

Abstract
The Abelson (Abl) family of non-receptor tyrosine kinases plays important role in cell morphogenesis, motility and proliferation. Although the function of Abl has been extensively studied in leukemia, its role in epithelial cell invasion remains obscure. Using the Drosophila wing epithelium as an in-vivo model system, we demonstrate that overexpression (activation) of Drosophila Abl (dAbl) causes loss of epithelial apical/basal cell polarity and secretion of matrix metalloproteinases, resulting in a cellular invasion and apoptosis. Our in vivo data indicate that dAbl acts downstream of the Src kinases, which are known regulators of cell adhesion and invasion. Downstream of dAbl, Rac GTPases activate two distinct MAPK pathways: JNK signaling (required for cell invasion and apoptosis) and ERK signaling (inducing cell proliferation). Activated Abl also increases the activity of Src members through a positive feedback loop leading to signal amplification. Thus targeting Src-Abl, using available dual inhibitors, could be of therapeutic importance in tumor cell metastasis.
Keywords: Drosophila, Abl, Src, kinase, cell invasive behavior
Introduction
Cell invasion is a critical process during the development and maintenance of multicellular organisms. It is important during many processes including gastrulation, axon guidance, border cell invasion in Drosophila, germ cell invasion in zebrafish, wound healing, and for normal functioning of the immune system. Deregulated cell invasion often results in dramatic consequences such as congenital brain defects, vascular disease, chronic inflammatory diseases, or and metastasis (Luster et al., 2005; Sanchez-Madrid and del Pozo, 1999; Yamaguchi et al., 2005). Cell invasion is an active process, with active remodeling of cell junctions along with actin cytoskeleton changes that modify contacts with neighboring cells. This Epithelial to Mesenchymal Transition (EMT) alters cell-cell and cell-matrix interactions leading to cell movement (Thiery, 2003; Yang et al., 2008). It is the aggressive and uncontrolled cell invasion that leads to tumor metastasis from the site of origin. Understanding of the molecular basis of cell invasion processes can be used effectively to target tumor metastasis in cancer patients.
Oncogenic activation of many genes not only results in tumor formation through growth but also leads to loss of cell polarity and subsequent metastasis. One gene that has been studied extensively for its oncogenic potential is the Abelson kinase (Abl) (Sawyers, 1999). Abl kinases form a conserved subfamily of non-receptor tyrosine kinases, including Drosophila Abl (dAbl) and mammalian c-Abl and Abl-related gene (Arg) (Lanier and Gertler, 2000). Mammalian Abl and dAbl differ in that the former shows both nuclear and cytoplasmic localization, while dAbl is primarily cytoplasmic (Taagepera et al., 1998). Nuclear Abl has been shown to regulate cell cycle progression, while cytoplasmic Abl mostly associates with actin cytoskeleton and is found at cellular junctions (Van Etten, 1999). Consititutive activation of Abl kinase (via a chimeric Bcr-Abl oncogenic isoform) in CML (chronic myelogenious leukemia) patients results in uncontrolled proliferation of blood cells (Sawyers, 1999).
Recent studies have begun to implicate its role in other forms of cancer, independent of the Bcr locus translocation mediated activation (Lin and Arlinghaus, 2008; Sirvent et al., 2007; Srinivasan and Plattner, 2006). These studies have suggested a role of c-Abl in solid tumors, including highly invasive breast and lung cancer cells. In order to study the cell biology and molecular mechanism of Abl mediated cell invasion in an intact organism, we used the Drosophila wing epithelium paradigm (Vidal et al., 2006). The Drosophila wing imaginal disc is a columnar pseudostratified monolayer epithelium and provides a very good model to study gene function in the contexts of cell polarity, growth, and invasive behavior.
Our study suggests that overexpression (activation) of dAbl induces cell migratory behaviour, associated with loss of epithelial polarity, secretion of matrix metalloproteinases (MMPs), and exclusion of cells from the epithelial sheet, which eventually die as evident by activation of Caspase 3. Cells expressing dAbl show increased cell proliferation by activation of ERK MAPK signaling. Our data suggest that dAbl serves as a general mediator of the cell invasion phenotypes caused by Src activation. Moreover, once activated dAbl leads to activation of Src kinases through unknown mechanism thus amplifying the signal in a positive feedback loop. This dAbl induced cell invasion behavior is mediated by Rac GTPAses and JNK activation. Thus, targeting Abl/Src family kinases using dual inhibitors could be of significant value in alleviating the cellular metastasis of cancer patients.
Results
dAbl activation results in cell invasion and apoptosis through a loss of epithelial cell polarity and MMP1 secretion
To explore the potential role of Abl in cellular invasion, we used the GAL4/UAS system (Brand and Perrimon, 1993) in a defined region of the Drosophila wing disc. Overexpression of dAbl (at 25°C) under decapentaplegic-Gal4 (dppGal4) control (along with UAS-GFP marking the expression domain, a stripe along the A/P compartment boundary) resulted in delamination of such cells from the normal epithelium and their migration away from the dpp expression stripe basally (Figures 1C-C″ and E-E″). Most of these cells undergo apoptosis as evident by staining for activated Caspase-3 (Figure 1C,C″; this is never observed in control discs expressing GFP alone, Figure 1A,A″). As the activity of the Gal4 protein is temperature sensitive, lower expression of dAbl at 18°C did not result in a cell migration phenotype (Figure 1B-B″), suggesting that a threshold level of dAbl activation is required to induce the migratory behavior. Accordingly, expression of dAbl at higher temperature (29°C) resulted in an increased cell migration phenotype and strong activation of Caspase 3 in many migrating cells. X/Z focal planes of such discs showed that upon dAbl expression, cells leave the normal epithelium through the basal membrane and move to areas distant to the dpp-domain (Figure 1E-E′). Similar results were obtained when dAbl was overexpressed with patchedGal4 (Figure S1A-A‴). Thus we conclude that overexpression of dAbl results in migration of epithelial cells along with a marked increase in cell death.
Figure 1. dAbl overexpression in the wing epithelium leads to cell exclusion, invasion and apoptosis by loss of cell polarity and secretion of MMP1.

Confocal projections of third instar larval wing discs where dppGal4 was used to express either GFP alone (control) or dAbl and GFP to mark cells. Temperatures are indicated. (A-J) Merge of all channels, (A′-D′, E″ and G′-J′) GFP (green channel), (A″-D″, E′) cleaved caspase-3 (red channel), (F″ and G″) Dlg and (H″-J″) MMP1 (blue channel). Here and in all subsequent figures anterior is to the left and dorsal is up.
