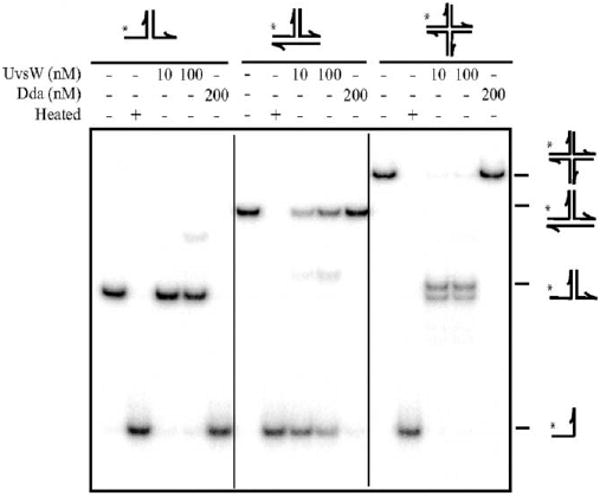
Phage T4 UvsW- and Dda-catalyzed unwinding of replication fork, duplex DNA Y substrate and Holliday junction substrates. The reaction products were resolved on a 7.5% PAGE gel shows the products of unwinding. (Reproduced with permission from Ref. [19]).
4.3. Annealing activity of UvsW helicase
UvsW was observed to have a strand annealing property [18]. UvsW does not unwind the duplex DNA substrates to completion when short oligonucleotides are used as DNA substrates. This activity was detected from the unwinding time courses, where a burst of ssDNA product was generated followed by a decrease in the observed unwound product (Fig. 5). Under the reaction conditions at 37 °C, the spontaneous annealing of the strands used for the substrates is slower with a half-life of ∼100 min (0.0069 min−1). Consequently UvsW contains a potent ssDNA annealing activity. It has been suggested that a strand annealing activity is probably involved in synthesis-dependent strand annealing (SDSA) and also replication fork regression during DNA repair pathways. Such an activity has been recently observed with other helicases as well and includes Hel112 from Sulfolobus solfataricus [34] and the HARP [35,36] and WRN [37] helicases from humans among others. The annealing activity of UvsW is enhanced by ATP hydrolysis but ATP binding alone is insufficient for the observed activity [18]. Furthermore, addition of the ssDNA-binding protein (gp32) of T4 phage that sequesters the ssDNA generated during the unwinding reaction should reduce the annealing activity. Indeed, the presence of gp32 protein significantly reduced the annealing activity of UvsW but did not completely abolish it [18]. The annealing reactions were performed under identical conditions as the unwinding assays with a substrate concentration of 0.2 nM to eliminate possible spontaneous annealing of the ssDNA strands. The reactions were quenched at various time points with a quench solution containing 20 mM EDTA, proteinase K and 0.2% SDS. The aliquots were mixed with loading buffer (50% glycerol, 1 μg/ml bromophenol blue, 1 μg/ml xylene cyanol FF). The products of the reactions were analyzed on a 10% PAGE ran in 1× TBE buffer at 25 mA for 3 h at room temperature. The gels were then exposed to a Phosphoimager screen overnight, imaged and quantitated identically to the above described unwinding reactions.
Fig. 5.
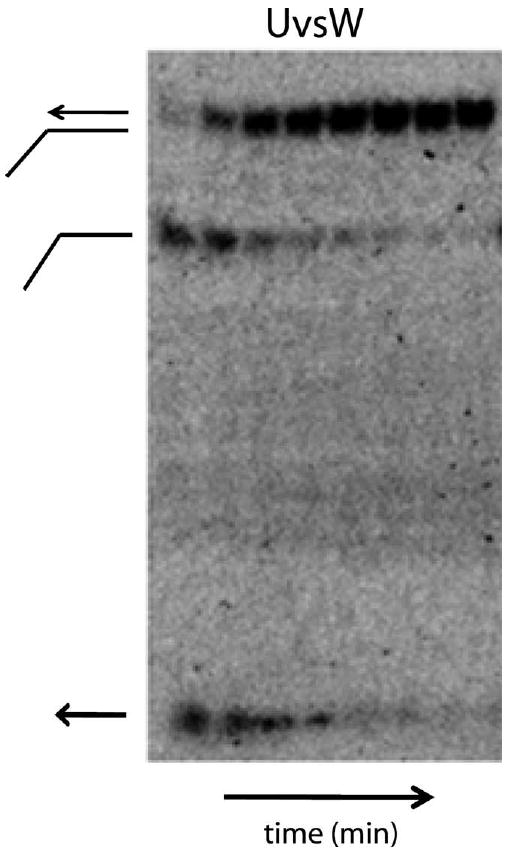
DNA annealing activity mediated by UvsW helicase. A typical time course (0.25–4 min) of ssDNA annealing in the presence of ATP catalyzed by UvsW.
5. Preparation and unwinding reactions of D-loop and R-loop structures
Substrates that resemble the replication and recombination intermediates such as R-loops and D-loop structures are relatively more complicated than the duplex DNA substrates used above. These D-loop and R-loop structures shown schematically in Fig. 4 were synthesized as described below.
Fig. 4.
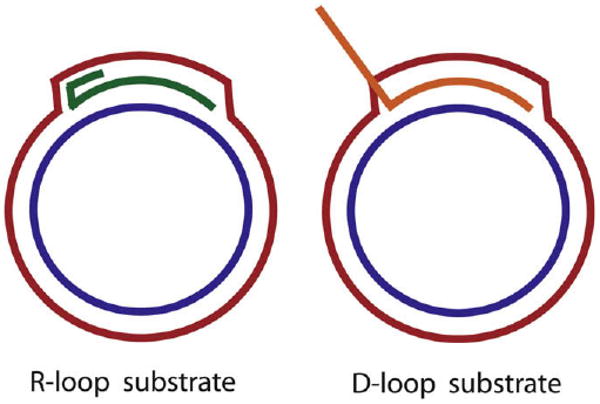
Schematic representation of mobile R-loop and D-loop substrates used in the study of helicases. R-loop structure was synthesized by T7 RNA polymerase, whereas the D-loop structure was produced by RecA mediated strand-invasion reaction.
