

Iron
Properties of iron
With few exceptions life on this planet is dependent on iron for most animal and plant species. The chemistry of iron in biological systems is quite complex since it takes place in aqueous solutions, close to neutral pH and in the presence of oxygen (Aisen et al., 2001; Greenwood and Earnshaw, 1998). Iron in aqueous solutions can exist in two oxidation states, ferrous (Fe2+) and ferric (Fe3+). These are interconvertible through single electron exchanges. Fe2+ is the preferred form for the membrane transport of iron, and for heme synthesis.
In acidic solution Fe2+ and Fe3+ exist as the complexes [Fe(H2O)6]2+ and Fe(H2O)6]3+, respectively. In the absence of oxygen, or at very low oxygen tensions, Fe2+ is estable in the physiological pH range; however, the Fe3+ complex undergoes a process of hydrolytic deprotonations, leading to the rapid appearance of very insoluble species of Fe3+ as the pH becomes less acidic. At neutral pH, for example, the total concentration of Fe3+ is 10-9 M, but only 10-17 M for some of those species. When oxygen is present, Fe2+ easily oxidizes to Fe3+ dramatically decreasing its water solubility. Iron dependent life, therefore, required the evolution of special iron binding proteins to maintain this element in soluble form and to assure its bioavailability.
Redox reactions are the cause of toxicity of iron in biological systems. The presence of iron automatically aggravates any possible situation of oxidative stress (Halliwell and Gutteridge, 1990; Sies, 1991) by catalyzing the conversion of oxygen into highly reactive free radicals (Figure 1).
Figure 1.
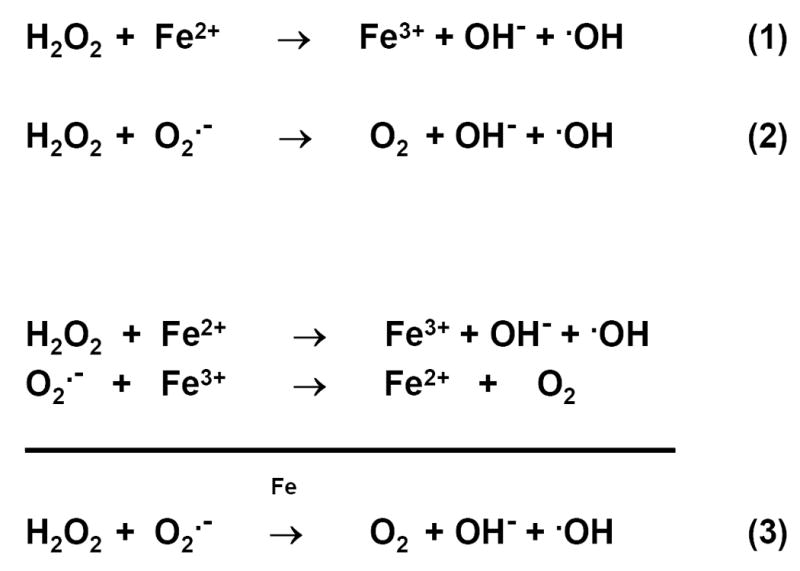
Reactive oxygen species and redox cycling of iron. Fenton reaction is represented by equation (1), Haber-Weiss reaction by equation (2), and iron-catalyzed Haber –Weiss reaction by equation (3), also known as superoxide-driven Haber-Weiss reaction (Halliwell and Gutteridge, 1990). Fenton reaction describes the decomposition of hydrogen peroxide to the highly reactive hydroxyl radical, in the presence of ferrous iron.
Several enzymes and metabolic substrates and intermediaries are present which function to neutralize these reactive molecules. It can be safely said that once a disease is shown to be caused by oxidative stress, it follows that any kind of iron overload will accelerate the disease progress, if conditions favor redox cycling of iron, as shown in Figure 1.
Iron homeostasis
Proteins that require iron have two major functions, (i) oxygen transport and storage, and (ii) electron transfer (Beard, 2006). Many additional enzymes and metabolic pathways also require iron (listed by Connor et al., 2001; He et al., 2007). To cover the needs of iron for erythropoiesis (Beard, 2006), plasma turnover of iron mostly represents recycling given the lack of renal excretion of iron. Iron homeostasis becomes almost exclussively a problem of regulating the intestinal absorption of ingested iron by hepcidin, to replenish the small amounts of iron lost by desquamation of skin and mucosas and by menstruation. As shown in studies performed over the last ten years hepcidin is a key regulator of systemic iron homeostasis explaining both the iron overload situations (hemochromatosis disorders) and the chronic anemias (Collins et al., 2008).
Cells incorporate iron through several different types of transporters (carriers). The polarized intestinal absorptive cells, for example, have transporters for the uptake of iron at their apical, brush-border membranes (DMT1, divalent metal transporter 1) (Gunshin et al., 1997) and transporters for the export of iron at the basolateral membranes (ferroportin) (Andrews and Schmidt, 2007; Mckie et al., 2000). Both use Fe2+ as substrates. Most other cells acquire iron through the transferrin receptor (TfR). This receptor controls the cellular uptake of iron by regulation at the translational level. Ferritin, the iron storage protein, and the iron transporters are also reciprocally regulated by iron regulatory proteins (IRP) and iron responsive elements (IRE), as described in detail by Rouault (2001).
This review will summarize our understanding of recent studies on iron metabolism in the retina and in the lens. Age-related macular degeneration (AMD) and age-related cataracts (ARC) will be considered under the light of photoxidative damage and iron homeostasis. With the support of data already available in the literature obtained by other authors and by our own work, a scheme will be proposed for the overall circulation of iron within and out of the eye, pumped from the retina to the vitreous body by ferroportin at the endfeet of Müller cells of the retina and from the vitreous to the aqueous humor by an endocytosis-mediated process at the anterior epithelium of the lens. Finally, the similarities of altered iron metabolism in AMD and ARC, and their apparent relationship to anemia of inflammation will be highlighted.
The retina
The blood-retinal barrier
Iron is not freely exchanged between the systemic circulation and the retina. In concordance with the two blood supplies that nourish the retina (see below) there are two distinct components that establish the blood-retina barrier: one, at the level of the retinal pigmented epithelium (RPE) in the outer retina and a second at the level of the retinal capillary endothelial cells in the inner retina (Oyster, 1999). The junctional complex between the RPE cells that form the outer component of the blood-retinal barrier has received thus far the most attention probably because of its obvious involvement in the pathogenesis of AMD, a highly prevalent, sight-threatening condition of the elderly (Jager et al., 2008).
The outer blood-retinal barrier
The RPE is a highly polarized epithelium, comprised of cells with long apical microvilli that interdigitate with the light-sensitive photoreceptor outer segments and a basolateral surface with elaborate basal infoldings that interact with Bruch's membrane. The RPE cells create an outer barrier to passive movement of polar solutes and large macromolecules that escape the choriocapillaris by sealing off the paracellular space at the level of a continuous band of tight junctions (zonula occludens) near the apical border of the RPE (Amasheh et al, 2009). This barrier prevents the passive movement of small and large hydrophilic molecules paracellularly and also allows creation of standing osmotic gradients between the apical and basolateral paracellular compartments. In addition the RPE cells phagocytose and degrade the shed tips of the photoreceptor outer segments each day and play an essential role in regenerating visual pigments (Newman, 1994).
The choriocapillaris located below the RPE-Bruch's membrane complex is highly permeable due to the presence of numerous fenestrae through the capillary wall that allow nutrients and macromolecules to easily reach the extracapillary compartment. Many of these molecules cross Bruch's membrane and reach the basolateral border of the RPE. Because of the barrier to passive diffusion near the apical surface of the RPE, the molecules that come from the choriocapillaris that are needed by the photoreceptors must enter the RPE cells through the basolateral membranes with the help of specific transport systems if they are to reach the subretinal space. The RPE thus controls the composition of the subretinal fluid that bathes the photoreceptor outer segments.
The transfer of Fe3+ bound to plasma transferrin (Tf) through the RPE was studied in cultured human retinal pigment epithelial cells (Hunt and Davis, 1992) with 55Fe- and and 125I-labeled Tf. The results obtained by these authors supported the model of receptor-mediated endocytosis favored by the investigations made in the preceding decade for the entry of iron in many cells, as the main mechanism of crossing this barrier. Their conclusion was based on the finding that inhibitors of endosome acidification (monensin, chloroquine) were very effective in blocking the uptake of radioactive iron by the RPE cells but were not effective in modifying the amount of iron released (recycled) to the medium by the cells.
Briefly, the initial event in the accepted model of receptor-mediated endocytosis of iron is the high affinity interaction of diferric Tf with the Tf-receptor (TfR) found in clathrin enriched coated pits of the cell membrane (in this case the basolateral plasma membrane of the RPE cells), leading to the internalization of the complex Fe3+/Tf/TfR into a sorting, early endosome. The acidic interior of the endosome (Yamashiro and Maxfield, 1984), created by a proton-dependent ATPase, facilitates the dissociation of Fe3+ from Tf and increases the interaction of ApoTf with the TfR (Aisen, 1992; Aisen et al., 2001). The iron dissociated from Tf must somehow be reduced to Fe2+ by a ferrireductase (McKie et al., 2001; Ohgami et al., 2005) before being transported by the proton-metal symporter DMT1 (Burdo et al., 2001; Fleming et al., 1997; Gunshin et al., 1997) of the endosomal membrane to the cytoplasm. The apoTf/TfR complex associated to the recycling, late endosome returns to the cell membrane where from, upon exposure of the endosomal interior to physiological pH, apoTf is released, leaving the TfR in the cell membrane ready to participate in a new uptake cycle. Within the context of this model, therefore, the inhibitors of endosomal acidification should be very effective in blocking the transport of iron into the cell, as found by Hunt and Davis (1992).
In their work Hunt and Davis (1992) also analyzed the nature of the cytoplasmic pool of iron released by the endosomes, to find that it was composed of a “transient” low MW fraction (<30kD by membrane filtration) and a higher MW fraction. Early in the release process, iron was mostly bound to low MW components. The low MW fraction consisted of an heterogeneous population of iron chelates which was rapidly converted to either intracellular ferritin-like material (as judged by its comigration with ferritin on non-denaturing PAGE) of higher MW for storage, or partially released to the medium. The authors further showed that most of the low MW iron, if exposed to apoTf, became bound to it. They also studied the release of radioactive iron to the culture medium (equivalent to the interphotoreceptor matrix) by RPE cells preloaded with 55Fe-Tf, to find out that at least 40% of the label was released if the mediun was frequently renewed, or if serum was added to the medium. In the in vivo situation several iron chelators including locally synthesized Tf, could be available in the interphotoreceptor matrix of the retina. This iron would thus probably be ready to enter the photoreceptors through TfR-mediated mechanisms to provide them with the high levels of iron that the extremely active metabolism of these cells demands. The supply of iron to the photoreceptors must be direct since Müller/glial cells are not present in the interphotoreceptor matrix.
A second alternative for the transfer of iron was also considered by Hunt and Davis (1992) in which the endosome vesicle is not recycled to the basolateral side of the RPE but travels across the cytoplasm towards its apical side, where it becomes fused with the apical membrane, releasing the complex Fe3+/Tf. This would constitute a Tf-mediated transcytosis mechanism. Its existence was suggested by the authors because (i) there was indeed transfer and release to the medium of intact iodine-labeled diferric-Tf and (ii) only 60% of that transfer was inhibited by monensin. However, against the quantitative relevance of this mechanism was the finding that iron-labeled Tf could not be detected in the HMW material released to the medium by the cells; this material instead had the mobility of ferritin in electrophoresis under denaturing conditions although, unexpectedly, did not react with antiferritin antibodies. Straightforward transcytosis also has the difficulty that it requires dissociation of the high affinity Fe3+/Tf complex from TfR under physiological pH conditions without the previous dissociation of Fe3+ (Aisen et al., 2001; Klausner et al., 1983). Consequently, Hunt and Davis (1992) cautiously conclude that transcytosis clearly “is not the mechanism by which the majority of the iron is released from RPE cells”.
The asymmetric presence of the TfR in the RPE cells would impart unidirectionality to the transfer of iron towards the photoreceptors. Its exclusive presence in the basal membrane, as mentioned in passing by Hunt and Davis (1992) is usually taken to mean unidirectional movement of iron towards the photoreceptors, its exclusive presence in the apical side would mean unidirectional movement of iron in the reverse direction. If symmetrically distributed in the plasma membrane (as described by He et al., 2007) there would be the possibility of transit in both directions.