(A-A″) dppGal4, UAS-GFP (dpp>GFP) at 29°C; note expression domain of dpp marking cells along the anterior-posterior boundary of wing discs, very low levels of caspase-3 staining are observed.
(B-B″) dpp>GFP, dAbl at18°C; looks indistinguishable from the wild-type control.
(C-C″) dpp>GFP, dAbl at 25°C, note that GFP labeled cells begin to migrate (arrowhead) away from the stripe of expression and show activation of caspase-3. Cells start to leave the dpp expression domain mostly from the hinge region of the wing disc.
(D-D″) dpp>GFP, dAbl at 29°C: many cells leave the anterior-posterior boundary expression region. The migrating cells are not restricted to the hinge region but invade throughout the dpp expression domain. Many of the migrating cells undergo apoptosis as evident by increased caspase 3 staining (D and D″).
(E-E″) X/Z section of the wing disc shown in D to reveal the location of the migrating cells below the basal membrane as they leave the epithelium basally (E and E″), which are also the cell that undergo apoptosis (E and E′).
(F-F″) Expression of dAbl with dppGal4 (dpp>GFP, Abl; GFP marks expressing cells in F and F′) stained with anti-Dlg (DLG. Magenta; in F and F″). Expression of DLG is reduced in the region where dAbl expression is induced, suggesting loss of apical-basal cell polarity.
(G-G″) Transverse (X/Z)-section of wing disc shown in (F); note reduction in DLG staining in the region where dAbl is primarily expressed (between arrows in G″); also note migrating cells below basal membrane in the GFP channel (panels G and G′)
(H-H″) Control discs dpp>GFP stained with MMP1 (H and H″). There is no expression of MMP1 in wild-type wing imaginal discs.
(I-I″) dppGal4, UAS-dAbl (dpp>GFP, dAbl) discs with anti-MMP1 (I and I″). Expression of dAbl results in strong activation of MMP1 expression in cells that are migrating away from the dpp-expression domain.
(J-J″) Expression of dAbl along with p35 (dpp>GFP, p35, dAbl) stained with anti-MMP1 (J and J″). Note suppression of cell death and expansion of width of dpp-expression domain and concomitant accumulation of “undead” cells with high expression of MMP1 (J and J″).
Loss of cell polarity has been linked to tumor growth and metastasis (Igaki et al., 2006) and downregulation of Cadherin-Catenin complexes has been linked to many types of malignant human cancers (Hirohashi, 1998). Furthermore, in many tumor cell models, overexpression of E-cadherin acts as a suppressor of cell invasion (Navarro et al., 1991; Vleminckx et al., 1991). To test whether dAbl mediated cell migration results in reduced cellular junctional complexes and associated cell adhesion, we stained wing discs for Discs large (Dlg, labeling septate junctions) and E-cadherin (a component of adherens junctions). Cells with overexpressed dAbl displayed a downregulation of Dlg from sub-apical areas (Figures 1F-F″ and 1G-G″). Similarly, we detected a downregulation of β-catenin/Armadillo and DE-cadherin (Figures S2A-A″, S2B-B″ and S2C-C″, S2D-D″ respectively) at the cell junctions of dAbl expressing cells. It is noteworthy that co-overexpression of E-Cadherin along with dAbl resulted in suppression of the dAbl mediated cell migration phenotypes, suggesting that loss of cell polarity is a hallmark of dAbl induced cell migration (Figure S2E-E″).
The basement membrane is a specialized sheet of Extra Cellular Matrix (ECM) on the basal side of epithelial tissues and essential for integrity of epithelial cells. Basement membrane remodeling is a critical process during the development of an organism and in progression of many types of cancer (Coussens and Werb, 2002; Srivastava et al., 2007). Its degradation by enzymes of the Matrix Metalloproteinase's (MMP) family, cleaving components of the basement membrane, has been studied extensively during tumor invasion (Deryugina and Quigley, 2006). To test whether dAbl activation involves upregulation of MMPs, we stained dppGal4>dAbl wing discs with Drosophila MMP1 antibody. Strikingly, in discs where dAbl was overexpressed, we saw robust expression of MMP1 in migrating cells mostly at the leading edge (Figure 1I-I″; compare to control GFP expressing discs, Figure 1H-H″). Furthermore, removing a genomic copy of dMMP1 and dMMP2 suppressed the dAbl-induced invasion phenotype, confirming a role of MMPs in this dAbl-mediated cell invasion (Figure S3A-A″).
To test whether cell invasion is a primary event mediated by dAbl (not a consequence of cell death and proliferation induced by dAbl expression), we co-expressed p35, which blocks Caspase mediated cell death, along with dAbl. Co-expression of p35 with dAbl blocked cell death (as evident by lack of Caspase 3 activation; not shown), leading to an increase of dAbl/GFP positive cells (Figure 1J-J′) and resulting in a widening of the dpp domain (Figure 1J′). Importantly, these “undead” cells (Huh et al., 2004; Ryoo et al., 2004) secrete large amounts of MMPs, which accumulate at the base of the epithelium (Figure 1J″), suggesting that cell invasion is a primary event induced by dAbl.
dAbl mediates cellular proliferation by activation of ERK signaling pathway
We next tested whether dAbl overexpression in wing discs can result in proliferation as observed in CML patients (Sawyers, 1999). To test this, we performed BrdU (5-Bromo-2′-deoxy-Uridine) labeling, which gets incorporated into newly synthesized DNA of replicating cells. Overexpression of dAbl resulted in an increase in BrdU incorporation within the dpp-stripe (Figure 2B-B″; quantified in Figure S1B) as compared to control discs (GFP only; Figure 2A-A″). Like with the migration behavior, this effect of dppGal4>dAbl on cellular proliferation is milder at 25°C (data not shown) as compared to 29°C. Interestingly, we also see an increase in BrdU positive cells outside of the dAbl expression domain. Thus, our experimental data indicate that dAbl has a direct effect on cell proliferation and also has a non-autonomous effect on cell proliferation in the surrounding cells via a “compensatory proliferation” mechanism (see below). As dAbl is cytoplasmic (Taagepera et al., 1998), cellular proliferation resulting from dAbl overexpression should be independent of any nuclear function (unlike that of mammalian Abl). The ERK/MAPK signaling pathway has been shown to promote cellular proliferation, differentiation, and survival in normal cells. Thus, we tested whether dAbl overexpression leads to activation of ERK, using anti-diphosphoERK antibodies (dpERK), which labels the active form of ERK. Activation of dAbl resulted in an increase of dpERK staining in the cells, which express dAbl (Figure 2D-D″ and 2E-E″). These results suggest that dAbl causes a cell-autonomous increase in cell proliferation via ERK signaling.
Figure 2. dAbl induces cell proliferation by activating ERK signaling pathway.