5.1. R-loop substrates
A 530 bp DNA sequence corresponding to positions 114,754–115,284 of the T4 phage genome was inserted into the 3 kb pGEM vector between NdeI and NcoI sites. The insert contains the T4 DNA origin OriF. The R-loop substrate was synthesized by combining 10 mg of pGEM-T4OriF vector with 200 mM rNTPs, 10 μCi of either [α-32P]CTP or [α-32P]ATP in the transcription buffer containing 40 mM Tris–HCl (pH 7.9), 6 mM MgCl2, 10 mM DTT and 2 mM spermidine. The transcription reaction was performed by adding T7 RNA polymerase (RNAP) at 37 °C for 30 min. The R-loop, thus, synthesized was purified on a Qiaquick spin column (Qiagen) following the manufacturer's protocol [18].
5.2. D-loop substrate
The D-loop invading strand ssDNA 80mer (300 nM) with a sequence of 5′-CGCGAATTTTAACAAAATATTAACGCTTACAATTTCC TGATGCGGTATTTTCTCCTTACGCA-3′ was radiolabeled at the 5′-end with [γ-32P]-ATP in kinase buffer catalyzed by T4 PNK for 1 h at 37 °C. The enzyme was inactivated at 65 °C for 10 min. The 5′-radiolabeled strand-invasion primer that was diluted 10-fold was incubated with 0.75 μM E. coli RecA obtained from NEB in a buffer containing 20 mM Tris–HCl (pH 7.8), 125 mM potassium acetate, 40 mM magnesium acetate, 2 mM ATP, 20 mM creatine phosphate and 2 U/ml creatine kinase for 5 min at 37 °C. To the above mixture, the pGEM-T4OriF vector (70 nM) described above was added to initiate the strand-invasion reaction. The reaction was allowed to proceed for 2 min after which the helicase was added to unwind the D-loop substrate [18].
5.3. Unwinding reactions
The unwinding reactions of R-loop and D-loop substrates were performed by the addition of UvsW (200 nM) [18]. Aliquots from the reactions were stopped at various times with quenching buffer containing 20 mM EDTA, proteinase K and 0.2% SDS. The products were analyzed on a 1% agarose gel which was run at 40 V in 0.5× TBE for 16 h at room temperature. Upon completion of the agarose gel electrophoresis, the gel was dried on a DE81 paper followed by vacuum drying the gel on DE81 (Whatman) paper for 1 h at 60 °C. The dried gel was exposed to a Phosphoimager screen overnight, imaged and analyzed using ImageQuant. The results obtained indicated that D-loop and R-loop DNA structures served as good substrates for unwinding by UvsW, whereas they are not substrates for Dda mediated unwinding [18,19].
6. Fluorescence-based assay for monitoring helicase activity
A continuous fluorescence-based assay has been used to study the unwinding activity of Dda helicase. This involves a duplex DNA that contains the fluorescent base 2-aminopurine (2-AP) which has been uniformly incorporated into one of the duplex strands. 2-Aminopurine is an adenine analog that can form a Watson–Crick base pair maintaining the overall structural integrity of duplex DNA. 2-Aminopurine fluorescence is sensitive to the surrounding environment. The fluorescence of the 2-AP is quenched 2-fold when it is annealed to its complementary strand. Upon unwinding of the duplex DNA catalyzed by Dda helicase the fluorescence of 2-AP is restored and its recovery was followed in real-time by a stopped-flow pre-steady-state or steady-state fluorescence spectroscopy. The forked substrate used for the unwinding assay of Dda is shown in Fig. 6 and the positions of 2-AP in the substrate are indicated by bold faced letters. The experiment was carried out on an Applied Photophysics stopped-flow instrument (Surrey, U.K.). Forked DNA (500 nM) and Dda helicase (100 nM) were incubated in the unwinding buffer containing 20 mM Tris acetate (pH 7.8), 125 mM potassium acetate in one syringe, and ATP (6 mM) and magnesium acetate (20 mM) were placed in the other syringe. The solutions were excited at 310 nm with a bandpass of 30 nm with a 330 nm cut-off filter to monitor the emission of 2-AP. The reaction was initiated by rapidly mixing the contents of the two syringes and the fluorescence enhancement was assessed as a drop in signal voltage by the instrument. The data obtained from this assay agreed very well with data from a radiometric assay with the same substrate. The fluorescence assay being continuous and observable in real-time is very attractive for the study of helicase activity. Another such assay from the Kowalczykowski laboratory employed the change in the intrinsic fluorescence of the E. coli ssDNA-binding protein (SSB) upon binding the DNA [38]. The ssDNA generated by the unwinding action of E. coli RecBCD helicase was bound by SSB producing a change in its fluorescence that enabled the determination of the kinetic properties of RecBCD helicase.
Fig. 6.
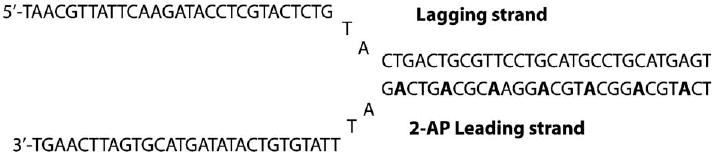
2-AP containing replication fork structure with regularly spaced seven 2-AP on the duplex region of the fork. This structure was employed in the fluorescence-based study the unwinding activity of helicase using the fluorescence signal change of 2-AP unwinding the duplex region.