It can also be hypothesized that a specific endocytic mechanism would tend to keep plasma Tf on the plasma side and locally synthesized Tf on the retina side. On the contrary, in a transcytotic mechanism the tendency would be for plasma Tf to eventually be mixed with retina Tf, unless specific recognition mechanisms are built in the Tf molecule, which is certainly possible given the potentially high microheterogeneity of Tf (DeJong et al., 1990).
The inner blood-retinal barrier
Two main capillary beds can be distinguished in the retina, the deep (outer) capillary plexus between the inner nuclear and outer plexiform layers, and the superficial (inner) plexus at the level of the nerve fiber and ganglion cell layers (Oyster, 1999). Retinal capillaries are non-fenestrated and their endothelial cells are sealed with zonula occludens junctions at all their contacts establishing a continuous capillary wall and constituting the inner retinal component of the blood-retinal barrier. The endothelial cells of these capillaries show a very low level of non-discriminating pinocytosis as revealed by the small number of pinocytic vesicles visible with electron microscopy (Kaur et al., 2008). This supports the idea that non-specific exchanges across the barrier must be highly restricted. The normal flow of iron in these cells is apical to basal, i.e., of opposed polarity to that occurring in the RPE. Other cellular elements also make important contributions to the inner blood retinal barrier. Müller glial cells (Newman, 1994) fill the space of the retina between the inner and the outer limiting membranes. The inner limiting membrane (ILM) is formed by the basement membranes of the endfeet of Müller cells at the vitreoretinal interface. The outer limiting membrane (OLM) is formed by the intermediate junctions between the apical border of these cells and the photoreceptor inner segments. Therefore, Müller cells do not interact directly with the RPE. Müller glial cells take over in the retina the role of astrocytes in other neural tissues, providing support for the electrical activity of the closely apposed neurons, controlling the composition and pH of the retinal interstitial fluid, and mediating metabolic transfers between retinal neurons and blood plasma within the lumen of the capillaries. Müller cells are also responsible for the induction of the expression of proteins forming the endothelial tight junctions, and thus of the maturation of the blood-retinal barrier itself (Kaur et al., 2008). Regular astrocytes migrate into the retina from the optic nerve and are limited to the innermost ganglion cell and the nerve fiber layers of the retina. Oligodendrocytes are absent from the retina since normally myelination is restricted to the retrolaminar portion of the optic nerve beyond the lamina cribrosa (Newman, 1994).
Pericytes, better known for their role in providing structural support for growing capillaries during angiogenesis, and for their ability to control regional blood flow because of their contractile activity, also provide coverage to the endothelial cells and are now thought to contribute to maintain the structural and functional integrity of the endothelial tight junctions, and to even participate in some of the fluid and metabolic exchanges taking place across the barrier (Kaur et al., 2008).
Burdo et al. (2003) made a valuable contribution to the knowledge of the inner retinal barrier because they worked with cultured bovine retinal endothelial cells as an in vitro model of the blood-brain barrier (BBB). In this system a monolayer of endothelial cells is grown on a special filter that separates an upper chamber (equivalent to the plasma in contact with the apical or luminal side of the endothelial cells) from a lower chamber (equivalent to the interstitial fluid in contact with the basal or abluminal side of the cells). The existence of a Tf-initiated endocytotic process was revealed in this case by the effect of ammonium chloride (NH4Cl), which prevents acidification of the endosomes (Galloway et al., 1983), on the transfer of iron from 59Fe/fluorescein-labeled Tf placed in the upper chamber. Relative to the control, NH4Cl decreased 59Fe flux into the basal chamber by 70%.
The fluid transferred to the basal chamber was separated in two fractions by size-exclusion chromatography using Sephadex G-25: an excluded high MW fraction (>5kD) and an included low MW fraction (<5kD). The first fraction was named Tf-bound iron (TBI), and the second non-Tf bound iron (NTBI), in spite that no attempt was made to positively identity the protein(s) actually present in the TBI fraction. The NTBI pool was assumed to be related to the Tf-initiated endocytosis of iron just mentioned, which implies the cytoplasmic separation of iron from Tf. The TBI pool was related to a second, apparently constitutive mechanism of crossing the barrier, the Tf-mediated transcytosis of iron. Indeed, the cells were able to transfer fluorescein-Tf to the basal chamber as revealed by the accumulation of fluorescence in that chamber, and the authors assumed that this represented a basal rate for Tf transcytosis. They acknowledged the difficulties that a straightforward, simple transcytotic transport or iron presents (previously discussed under the outer blood-retinal barrier) and considered incomplete the scheme of transcytosis depicted in their own figure 5. It should be noted, however, that the fact that endothelial cells lack ferroportin (Burdo et al., 2001) favors the involvement of some sort of transcytotic process in the crossing of the inner blood-retinal barrier, a process that should not require ferroportin to be completed.
Burdo et al. (2003) also proposed that the need for iron of the endothelial cells themselves is in principle met with the NTBI pool of iron, although mention is made that some of this iron might under normal conditions end up transferred to the brain (retina).
The influx of radioactive iron from double-labeled Tf into the basal chamber significatively decreased by 60% after overloading the cells with ferric ammonium citrate (FAC). This change was mostly the result of a reduction in the NTBI pool. TBI did not significatively change with pretreatment of the cells with FAC or NH4Cl, nor with other manipulations to which the cells were submitted. The flux of fluorescein-Tf increased significatively upon loading the cells with FAC but was insensitive to NH4Cl. FAC also increased the amount of iron detected within the endothelial cells, where presumably was stored as ferritin.
The three effects of FAC just described were interpreeted by the authors as indicative that the iron status of the endothelial cells influenced the amount of Tf and iron that is transported through the barrier. When exposed to an overload of iron, more iron was safely stored intracellularly, more apoTf was somehow made available to the process and less iron was detectable in the NTBI pool. These effects are all of a protective nature both to the endothelial cells themselves, and to the cells across the barrier, and under this light were interpreted by the authors.
Burdo et al. (2003) also found that filipin, a blocker of caveolin–mediated, pinocytotic transport (Stremmel et al., 2001) did not have any effect on iron transfer in their model, and thus disregarded the possibility of non specific pinocytosis as a mechanism of transfer through the retinal capillaries endothelial cells.
In summary, for Burdo et al. (2003) the main mechanism for iron to cross the inner blood-retina barrier, and the source of iron for the inner retina under normal conditions, would be TfR-mediated transcytosis, while an endocytosis-initiated mechanism that depends on the iron status of the endothelial cells would play only a regulatory role. The reverse seems to be implied by the results of Hunt and Davis (1992) who interpreted them as evidence that a TfR-initiated endocytosis of iron would be the main way for iron to cross the outer blood-retina barrier, thus becoming the normal source of iron for the retina photoreceptors. A valid question to be asked would then be, do these apparent differences between the outer and the inner components of the blood-retina barrier truly represent intrinsically different mechanisms of transport, or are they mainly dependent on the different experimental set-ups used to study them?
Considering that the retina is part of the central nervous system the unraveling of iron handling by the retina and the brain will probably be closely interwoven. Mention should be made in this respect of two controversial issues relative to the BBB: (1) the direct delivery of iron from brain capillary endothelial cells to the neuroglia by low MW iron-binding components known to be present in the brain interstitium in high concentrations: citrate and ATP produced by glial cells, and ascorbate incorporated to this space by active transport at the choroid plexus. This would be to circumvent the finding that brain neuroglia does not posses TfRs nor DMT1, according to Moos et al. (2007); and, (2) alternative ways for iron to cross the BBB since the brain capillary endothelial cells do not posses sufficient DMT1 for that purpose, according to the same authors.
Oxygen and the retina
The retina is the most metabolically active tissue of the body, with the photoreceptors consuming oxygen at rates higher than neurons in the brain (Berman, 1991). The source of oxygen for the metabolism of the retina is from two vascular supplies: the choroidal circulation behind the RPE and the retinal circulation derived from the central retinal artery (Wangsa-Wirawan and Linsenmeier, 2003). The choroidal circulation basically nourishes the RPE and the photoreceptors, while the retinal circulation provides oxygen and nutrients to the inner retina. At the level of the avascular foveola (covering a radius of about 0.18 mm eccentricity in the human eye) all the layers of the retina present there receive nutrition from the choroidal circulation: the RPE, the photoreceptors, the OLM, the outer nuclear layer and a fraction of the outer plexiform layer (fibers of Henle), directly followed by the ILM (Oyster, 1999; Provis et al., 2005). The other retinal layers have been radially displaced and thus are absent in the foveola, and only start to appear as one moves out towards the rim of the foveal depression beyond which the retina recovers its classical 10-layer appearance.
Blood moves very rapidly through the choroidal capillaries, maintaining a high choroidal pO2 of about 60 mmHg. Only the retinal circulation exhibits autoregulation and responds with vasodilatation and increased blood flow, for example, to both hypoxemia and increased intraocular pressure (IOP). This does not occur in the choroidal circulation where the high blood flow is insensitive to oxygen levels, and hypoxemia produces a marked decrease in choroidal pO2 (as it does with an increase in IOP), decreasing the O2 consumption by photoreceptors (Oyster, 1999; Provis et al, 2005).
Iron-related immunochemistry of the retina
The immunochemistry of the retina, as related to iron homeostasis, has been thoroughly reviewed by several investigators (Dunaief, 2006; He et al., 2007; Yefimova et al., 2000). I shall highlight here only some of their contributions. Earlier work led to establish the basic model of iron transfer through the blood-retinal barrier described above.
Yefimova et al. (2000), based on the ample distribution of iron, ferritin, Tf and TfR in the retina of the adult rat stressed the importance of Tf- and TfR-mediated movements of iron throughout the retina. Transferrin mRNA expression, followed through in situ hybridization, was identified in the RPE, indicating that the RPE is the main site of local transferrin synthesis (Yefimova et al., 2000). According to these authors, local RPE transferrin could carry iron from the RPE to the photoreceptors and the rest of the retina via Tf/TfR-dependent mechanisms instead of by iron binding to low MW species. In the RPE, TfRs were detectable both in the basolateral and the apical poles of the cells (He et al., 2007) thus providing support to a bidirectional flow of iron through the outer blood-retina barrier.
Strong DMT1 immunolabel was detected in rod bipolar cell bodies and axons, horizontal cell bodies, and photoreceptor inner segments (He et al., 2007). It was not possible from this distribution pattern to determine whether retinal DMT1 serves to export iron from cells, export iron from endosomes, or import iron.
Immunohistochemistry identified ceruloplasmin localization throughout the mouse retina, with the strongest label associated with Müller cells; its mRNA appeared associated with the RPE in both mice and humans eyes (Hahn et al., 2004; He et al, 2007). Hephestin mRNA was detected in the RPE of mouse and human retinas and in RPE cell lines. The greatest density of hephestin immunoreactivity was found in the endfeet of Müller cells of the retina. Double labeling experiments with a marker for Müller cells confirmed the localization of hephestin to Müller cells (He et al., 2007).
Ferroportin was present in normal retina at high levels in the Müller cells endfeet and in the RPE (He et al., 2007), in addition to other less conspicuous locations. In the RPE the immunoreactivity was primarily basolateral, suggesting that ferroportin functions there to facilitate export of iron, along with ceruloplasmin and hephestin, towards the choroidal capillaries (He et al., 2007). As proposed later in this work, the localization of ferroportin in Müller cells endfeet is highly suggestive that iron could easily move into the vitreous with the help of the ferroxidases ceruloplasmin and hephestin.
Hepcidin mRNA was detected in the mouse retina by in situ hybridization in the inner nuclear layer that contains the nuclei of Müller cells, in the inner segments of photoreceptors and in the RPE (Gnana-Prakasan et al., 2008). According to the authors, hepcidin was abundantly expressed in those same cells that express ferroportin, the transporter to which hepcidin binds. Low levels of immunoreactivity were detectable throughout the retina. The authors also showed that primary cultures of RPE and Müller cells, as well as a human line of RPE and a rat line of Müller cells, were positive for hepcidin. Moreover, hepcidin expression increased after the intravitreal injection of lipopolysaccharide (LPS), mimicking a bacterial infection.
Some basic photochemistry concepts
One of the potential consequences of absorption of sufficiently energetic light by a chromophore is the initiation of a photochemical reaction. The excess energy accumulated in an excited chromophore as the result of the absorption of photons may provoke the rearrangement of bonds in the absorbing chromophore or its participation in a chemical reaction. If a covalent bond is disrupted in such a way as to leave orbitals with one unpaired electron, a free radical is formed, which can be very reactive. Non-radical, but still reactive species can also be formed.