All panels show third instar wing discs either expressing GFP alone (A and C; controls), or dAbl and GFP (B and D) under dppGal4 control at the indicated temperatures. The discs are stained for BrDU (red in A-B) or di-phosphoERK (activated ERK, magenta channel in C-D). (A-D) Merged channels, (A′-D′) green channel-GFP, and (A″-D″) either red (BrDU) channel or magenta (dpERK) channel.
(A-A″) dpp>GFP control wing discs
(B-B″) dpp>GFP, dAbl expression at 29°C. Note an increase in BrDU staining in the domain where dAbl is overexpressed (arrow in B″).
(C-C″) dpp>GFP wing discs.
(D-D″) dpp>GFP, dAbl expressed at 25°C. Note increase in the activation of ERK/MAPK (anti-dpERK; D and D″), as compared to control, in the dAbl over expressing cells.
(E-E″) XZ-section of wing disc shown in (D) to highlight anti-dpERK expression (E and E′) in migrating cells (E and E″). Most of dpERK labeling is detected in cells that also express dAbl (marked by GFP) suggesting its role in cell proliferation.
dAbl is a primary mediator of dCsk induced cell invasion
C-terminal Src kinase (Csk), and its paralog Chk, act as tumor suppressors by inhibiting the activity of Src family members, and Csk itself has been implicated in a number of tumor types (Biscardi et al., 2000; Irby and Yeatman, 2000; Sugimura et al., 2000). Local inhibition of dCsk (using inhibitory RNA transgenic expression; UAS-dCsk-IR) in cells surrounded by wild-type cells, leads to the extrusion of these cells through the base of the epithelium, migration of these cells away from the dCsk mutant area, and subsequent cell death by activation of caspases (Vidal et al., 2006). As overexpression/activation of dAbl results in similar phenotypes, we hypothesized that dAbl mediates the dCsk phenotype. Expression of UAS-dCsk-IR with dppGal4 resulted in migration of GFP positive cells away from the dpp-expression domain (Figure 3A-A), similar to the dAbl overexpression phenotype (Figure 1C-C′). Strikingly, co-expression of UAS-dCsk-IR along with dAbl resulted in a synergistic enhancement of the phenotype even at low temperature (18°C; Figure 3B-B′), where neither dAbl nor dCsk-IR showed any effects alone (Figure 1B-B′ and not shown). This was accompanied by strong activation of MMP1 (Figure 3A,A″), expressed at the leading edge of the migrating cells. Importantly, the cell migration phenotype induced by dCsk-IR was suppressed by reducing dAbl function (co-expressing dCsk-IR with dAbl-RNAi [dAbl-IR]), suggesting that dAbl acts downstream of dCsk (Figure 3C-C″).
Figure 3. dAbl is a primary mediator of dCsk-IR induced cell invasion behavior.
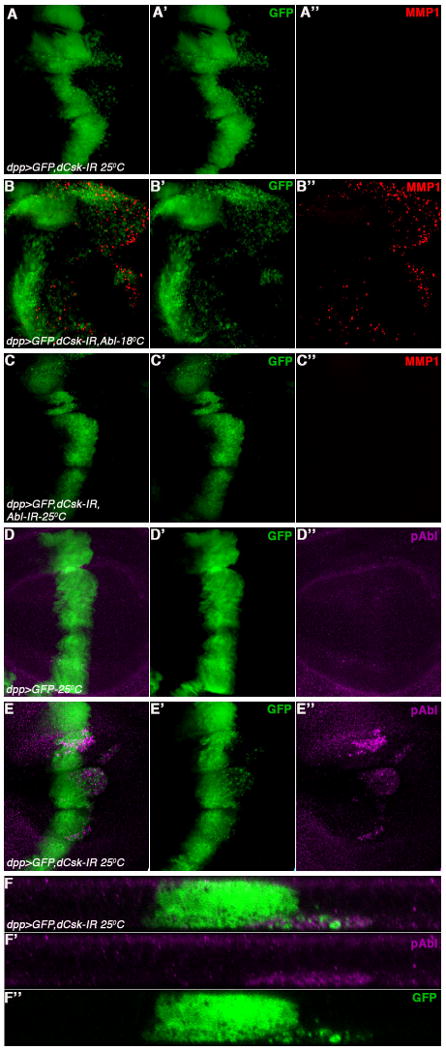
All panels show third instar wing discs expressing GFP with other genotypes and temperatures indicated under dppGal4 control. The discs are stained with anti-MMP1 (red in A-C) or anti-phosphoAbl (pAbl, activated dAbl; magenta in D-E).
(A-F) Merged channels, (A′-E′ and F″) green channel-GFP, and (A″-E″) either red (anti-MMP1) channel or magenta channel (anti-pAbl).
(A-A″) dpp>GFP, dCsk-IR (UAS-dCskRNAi, with UAS-Dcr2 to increase RNAi efficiency) results in a cell invasion phenotype (A and A′) with only low MMP1 upregulation (A and A″).
(B-B″) Co-expression of dAbl along with dCsk-IR (dpp>GFP, dCsk-IR, Abl) at 18°C results in a massive cell invasion phenotype (B and B′) along with strong MMP1 upregulation in migrating cells (B and B″). The number of migrating cells is much higher than when dCsk-IR (A′) or dAbl (Fig. 1B) are expressed individually, suggesting a synergistic interaction between dCsk knock-down and dAbl overexpression.
(C-C″) Co-expression of dAbl-IR along with dCsk-IR results in a suppression of the cell invasion phenotype (C and C′; note that integrity of dpp-expression domain is largely restored) suggesting that dAbl is acting downstream of dCsk. There is also no detectable MMP1 upregulation (C, C″).
(D-D″) dpp>GFP control wing discs stained with anti-pAbl (D and D″). Note low level staining of pAbl throughout wing discs.
(E-E″) dpp>GFP, dCsk-IR expression (E-E′) causes activation of Abl as detected by anti-pAbl (E and E″), suggesting that dCsk negatively regulates dAbl activation.
(F-F″) XZ-section of wing disc shown in (E) to highlight anti-pAbl expression (F and F″) in migrating cells (F and F′). Most of pAbl is detected in cells that have migrated below the basal membrane, suggesting its role in Csk-IR mediated cell invasion.
As the effect caused by reduction of dCsk can be suppressed by dAbl-IR, we tested whether reduced dCsk function (Csk-IR) leads to activation of dAbl (using anti-phosphoAbl [pAbl], raised against an evolutionary conserved tyrosine residue and indicative of active dAbl (Stevens et al., 2008)). Expression of dCsk-IR in wing discs led to strong activation of dAbl as detected with this antibody (Figure 3E-E″, 3F-F″; compare to control discs with a flat basal anti-pAbl staining throughout the disc, Figure 3D-D″). Taken together, these data indicate that dAbl acts downstream of dCsk.