7. Functional form of Dda during unwinding reactions
Many helicases are known to function as hexamers forming a ring around the loading strand that they use for unwinding the duplex DNA. Examples of these oligomeric helicases include T4 gp41, T7 gp4, E. coli DnaB and other enzymes that belong to the DnaB-like helicase superfamily [39,1]. These helicases are generally highly processive, meaning that they unwind hundreds to thousands of base pairs in a single binding event. The ring-shape structure allows multiple DNA binding sites as well as topological binding, which contributes to the processive unwinding of DNA. Hence, it is important to understand the oligomeric form of these enzymes and therefore, the nature of the functional form of Dda involved in its unwinding reactions was examined with pre-steady-state kinetic analysis [40]. If a helicase functions as a processive monomer, then the pre-steady-state burst amplitude of the unwinding reaction should be similar to the enzyme concentration provided a stoichiometric tight 1:1 complex was formed from the enzyme and the DNA substrate. However, if the enzyme functions as a higher order oligomer or is distributive, then the observed burst amplitude of the helicase activity would be significantly less than the enzyme concentration used in the reaction. The unwinding reactions were performed in a Kintek rapid chemical quench flow apparatus (Austin, TX). Varying concentrations of Dda helicase incubated with duplex DNA substrates (12 bp in length) where the 5′-end of the loading strand was radiolabeled with [γ-32P]ATP by T4 PNK in a buffer containing 25 mM Tris acetate (pH 7.8), 125 mM potassium acetate was placed in the first reaction syringe. The second reaction syringe contained a mixture of ATP (10 mM) and magnesium acetate (20 mM) in the same buffer. The reaction was initiated by mixing the contents of the reaction syringes and quenched at various times with 400 mM EDTA introduced from the quench syringe. An annealing trap of a complementary oligonucleotide was either included in the quench buffer or the reaction syringe containing ATP to sequester the unwound product and prevent the rehybridization of the oligonucleotides. Samples were analyzed by 20% native PAGE (Fig. 7A) and quantitated with a Phosphoimager as discussed above.
Fig. 7.
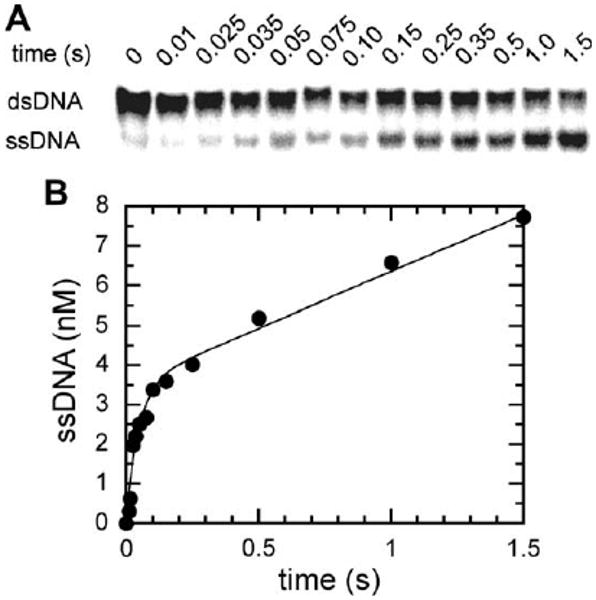
Duplex DNA unwinding catalyzed by Dda helicase under pre-steady-state conditions. (A) Products of the helicase reaction showing the separation of the reactants and products on a 20% native PAGE. (B) The data from A with the ssDNA product under pre-steady-state conditions plotted against time. The data were fit to a single exponential followed by a steady-state rate (Eq. (1)). A burst amplitude of 3.5 ± 0.2 nM was obtained when 4 nM Dda helicase was used, implying that the enzyme functions as a monomer during unwinding of the duplex DNA. (Figure reproduced from Ref. [40].)
| (1) |
| (2) |
The product formed during the reaction was plotted against time and the data were fit to the equation describing a single exponential followed by a steady-state rate (Eq. (1)) using Kaleidagraph software (Fig. 7B). When a protein trap (5 μM poly(dT)) was included in the reaction, data were fit to a single-exponential (Eq. (2)) because the steady-state phase was prevented by the trap. It was observed that the burst phase amplitude corresponded to 3.5 nM when 4 nM enzyme was used in the unwinding reactions (Fig. 7). The rate of the burst amplitude is unaltered over a range of 4–100 nM enzyme concentrations examined. Another assay tested for DNA-independent oligomerization of Dda with varying concentration of Dda (20–150 nM) under constant DNA concentration (200 nM) again confirmed that the burst amplitude observed correlated stoichiometrically to the enzyme concentration i.e., the amplitude remained >75 % of the total enzyme. Furthermore, when the ratio of protein to DNA was changed, the burst amplitude still corresponded well to the enzyme concentration and the rate of the burst phase remained constant. All these results indicate that high-order oligomers are not necessary for Dda mediated DNA unwinding so the enzyme functions as a monomeric helicase.
8. Translocation activity of UvsW and Dda helicases
The duplex DNA unwinding mechanism of helicases is thought to be coupled to the mechanism of translocation by these enzymes on ssDNA. The theoretical basis for the unwinding activity of helicases is, in fact, dependent upon their ability to translocate on ssDNA. Based upon the theory developed by von Hippel, the steady-state kinetic parameters kcat and KM-DNA can be utilized in the determination of their translocation property, through the relationship dependence of the ATPase activity and the length of DNA lattice [41]. Accordingly the DNA length dependent ATPase activities of Dda and UvsW helicases during translocation were investigated [12,42]. In these studies ssDNA oligonucleotides of lengths varying from 10 to 150 bases were rationally designed so that secondary hairpin and intrastrand duplex structures were eliminated. The sequences of the oligonucleotides used for the translocation assay are listed in Table 2.
Table 2.
Oligonucleotides used in the analysis of the translocation activity of UvsW.
| Substrate | Oligonucleotide sequence |
|---|---|
| TS10 | 5′-ATGACACA GT-3′ |
| TS15 | 5′-AGATGACACAGTAAG - 3′ |
| TS20 | 5′-ACACAGTAAGAGATGACACA - 3′ |
| TS30 | 5′-AGATGACACAGTAAGAGATGACACAGTAAG-3′ |
| TS45 | 5′-ACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGAC-3′ |
| TS60 | 5′-AGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAG-3′, |
| TS90 | 5′-AGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAG-3′ |
| TS150 | 5′-AGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGACA CAGTAAGAGATGACACAGTAAGAGATGACACAGTAAGAGATGA CACAGTAAG-3′ |
The rates of ATPase activity at various total concentrations of the bases for an oligonucleotide of a defined length were measured employing a well established coupled enzymatic assay that monitors the consumption of ATP spectrophotometrically during the reaction (Scheme 1). Steady-state and transient state kinetic approaches to study the ATP-dependent molecular motors have been reviewed in detail by Gilbert [43]. The coupled enzymatic NADH oxidation assay is attractive because it avoids the use of radiolabeled substrates, and monitors ATP hydrolysis in a continuous manner through redox changes in the chromophoric NADH cofactor.