If chemical modification involves neighboring molecules instead of the absorbing chromophore the result is a photosensitized reaction (Glickman, 2002; Roberts, 1996; Wu et al., 2006). Such a chromophore is referred to as a photosensitizer. The photosensitizer may interact with a biomolecule, specifically directing light energy towards the interacting biomolecule. There are two types of photosensitized reactions. In type I reactions the photosensitizer induces the production of free radicals by exchanging electrons or protons. In type II reactions, the excited state of the photosensitizer interacts with oxygen to produce singlet oxygen, a very reactive species. Both UV and short wavelength visible light are able to drive direct photochemical, or photosensitized reactions.
Singlet oxygen and free radicals can initiate self-propagating, lipid peroxidation chain reactions in membranes that will ultimately destroy fatty acids and the membrane itself. Nucleic acids and proteins can also be damaged. Photochemical processes thus have a serious destroying potential in biological systems.
Susceptibility of the retina to photochemical damage
The retina is particularly susceptible to oxidative damage for several reasons indicated below. The choroidal circulation provides abundant oxygen, as it was already described. Within the retina, the region with the highest sensitivity to photoxidation is the macular area, where the light is focused and where photoreceptors are at their highest density and light is thus more efficiently captured. In the foveola, light reaches the external segments of the photoreceptors with very little impediment from tissue structures in front of them. Only protective macular pigments lutein and zeaxanthin (reviewed by Whitehead et al., 2006) stand strategically in the middle of the lightpath to absorb damaging blue light, and directly or indirectly act as antioxidants. Damage inflicted to the transducing machinery by light is probably of an irreversible nature given the fact that the external segments of rod and cone photoreceptors are being constantly renewed. The pieces of external segments released at the tips of the photoreceptors are phagocytized by adjacent RPE cells imposing a heavy metabolic load on these cells, as reflected by the abundance of their mitochondria. Residues tend to accumulate with age, forming lipofuscin granules in the RPE (Newman, 1994). Lipofuscin granules are toxic to RPE cells when incubated together in culture (Ng et al., 2008). The lipofuscin content of RPE rapidly increases during the first two decades of life, perhaps due in part to the relatively high transmission to blue light and UV-A of the lens during this period, until the lens itself becomes a more effective yellow filter (Wood and Truscott, 1993; Wu et al., 2006).
Characteristically, there is an elevated proportion of polyunsaturated fatty acids (PUFAs) in the photoreceptor outer segments (85-90 mol% of total lipids), and low cholesterol levels (10 mol%), in several vertebrate animal species including human retinas (Fliesler and Anderson, 1983). Purified rods outer segments (ROS) contain a high proportion of PUFAs (docosahexaenoic acid (DHA) 22:6ω3 ⋙ arachidonic acid 20:4ω6), typically representing 50-60 mol% of total fatty acids. A high level of PUFAs is maintained in the phospholipids (43.1mol% in phosphatidylethanolamine, 58.1mol% in phosphatidylserine), the most abundant of which was always DHA (up to 79% in phosphatidylethanolamine). Interestingly, phosphatidylserine contained 20.1% of 22C PUFAs other than DHA and 17.4% of 24C PUFAs.
The combination of high levels of PUFAs with low levels of cholesterol relative to phospholipids results in a fluidity of ROS membranes, which is one of the highest, compared with other membranes (Berman, 1991). This allows fast movements of integral proteins in the membrane as required by the visual transducing system. The inconvenient part of it is that PUFAs easily start lipid peroxidation self-propagating chains. Carbonyl 4-hydroxy-7-oxohept-5-enoic acid, a fragment derived from lipid peroxidation of DHA-containing phospholipids, reacts with amino groups of proteins to produce 2-(ω-carboxyethyl)pyrrole adducts (CEP adducts) (Crabb et al., 2002). These products thus appear in the lipofuscin granules of RPE (Ng et al, 2008). Lipofuscin granules also contain fluorescent bisretinoid derivatives: A2E (2-[2,6-dimethyl-8-(2,6,6-trimethyl-1-cyclohexen-1-yl)-1E,3E,5E,7E-octatetraenyl]-1-(2-hydroxyethyl)-4-[4-methyl-6-(2,6,6-trimethyl-1-cyclohexen-1-yl)-1E,3E,5E-hexatrienyl]pyridinium), isoA2E, and all-trans-retinal dimer-phosphatidylethanolamine (Ng et al., 2008). Bis-retinoids are toxic to RPE cells in culture. All these derivatives will eventually become part of drusen in macular degeneration, and anti-CEP autoantibodies will appear in circulation, when the RPE is damaged and the retinal-RPE barrier breaks down (Crabb et al., 2002). Mice immunized with the CEP hapten linked to serum albumin develop retinal lesions similar to those of age-related macular degeneration probably by sensitization to the generation of CEP adducts in the outer retina (Hollyfield et al., 2008).
In the retina the action spectrum for photochemical damage shows two maxima, one coincident with the absorption spectra of the photosensitive pigments in the photoreceptors (class I damage) and the other coincident with short wavelength (blue) visible absorption (class II damage). The latter mainly affects the RPE and eventually leads to breakdown of the blood-retinal barrier (Ham et al., 1979).
Pathology of iron homeostasis in the retina
Age-related macular degeneration (AMD) is a progressive, multifactorial disease that targets the central few millimeters of retina that are critical for high acuity vision, causing degeneration of photoreceptors and the RPE (“dry” form) or abnormal growth of choroidal vessels through Bruch's membrane (“wet” form) (Gu et al., 2009).
As the result of age-related photochemical oxidative mechanisms described above for the retina, including direct and photosensitized damage to the RPE and given the vulnerability of the macular region, a slow deposition of a large variety of RPE-generated and blood-borne substances diffusing out of the choriocapillaris occurs at the macular level in Bruch's membrane and the sub-RPE space. These deposits (drusen) are one main, early characteristic of AMD. They are able to create local inflammatory responses including leukostasis (Provis et al., 2005) that are likely to reduce choroidal flow and create an obstacle to the diffusion of metabolic substrates and induce finally degenerative changes in the photoreceptors and RPE cells. The presence of autoantibodies, against antigens inside the blood-retinal barrier (CEP adducts, for example), suggests barrier breakdown. There is strong evidence of increased permeability of the outer retinal barrier in both the dry and wet forms of AMD (Provis et al., 2005). An inflammatory response stimulates additional oxidative stress and contributes to progressive deterioration. There is also evidence that CEP adducts may stimulate neovascularization in vivo (Ebrahem et al., 2006).
Iron could play a role in the pathogenesis of AMD by exacerbating free radical damage and even by directly provoking inflammatory reactions through the activation of the complement cascade (Wong et al., 2007). Indeed, postmortem AMD-affected eyes were found to have an excess of both chelatable and nonchelatable iron in the RPE and Bruch's membrane (Hahn et al., 2003) including drusen, and macular iron levels were found to increase with age in human postmortem specimens (Hahn et al., 2006); combined deficiency of the iron ferroxidases ceruloplasmin and hephaestin in mice (that interferes with efficient export of iron) leads to a retinal degeneration by iron overload with some features of AMD, including sub-RPE deposits and subretinal neovascularization (Hadziahmetovic et al., 2008; Hahn et al., 2004); human aceruloplasminemia, that also leads to retinal iron overload, produced early onset macular drusen-like opacities (Wong et al., 2007); finally, transferrin is upregulated at the mRNA and protein levels in patients with AMD compared to age-matched controls (Chowers et al., 2006). The authors Wong et al. (2007), recognizing the chronic inflammatory nature of AMD, suggested that upregulation of hepcidin could be the cause of the intracellular iron overload in AMD, as it is known to occur in chronic inflammation in other tissues (Ganz, 2003). Additional comments will be made on this topic later in the review.
The lens
The avascular lens of the eye is suspended in a strategic position within the eye between the aqueous humor anteriorly, and the gel vitreous posteriorly. Fresh aqueous humor is continuously delivered to the lens equatorial region by the ciliary body (pars plicata) encircling the lens (Oyster, 1999), providing the latter with the proper environment to grow and to maintain its transparency to light. The circulation of the aqueous humor is at the same time a useful vehicle for the removal of metabolic waste produced by the lens and other metabolically active ocular avascular tissues.
Development and growth of the lens
The adult lens consists of an anterior monolayer of polarized cuboidal ectodermal cells, the lens epithelium, and a much larger mass of cells behind the epithelial monolayer undergoing a slow process of elongation and differentiation that ends with the formation of secondary, mature fiber cells.
Lens cells are never shed but keep accumulating in concentrical layers around the central space of the lens. Briefly, new cells generated in the germinative region of the anterior epithelium are displaced towards the lens equator. Here they start to elongate at the same time that undergo a rotation of 180°, their apical pole becoming anterior and their basal pole becoming posterior. Once rotated, these cells are committed to be stacked layer over layer in the nuclear region of the lens, as new cells arrive to the lens equator from the germinative region of the epithelium.
While the young fiber cells elongate, their apical poles interact with the apical poles of the anterior epithelium until they form a suture and stop growing, detaching from the epithelium to become part of the lens cortex, first, and eventually of the lens nucleus (Kuszak and Brown, 1994).
As part of their differentiation the immature elongating fibers become loaded with lens crystallins, the characteristic structural proteins of the lens, and lose all intracellular organelles. Only polysomes, remain in the cytoplasm of mature fibers. This is very advantageous for lens transparency but leaves the bulk of lens cells in a precarious metabolic situation, with a metabolism severely limited by the lack of organelles and able to exclusively produce energy through anaerobic glycolysis, an activity that solely requires soluble cytoplasmic proteins to proceed. The nuclear fiber cells thus become very dependent on the cortical fibers still containing mitochondria, and the anterior epithelium, for nutrient supply and maintenance. In compensation, they acquire an extensive network of gap junctions (cell-to-cell connections built with the proteins connexins) and aqueous pores (cell-to-extracellular space connections built with aquaporins) in order to facilitate the distribution of nutrients, and the elimination of metabolic waste. This converts the lens in a functional syncytium that permits ample electrical and metabolic coupling among cells (Rae, 1994). The aqueous pores, by allowing fast dissipation of osmotic gradients that might be created within the lens are especially important for the maintenance of lens transparency.
The fact that no cells are lost during growth of the lens does not mean that nothing is lost from the cells. On the contrary, there is a continuous leakage of fragments of crystallins that can be detected immunologically for life in the blood plasma of all individuals (unpublished observations). Lens fibers may thus shrink (compact) with age without changing their cytoplasmic protein concentration, giving the type of flat protein concentration profile across the nucleus described for the human lens (Brown and Bron, 1996; Jaffe and Horwitz, 1992).
The anterior epithelium of the lens
Cells in the lens epithelium contain junctional complexes at the apical border, but the existence of zonula occludens (tight junctions) in these complexes has been a controversial issue, because of marked structural species differences, a relative low level of interlaced strands apparently unable to seal off the paracellular route, or the atypical presence of small gap junctions (Kuszak and Brown, 1994).
The fact is, however, that the apical complexes of the lens epithelium actually impede the movement of the tracers horseradish peroxidase (MW = 40,000) and lanthanum (MW = 430) through the barrier in several species: chick, frog, rat, and human (Lo, 1987). In bovine lenses, Sugiyama et al. (2008) found the unequivocal expression of tight junction proteins in the epithelium, although with a characteristic lack of segregation of E-cadherin (a protein of adherens junctions) from ZO-1, something that does not occur in tight junctions of well-polarized cells.
Barriers with a low resistance to fluid movement as that present in the lens epithelium are classified as “leaky”, but they still have the important physiological role of facilitating the unidirectional movement of substances and fluid through the barrier, and contain tight junction proteins in very specific proportions (Amasheh et al., 2009; Förster, 2008).
Na/K-ATPase pumps located at the apical membrane of the epithelial cells (Unakar and Tsui, 1980) initiate osmotic, Na-driven fluid movement into the lens through the paracellular route that may help to distribute solutes within the lens (Fischbarg et al., 1999; Mathias and Rae, 2004). At the same time, a sodium gradient is created (low intracellular Na+) that drives the Na-dependent, active incorporation of aqueous humor components like aminoacids (Jaffe and Horwitz, 1992) and GSH (Mackic et al., 1996) through the basal pole into the cells. Once inside the cell, all those substances need additional passive transport carriers at the apical pole to exit the cells and enter the extracellular space between the epithelium and the fibers (EFI, see below). Glucose and dehydroascorbate (Garland, 1991; Kern and Zolot, 1987) are incorporated to the lens by Na-independent mechanisms at the basal and the apical membranes of the epithelial cells.