Activation of Drosophila Src kinases leads to cell invasion and death phenotypes by activating dAbl
Src family kinases (SFKs) play key roles in signal transduction of cellular growth, differentiation, invasion and survival. In wild-type, SFKs are maintained in an inactive state by tyrosine phosphorylation of their C-terminal tail by Csk. In Drosophila, dCsk has been shown to antagonize the two Drosophila Src family members Src42A and Src64B (Pedraza et al., 2004). To test whether dCsk causes the cell invasion and dAbl activation phenotype via Src, we analyzed the role of Src in cell invasion directly and its relationship with dAbl activation. Overexpression of either Src with dppGal4 is lethal. To circumvent this problem, we used the temperature-sensitive GAL4/tubulinGAL80ts system to control expression levels of Src42A and Src64B using dppGal4. Overexpression of either Src42A or Src64 resulted in a strong cell invasion phenotype (Figure 4A-A′ and 4B-B′) along with activation of Caspase 3 and upregulation of MMP1 (Figure 4A-A‴ and 4B-B‴), suggesting that both Src42A and Src64B can function in the cell invasion process.
Figure 4. Src42A and Src64B overexpression leads to cell invasion and dAbl activation.

Third instar wing discs expressing GFP with other UAS-transgenes under dppGal4 control. The discs are stained for cleaved Caspase 3 (red in A-C) and anti-MMP1 (blue in A-C) or anti-pAbl (activated dAbl; magenta in D). (A-D) Merged channels, (A′-D′) GFP, (A″-C″) red (anti-cleaved Caspase 3), magenta (D″) (anti-pAbl) or blue (anti-MMP1) (A‴-C‴)
(A-A‴) dpp>GFP, Src42A (with tubGal80ts/+ in the background; Gal80ts expression and temperature shifting is required as dppGal4>Src42A is otherwise early larval lethal) leads to a strong cell invasion phenotype (A-A′) along with activation of cleaved caspase 3 (A, A″) and MMP1 expression (A, A‴).
(B-B‴) dpp>GFP, Src64B (with tubGal80ts/+ in the background) causes a strong cell invasion behavior (B-B′) along with activation of cleaved Caspase 3 (B, B″) and MMP1 expression (B, B‴).
(C-C‴) Genetic suppression of the cell invasion phenotype induced by overactivated Src42A by co-expression of dAbl-IR (C, C′). Reducing dAbl levels also suppresses the activation of cleaved Caspase 3 (C, C″) and reduces MMP1 expression (C, C‴), suggesting that dAbl is a primary mediator of Src42 mediated cell invasion.
(D-D″) Overexpression of dSrc42A (same genotype as in A-A‴) causes activation of dAbl as evident by staining with anti-phosphoAbl (pAbl; D, D″) in migrating cells. This is similar to the activation of dAbl seen by reducing the levels of dCsk (see Figure 2E).
(E-E″) XZ-section of wing disc shown in (D) to highlight anti-pAbl expression (E and E′) in migrating cells (E and E″). Note that the expression of pAbl is increased in the cells, which express Src42A (marked by GFP) and also in cells that have migrated below the basal membrane, suggesting its role in Src42A mediated cell invasion.
As loss of dCsk leads to the activation of dAbl and overexpression of Src exhibits very similar phenotypes to dAbl, we reasoned that the link between dCsk and dAbl could be mediated through the Src kinases. Further, as the dCsk-IR cell invasion phenotype (presumably due to activation of Src) can be suppressed by reducing dAbl function (dAbl-IR; Figure 3C), we hypothesized that the same relationship exists between the Src kinases and dAbl. We thus expressed dAbl-IR in the Src42A and Src64B overexpression background. Strikingly, expression of dAbl-IR suppressed most of the Src42A and Src64B cell invasion phenotype (Figure 4C-C′ and S3C-C″) along with reduction of cell death and MMP1 upregulation (Figure 4C-C″ and S3C-C″). These data suggest that dAbl acts downstream of Src in this process. Next we tested whether overexpression of Src can also lead to activation of dAbl similar to dCsk-IR (Figure 3E). Like dCsk-IR, overexpression of Src42A and Src64B resulted in the activation of dAbl (Figure 4D-D″, 4E-E″ and Figure S3B-B″), suggesting that dAbl is a downstream target of the Src kinases. It is interesting to note that basal levels of pAbl (in the apical region) is seen in most wing epithelial cells, but its expression is upregulated in Src42A expressing cells not only in the apical region but also in the cells which are migrating at the base of epithelium. Taken together, these data indicate that Csk controls the activation of the Src kinases, and Src in turn activates dAbl, regulating cell migratory behavior.
A Src-dAbl signal amplification loop
Cellular signaling cascades often display feedback loops either of signal amplification or attenuation depending on the context in which they are activated. We explored the possibility of a feedback loop, where dAbl could activate Src for signal amplification. To monitor Src activation, we used phospho-Src (pSrc) antibody directed against an evolutionary conserved residue in the activation loop, known to recognize the activated form of Src42A in Drosophila (Shindo et al., 2008). In control discs (dpp-Gal4, UAS-GFP) basal levels of anti-pSrc were detected throughout the disc (Figure 5A-A″), suggesting a requirement of Src42A in normal wing development. Strikingly, overexpression of dAbl resulted in robust activation of Src42A as detected by anti-pSrc staining (Figure 5B-B″ and 5C-C″) in cells where dAbl was expressed (activation of Src64B cannot be tested as anti-pSrc does not recognizes activated Src64B (Shindo et al., 2008)). Taken together with the data documenting that dAbl acts downstream of Src (the rescue of Src42A and Src64B overexpression phenotypes by co-expression of dAbl-IR; see above and Figure S3C-C‴) this result argues for a role of dAbl in a positive feedback loop leading to signal amplification. This interpretation is consistent with the observation that expression of dAbl-IR did not affect the basal levels of anti-pSrc or Src activation (Figure 5D-D″) and is further supported by the rescue/suppression of the lethality associated with Src42A-IR (under dppGal4 control) by coexpression of dAbl (not shown). This mechanism not only explains signal amplification but also some of the observations in cancer treatment/cancer cells where dual Abl/Src inhibitors in clinical trials have been shown to be more effective than individual inhibitors (Araujo and Logothetis, 2010; Das et al., 2006; Golas et al., 2003)
Figure 5. dAbl overexpression leads to Src activation.