Scheme 1.
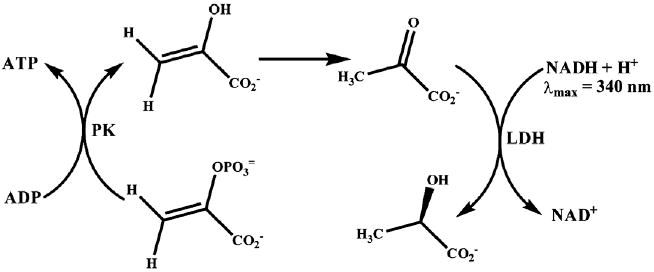
Coupled enzymatic NADH oxidation assay.
The ssDNA substrate used for the translocation assay was dissolved in the helicase buffer (25 mM Tris acetate (pH 7.8), 125 mM potassium acetate, 10 mM magnesium acetate) containing 1 mM ATP, 2 mM PEP, 0.2 mM NADH, 5 U/ml LDH and 10 U/ml PK. ATP hydrolysis rates were followed by monitoring the oxidation of NADH to NAD+ at 340 nm and the rates obtained in AU/min were converted to M/min by dividing the rates by 6220 M−1 cm−1, the extinction coefficient of NADH at 340 nm. The measured rates were plotted as a function of the concentration of bases and were fit to the Michaelis–Menten equation to obtain the kinetic parameters KM-DNA and kcat. The affinity of UvsW for the ssDNA translocation substrate is represented by the KM-DNA that in turn were plotted as a function of varying ssDNA length. For UvsW helicase the values of KM-DNA displayed a sharp decrease until the length of the DNA substrate was 30 nt long and remained relatively constant thereafter. This suggests an optimal binding site size of 20–30 bases for translocation of UvsW helicase. Likewise, the kcat values also remained unchanged until a DNA length of at least 20 bases was reached. Upon reaching this optimal DNA length, kcat increased hyperbolically from 20 to 150 bases. Although the binding affinity had reached its maximum after a DNA length of 30 bases, the rate of ATP hydrolysis increased with increasing ssDNA length consistent with UvsW being an ATP-driven ssDNA translocase. The data best fit to the following kinetic scheme (Scheme 2) and Eqs. (3)–(5).
Scheme 2.
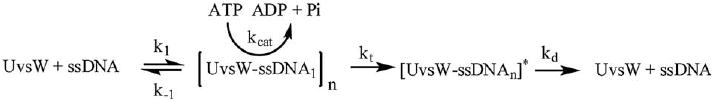
Kinetic scheme for ATP-dependent ssDNA translocation of UvsW.
| (3) |
| (4) |
| (5) |
The kcat dependence on the DNA length is represented by Eq. (3) where KG is defined by Eq. (4). In the above equations, V represents the value of kcat on indefinitely long ssDNA, λ is the length of the ssDNA lattice, n is the minimal length of ssDNA required for optimal translocation activity (maximal binding), kt represents the rate constant for the translocation step of the ATP-driven process, kd represents the dissociation of UvsW enzyme from the end of the ssDNA substrate, k1 and k−1 represent the rate constants of binding and dissociation from the internal regions of the DNA. Furthermore, kt is related to the intrinsic rate of translocation of a single nucleotide (ki) as shown in Eq. (5). Fitting the kcat data from different lengths of DNA to Eq. (3) yielded values of V = 120 ± 16 s−1, KG = 65 ± 9 bases and n = 23 ± 4 bases.
8.1. Processivity of UvsW translocase
The processivity of UvsW while translocating on ssDNA was determined by utilizing the ability of heparin to bind UvsW competitively in the presence of ssDNA. Heparin is a commonly used passive trap for proteins that bind nucleic acids. The dissociation rate of UvsW was measured by preincubating UvsW (47 nM) with ssM13 DNA(25 μM) in helicase buffer containing PEP (2 mM), NADH (0.2 mM), PK/LDH (10 U/ml and 5 U/ml, respectively) for several minutes followed by the simultaneous addition of ATP (1 mM) and heparin (0.5 or 1 mg/ml). The time-dependent decrease in ATPase activity monitored at 340 nm employing the coupled NADH oxidation assay can be used to measure the dissociation of UvsW from the ssDNA. Control experiments that involved the simultaneous addition of UvsW and ATP to a reaction mixture containing ssDNA and heparin (0.5 or 1 mg/ml) demonstrated the ability of heparin to successfully outcompete ssDNA for UvsW binding and that the ATP hydrolysis rate of the heparin-bound form of UvsW was negligible. The time-dependent reduction in the rate of ATPase activity at heparin concentrations of 0.5 and 1.0 mg/ml was fit to a single-exponential equation yielding rate constants for dissociation of 0.062 and 0.053 min−1, respectively. The fact that the dissociation rate constants at both these concentrations of heparin are similar indicates that heparin is unable to trap UvsW that is actively translocating along the ssDNA. To ensure that UvsW was indeed translocating on the ssDNA, rather than merely hydrolyzing ATP while bound to DNA, the same order of addition reactions were carried out on the TS60 substrate. In the presence of this short linear ssDNA substrate, UvsW, in theory, should very rapidly translocate off the 5′-end of the DNA and be trapped by the heparin with minimal ATP hydrolysis and indeed no ATP hydrolysis was observed in the presence of heparin when the d(N)60 oligonucleotide was used irrespective of the order of addition.