Lens epithelial cells show abundant gap junctions in their lateral membrane infoldings, suggesting that they may be electrically and metabolically coupled under some circumstances.
The epithelial-fiber cell interface (EFI)
The extracellular space immediately posterior to the apical poles of the lens epithelium represents the obliterated lumen of the embryonic lens vesicle. This space is posteriorly delimited in the adult lens by the apical poles of the elongating fiber cells behind. The whole arrangement is called the epithelial-fiber cell interface (EFI) (Kuszak and Brown, 1994; Kuszak et al., 1995), or simply the apical interface (Zampighi et al., 2000). At the EFI gap junctions are rare and the fiber cells lack apical tight junctions (Kuszak et al., 1995). Instead, both the fiber cells and the epithelial cells show unequivocal electron microscopic signs of marked receptor-mediated endocytotic activity, including the presence of clathrin coated pits, and pinocytic vesicles, an indication of the importance of transepithelial movements across the EFI.
The EFI also represents a pathway by which materials can be short-circuited to the lens directly from the ciliary body, because of the physical proximity of the lens equator to the ciliary body and the abundance of gap junctions in the young cortical fibers of the lens equatorial region. Gap junctions in these cells are at their highest density in the middle region of the fibers. Several authors (Kuszak et al., 1995; Lo, 1987) have thus proposed that there may exist a preferential diffusion pathway of aqueous humor components into the lens near the equator. Moreover, given the lack of apical tight junctions in those young cortical fibers (Kuszak and Brown, 1994) the EFI intercellular space must be amply accessible to the extracellular space of the lens at the equatorial level. It has been shown, for example, that the vitreal injection of the marker horseradish peroxidase (Lo, 1987) near the region of the ciliary body leads to the rapid appearance of the protein in the EFI. On the other hand, using an in vitro chamber model of barrier consisting of a confluent monolayer of cultured rabbit lens epithelial cells, Sabah et al. (2007) demonstrated the transference of albumin towards the aqueous humor side of the chamber through two mechanisms: receptor mediated transcytosis and nonspecific pinocytosis. It is therefore likely that proteins in the vitreous body could be directly incorporated into the lens EFI through the equatorial extracellular pathway and then transported into the aqueous humor through the lens epithelium by receptor-mediated endocytosis, as will be proposed later for transferrin.
Oxygen and the lens
Using fiber optic probes sensitive to oxygen Barbazetto et al. (2004) found in vivo in the rabbit that along the visual axis pO2 goes from about 29 mmHg in the aqueous humor in the middle of the anterior chamber to about 22 mmHg under the capsule of the lens. The pO2 progressively dropped inside the lens, until a minimum of 9-10 mmHg was reached in the nucleus. The posterior region of the lens and anterior vitreous had about 11 mmHg. Then, pO2 started to increase, slowly at the beginning and then faster in the posterior vitreous as the retina was approached, reaching about 26 mmHg near the retina and 40-60 mmHg touching it. Although the absolute values of pO2 within the eye may vary according to authors, animal species and detection methods, the avascular lens and particularly its nuclear region, always shows the lowest values.
Photoxidative damage to the lens
The lens is vulnerable to photochemical damage and age-related degeneration for physiological circumstances very different from those affecting the retina. The lens is a tissue much simpler that the retina from the cellular point of view, having only one type of cell in several stages of differentiation, lacks the metabolic oxidative overload of the retina, and is entirely avascular. Mitochondria are limited in the lens to the epithelial monolayer and the superficial cortex and, relative to the retina, the concentration of oxygen to which the lens is exposed is very low, as just noted. Protein concentration in the lens is higher than normal, and the levels of the intracellular antioxidant GSH are maintained very high mainly by local synthesis (Reddy, 1990) but also through active transport from the aqueous humor (Mackic et al., 1996). All these circumstances tend to substantially lower the level of photoxidative stress to which the lens is exposed. In comparison to the retina, however, the lens not only absorbs the more energetic UV-B light but, most typically, contains very long-lived proteins that stay exposed for life to the cumulative direct or photosensitized photoxidative effects of UV light and other posttranslational modifications mechanisms (Dillon, 1991; Garland, 1990; Harding, 1991; Pitts et al., 1986; Truscott, 2005). Not even the part of the lens covered by the iris is spared from the harmful effects of light because rays obliquely hitting the cornea are refracted onto opposite corticoequatorial regions of the lens (Merriam, 1996). The lens is therefore a tissue extremely enriched in age-dependent posttranslational modifications of diverse nature leading to both oxidative and non-oxidative changes. Some of them may have a stabilizing effect on its structure favoring transparency, but others will in the long run compromise lens transparency, causing aggregation and insolubilization of structural proteins, progressively inactivating the enzymes involved in metabolism, damaging transporters of electrolytes and nutrients, interfering with the synthesis of antioxidants needed in increasing demand by the aging lens, and accelerating the accumulation of brown pigments, to mention but the main functional consequences of lenticular posttranslational modifications.
The lens, by increasingly absorbing UV-A and blue light as it ages, contributes to protect the retina from photochemical damage. The young human lens synthesizes from tryptophan UV filters (Wood and Truscott, 1993) with absorption centered at about 365nm which temporarily protect the retina from damaging light while the more definitive adult pigments are generated and incorporated into lens proteins.
The adult lenticular pigments are derived from two main mechanisms: (i) photoxidation of the aromatic aminoacids tryptophan and tyrosine, primary absorbers of UV-B light (Cuevas and García-Castiñeiras, 1993; Dillon et al., 1984; García-Castiñeiras et al., 1978; Rousseva et al., 2007; Truscott, 2005; Truscott and Augusteyn, 1977); and, (ii) spontaneous glycosylation of lens proteins (Maillard reaction) (Frye et al., 1998). Despite lacking tyrosinase (Carrasquillo et al., 1993), DOPA can be occasionally formed in the lens probably through hydroxylation of tyrosine by the hydroxyl radical (Fu et al., 1998), a reaction that initiates the formation of pheomelanin pigments (SH-containing melanins), as suggested by the finding of thiazoledicarboxylic acid after digestion of nuclear cataract extracts with potassium permanganate (Cuevas and García-Castiñeiras, 1993). The spontaneous glycation of lens proteins is initiated by the non-oxidative mechanism of Schiff's base adduct formation between glucose and free amino groups of the protein. At later stages the chemistry involved becomes extremely complex with intermediaries undergoing oxidative and non-oxidative degradation and generating new glycosylating fragments that are recycled through the basic reactions. Reactive oxygen species including free radicals are formed as the result of the oxidative degradation of intermediaries, adding further complexity to the system. The final products are the advanced glycation end-products (AGEs) that include deeply pigmented molecules. Some of the AGEs that have thus far been characterized clearly constitute crosslinks (Frye et al., 1998). Ascorbate, a quantitatively important antioxidant component of the lens may eventually participate in these reactions (Nagaraj et al., 1991).
The lens and iron homeostasis
In spite of the apparent simplicity of lens structure, knowledge of iron homeostasis in the lens still lags behind in the assignment of iron related proteins to cell and subcellular structures. The lens has to secure that the metabolically active epithelium and cortical region receive enough iron to cover their own metabolic needs including those associated to differentiation and growth. For this reason it is likely that the lens contains all the proteins involved in iron metabolism or is able to synthesize them if need be. Thus far, this seems to be true as very recently reviewed by Goralska et al (2009).
The lens contains an iron average of 0.18 ng/mg wet weight (rabbit) and maintains these levels under close control (McGahan, 1992). In humans Garner et al. (2000) report levels of 7 ng/mg dry weight and Levi et al. (1998) report 10 ng/mg protein. Vázquez-Quiñones and García-Castiñeiras (2007) reported averages of about 0.4, 0.3, and 0.9 ng/mg protein in the cow, pig, and rat, respectively.
Ferritin is also present in the human lens at the level of about 137 ng/mg protein (Levi et al., 1998). Garner et al. (2000) reported 120 ng/mg protein and immunochemical techniques showed highest density of ferritin in the epithelium, with diffuse staining throughout the cortex and nucleus of normal lenses. McGahan et al. (1994) reported ferritin levels of 60-200 ng/mg protein in cultured lens epithelial cells (dog), with much lower levels in the fiber cells (0.08 ng/mg protein) (Goralska et al., 2009).
Dog lens epithelial cells in culture were able to synthesize and secrete ceruloplasmin and the effects of added ceruloplasmin on efflux of iron from lens cells were particularly conspicuous in conditions mimicking iron overload with ferric ammonium citrate (Harned et al., 2006).
Relative to Tf several arguments were used in the 80's to conclude that a fraction of aqueous humor Tf was not derived from bodily plasma but from local synthesis within the eye (Dernouchamps, 1982). The work of McGahan et al. (1995) further and definitively established that indeed many tissues within the eye express Tf including the lens. In some cases Tf was also secreted to extracellular spaces or into culture media. Synthesis and liberation of Tf was exhibited by rabbit lens capsular bags, by dog lens cells in culture and by human lens capsular bags from autopsy specimens (Davidson et al., 1998). In all these cases Tf synthesis was upregulated. The presence of Tf in the lens was later confirmed in several animal species in the range from 1.2 to 7 μg/mg protein (Vázquez-Quiñones, 2005)
An interesting connection was recently made (Lall et al., 2008) between cytosolic aconitase (which functions as an iron regulatory protein (IRP) when iron levels are low) and synthesis of GSH, through a glutamate-cystine antiporter, thus linking iron homeostasis with redox levels in lens epithelial (and RPE) cells.
No studies were available at the time of this writing about the presence and localization, or expression in the lens of the iron transporters DMT1 and ferroportin, the ferroxidase hephestin, or the iron regulatory hormone hepcidin.
Comparative iron homeostasis in eye tissues and fluids
In order to obtain an overall picture of iron metabolism in the eye, and an idea of the relative susceptibility of ocular tissues and fluids to oxidative stress, we compared a limited number of iron related parameters (total iron (TI), total iron binding capacity (TIBC), % saturation of TIBC, and Tf) in blood serum and eye fluids and tissues of three animal species (pig, cow, and rat) (Vázquez-Quiñones, 2005; Vázquez-Quiñones and García-Castiñeiras, 2007). Previously, McGahan and Fleisher (1986) had provided for the first time simultaneous values for TI, TIBC and TIBC % saturation in blood plasma and intraocular fluids of several animal species (dog, cat, pony, pig and rabbit) using an electrothermal atomic absorption spectroscopic (EAAS) microprocedure for the determination of iron.
After conversion to the same concentration units, and excluding rats from our series because of their nocturnal living habits, comparable results were in general obtained by both groups of investigators regarding plasma and intraocular fluids, including identical plasma values for TI. Saturation of TIBC was very low in the aqueous humor of the cow and the pig in our series. In the McGahan and Fleisher (1986) series which also included the pig, saturation of TIBC in the aqueous humor of this animal was also below 10%, suggesting that our finding in the cow and pig may represent a truly species-related difference. Two important coincidences were also noted in both series that were species-independent: (i) TI and TIBC concentration values (mg/L) were much higher in plasma than in intraocular fluids; and, (ii) % TIBC saturation values were highest in the vitreous.
Our results, all expressed relative to protein, are condensed in figure 2. Vulnerability to oxidative stress was considered higher in association with high relative concentrations of TI, low relative concentrations of Tf and TIBC, and high % saturation TIBC.
Figure 2.