All panels show third instar wing discs expressing GFP with other UAS-trasgenes as indicated under dppGal4 control (at 25°C). The discs are stained for anti-phosphoSrc (pSrc, activated Src42A; red in A-C). (A-C) Merged channels, (A′-C′) green channel-GFP, (A″-C″) anti-pSrc (red).
(A-A″) Control: dpp>GFP, showing endogenous staining of anti-pSrc (A-A″). Note there is a basal level of activation of Src42A throughout the wing disc tissue.
(B-B″) dpp>GFP, dAbl: Overexpression of dAbl leads to induction of anti-pSrc staining (activation of Src42A; B- B″) in the dpp-expression domain.
(C-C″) XZ-section of wing disc shown in (B) to highlight anti-pAbl expression (C and C″) in migrating cells (C and C′). Increase in the levels of pSrc can be seen in cells that express Abl (marked by GFP).
(D-D″) dpp>GFP, dAbl-IR: Decrease in dAbl levels (using Abl-IR) does not affect pSrc levels, suggesting that dAbl is not required for basal Src activation and thus likely downstream of the Src kinases. A likely feed back loop of active dAbl back to Src results in the amplification of signal as seen in panel B.
A role of Rac and JNK signaling in dAbl mediated cell invasion
The Rho family of GTPases, which in Drosophila include RhoA, three Rac genes (Rac1, Rac2, and Mtl) and Cdc42, are required for many aspects of actin organization and dynamics during cell movement (Hall, 1998). A potential role of Abl kinases in the regulation of Rac activation has been suggested (Boureux et al., 2005). Furthermore, the BCR-Abl mediated oncogenic function requires the activity of Rac (Skorski et al., 1998). We thus tested the requirement(s) of the dRacs in dAbl-mediated cell invasion. Reducing a genomic copy of all three Drosophila Rac genes, dRac1-/+, dRac2-/+ and dMtl-/+, strongly suppressed all aspects of the dppGal4, UAS-dAbl cell invasion phenotype (Figure 6A-A″), suggesting that Rac family members act downstream of dAbl in this context.
Figure 6. Rac and JNK are required for Abl mediated cell invasion.

All panels (except E) show third instar wing discs expressing GFP with other UAS-transgenes as indicated under dppGal4 control (at 25°C). The discs are stained for anti-MMP1 (red in A-B) or anti-lacZ (magenta in C-D), which is monitoring JNK activation via puc-lacZ. (A-D) Merged channels, (A′-D′) GFP, (A″-D″) either red (anti-MMP1) or magenta (anti-lacZ).
(A-A″) The cell invasion phenotype of dpp>GFP, dAbl is suppressed (A-A′) by removing a genomic copy of each of the three rac genes (rac1J10/+, rac2Δ/+, mtlΔ/+) along with suppression of MMP1 expression (A, A″) and reduction of cell growth.
(B-B″) Co expression of BskDN along with dAbl results in suppression of cell invasion (B- B′) along with suppression of MMP1 expression (B, B″). Note the dpp-expression domain becomes enlarged suggesting that Bsk suppresses cell invasion and cell death (data not shown) but not cell proliferation as a result of Abl expression.
(C-C″) dpp>GFP, dAbl causes an activation of JNK signaling as detected by upregulation of the pucLacZ reporter(C, C″).
(D-D″) XZ-sections of wing disc shown in C to highlight puc-lacZ expression (D, D″) in migrating cells (D and D′). Most of the expression of pucLacZ is detected in cells, which have migrated below basal membrane, suggesting a role of JNK signaling in cell invasion downstream of dAbl.
(E) Activation of JNK as detected by Western blot analysis. Third instar larval wing discs of indicated genotype, dpp>GFP (control) or dpp>GFP, dAbl, were probed with anti-phosphoJNK antibody (pJNK). Expression of dAbl results in pJNK activation as compared to control lane.
(F-F″) Expression of Bsk-DN and removing a genomic copy of ERK completely suppresses the cell proliferation, invasion and cell death associated with dAbl expression in the wing imaginal disc.
Experiments in cell culture and in model organisms have shown that Rho family GTPase, in particular the Racs, can activate JNK (c-Jun N-terminal kinase) signaling (Coso et al., 1995; Harden et al., 1995). To test whether Drosophila JNK, Basket (Bsk), is required for dAbl mediated cell invasion downstream of Rac, we co-expressed a Bsk dominant negative (UAS-Bsk-DN) isoform along with dAbl (expression of Bsk-DN alone does not show any cell invasion phenotypes in wing discs; not shown). When co-expressed with dAbl, Bsk-DN suppressed the dAbl mediated cell invasion and MMP1 upregulation phenotypes along with suppression of caspase activation (Figure 6B-B″ and not shown). Interestingly the dppGal4 domain became enlarged (as observed by the width of the GFP-expressing stripe of cells, Figure 6B, and increased incorporation of BrdU within the dpp-expression domain, Figure S4A-A″) as compared to dAbl alone (cf. Figure 1C′ and 6B′). This suggests that Bsk-DN can suppress the cell migration and apoptosis phenotypes associated with dAbl overexpression, but does not suppress cell proliferation, which is likely associated with ERK signaling (see above; Figure 2D-D″). To directly monitor activation of JNK signaling, we used the puc-LacZ reporter. puckered (puc) encodes a dual specificity JNK phosphatase, whose expression is regulated by JNK itself, and thus serves as a faithful read-out of JNK activation (Martin-Blanco et al., 1998). In wild-type Drosophila wing discs puc-LacZ expression is restricted to the notum region (not shown). Upon dAbl overexpression with dppGal4, puc-LacZ expression was activated in the dpp-stripe (Figure 6C-C″), and in particular detected in the migrating cells (Figure 6D-D″). Furthermore, activation of JNK was confirmed using phospho-JNK Western blots, where an increase in pJNK was seen in dAbl expressing discs (Figure 6E). Taken together, these data indicate that dAbl activates JNK signaling, which promotes the cell invasion and cell death phenotypes, and that activation of cell proliferation downstream of dAbl is likely mediated through a distinct MAPK pathway (ERK, see above).
Furthermore, we tested whether JNK and ERK are indeed the main effectors, which mediate the dAbl phenotype(s). To address this we simultaneously removed a copy of ERK (using Df(2R)Rl-10a) and expressed Bsk-DN in the dAbl expression background. Strikingly, reducing both ERK and Bsk/JNK function completely rescued all aspects of the dAbl-induced phenotypes (Fig. 6F-F″; note that proliferation within the dppGal4 domain is also suppressed as evident by normal BrdU staining and a regular width dpp-stripe, cf to Figure 6B). These data indicate that the two MAPKs mediate all phenotypic aspects associated with dAbl expression: cell invasion, proliferation, and cell death.