Thus, it appears that heparin also binds to a ssDNA-bound form of UvsW prompting the use of an active-site K141A mutant to trap the ssDNA substrate in competition with the wild-type enzyme. The K141A mutant lacks the amino group that is believed to interact with the γ-phosphate group of ATP and promote ATP hydrolysis. The mutant does not alter the binding affinity of the enzyme to ssDNA substrates. When the K141A mutant and wild-type UvsW in a molar ratio of 10:1 were simultaneously added to a reaction containing ATP and ssDNA, no ATP hydrolysis is observed. However, when K141A and ATP are added simultaneously to a preincubated complex of wild-type UvsW and ssDNA, the rate of ATP hydrolysis over the first ∼60 s of the reaction was nearly equivalent to a reaction performed without K141A. The subsequent decrease in the ATP hydrolysis rate was fit to a single-exponential equation to obtain a dissociation rate constant of 0.162 min−1 or a half-life of actively translocating UvsW of >6 min. With knowledge of this dissociation rate constant and further assuming that ATPase hydrolysis is strictly coupled to translocation, the average distance translocated in a single binding event is 38 kb. To ensure that ATP hydrolysis is linked to UvsW translocation, the same order of addition experiment was carried out using a shorter d(N)60 oligonucleotide. No ATP hydrolysis was noted even with a preincubated enzyme–DNA substrate complex. This result is expected if UvsW quickly translocates off the ssDNA, which is then bound by the excess K141A in solution. All these data suggest that UvsW is a processive translocase. This probably also suggests that UvsW might be a processive helicase, which remains to be tested.
8.2. Oligomeric state of the enzyme during translocation
The steady-state ATPase assay mentioned above can also be utilized to evaluate the oligomeric state of the protein during translocation. To determine their oligomeric form during the ATP-dependent translocation, the ATPase activity was measured with varying concentrations of the enzymes. The TS60 ssDNA substrate was used as substrate for UvsW in this assay. The concentration of the substrate was 10 μM, approximately 10-fold higher than the KM-DNA value and the enzyme concentration was varied from 2 to 200 nM, thus maintaining the concentration of the substrate in excess. The specific ATPase activity was calculated by dividing the observed ATP hydrolysis rate by the concentration of the enzyme and plotted against the concentration of the enzyme species. For an enzyme whose functional form remains the same over this concentration range, the specific activity for ATP hydrolysis should remain constant with increasing enzyme concentration. However, for an enzyme that forms functional high-order oligomeric structures, the specific activity will increase with increasing enzyme concentrations until it reaches the Kd of subunit association and will remain constant thereafter. It was observed that the specific activity of UvsW remained unaltered over the concentration range used in the assay suggesting that the functional form was the same at all concentrations. Thus UvsW is a monomer under these conditions or the Kd of the subunit association is less than the lowest concentration tested in the assay.
The latter possibility was ruled out by using the catalytically inactive mutant trap in a modified version of the assay. The effect of different ratios of the UvsW-K141A catalytic mutant to UvsW wild-type enzyme on the specific activity for ATP hydrolysis was examined. The concentration of TS60 ssDNA substrate used for this assay was maintained at 10 μM and the ratio of K141 mutant to wild-type enzyme ranged from 0.5 to 5. The specific ATPase activity was calculated as before and plotted against the ratio of the K141A mutant to wild-type enzyme. If the UvsW helicase forms high-order oligomeric structures, then heterooligomers of UvsW wild-type and UvsW-K141A will form upon mixing these proteins together. Consequently a reduction in the ATPase activity is expected upon increasing the ratio of K141A to wild-type enzyme, if the enzyme exists as an oligomer in these assays. The assumption for such an analysis is that the individual subunits within such oligomeric structures are not functionally active independent of each other. On the other hand if the enzyme were to exist as a monomer, the specific activity would remain constant over the different ratios of K141A mutant to the wild-type enzyme. Indeed, the specific activity of UvsW helicase remained unaltered further confirming that UvsW functions as an ATP-dependent monomeric ssDNA translocase.
8.3. Directionality of UvsW helicase translocation
The directionality of unwinding of a helicase is generally determined by employing substrates B and C in Fig. 2, and the data analyzed to determine which of those two served as substrates. In the case of some helicases, the translocation direction has been described by a fluorescent assay for DNA helicases. This assay involves fluorescent probes placed either on a 5′-end or a 3′-end of a ssDNA molecule and monitoring the change in the intensity of the fluorescent dye upon arrival of the translocating enzyme in close proximity to the dye. The requisites for this assay are that the protein binding to the dye should give rise to a change in the fluorescence intensity. Examples in the literature include UvrD and PcrA helicases where the directionality of these enzymes was determined by monitoring the change in the fluorescent signal of the probes [44,45]. However, this is not always the case with other enzymes, especially for UvsW that has a strong affinity for the bulky hydrophobic dyes. Therefore, a method that uses a protein block to study the DNA replication in T4 phage developed in our laboratory was utilized [12].
This involves the use of a biotin-streptavidin block at the 5′-end or the 3′-end of the oligonucleotide. In the presence of a streptavidin block, there will be a reduction in the translocation of UvsW off the end of the DNA due to the presence of the block. Consequently, the kinetic parameters of ATPase activity of the enzyme will be altered leading to a decreased KM-DNA due to an increased residence time. These scenarios are schematically shown in Fig. 8 A1 and B1. Analogous to the determination of the kinetic parameters for DNA substrates of varying lengths, the kinetic parameters for a DNA with a biotin at either the 5′-terminus or the 3′-terminus in the presence or the absence of streptavidin were determined and are shown in Fig. 8 A2 and B2. In the case of UvsW, a 9-fold reduced KM-DNA was obtained with the DNA carrying a biotin/streptavidin complex on the 5′-end of ssDNA indicative of a 3′-to-5′ unidirectional translocation of UvsW on ssDNA lattice [42]. With Dda a 8-fold reduction in the KM-DNA was observed with a 3′-biotinylated ssDNA, which suggests that the Dda helicase translocates unidirectionally from 5′-to-3′ on ssDNA substrates [12].