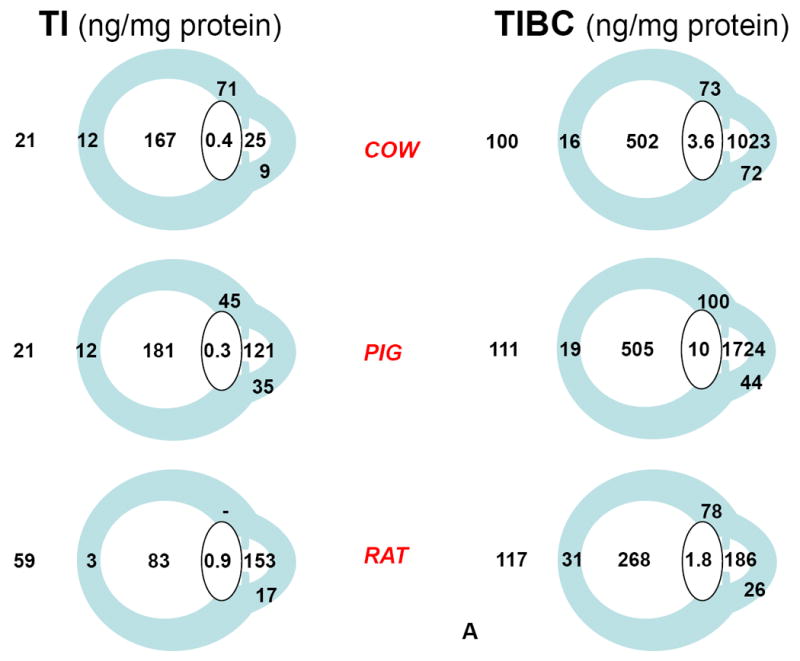
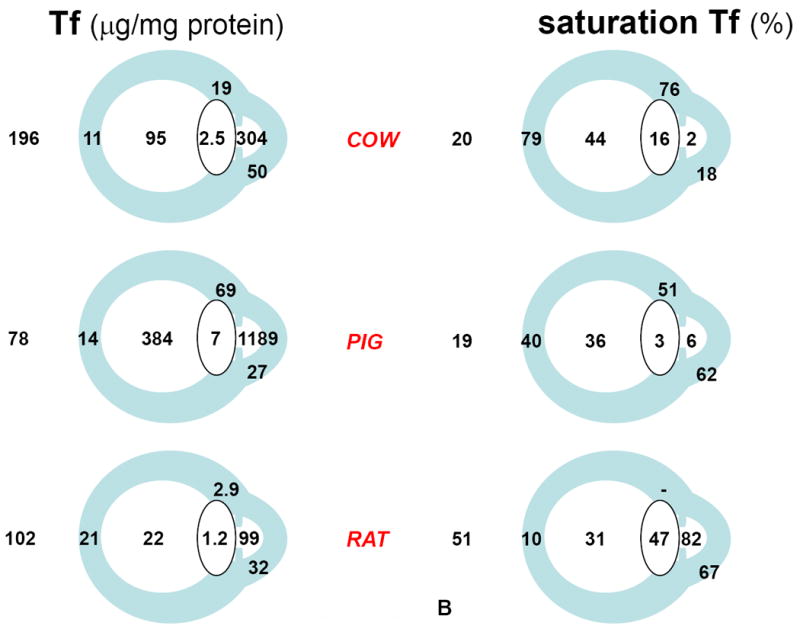
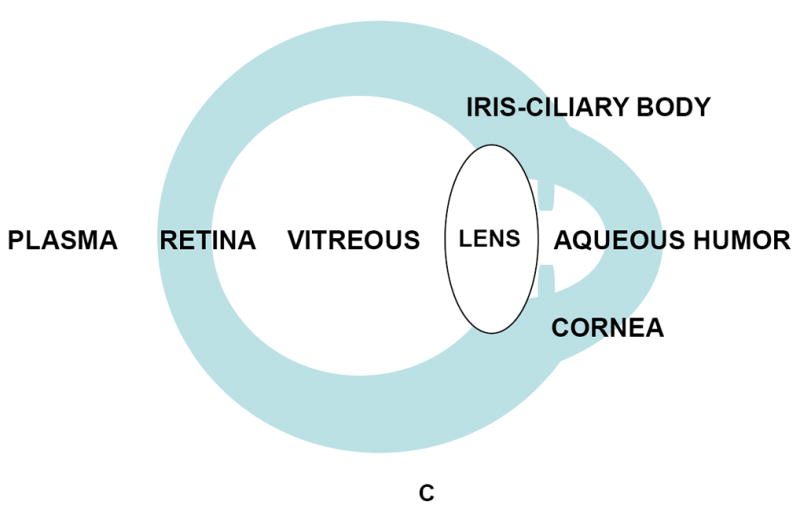
Iron and iron-related parameters in three animal species, cow, pig, and rat (Vázquez-Quiñones and García-Castiñeiras, 2007). A: Total iron (TI) and Total iron binding capacity (TIBC). B: Transferrin (Tf) and % saturation of TIBC(Tf). C: anatomical identification key. A and B are reproduced with permission from the Puerto Rico Health Sciences Journal.
The main conclusions derived from our work can be summarized as follows: (1) the aqueous humor of diurnal species (cow, pig) exhibited the highest relative concentration of Tf, followed by the vitreous body. Among ocular tissues, the lens always exhibited the lowest relative concentration of Tf; (2) TI relative concentration was highest in the vitreous body of diurnal species and lowest in the lens; (3) also in diurnal species, saturation of Tf was in the 20% range expected for plasma, increased several times in the retina, was maintained at about 40% in the vitreous, and decreased to less than 10% in the aqueous humor; (4) results suggested a high degree of oxidative stress in the rat, interpreted on the basis that, being a nocturnal animal, the rat does not require significant protection against photochemical effects of light on the eye; (5) it was also noted in the diurnal species that the back of the lens is exposed to the high TI relative concentration (and high % Tf saturation) of the vitreous body, which could contribute to the damaging effects on the lens of increased oxygen levels after vitrectomy (Barbazetto et al., 2004), and thus to postvitrectomy cataract.
Very relevant to the model considered below is the finding of the highest TI relative concentration (and high % Tf saturation) in the vitreous body, and the highest TIBC (and Tf) relative concentration (with % saturation below 10%) in the aqueous humor of the eye in both diurnal species.
A model of iron outflow from the eye
Although it is still premature to propose detailed mechanisms on the handling of iron by eye tissues and cells, I believe that there is already sufficient data in the literature to propose a testable hypothesis about the overall movement of iron within the eye and out into systemic circulation where this iron will be recycled (Figure 3).
Figure 3.
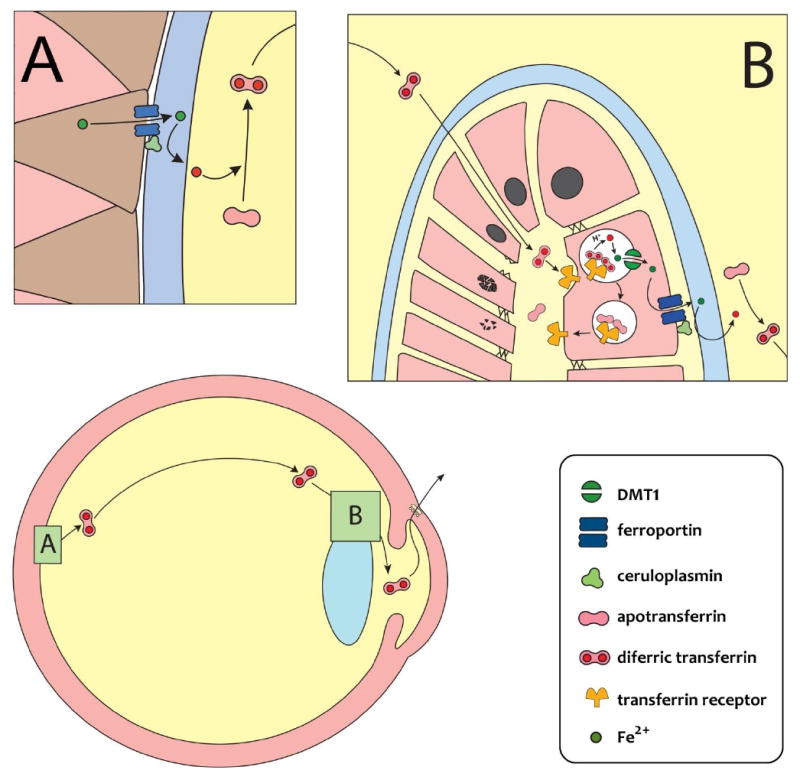
A model of the iron outflow pathway in the eye. The scheme represents the proposed movement of iron within the eye, starting at the endfeet of Müller cells in the retina (insert A) where a strong immunoreactivity for ferroportin and the ferroxidases ceruloplasmin/hephestin has been described (He et al., 2007) that permits the export of Fe2+ as Fe3+ towards the vitreous cavity. In the vitreous body, Fe3+ binds to Tf, and diffuses forward towards the lens. At the equatorial region of the lens (insert B) diferric transferrin could preferentially reach the apical interface (EFI) of the lens given the lack of tight junctions between fibers cells in that region (Kuszak and Brown, 1994), to be endocytosed by Tf receptors in the apical membranes of the lens epithelium. Intracellular events could be as described for many cells (Aisen, 1992): after acidification of the endosomes with a proton-ATPase, Fe3+ dissociates from Tf, is reduced to Fe2+ by some ferrireductase activity, and is transferred to a labile cytoplasmic pool through the H+/Fe2+ symporter DMT1. This leaves apoTf ready to move back to the apical pole of the cells and restart the cycle. Cytoplasmic iron moves towards the basal pole of the cells by not yet characterized mechanisms. A second ferroportin/ferroxidase system in the basal membranes is being proposed that pumps Fe3+ into the aqueous humor, where it is bound again to Tf and then recycled into systemic circulation through the trabecular meshwork and Schlemm’s canal with the aqueous humor bulk flow at the iridocorneal angle. Other destinations of the cytoplasmic pool of iron to, for example, meet the epithelial cells own need for iron or to store iron as ferritin, are not represented in this diagram. Also undepicted is a potential backflow of iron through the blood-retinal barrier.
The localization of ferroportin/ceruloplasmin(hephestin) in the retina, at the endfeet of glial Müller cells (He et al., 2007), critically positions export of retinal iron at the retinovitreal interface, towards the vitreous (Figure 3, insert A). Once in the vitreous, it can be assumed that Fe3+ binds to Tf, including locally synthesized Tf, the complex diffusing anteriorly towards the lens. As already pointed out, it is likely that iron loaded Tf preferentially enters the extracellular space of the lens at its equatorial region, and directly reaches the apical interface (EFI), given the lack of apical tight junctions in the elongating lens cells of the region (Kuszak and Brown,1994) (Figure 3, insert B). Once within EFI, diferric Tf could enter the anterior epithelial cells by TfR-initiated endocytosis at the apical pole of these cells. Inside lens cells, the fate of Fe3+ could be as described for many other iron-handling cells (Aisen, 1992; Andrews and Schmidt, 2007): (i) dissociation from Tf; (ii) reduction to Fe2+; (iii) release to the cytoplasm through the endosomal symporter DMT1; and, (iv) move towards the basal pole of the cells by mechanisms that remain mostly unknown. The transfer or iron to the aqueous humor could then be effected by a ferroportin/ferroxidase export system presumably located at the basal pole of the cells (Figure 3, insert B). Although not depicted in Figure 3, the labile cytoplasmic pool of iron could also provide iron for the own metabolic needs of the lens epithelial cells, or to safely store the excess as ferritin. In our model, Fe3+ exported by the lens would be picked up by aqueous humor Tf and the high affinity complex would leave the eye with the bulk, pressure-dependent flow of aqueous humor across the trabecular meshwork and Schlemm's canal, towards the systemic blood circulation (Figure 3).
At the level of the vitreoretinal interface the ferroportin/ceruloplasmin(hephestin) system creates a favorable gradient for the transport of iron through the vitreous. The presumed ferroportin/ferroxidase transport at the basal membranes of the lens epithelial cells would provide the final impulse for the transfer of iron to the aqueous humor. The intraocular gradients of Tf relative concentration increasing towards the aqueous humor, and of Tf % saturation decreasing towards the aqueous humor, that we detected, would tend to facilitate the movement of iron towards the aqueous humor, giving additional internal consistency to the proposed model.
Our model may also provide the basis of the apparent regulatory mechanism put forward by McGahan (1992) underlying the “sink” effect of the lens for iron under pathological conditions, i.e., after inducing ocular inflammation in rabbits by the intravitreal injection of an E. coli lipopolysaccharide endotoxin. The situation created by this uveitis could be very complex, however, since it affects the permeability of all blood-ocular, epithelial and endothelial barriers of the eye, and may result in intraocular bleeding and tissular necrosis (McGahan, 1992). Following the injection of the toxin a marked increase of iron and TIBC in intraocular fluids occurred, as well as an increase in the saturation of TIBC with iron. These changes were attributed to the flooding of the eye with plasma transferrin through the disrupted barriers and the extravasation and degradation of iron containg blood proteins. According to the author, the lens absorbed a significant amount of the intraocular iron overload, because of the temporary and specific lenticular uptake of iron during the inflammatory episode (Cu concentration remained normal within the lens). The injection of the toxin also triggered an inflammatory response consisting of a dose-dependent increase in cells and in the concentration of protein of the intraocular fluids.
All those changes peaked within 24 hours (later in the vitreous body), then slowly subsided, and after 15 days there were no longer significant differences between the eye exposed to the endotoxin and the contralateral, control eye. At the lowest dose used (0.25 ng) no increase in iron in 24 hours was detected within the lens.