Discussion
Tumor metastasis is the leading cause of cancer related deaths. Mechanisms by which tumor cells acquire increased motility and invasive behavior are, however, still poorly understood. To our knowledge this is the first study to provide in vivo evidence for the role of Abl in cell invasion. Cells expressing dAbl (in the dpp-domain) become invasive and migrate into the area of the posterior compartment, where they are located basally to the basement membrane. Although during this process many cells die, those that “resist” cell death can be visualized by the presence of GFP at the base of the epithelium in either compartment (Fig 1E). Further we provide mechanistic evidence for a Src-Abl signaling cascade and a Abl/Src signal amplification loop in epithelial cell invasion. Targeting both kinase types using dual Abl/Src inhibitors in cancer patients could thus be of clinical significance. We also show that increased cell proliferation associated with Abl can be separated from its cell invasion function by distinct downstream effectors. Different MAPKs are activated downstream of dAbl and Rac, and mediate the cell proliferation and cell invasion phenotypes, respectively (see model in Figure 7).
Figure 7. Model depicting mechanism of dAbl mediated cell invasion and proliferation.
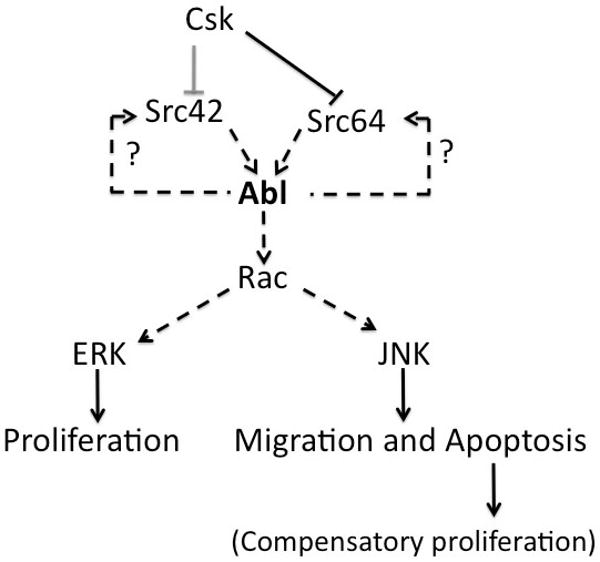
Schematic illustration of proposed model where Abl act as a key mediator of the Src activation induced cell invasion effect. Abl once activated can feedback on Src for signal amplification. Downstream, Abl acts as an activator of MAPK signaling pathways (mediated by Rac) to control cell proliferation, invasion and death (see text for details).
Abl induces metastatic behavior in epithelial tissues
Loss of cell polarity has been linked to tumor growth and cell invasion (Igaki et al., 2006). The mechanism(s) by which dAbl downregulates cell adhesion/polarity genes like DE-Cadherin, β-Catenin/Armadillo and Dlg are not known. This could be a direct effect of dAbl on junctional complexes or a consequence of the cell invasive behavior. Downregulation of E-cadherin has been linked to several types of tumors. Furthermore, Src family members have been shown to increase the turnover of AJs, which in turn would cause an increase in cell mobility, a possible mechanism by which Abl can mediate loss of cell polarity. This hypothesis is further supported by the observation that overexpression of DE-cadherin suppresses the effects induced by Src upregulation (via Csk reduction, UAS-dCsk-IR) in the retina (Vidal et al., 2006). Consistent with this notion, overexpression of DE-Cadherin rescues the dAbl induced cell invasion phenotype (Figure S2C-C″). Also, removing a genomic copy of mmp1 and mmp2 results in suppression of the dAbl cell invasion phenotype (Figure S3A-A″). Based on these data we conclude that loss of cell polarity and MMP secretion are the key factors in contributing to cell invasive behavior of dAbl expressing cells. However, we cannot completely rule out the possibility of minor contributions of unknown factors in this process
Cell proliferation mediated by dAbl expression
A complicated question is how dAbl causes cell proliferation in epithelial cells (Drosophila Abl lacks nuclear localization signals). The effect of dAbl expression results in cell autonomous and non-autonomous cell proliferation. In Drosophila, cells destined to undergo apoptosis express specific growth factors (Wingless and Dpp; their upregulation is mediated by JNK activation), inducing non-autonomous compensatory proliferation in neighboring cells (Huh et al., 2004; Ryoo et al., 2004). This compensatory proliferation is important for maintaining proper tissue homeostasis and may also be relevant for the induction of tumor cell proliferation. As dAbl activation results in cell death in migrating cells, one argument could be that cell proliferation associated with dAbl activation is a consequence of compensatory proliferation. Interestingly, dAbl expression results in an increase in Wg expression, suggesting that compensatory proliferation takes place in response to dAbl (not shown). Taken together, our data suggest that at least some aspects of dAbl mediated cell proliferation (mediated by activation of ERK) are cell-autonomous independent of such compensatory proliferation, as Bsk-DN co-expression in a dAbl overexpression background (which blocks JNK signaling and thus induction of Wg and Dpp expression (Ryoo et al., 2004)), does not block excessive proliferation within the dAbl expression domain (Figure 6B and Figure S4A-A″).
Role of dAbl in Src mediated cell invasion
The cell invasion phenotype of dAbl overexpression is similar to Csk reduction (dCsk-IR; Figure 3A) and our data indicate that dAbl acts downstream of dCsk. As Csk negatively regulates Src family kinases, this suggested that Src mediates the effect of dCsk on dAbl. Previous studies have shown that Abl can act as a substrate of Src family kinases, though other studies have shown that the opposite can also be true (Pendergast, 2002). Our data indicate that Src acts upstream of Abl and that Abl can feed back and amplify the signal through its positive effect on Src (see Results). How is the dAbl-Src feedback loop working mechanistically? From the in vivo experiments it is not possible to conclude whether dAbl acts directly on Src, dCsk, or unknown upstream components. dAbl does not co-immunoprecipitate either dCsk or the Src kinases in Drosophila S2 cells (not shown). As binding between kinases can be of very transient nature, it is possible that even if dAbl would bind Src or dCsk in vivo, we may not be able to detect it. However, our in vivo data suggest that dCsk does not mediate the dAbl effect: if dAbl would act through dCsk (by inhibiting it), phospho-Src (pSrc) levels should be similar with dCsk-IR or dAbl expression, which is not the case. dAbl expression results in a much more robust activation Src with pSrc detected in all dAbl/GFP positive cells, while dCsk-IR does not result in such strong activation. This observation suggests that dAbl does not act through dCsk in this process. Although we cannot exclude the possibility that dAbl could modulate an unknown component upstream of dCsk, the fact that co-expression of dCsk-IR and dAbl (dCsk-IR; UAS-Abl at 18°C, Figure 3B) shows a synergistic effect (even at 18°C, where neither individual transgene has a phenotype on its own) suggests that dAbl and Csk act in parallel on Src. As Abl can phosphorylate Src kinases (Pendergast, 2002), we favor a direct effect of dAbl on the Src kinases (as it is impossible to prove this in vivo this relationship is indicated by a dotted line in Figure 7).