Fig. 8.
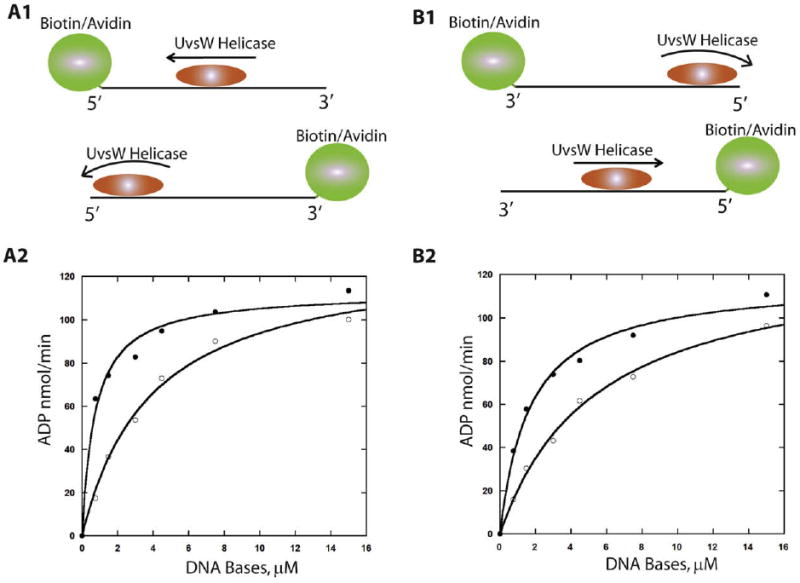
Biotin–streptavidin block assay in the determination of UvsW translocation directionality. (A1 and B1) Schematic representation of the translocation of UvsW helicase on the biotin–streptavidin bearing DNA. Translocation of the enzyme away from the biotin–streptavidin block leads to rapid dissociation from the end of the DNA, whereas dissociation is prevented when translocation occurs in the direction of the block. ATPase activity of UvsW as a function of the 5′-biotin-labeled 30mer (A2) and 3′-biotin-labeled 30mer (B2) in the presence (closed circles) and absence (open circles) of streptavidin (1 μM).
Additional evidence for the 3′-to-5′ unidirectional translocation of UvsW was also provided by using a homopolynucleotide trap that binds UvsW with the same affinity as the mixed sequence oligonucleotide but differs in its significantly reduced induction of ATPase activity. This property of UvsW was exploited to determine the translocation directionality by placing d(T)30 on either the 3′ or 5′-end of a d(N)30 ssDNA, where N represents a mixed sequence of oligonucleotides. The kinetic parameters of these modified substrates were again determined using the enzymatic coupled spectrophotometric assay described above. It was observed that the ATPase activity is 3-fold greater when the d(T)30 moiety was placed on the 3′-end than the 5′-end of DNA [42]. However, no change in KM-DNA was observed, but only a higher activity due to an increase in the ATP turnover rate [42]. This increased ATP turnover can be rationalized in terms of the enyzme's binding being distributed approximately equally between the d(T)30 on the 3′-end and the mixed sequence from which it would translocate in an ATP-dependent manner and dissociate from the DNA. However, binding of the enzyme on a substrate with d(T)30 on the 5′-end would trap the enzyme on the d(T)30 part of the substrate and thus displaying reduced turnover [42]. Internal dissociation would account for the residual ATP hydrolysis. This further supports that UvsW translocates unidirectionally from 3′-to-5′ on a ssDNA substrate. This assay can be employed for any helicase translocation provided there is a polymer that supports only binding of the protein but not its translocase activity. Examples of such polymers include peptide-nucleic acids (PNA) that carry a neutral backbone composed of peptide bonds and RNA oligonucleotides as probes for DNA helicases and, PNA and DNA as probes for RNA translocases.
9. Protein displacement activity of Dda helicase
During the physiological function of helicases and translocases, these enzymes frequently encounter protein blocks on their path. Such protein blocks prevent the translocation and unwinding activities of these enzymes and have to be removed for the restoration of the normal function of these molecular motor proteins. These helicases may possess the ability to displace these protein blocks. Hence, it is important to test the ability of these helicases involved in DNA replication, repair and recombination for their protein displacement activity. This activity of T4 helicase gp41 and Dda has been extensively reviewed earlier [14]. A brief description is presented here for completeness. The first protein displacement activity measured was that of streptavidin displacement from a biotinylated oligonucleotide that determined the ability of helicases to increase the rate of dissociation of streptavidin from biotin [14,15]. A unidirectionally translocating enzyme on ssDNA will displace the streptavidin only from one end of the DNA. The reaction progress is monitored by employing a radiolabeled oligonucleotide and analyzed by gel separation of remaining streptavidin bound DNA from free-DNA molecules. A typical protocol uses 10 pmol of biotin-labeled DNA radiolabeled at the 5′-end with [γ-32P]ATP as described earlier. The labeled oligonucleotides were purified by passage through a Sephadex G-25 spin column to remove the free [γ-32P]ATP. The concentration of the purified radiolabeled DNA was brought up to 100 nM in stock solutions. A 100 μM solution of streptavidin was prepared by dissolving the protein in a buffer containing 25 mM HEPES (pH 7.4), 10 mM NaCl and 20% glycerol. A typical protein displacement assay of gp41, Dda or UvsW involved incubating 10 nM oligonucleotide at 37 °C for 2–3 min in helicase buffer (25 mM Tris acetate (pH 7.8), 125 mM potassium acetate, 10 mM magnesium acetate) containing 5 mM ATP, 300 nM streptavidin, 2 mM PEP, PK/LDH (10 U/ml and 16 U/ml, respectively), 0.1 mg/ml BSA, 1 mM BME. To the above incubated mixture was added 6 μM biotin followed by the addition of the helicase (gp41 or UvsW) to initiate the reaction. The streptavidin displaced during the reaction was trapped by the excess of biotin added to the reaction mixture.