The results just described can be interpreted under the light of the model now being proposed for the normal movement of iron within the eye. It can be assumed that initially, and with the lowest dose of endotoxin, the anterior lens epithelium is able to handle all the iron reaching the EFI, perhaps by simply accelerating the recycling of endosomes, without iron being accumulated within the lens. Once the lenticular transport is overwhelmed, the excess labile iron within the epithelial cells would be safely stored as ferritin, particularly after ferritin synthesis becomes upregulated. De novo synthesis of local Tf may also occur, in an effort to both decrease the damaging effects of labile cytoplasmic iron, and increase its transfer towards the aqueous humor and out of the eye. As inflammation subsides all parameters of iron metabolism in the lens slowly return back to normal until control levels are achieved in about two weeks.
Our model provides an important function for locally synthesized Tf in the outflow of iron from the eye, but it would be premature at this time to try to provide specific details on the fate of local and systemic Tf along the pathway. The model does not exclude that iron may exit the eye through other routes involving the blood-retinal barrier of the RPE (He et al., 2007) and the retinal capillaries. The model requires that ferroportin (or an iron transporter sensitive to hepcidin) be located in the basal side of the lenticular anterior epithelial cells in order to explain the epidemiological link of age-related cataracts with ferropenic anemia (see below).
Age-related cataracts and anemia
With advancing age, the lens progressively loses its two main functional characteristics: first, accomodation to near vision, giving rise to presbyopia; and, at a later age, transparency to light, giving rise to cataracts. Age-related cataracts (ARC) is a multifactorial disease, best described in terms of epidemiological risk factors leading to lens opacification. ARC is thought to be the result of the accumulation of an excess of posttranslational changes in cytoplasmic and plasma membrane lens proteins, many of which are of a well-documented oxidative nature (Augusteyn, 1981; Cuevas and García-Castiñeiras, 1993; Dillon, 1991; Garland, 1990; Spector, 1991; Truscott, 2005). Depending on the lenticular target affected three main types of ARC are usually defined: nuclear, cortical and posterior subcapsular cataracts. The fact that most cataracts are of the mixed type (Cataract Management Guideline Panel, 1993) underlines the idea that the different types of cataract may depend more on the nature of the target affected within the lens than on entirely different ethiopathogenic risk factors.
The role of sunlight exposure was always considered important among cataract risk factors to explain the geographical factor in the prevalence of ARC and as a source of photoxidative stress (Pitts et al, 1986). Unexpectedly, however, epidemiological studies systematically failed to detect an association between sunlight exposure and nuclear cataracts. The only association detected was between sunlight exposure to UV-B light and cortical and posterior subcapsular cataracts (reviewed by McCarty and Taylor, 1999). Can this be taken to mean that nuclear cataracts respond to an entirely different ethiopathogenetic mechanism? This latter possibility is not very likely, as just noted above. It is easier to assume that some confounding factor is obscuring a valid epidemiological relationship between light exposure and opacification. The simplest confounding would occur if there were photoxidative posttranslational modifications that simultaneously have beneficial and damaging effects on lens transparency. For example, some type(s) of crosslinks could be involved in the long term structural stabilization of the nuclear lens proteins and thus promote long term lens transparency, while certain other type(s) of crosslinks could promote lens opacification by the uncontrolled generation of dark pigments and insolubilization of nuclear lens proteins. We can speculate that such a scenario is probably much closer to what really happens in the lens than the assumption of entirely different causative mechanisms.
Having commented on that, space limitations do not allow us to enter in the analysis of additional individual risk factors for ARC, but there is one special risk which caught our attention years ago (see Supplemental Material) and is very relevant to this review: the epidemiological association of age-related cataracts with anemia.
There exist two epidemiological studies in which the iron status of the population studied was considered: the lens opacities case/control study (LOS) (Leske et al., 1991) and the case/control study carried out at the Instituto de Oftalmobiología Aplicada (IOBA, Institute of Applied BioOphthalmology) of the University of Valladolid, Spain (Villaroya Lequericaonandia, 1994). In LOS it was found that iron represented a protective factor against cataract. In the IOBA study it was found that lack of iron represented an increased risk factor for cataracts. Both sets of results thus appear internally consistent. In LOS the parameter studied was the intake of iron, as judged by the amount and type of food ingested, while in the IOBA study the parameter reflecting iron status was if the patient had a diagnosis of anemia, specifically iron-deficiency anemia. It was difficult to rationalize at that time why increased systemic iron availability could represent a protective factor, and lack of it a risk factor for cataracts, given the role of iron in aggravating oxidative stress. There was also the possibility of unrecognized coincidences acting as confounding factors to explain the epidemiological association. I believed then that a deeper study of iron metabolism was granted, particularly under the light of our finding that transferrin was specifically enriched in the aqueous humor of senile cataract patients, in proportion to the severity of the cataract (Cortés-Velázquez and García-Castiñeiras, 1997; see also Supplemental Material).
Progress done in the last decade in systemic iron homeostasis as related to erythropoiesis and its regulation by the peptide hormone hepcidin (Ganz, 2003), now provides an attractive explanation for the epidemiological finding described above. The so called anemia of inflammation (also known as anemia of chronic disease) (Roy and Andrews, 2005) was classically interpreted as a way to restrict the availability of iron to pathogenic bacteria in an attempt to inhibit bacterial growth associated to chronic infection. Previously to the discovery of hepcidin, the pathogenesis of this anemia was attributed to the effects of interleukin 6 (IL-6) as part of the inflammatory response. It was later shown that one of the effects of IL-6 was to stimulate the liver to produce the antimicrobial peptide hepcidin (Nemeth et al., 2004). The mechanism of action of hepcidin was to induce the internalization and lysosomal destruction of the iron exporter (to plasma) ferroportin located in the basolateral membranes of the intestinal absorptive cells (Nemeth, 2008). One main consequence of the disappearance of ferroportin could then be the accumulation of iron in the cytoplasm of cells exporting iron through this transporter, accumulation that could in turn cause increased intracellular oxidative stress in the cells. Hepcidin is now known to be synthesized by non-hepatic cells (Collins et al., 2008) and direct evidence of iron retention has been obtained in monocytes, macrophages and retinal cells (Gnana-Prakasam et al., 2008; Theurl et al., 2008). Although still has to be shown that this is what actually happens in the lens, we could have here the explanation for the apparently contradictory finding in cataracts of an increased epidemiological risk due to anemia. In our proposed model, all we need to provoke intracellular oxidative stress of the lens would be the localization of ferroportin in the basal membranes of the anterior epithelium of the lens, exporting iron to the aqueous humor.
On the other hand, it is well known that ferropenic anemia is the main stimulus for a specific increase in the plasma concentration of transferrin (Aisen, 1984; Ponka et al., 1998). A specific increase in the aqueous humor level of Tf in cataracts as the one described by us (see Supplemental Material), could also be explained on the basis of an increased local synthesis of Tf. This would be a very convenient way to try to neutralize an increased intracellular concentration of iron secondary to a ferroportin blockade. How the local signal might be generated, however, is unknown at the present time.
The same situation is apparently occurring in AMD as proposed by Wong et al. (2007). For these authors the link did not come from epidemiological data but, probably, simply from the possibility of an intracellular iron overload in the retina secondary to the chronic inflammation likely associated to AMD progression, as indicated before.
Table 1 is a comparison of the two age-related diseases, macular degeneration and cataracts, concerning iron-related parameters underlying both diseases. Even with the partial data available the coincidences are remarkable. Why the similitude between both conditions?
Table 1.
Iron homeostasis parameters in age-related macular degeneration (AMD) and cataracts (ARC)
| parameter | AMD | ARC |
|---|---|---|
| local transferrin synthesis | increased mRNA expression in retina (Chowers et al, 2006) | increased ratio Tf/total protein in aqueous humor (Cortés-Velázquez and García-Castiñeiras, 1997) |
| intracellular chelatable iron | increased levels (Hahn et al, 2003) | increased levels (Garner et al, 2000) |
| hepcidin | expressed in retina (Gnana-Prakasam et al, 2008) | ? |
| ferroportin | abundant in Müller cells endfeet (He et al, 2007) | ? |
| epidemiological association to anemia | ? | yes (Leske et al, 1991; Villarroya Lequericaonandia, 1994) |
| plasma transferrin, other iron status parameters | ? | ? |
It is not very difficult to visualize in AMD compelling evidence of chronic inflammatory phenomena. Is there also a chronic condition of inflammatory nature in cataracts? The idea that autoantibodies might be involved in cataractogenesis is not new (Angunawella, 1987) but it never was unequivocally demonstrated that they had a role in the disease. There is little doubt that circulating anticrystallins antibodies are detectable in cataractous patients (Chylack, 1999), that they increase after endocapsular cataract surgery (García-Castiñeiras et al., 1993) and that they can also be detected for life in normal persons, as already commented. We found evidence (see Supplemental Material) that suggests that there is a slow, progressive increase in the permeability of the blood-aqueous humor barrier to protein, revealed by the tendency for total protein to increase in the aqueous humor, as a function of age and severity of the cataract. Antibodies could therefore reach back to the aqueous humor and then to the lens, through an increasingly permeable blood-aqueous humor barrier, creating a background of chronic inflammation in cataracts. An alternative to such explanation is that chronic inflammatory conditions of old age, coincidental with, but unrelated to cataracts, could also explain an apparent epidemiological association with anemia, instead of being the cataracts themselves the cause of chronic inflammation. The problem is far from settled and deserves further attention.