Rac and JNK act downstream of dAbl mediating the cell invasion behavior
JNK signaling is activated in response to environmental stress and by several classes of cell surface receptors, including cytokine receptors and receptor tyrosine kinases. In mammalian cells, JNK has been implicated in oncogenic transformation in fibroblasts and hematopoietic cells (Raitano et al., 1995), apoptosis (Xia et al., 1995), and in cell invasion (Davis, 2000). In oncogenic transformation, JNK signaling can promote tumor growth, while it can also act as a tumor suppressor (Kennedy and Davis, 2003). It also functions in basement membrane remodeling during imaginal disc eversion and tumor invasion (Pastor-Pareja et al., 2004; Srivastava et al., 2007). Here, we provide evidence for a link between Src and JNK during cell invasion, mediated via dAbl. The cell invasion and apoptosis phenotypes induced by dAbl require JNK activity, while the cell proliferation function of dAbl appears to be mediated by ERK signaling. dAbl does not affect expression levels of JNK (not shown) but instead causes an increase in active JNK (phospho-JNK; Figure 6E). It is worth noting that removing a genomic copy of each of the Drosophila Rac genes suppresses all phenotypes associated with dAbl overexpression (cell invasion, death and proliferation). These data are consistent with the study of BCR-Abl mediated cell growth, which requires Rac function (Skorski et al., 1998), suggesting a general relevance of Rac GTPases as Abl effectors.
It is not established how Abl mediates Rac activation. A possible link can be Crk, which primarily consists of SH2 and SH3 domains, serving as an adaptor. Crk-I can associate with and be phosphorylated by c-Abl (Ren et al., 1994). Furthermore, ectopic expression of Crk can result in JNK activation (Dolfi et al., 1998). As overexpression/activation of dAbl results in JNK activation, Crk may provide a missing link between dAbl and Rac for JNK activation. Another candidate to mediate an interaction between dAbl and Rac GTPases can be Trio, a guanine exchange factor (GEF). Trio has two putative Rac and Rho binding domains (Debant et al., 1996). In Drosophila, trio function has been studied extensively in the context of axon guidance where it has been shown to interact with dAbl (Lanier and Gertler, 2000). Interestingly, a recent report has identified Trio as one of the GEFs responsible for invasive behaviour of glioblastoma (Salhia et al., 2008). Thus a potential role of Trio in the context of Abl mediated cell invasion warrants further investigation.
Materials and Methods
Drosophila Stocks
Flies were reared at indicated temperatures using standard procedures. The following stocks were used: UAS-Abl (E. Giniger), UAS-dCsk-IR (R. Cagan), UAS-dAbl-IR (VDRC), UAS-Src42ACA, Bsk-DN, UAS-p35 and UAS-Src64B (Bloomington stock centre), rac1, rac2, mtl (N. Paricio) and puc-lacZ (C. Pfleger), Experiments with UAS-Abl at 29°C were carried out by rearing the flies at 25°C until the second instar stage and then shifted to 29°C for 2 days before dissection and staining. The Gal4/Gal80ts system was used to induce timed expression of UAS-Src42 and UAS-Src64 transgenes under dppGal4 control.
Immunostaining and BrdU Labeling
Third instar wing imaginal discs were dissected in PBS and fixed in 4 % paraformaldehyde for 20 mins. Discs were washed in PBT (0.1% Triton X-100) followed by over night staining at 4°C with the following primary antibodies: anti-cleaved Caspase 3 (1:100, Cell Signaling), anti-dpERK (1:50, Sigma), anti-Dlg (1:200), E-Cadherin (1:20), anti-Arm (1:10) and anti-MMP1 (1:50) were all from Developmental Studies Hybridoma bank, pAbl (1:50, Biosource), anti-pSrc (1:50, Invitrogen), anti-βGal (1:1000, Cappel). After primary antibody incubation, discs were washed with PBT several times and incubated with secondary fluorescent antibodies for 2 hrs followed by washing with PBT. Mounted discs were analyzed with a Zeiss LSM510 Meta Confocal microscope. Images were processed with NIH ImageJ and Adobe Photoshop software. For BrdU labeling, larvae were dissected in S2 medium and incubated with BrdU at 29°C for 45 mins. Discs were washed in PBTW (PBS with 0.1% Tween 20) 3 times followed by DNAase (Promega) treatment for 1 hr at 37°C. Discs were washed with PBTW 3 times and stained with BrdU antibody (DSHB).
Immunoprecipitation and Western blotting
Serum starved cancer cell lines were lysed in ice cold lysis Buffer (50mM Tris pH 8.0, 150mM NaCl, 1% Triton X-100, 1mM EDTA, 5mM betaglycerophosphosphate, protease and phosphotase inhibitor cocktail, Sigma) for 20 mins. Samples were cleared in ice cold centrifuge at 13,000 rpm followed by protein estimation on the supernatant. Samples were stored at -80°C untill further processing. Frozen samples were thawed on ice and equal amounts of protein was subjected to immunoprecipitation with either anti-Abl (Rabbit K12, Santa Cruz) or with anti-Src (GD11, Upstate) antibodies for 2 hrs at 4°C. Next, samples were incubated with either Protein A or G Agarose for 1 hr followed by washing of the beads with ice cold lysis buffer. Sample buffer was added to the beads followed by boiling at 95°C for 5 mins. Proteins were visualized by immunoblotting with the following antibodies: mouse anti-phospho-tyrosine (4G12, Upstate), anti-Abl (Santacruz), anti-pSrc (Invitrogen) and anti-Src (GD11, Millipore).
Supplementary Material
Acknowledgments
We thank the Bloomington Stock Center, DSHB, Ed Giniger, and Ross Cagan for fly stocks and antibodies. We are grateful to members of the Mlodzik lab for helpful suggestions, discussions and criticism, Ross Cagan and members of the Cagan lab for discussion and reagents, in particular Tirtha Das. We also thank William Gault for critical reading of the manuscript, and Nadinath Nillegoda and Maneesha Chhikara for helpful comments and suggestions. We thank Joyce Lau, Sophy Okello and Andrea Blitzer for their technical help. Confocal laser microscopy was performed at the MSSM Microscopy SRF, supported by a NIH/National Cancer Institute shared instrumentation grant. This research was supported by NIH/National Eye Institute grant R01 EY13256 to MM.