The streptavidin displacement with Dda helicase required a rapid chemical quench flow method to measure the streptavidin displacement by Dda [15,14]. The assay was carried out in two ways. The first method involved incubation of Dda with 12 μM biotin, 2 mM PEP and PK/LDH (21 U/ml and 33 U/ml, respectively) in one reaction syringe and 20 nM biotinylated oligonucleotide incubated with 0.6 μM streptavidin and 10 mM ATP in the other syringe. The quench syringe contained 0.6 % SDS, 200 mM EDTA in the helicase buffer. The other method involved incubation of Dda, biotinylated oligonucleotide, streptavidin, PEP and PK/LDH that preforms the enzyme–DNA complex in one syringe and, ATP and biotin trap in the other reaction syringe. The reactions were initiated by rapidly mixing the solutions from the reaction syringes followed by rapidly mixing the quench solution at various times. Aliquots of the reactions were mixed with gel loading buffer containing 5% glycerol, 0.1% xylene cyanol, 0.1% bromophenol blue and 5 μl was loaded onto a 15% PAGE and analyzed identically to that of the unwinding reactions described above (Fig. 9). The results showed that gp41 and Dda were able to displace the streptavidin blocks when biotin was placed at the 3′-end of the oligonucleotides [15]. However, no detectable streptavidin displacement activity was observed with UvsW suggesting that the enzyme was completely blocked by the biotin/streptavidin complex (Perumal and Benkovic, unpublished results). Thus some helicases have the ability to displace protein blocks on their path whereas others such as UvsW do not.
Fig. 9.
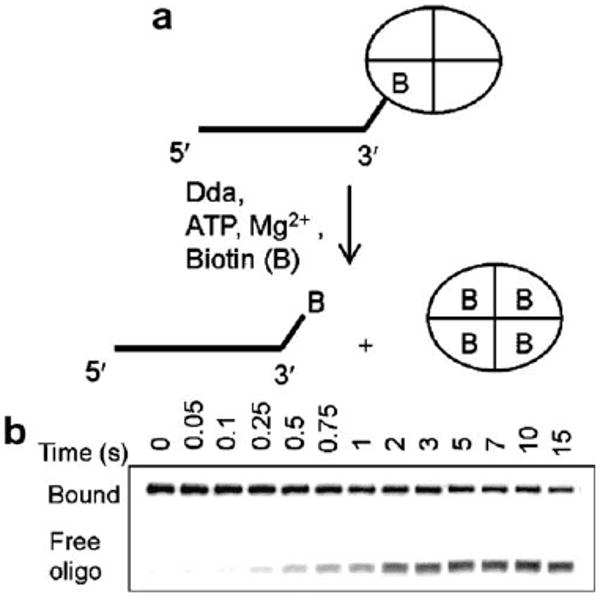
(A) Schematic representation of the streptavidin displacement assay of T4 Dda helicase. (B) The products of the assay resolved on a native PAGE. (Reproduced with permission from Ref.[13]).
The type of protein block can have different effects on protein displacement. Helicase-catalyzed protein displacement activity has been examined for removing the site-specific DNA-binding protein yeast GAL4 and the Lac repressor protein. The presence of Lac repressor protein did not inhibit the unwinding activity of Dda, but a DNA substrate containing the GAL4 binding site completely blocked the unwinding activity of Dda in the presence of GAL4 protein [46]. This emphasizes the fact that protein displacement varies in a way that depends on the specific helicase and the type of block.
10. Conclusion
Understanding the biochemical properties of helicases is key to the understanding the mechanochemistry of these molecular motors. These details of the translocase and helicase mechanism will be critical in the understanding how the energy derived from ATP hydrolysis hydrolysis is coupled to the directional movement of these enzymes. The methods presented here have been successfully applied to the superfamily 1 (Dda) and superfamily 2 (UvsW) helicases from bacteriophage T4, but will provide the necessary details to study the translocation activity and unwinding activity of other helicases. Through steady-state and pre-steady-state kinetic analyses, the oligomeric functional form of these enzymes during translocation and helicase activity can be determined. Furthermore, an estimate of the processivity of these enzymes can be measured from the protein trap or heparin trap experiments detailed above. The protein displacement activity of the helicases is likely to be important in an in vivo situation where these enzymes encounter protein blocks during DNA metabolism. Protein displacement activity can be determined easily with protein blocks such us biotin–streptavidin block or site-specific DNA-binding proteins.
Acknowledgments
This work was supported by the National Institutes of Health (GM013306) to S.J.B. and (GM059400) to K.D.R.