Supplementary Material
Acknowledgments
The author highly values the help of Amarilys Irizarry Hernández, Graphic Designer of the Medical Sciences Campus of the University of Puerto Rico in the development of the artwork of Figure 3. I am deeply grateful to Freddie Hernández Rivera of the Interlibrary Loan Section of the Medical Sciences Campus Library for his diligence in systematically providing me all the references requested for this review. I am indebted to the graduate and undergraduate students, medical students and residents of Ophthalmology who in some way or another collaborated with the research herein described.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen P. Transferrin Metabolism and the Liver. Semin Liver Dis. 1984;4:193–206. doi: 10.1055/s-2008-1041770. [DOI] [PubMed] [Google Scholar]
- Aisen P. Entry of Iron into Cells: A New Role for the Transferrin Receptor in Modulating Iron Release from Transferrin. Ann Neurol. 1992;32:S62–S68. doi: 10.1002/ana.410320711. [DOI] [PubMed] [Google Scholar]
- Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. 2001:940–959. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- Amasheh S, Milatz S, Krug SM, Markov AG, Günzel D, Amasheh M, Fromm M. Tight Junction Proteins as Channel Formers and Barrier Builders. Ann N Y Acad Sci. 2009;1165:211–219. doi: 10.1111/j.1749-6632.2009.04439.x. [DOI] [PubMed] [Google Scholar]
- Andrews NC, Schmidt PJ. Iron Homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- Angunawella II. The role of autoimmune phenomena in the pathogenesis of cataract. Immunol. 1987;61:363–368. [PMC free article] [PubMed] [Google Scholar]
- Augusteyn RC. Protein Modifications in Cataract: Possible Oxidative Mechanisms. In: Duncan G, editor. Mechanisms of Cataract Formation in the Human Lens. Academic Press; London: 1981. pp. 71–115. [Google Scholar]
- Barbazetto IA, Liang J, Chang S, Zheng L, Spector A, Dillon JP. Oxygen tension in the rabbit lens and vitreous before and after vitrectomy. Exp Eye Res. 2004;78:917–924. doi: 10.1016/j.exer.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Beard J. Iron. In: Bowman BA, Russell RM, editors. Present Knowledge in Nutrition. 9. Ch. 34. Vol. 1. ILSI Press; Washington, DC: 2006. pp. 430–444. [Google Scholar]
- Berman ER. Biochemistry of the eye. Ch. 7. Plenum Press; NY: 1991. pp. 309–467. Retina. [Google Scholar]
- Brown NAP, Bron AJ. A Clinical Manual of Cataract Diagnosis. Ch. 3. Butterworth-Heinemann Ltd.; Oxford: 1996. Lens Disorders; pp. 17–31. Lens growth. [Google Scholar]
- Burdo JR, Menzies SL, Simpson IA, Garrick LM, Garrick MD, Dolan KG, Haile DJ, Beard JL, Connor JR. Distribution of divalent metal transporter 1 and metal transport protein 1 in the normal and Belgrade rat. J Neurosci Res. 2001;66:1198–1207. doi: 10.1002/jnr.1256. [DOI] [PubMed] [Google Scholar]
- Burdo JR, Antonetti DA, Wolpert EB, Connor JR. Mechanisms and Regulation of Transferrin and Iron Transport in a model Blood-Brain Barrier System. Neuroscience. 2003;121:883–890. doi: 10.1016/s0306-4522(03)00590-6. [DOI] [PubMed] [Google Scholar]
- Carrasquillo E, Rodríguez-Medina J, Flores MS, García-Castiñeiras S. Tyrosinase is not expressed in the normal human lens. Int Coop Cataract Res Group. 1993:99. Program and Abstracts. [Google Scholar]
- Cataract Management Guideline Panel. Clinical Practice Guideline, Number 4. U.S. Department of Health and Human Services, Public Health Service, Agency for Health Care Policy and Research; Rockville, MD: 1993. Cataract in Adults: Management of Functional Impairment; pp. 5–28. AHCPR Pub No 93-0542. [Google Scholar]
- Chowers I, Wong R, Dentchev T, Farkas RH, Iacovelli J, Gunatilaka TL, Medeiros NE, Presley JB, Campochiaro PA, Curcio CA, Dunaief JL, Zack DJ. The Iron Carrier Transferrin Is Upregulated in Retinas from Patients with Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2006;47:2135–2140. doi: 10.1167/iovs.05-1135. [DOI] [PubMed] [Google Scholar]
- Chylack LT., Jr . Function of the lens and methods of quantifying cataract. In: Taylor A, editor. Nutritional and Environmental Influences on the Eye. Ch 3. CRC Press LLC; Boca Raton, FL: 1999. pp. 25–52. [Google Scholar]
- Collins JF, Wessling-Resnick M, Knutson MD. Hepcidin Regulation of Iron Transport. J Nutr. 2008;138:2284–2288. doi: 10.3945/jn.108.096347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, Burdo JR, Boyer PJ. Iron and Iron Management Proteins in Neurobiology. Pediatr Neurol. 2001;25:118–129. doi: 10.1016/s0887-8994(01)00303-4. [DOI] [PubMed] [Google Scholar]
- Cortés-Velázquez A, García-Castiñeiras S. Preliminary screening of aqueous humor proteins in human senile cataracts. Invest Ophthalmol Vis Sci. 1997;38:S294. [Google Scholar]
- Crabb JW, Miyagy M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci USA. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas AA, García-Castiñeiras S. Melanins and Lens Pigments: A Comparative Study. PR Health Sci J. 1993;12:129–135. [PubMed] [Google Scholar]
- Davidson MG, Harned J, Grimes AM, Duncan G, Wormstone IM, McGahan MC. Transferrin in after-cataract and as survival factor for lens epithelium. Exp Eye Res. 1998;66:207–215. doi: 10.1006/exer.1997.0413. [DOI] [PubMed] [Google Scholar]
- DeJong G, vanDijk JP, vanEijk HG. The biology of transferrin. Clin Chim Acta. 1990;190:1–46. doi: 10.1016/0009-8981(90)90278-z. [DOI] [PubMed] [Google Scholar]
- Dernouchamps JP. The proteins of the aqueous humor. Docum Ophthalmol. 1982;53:193–248. doi: 10.1007/BF00140422. [DOI] [PubMed] [Google Scholar]
- Dillon J. The photophysics and photobiology of the eye. J Photochem Photobiol B Biol. 1991;10:23–40. doi: 10.1016/1011-1344(91)80209-z. [DOI] [PubMed] [Google Scholar]
- Dillon J, García-Castiñeiras S, Santiago MA, Spector A. The endopeptidase-resistant protein fraction from human cataractous lenses. Exp Eye Res. 1984;39:95–106. doi: 10.1016/0014-4835(84)90118-0. [DOI] [PubMed] [Google Scholar]
- Dunaief JL. Iron Induced Oxidative Damage As a Potential Factor in Age-Related Macular Degeneration: The Cogan Lecture. Invest Ophthalmol Vis Sci. 2006;47:4660–4664. doi: 10.1167/iovs.06-0568. [DOI] [PubMed] [Google Scholar]
- Ebrahem Q, Renganathan K, Sears J, Vasanji A, Gu X, Lu L, Salomon RG, Crabb JW, Anand-Apte B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: implications for age-related macular degeneration. Proc Natl Acad Sci USA. 2006;103:13480–13484. doi: 10.1073/pnas.0601552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbarg J, Diecke FPJ, Kuang K, Yu B, Kang F, Iserovich P, Li Y, Rosskothen H, Konariek JP. Transport of fluid by lens epithelium. Am J Physiol. 1999;276:548–557. doi: 10.1152/ajpcell.1999.276.3.C548. [DOI] [PubMed] [Google Scholar]
- Fleming MD, Trenor CC, III, Su MA, Foernzler D, Beier DR, Dietrich WF, Andrews NC. Microcytic anaemia mice have a mutation in Nramp2, a candidate iron transporter gene. Nat Genet. 1997;16:383–386. doi: 10.1038/ng0897-383. [DOI] [PubMed] [Google Scholar]
- Fliesler SJ, Anderson RE. Chemistry and Metabolism of Lipids in the Vertebrate Retina. Prog Lipid Res. 1983;22:79–131. doi: 10.1016/0163-7827(83)90004-8. [DOI] [PubMed] [Google Scholar]
- Förster C. Tight junctions and the modulation of barrier function in disease. Histochem Cell Biol. 2008;130:55–70. doi: 10.1007/s00418-008-0424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye EB, Degenhardt TP, Thorpe SR, Baynes JW. Role of the Maillard reaction in aging of tissue proteins. Advanced glycation end product-dependent increase in imidazolium cross-links in human lens proteins. J Biol Chem. 1998;273:18714–18719. doi: 10.1074/jbc.273.30.18714. [DOI] [PubMed] [Google Scholar]
- Fu S, Dean R, Southan M, Truscott R. The hydroxyl radical in lens nuclear cataractogenesis. J Biol Chem. 1998;273:28603–28609. doi: 10.1074/jbc.273.44.28603. [DOI] [PubMed] [Google Scholar]
- Galloway CJ, Dean GE, Marsh M, Rudnick G, Mellman I. Acidification of macrophage and fibroblast endocytic vesicles in vitro. Proc Natl Acad Sci USA. 1983;80:3334–3338. doi: 10.1073/pnas.80.11.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood. 2003;102:783–788. doi: 10.1182/blood-2003-03-0672. [DOI] [PubMed] [Google Scholar]
- García-Castiñeiras S, Andino Moreno ME, Pérez R, Vázquez LA. Antibodies to Lens Crystallins after Endocapsular Cataract Surgery. PR Health Sci J. 1993;12:123–128. [PubMed] [Google Scholar]
- García-Castiñeiras S, Dillon J, Spector A. Non-tryptophan fluorescence associated with human lens protein; apparent complexity and isolation of bityrosine and anthranilic acid. Exp Eye Res. 1978;26:461–476. doi: 10.1016/0014-4835(78)90132-x. [DOI] [PubMed] [Google Scholar]
- Garland D. Role of site-specific, metal-catalyzed oxidation in lens aging and cataract: a hypothesis. Exp Eye Res. 1990;50:677–682. doi: 10.1016/0014-4835(90)90113-9. [DOI] [PubMed] [Google Scholar]
- Garland DL. Ascorbic acid and the eye. Am J Clin Nutr. 1991;54:1198–1202. doi: 10.1093/ajcn/54.6.1198s. [DOI] [PubMed] [Google Scholar]
- Garner B, Roberg K, Qian M, Eaton JW, Truscott RJ. Distribution of ferritin and redox-active transition metals in normal and cataractous human lenses. Exp Eye Res. 2000;71:599–607. doi: 10.1006/exer.2000.0912. [DOI] [PubMed] [Google Scholar]
- Glickman RD. Phototoxicity to the Retina: Mechanisms of Damage. Int J Toxicol. 2002;21:473–490. doi: 10.1080/10915810290169909. [DOI] [PubMed] [Google Scholar]
- Gnana-Prakasam JP, Martin PM, Mysona BA, Roon P, Smith SB, Ganapathy V. Hepcidin expression in mouse retina and its regulation via lipopolysaccharide/Toll-like receptor-4 pathway independent of Hfe. Biochem J. 2008;411:79–88. doi: 10.1042/BJ20071377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goralska M, Ferrell J, Harned J, Lall M, Nagar S, Fleisher LN, McGahan MC. Iron metabolism in the eye: A review. Exp Eye Res. 2009;88:204–215. doi: 10.1016/j.exer.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood NN, Earnshaw A. Chemistry of the elements. Ch. 25. Butterworth Heinemann; Oxford, UK: 1998. pp. 1070–1112. [Google Scholar]
- Gu J, Pauer GJT, Yue X, Narendra U, Sturgill GM, Bena J, Gu X, Peachey NS, Salomon RG, Hagstrom SA, Crabb JW Clinical Genomic and Proteomic AMD Study Group. Assessing Susceptibility to Age-related Macular Degeneration with Proteomic and Genomic Biomarkers. Molec Cell Proteomics. 2009;8:1338–1349. doi: 10.1074/mcp.M800453-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- Hadziahmetovic M, Dentchev T, Song Y, Haddad N, He X, Hahn P, Pratico D, Wen R, Harris ZL, Lambris JD, Beard J, Dunaief JL. Ceruloplasmin/Hephaestin Knockout Mice Model Morphologic and Molecular Features of AMD. Invest Ophthalmol Vis Sci. 2008;49:2728–2736. doi: 10.1167/iovs.07-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn P, Milam AH, Dunaief JL. Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigmented epithelium and Bruch’s membrane. Arch Ophthalmol. 2003;121:1099–1105. doi: 10.1001/archopht.121.8.1099. [DOI] [PubMed] [Google Scholar]
- Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL, Dunaief JL. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci USA. 2004;101:13850–13855. doi: 10.1073/pnas.0405146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn P, Ying GS, Beard J, Dunaief JL. Iron levels in human retina: sex difference and increase with age. Neuroreport. 2006;17:1803–1806. doi: 10.1097/WNR.0b013e3280107776. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Role of free radicals and catalytic metal ions in human disease: an overview. In: Packer L, Glazer AN, editors. Methods Enzymol. Vol. 186. Academic Press; NY: 1990. pp. 1–85. [DOI] [PubMed] [Google Scholar]
- Ham WT, Jr, Mueller HA, Ruffolo JJ, Jr, Clarke AM. Sensitivity of the retina to radiation damage as a function of wavelength. Photochem Photobiol. 1979;29:735–743. doi: 10.1111/j.1751-1097.1979.tb07759.x. [DOI] [PubMed] [Google Scholar]
- Harding J. Biochemistry, epidemiology and pharmacology. first. Chapman & Hall; London: 1991. Cataract. [Google Scholar]
- Harned J, Fleisher LN, McGahan MC. Lens epithelial cells synthesize and secret ceruloplasmin: Effects of ceruloplasmin and transferrin on iron efllux and intracellular iron dynamics. Exp Eye Res. 2006;83:721–727. doi: 10.1016/j.exer.2006.01.018. [DOI] [PubMed] [Google Scholar]