References
- Araujo J, Logothetis C. Dasatinib: A potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 2010 doi: 10.1016/j.ctrv.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscardi JS, Ishizawar RC, Silva CM, Parsons SJ. Tyrosine kinase signalling in breast cancer: epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000;2:203–10. doi: 10.1186/bcr55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boureux A, Furstoss O, Simon V, Roche S. Abl tyrosine kinase regulates a Rac/JNK and a Rac/Nox pathway for DNA synthesis and Myc expression induced by growth factors. J Cell Sci. 2005;118:3717–26. doi: 10.1242/jcs.02491. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, et al. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–46. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, et al. 2-aminothiazole as a novel kinase inhibitor template. Structure-activity relationship studies toward the discovery of N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1- piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (dasatinib, BMS-354825) as a potent pan-Src kinase inhibitor. J Med Chem. 2006;49:6819–32. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Debant A, Serra-Pages C, Seipel K, O'Brien S, Tang M, Park SH, et al. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci U S A. 1996;93:5466–71. doi: 10.1073/pnas.93.11.5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34. doi: 10.1007/s10555-006-7886-9. [DOI] [PubMed] [Google Scholar]
- Dolfi F, Garcia-Guzman M, Ojaniemi M, Nakamura H, Matsuda M, Vuori K. The adaptor protein Crk connects multiple cellular stimuli to the JNK signaling pathway. Proc Natl Acad Sci U S A. 1998;95:15394–9. doi: 10.1073/pnas.95.26.15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golas JM, Arndt K, Etienne C, Lucas J, Nardin D, Gibbons J, et al. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63:375–81. [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Harden N, Loh HY, Chia W, Lim L. A dominant inhibitory version of the small GTP-binding protein Rac disrupts cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development. 1995;121:903–14. doi: 10.1242/dev.121.3.903. [DOI] [PubMed] [Google Scholar]
- Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998;153:333–9. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–6. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16:1139–46. doi: 10.1016/j.cub.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle. 2003;2:199–201. [PubMed] [Google Scholar]
- Lanier LM, Gertler FB. From Abl to actin: Abl tyrosine kinase and associated proteins in growth cone motility. Curr Opin Neurobiol. 2000;10:80–7. doi: 10.1016/s0959-4388(99)00058-6. [DOI] [PubMed] [Google Scholar]
- Lin J, Arlinghaus R. Activated c-Abl tyrosine kinase in malignant solid tumors. Oncogene. 2008;27:4385–91. doi: 10.1038/onc.2008.86. [DOI] [PubMed] [Google Scholar]
- Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–90. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12:557–70. doi: 10.1101/gad.12.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Gomez M, Pizarro A, Gamallo C, Quintanilla M, Cano A. A role for the E-cadherin cell-cell adhesion molecule during tumor progression of mouse epidermal carcinogenesis. J Cell Biol. 1991;115:517–33. doi: 10.1083/jcb.115.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Pareja JC, Grawe F, Martin-Blanco E, Garcia-Bellido A. Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell. 2004;7:387–99. doi: 10.1016/j.devcel.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Pedraza LG, Stewart RA, Li DM, Xu T. Drosophila Src-family kinases function with Csk to regulate cell proliferation and apoptosis. Oncogene. 2004;23:4754–62. doi: 10.1038/sj.onc.1207635. [DOI] [PubMed] [Google Scholar]
- Pendergast AM. The Abl family kinases: mechanisms of regulation and signaling. Adv Cancer Res. 2002;85:51–100. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A. 1995;92:11746–50. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren R, Ye ZS, Baltimore D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994;8:783–95. doi: 10.1101/gad.8.7.783. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Salhia B, Tran NL, Chan A, Wolf A, Nakada M, Rutka F, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol. 2008;173:1828–38. doi: 10.2353/ajpath.2008.080043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Madrid F, del Pozo MA. Leukocyte polarization in cell migration and immune interactions. EMBO J. 1999;18:501–11. doi: 10.1093/emboj/18.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330–40. doi: 10.1056/NEJM199904293401706. [DOI] [PubMed] [Google Scholar]
- Shindo M, Wada H, Kaido M, Tateno M, Aigaki T, Tsuda L, et al. Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Development. 2008;135:1355–64. doi: 10.1242/dev.015982. [DOI] [PubMed] [Google Scholar]
- Sirvent A, Boureux A, Simon V, Leroy C, Roche S. The tyrosine kinase Abl is required for Src-transforming activity in mouse fibroblasts and human breast cancer cells. Oncogene. 2007;26:7313–23. doi: 10.1038/sj.onc.1210543. [DOI] [PubMed] [Google Scholar]
- Skorski T, Wlodarski P, Daheron L, Salomoni P, Nieborowska-Skorska M, Majewski M, et al. BCR/ABL-mediated leukemogenesis requires the activity of the small GTP-binding protein Rac. Proc Natl Acad Sci U S A. 1998;95:11858–62. doi: 10.1073/pnas.95.20.11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan D, Plattner R. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells. Cancer Res. 2006;66:5648–55. doi: 10.1158/0008-5472.CAN-06-0734. [DOI] [PubMed] [Google Scholar]
- Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci U S A. 2007;104:2721–6. doi: 10.1073/pnas.0611666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens TL, Rogers EM, Koontz LM, Fox DT, Homem CC, Nowotarski SH, et al. Using Bcr-Abl to examine mechanisms by which abl kinase regulates morphogenesis in Drosophila. Mol Biol Cell. 2008;19:378–93. doi: 10.1091/mbc.E07-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura M, Kobayashi K, Sagae S, Nishioka Y, Ishioka S, Terasawa K, et al. Mutation of the SRC gene in endometrial carcinoma. Jpn J Cancer Res. 2000;91:395–8. doi: 10.1111/j.1349-7006.2000.tb00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, et al. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci U S A. 1998;95:7457–62. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Van Etten RA. Cycling, stressed-out and nervous: cellular functions of c-Abl. Trends Cell Biol. 1999;9:179–86. doi: 10.1016/s0962-8924(99)01549-4. [DOI] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL. Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell. 2006;10:33–44. doi: 10.1016/j.devcel.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Vleminckx K, Vakaet L, Jr, Mareel M, Fiers W, van Roy F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell. 1991;66:107–19. doi: 10.1016/0092-8674(91)90143-m. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr Opin Cell Biol. 2005;17:559–64. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Yang JH, Wylie-Sears J, Bischoff J. Opposing actions of Notch1 and VEGF in post-natal cardiac valve endothelial cells. Biochem Biophys Res Commun. 2008;374:512–6. doi: 10.1016/j.bbrc.2008.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.