References
- 1.Singleton MR, Dillingham MS, Wigley DB. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 2.Ellis NA. Curr Opin Genet Dev. 1997;7:354–363. doi: 10.1016/s0959-437x(97)80149-9. [DOI] [PubMed] [Google Scholar]
- 3.Nelson SW, Zhuang Z, Spiering MM, Benkovic SJ. T4 Phage Replisome. In: Cameron C, editor. Viral Genome Replication. Springer; New Jersey: 2009. pp. 349–376. [Google Scholar]
- 4.Liu CC, Alberts BM. J Biol Chem. 1981;256:2813–2820. [PubMed] [Google Scholar]
- 5.Young MC, Schultz DE, Ring D, von Hippel PH. J Mol Biol. 1994;235:1447–1458. doi: 10.1006/jmbi.1994.1100. [DOI] [PubMed] [Google Scholar]
- 6.Venkatesan M, Silver LL, Nossal NG. J Biol Chem. 1982;257:12426–12434. [PubMed] [Google Scholar]
- 7.Cha TA, Alberts BM. J Biol Chem. 1989;264:12220–12225. [PubMed] [Google Scholar]
- 8.Barry J, Alberts BM. J Biol Chem. 1994;269:33063–33068. [PubMed] [Google Scholar]
- 9.Gauss P, Park K, Spencer TE, Hacker KJ. J Bacteriol. 1994;176:1667–1672. doi: 10.1128/jb.176.6.1667-1672.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Formosa T, Burke RL, Alberts BM. Proc Natl Acad Sci USA. 1983;80:2442–2446. doi: 10.1073/pnas.80.9.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodadek T, Alberts BM. Nature. 1987;326:312–314. doi: 10.1038/326312a0. [DOI] [PubMed] [Google Scholar]
- 12.Raney KD, Benkovic SJ. J Biol Chem. 1995;270:22236–22242. doi: 10.1074/jbc.270.38.22236. [DOI] [PubMed] [Google Scholar]
- 13.Byrd AK, Raney KD. Nat Struct Mol Biol. 2004;11:531–538. doi: 10.1038/nsmb774. [DOI] [PubMed] [Google Scholar]
- 14.Morris PD, Tackett AJ, Raney KD. Methods. 2001;23:149–159. doi: 10.1006/meth.2000.1116. [DOI] [PubMed] [Google Scholar]
- 15.Morris PD, Raney KD. Biochemistry. 1999;38:5164–5171. doi: 10.1021/bi9822269. [DOI] [PubMed] [Google Scholar]
- 16.Dudas KC, Kreuzer KN. Mol Cell Biol. 2001;21:2706–2715. doi: 10.1128/MCB.21.8.2706-2715.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carles-Kinch K, George JW, Kreuzer KN. EMBO J. 1997;16:4142–4151. doi: 10.1093/emboj/16.13.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson SW, Benkovic SJ. J Biol Chem. 2007;282:407–416. doi: 10.1074/jbc.M608153200. [DOI] [PubMed] [Google Scholar]
- 19.Webb MR, Plank JL, Long DT, Hsieh TS, Kreuzer KN. J Biol Chem. 2007;282:34401–34411. doi: 10.1074/jbc.M705913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreuzer KN. Annu Rev Microbiol. 2005;59:43–67. doi: 10.1146/annurev.micro.59.030804.121255. [DOI] [PubMed] [Google Scholar]
- 21.Derr LK, Drake JW. Mol Gen Genet. 1990;222:257–264. doi: 10.1007/BF00633826. [DOI] [PubMed] [Google Scholar]
- 22.Conkling MA, Drake JW. Genetics. 1984;107:505–523. doi: 10.1093/genetics/107.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamlett NV, Berger H. Virology. 1975;63:539–567. doi: 10.1016/0042-6822(75)90326-8. [DOI] [PubMed] [Google Scholar]
- 24.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning – A laboratory Manual. Cold Spring Harbour Press; 1989. [Google Scholar]
- 25.Morris PD, Tackett AJ, Babb K, Nanduri B, Chick C, Scott J, Raney KD. J Biol Chem. 2001;276:19691–19698. doi: 10.1074/jbc.M010928200. [DOI] [PubMed] [Google Scholar]
- 26.Bachrati CZ, Hickson ID. Methods Enzymol. 2006;409:86–100. doi: 10.1016/S0076-6879(05)09005-1. [DOI] [PubMed] [Google Scholar]
- 27.Cui S, Klima R, Ochem A, Arosio D, Falaschi A, Vindigni A. J Biol Chem. 2003;278:1424–1432. doi: 10.1074/jbc.M209407200. [DOI] [PubMed] [Google Scholar]
- 28.Imamura O, Fujita K, Shimamoto A, Tanabe H, Takeda S, Furuichi Y, Matsumoto T. Oncogene. 2001;20:1143–1151. doi: 10.1038/sj.onc.1204195. [DOI] [PubMed] [Google Scholar]
- 29.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Mohaghegh P, Karow JK, Brosh RM, Jr, Hickson ID. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozsoy AZ, Ragonese HM, Matson SW. Nucleic Acids Res. 2003;31:1554–1564. doi: 10.1093/nar/gkg243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ralf C, Hickson ID, Wu L. J Biol Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- 32.Machwe A, Xiao L, Groden J, Orren DK. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 33.Kerr ID, Sivakolundu S, Li Z, Buchsbaum JC, Knox LA, Kriwacki R, White SW. J Biol Chem. 2007;282:34392–34400. doi: 10.1074/jbc.M705900200. [DOI] [PubMed] [Google Scholar]
- 34.De Felice M, Aria V, Esposito L, De Falco M, Pucci B, Rossi M, Pisani FM. Biochem J. 2007;408:87–95. doi: 10.1042/BJ20070134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yusufzai T, Kadonaga JT. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Baumann P. Mol Cell. 2008;31:463–473. doi: 10.1016/j.molcel.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 38.Roman LJ, Kowalczykowski SC. Biochemistry. 1989;28:2863–2873. doi: 10.1021/bi00433a018. [DOI] [PubMed] [Google Scholar]
- 39.Patel SS, Picha KM. Annu Rev Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 40.Nanduri B, Byrd AK, Eoff RL, Tackett AJ, Raney KD. Proc Natl Acad Sci USA. 2002;99:14722–14727. doi: 10.1073/pnas.232401899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young MC, Kuhl SB, von Hippel PH. J Mol Biol. 1994;235:1436–1446. doi: 10.1006/jmbi.1994.1099. [DOI] [PubMed] [Google Scholar]
- 42.Nelson SW, Perumal SK, Benkovic SJ. Biochemistry. 2009;48:1036–1046. doi: 10.1021/bi801792q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert SP, Mackey AT. Methods. 2000;22:337–354. doi: 10.1006/meth.2000.1086. [DOI] [PubMed] [Google Scholar]
- 44.Fischer CJ, Maluf NK, Lohman TM. J Mol Biol. 2004;344:1287–1309. doi: 10.1016/j.jmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 45.Dillingham MS, Wigley DB, Webb MR. Biochemistry. 2002;41:643–651. doi: 10.1021/bi011137k. [DOI] [PubMed] [Google Scholar]
- 46.Byrd AK, Raney KD. Nucleic Acids Res. 2006;34:3020–3029. doi: 10.1093/nar/gkl369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong F, Weitzel SE, von Hippel PH. Proc Natl Acad Sci USA. 1996;93:14456–14461. doi: 10.1073/pnas.93.25.14456. (1445) [DOI] [PMC free article] [PubMed] [Google Scholar]