- He X, Hahn P, Iacovell J, Wong R, King C, Bhisitkul R, Massaro-Giordano M, Dunaief JL. Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res. 2007;26:649–673. doi: 10.1016/j.preteyeres.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008;14:194–198. doi: 10.1038/nm1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RC, Davis AA. Release of Iron by Human Retinal Pigment Epithelial Cells. J Cell Physiol. 1992;152:102–110. doi: 10.1002/jcp.1041520114. [DOI] [PubMed] [Google Scholar]
- Jaffe NS, Horwitz J. Lens Biophysics. In: Podos SM, Yanoff M, editors. Textbook of Ophthalmology. ch. 2. Vol. 3. Gower Med. Pub.; New York: 1992. pp. 2.1–2.7. [Google Scholar]
- Jager RD, Mieler WF, Miller JW. Age-Related Macular Degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- Kaur C, Foulds WS, Ling EA. Blood-retinal barrier in hypoxic ischaemic conditions: basic concepts, clinical features and management. Prog Retin Eye Res. 2008;27:622–647. doi: 10.1016/j.preteyeres.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Kern HL, Zolot SL. Transport of vitamin C in the lens. Curr Eye Res. 1987;6:885–896. doi: 10.3109/02713688709034857. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Ashwell JV, vanRenswoude JB, Harford J, Bridges K. Binding of apotransferrin to K562 cells: explanation of the transferrin cycle. Proc Natl Acad Sci USA. 1983;80:2263–2266. doi: 10.1073/pnas.80.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszak JR, Novak LA, Brown HG. An Ultrastructural Analysis of the Epithelial-Fiber Interface (EFI) in Primate Lenses. Exp Eye Res. 1995;61:579–597. doi: 10.1016/s0014-4835(05)80052-1. [DOI] [PubMed] [Google Scholar]
- Kuszak JR, Brown HG. Embriology and Anatomy of the Lens. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Ch. 5. W.B. Saunders Co.; Philadelphia: 1994. pp. 82–96. [Google Scholar]
- Lall MM, Ferrell J, Nagar S, Fleisher LN, McGahan MC. Iron Regulates L-Cystine Uptake and Glutathione Levels in Lens Epithelial and Retinal Pigment Epithelial Cells by its Effect on Cytosolic Aconitase. Invest Ophthalmol Vis Sci. 2008;49:310–319. doi: 10.1167/iovs.07-1041. [DOI] [PubMed] [Google Scholar]
- Leske MC, Chylack LT, Jr, Wu S-Y Lens Opacities Case-Control Study Group. The Lens Opacities Case-Control Study. Risk factors for Cataract. Arch Ophthalmol. 1991;109:244–251. doi: 10.1001/archopht.1991.01080020090051. [DOI] [PubMed] [Google Scholar]
- Levi S, Girelli D, Perrone F, Pasti M, Beaumont C, Corrocher R, Albertini A, Arosio P. Analysis of Ferritins in Lymphoblastoid Cell Lines and in the Lens of Subjects With Hereditary Hyperferritinemia-Cataract Syndrome. Blood. 1998;91:4180–4187. [PubMed] [Google Scholar]
- Lo WK. In Vivo and In Vitro Observations on Permeability and Diffusion Pathways of Tracers in Rat and Frog Lenses. Exp Eye Res. 1987;45:393–406. doi: 10.1016/s0014-4835(87)80126-4. [DOI] [PubMed] [Google Scholar]
- Mackic JB, Jinagouda S, McComb JG, Weiss MH, Kannan R, Kaplowitz N, Zlokovic BV. Transport of Circulating Reduced Glutathione at the Basolateral Side of the Anterior Lens Epithelium: Physiologic Importance and Manipulations. Exp Eye Res. 1996;62:29–37. doi: 10.1006/exer.1996.0004. [DOI] [PubMed] [Google Scholar]
- Mathias RT, Rae JL. The Lens: local transport and global transparency. Exp Eye Res. 2004;78:689–698. doi: 10.1016/j.exer.2003.07.001. [DOI] [PubMed] [Google Scholar]
- McCarty C, Taylor HR. Light and risk for age-related eye diseases. In: Taylor A, editor. Nutritional and environmental influences on the eye. CRC Press LLC; Boca Raton, FL: 1999. pp. 135–150. [Google Scholar]
- McGahan MC. Does the Lens Serve as a “Sink” for Iron During Ocular Inflammation? Exp Eye Res. 1992;54:525–530. doi: 10.1016/0014-4835(92)90131-b. [DOI] [PubMed] [Google Scholar]
- McGahan MC, Fleisher LN. A Micromethod for the Determination of Iron and Total Iron-Binding Capacity in Intraocular Fluids and Plasma Using Electrothermal Atomic Absorption Spectroscopy. Anal Biochem. 1986;156:397–402. doi: 10.1016/0003-2697(86)90271-x. [DOI] [PubMed] [Google Scholar]
- McGahan MC, Harned J, Goralska M, Sherry B, Fleisher LN. Transferrin secretion by lens epithelial cells in culture. Exp Eye Res. 1995;60:667–673. doi: 10.1016/s0014-4835(05)80008-9. [DOI] [PubMed] [Google Scholar]
- McGahan MC, Harned J, Grimes AM, Fleisher LN. Regulation of ferritin levels in cultured lens epithelial cells. Exp Eye Res. 1994;59:551–556. doi: 10.1006/exer.1994.1140. [DOI] [PubMed] [Google Scholar]
- McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager E, Mudaly E, Mudaly C, Richardson D, Barlow A, Bomford A, Peters TJ, Raja KB, Shirali MA, Hediger MA, Farzaneh F, Simpson RJ. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- Merriam JC. The concentration of light in the human lens. Tr Am Ophth Soc. 1996;94:803–918. [PMC free article] [PubMed] [Google Scholar]
- Moos T, Nielsen TR, Skjørringe T, Morgan EH. Iron trafficking inside the brain. J Neurochem. 2007;103:1730–1740. doi: 10.1111/j.1471-4159.2007.04976.x. [DOI] [PubMed] [Google Scholar]
- Nagaraj RH, Sell DR, Prabhakaram M, Orthwerth BJ, Monnier VM. High correlation between pentosidine protein crosslinks and pigmentation implicates ascorbate oxidation in human lens senescence and cataractogenesis. Proc Natl Acad Sci USA. 1991;88:10257–10261. doi: 10.1073/pnas.88.22.10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E. Iron regulation and erythropoiesis. Curr Opin Hematol. 2008;15:169–175. doi: 10.1097/MOH.0b013e3282f73335. [DOI] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward D, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Newman EA. Müller Cells and the Retinal Pigment Epithelium. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthalmology. Ch. 24. W.B. Saunders Co.; Philadelphia: 1994. pp. 398–419. [Google Scholar]
- Ng K-P, Gugiu B, Renganathan K, Davies MW, Gu X, Crabb JS, Kim SR, Rózanowska MB, Bonilha VL, Rayborn ME, Salomon RG, Sparrow JR, Boulton ME, Hollyfield JG, Crabb JW. Retinal Pigment Epithelium Lipofuscin Proteomics. Molec Cell Proteomics. 2008;7:1397–1405. doi: 10.1074/mcp.M700525-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37:1264–1269. doi: 10.1038/ng1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyster CW. The human eye Structure and function. Sinauer Assoc Inc Pub.; Sunderland, MA: 1999. [Google Scholar]
- Pitts DG, Cameron LL, Jose JG, Lerman S, Moss E, Varma SD, Zigler S, Zigman S, Zuclich J. Optical radiation and cataracts. In: Waxler M, Hitchins VM, editors. Optical Radiation and Visual Health. CRC Press Inc.; FL: 1986. pp. 5–41. [Google Scholar]
- Ponka P, Beaumont C, Richardson DR. Function and Regulation of Transferrin and Ferritin. Semin Hematol. 1998;35:35–54. [PubMed] [Google Scholar]
- Provis JM, Penfold PL, Cornish EE, Sandercoe TM, Madigan MC. Anatomy and development of the macula: specialization and the vulnerability to macular degeneration. Clin Exp Optom. 2005;88:269–281. doi: 10.1111/j.1444-0938.2005.tb06711.x. [DOI] [PubMed] [Google Scholar]
- Rae J. Physiology of the lens. In: Albert DM, Jakobiec FA, editors. Principles and Practice of Ophthtalmology. Ch. 8. W.B. Saunders Co.; Philadelphia: 1994. pp. 123–146. [Google Scholar]
- Reddy VN. Glutathione and its function in the lens - an overview. Exp Eye Res. 1990;50:771–778. doi: 10.1016/0014-4835(90)90127-g. [DOI] [PubMed] [Google Scholar]
- Roberts JE. Ocular phototoxicity. In: Marzuli FN, Maibach HI, editors. Dermatotoxicology. 5. Taylor & Francis; Washington, DC: 1996. pp. 307–313. [Google Scholar]
- Rouault TA. Systemic Iron Metabolism: A Review and Implications for Brain Iron Metabolism. Pediatr Neurol. 2001;25:130–137. doi: 10.1016/s0887-8994(01)00260-0. [DOI] [PubMed] [Google Scholar]
- Rousseva LA, Gaillard ER, Paik DC, Merriam JC, Ryzhov V, Garland DL, Dillon JP. Oxindolealanine in age-related human cataracts. Exp Eye Res. 2007;85:861–868. doi: 10.1016/j.exer.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Roy CN, Andrews NC. Anemia of inflammation: the hepcidin link. Curr Opin Hematol. 2005;12:107–111. doi: 10.1097/00062752-200503000-00001. [DOI] [PubMed] [Google Scholar]
- Sabah JR, Schultz BD, Brown ZW, Nguyen AT, Reddan J, Takemoto LJ. Transcytotic Passage of Albumin through Lens Epithelial Cells. Invest Ophthalmol Vis Sci. 2007;48:1237–1244. doi: 10.1167/iovs.06-0620. [DOI] [PubMed] [Google Scholar]
- Sies H. Oxidative Stress: introduction. In: Sies H, editor. Oxidative Stress: oxidants and antioxidants. Academic Press; London: 1991. pp. xv–xxii. [Google Scholar]
- Spector A. The Lens and Oxidative Stress. In: Sies H, editor. Oxidative Stress: Oxidants and Antioxidants. Academic Press; London: 1991. pp. 529–558. [Google Scholar]
- Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001:981–989. doi: 10.1007/s11745-001-0809-2. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Prescott AR, Tholozan FMD, Ohno S, Quinlan RA. Expression and localization of apical junctional complex proteins in lens epithelial cells. Exp Eye Res. 2008;87:64–70. doi: 10.1016/j.exer.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, Zoller H, Bellmann-Weiler R, Niederegger H, Talasz H, Weiss G. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood. 2008;111:2392–2399. doi: 10.1182/blood-2007-05-090019. [DOI] [PubMed] [Google Scholar]
- Truscott RJW. Age-related nuclear cataract – oxidation is the key. Exp Eye Res. 2005;80:709–725. doi: 10.1016/j.exer.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Truscott RJW, Augusteyn RC. The identification of anthranilic acid in proteolytic digests of cataractous lens proteins. Ophthalmic Res. 1977;9:263–268. [Google Scholar]
- Unakar NJ, Tsui JY. Sodium-potassium-dependent ATPase. I. Cytochemical localization in normal and cataractous rat lenses. Invest Ophthalmol Vis Sci. 1980;19:630–641. [PubMed] [Google Scholar]
- Vázquez-Quiñones LE. PhD Thesis. Associate Deanship for Biomedical Sciences and Graduate Program, School of Medicine, University of Puerto Rico; San Juan, PR: 2005. Comparative iron metabolism in ocular tissues and fluids of several species. [Google Scholar]
- Vázquez-Quiñones LE, García-Castiñeiras S. A comparative study of iron-related metabolic parameters in the eye of three animal species. PR Health Sci J. 2007;26:373–383. [PubMed] [Google Scholar]
- Villarroya Lequericaonandia ME. PhD Thesis. Department of Cell Biology and Pharmacology, School of Medicine, Univ of Valladolid; Valladolid, Spain: 1994. [Risk factors in senile cataractogenesis A study of cases and controls] Factores de riesgo en el desarrollo de catarata senil Estudio de casos y controles. [Google Scholar]
- Wangsa-Wirawan ND, Linsenmeier RA. Retinal Oxygen. Fundamental and Clinical Aspects. Arch Ophthalmol. 2003;121:547–557. doi: 10.1001/archopht.121.4.547. [DOI] [PubMed] [Google Scholar]
- Whitehead AJ, Mares JA, Danis RP. Macular Pigment. A Review of Current Knowledge. Arch Ophthalmol. 2006;124:1038–1045. doi: 10.1001/archopht.124.7.1038. [DOI] [PubMed] [Google Scholar]
- Wong RW, Richa DC, Hahn P, Green WR, Dunaief JL. Iron toxicity as a potential factor in AMD. Retina. 2007;27:997–1003. doi: 10.1097/IAE.0b013e318074c290. [DOI] [PubMed] [Google Scholar]
- Wood AM, Truscott RJW. UV filters in human lenses: tryptophan catabolism. Exp Eye Res. 1993;56:317–325. doi: 10.1006/exer.1993.1041. [DOI] [PubMed] [Google Scholar]
- Wu J, Seregard S, Algvere PV. Photochemical Damage of the Retina. Surv Ophthalmol. 2006;51:461–481. doi: 10.1016/j.survophthal.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Yamashiro DJ, Maxfield FR. Acidification of endocytic compartments and the intracellular pathways of ligands and receptors. J Cell Biochem. 1984;26:231–246. doi: 10.1002/jcb.240260404. [DOI] [PubMed] [Google Scholar]
- Yefimova MG, Jeanny J-C, Guillonneau X, Keller N, Nguyen-Legros J, Sergeant C, Guillou F, Courtois Y. Iron, Ferritin, Transferrin, and Transferrin Receptor in the Adult Rat Retina. Invest Ophthalmol Vis Sci. 2000;41:2343–2351. [PubMed] [Google Scholar]
- Zampighi GA, Eskandari S, Kreman M. Epithelial Organization of the Mammalian Lens. Exp Eye Res. 2000;71:415–435. doi: 10.1006/exer.2000.0895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.